

Abstract
The acidic environment inside secretory vesicles ensures that neuropeptides and peptide hormones are packaged in a concentrated condensed form. Although this is optimal for storage, decondensation limits release. Thus, it would be advantageous to alter the physical state of peptides in preparation for exocytosis. Here, we report that depolarization of the plasma membrane rapidly increases enhanced green fluorescent protein (EGFP)-tagged hormone fluorescence inside secretory vesicles. This effect requires Ca2+ influx and persists when exocytosis is inhibited byN-ethylmaleimide. Peptide deprotonation appears to produce this response, because it is not seen when the vesicle pH gradient is collapsed or when a pH-insensitive GFP variant is used. These data demonstrate that Ca2+ evokes alkalinization of the inside of secretory vesicles before exocytosis. Thus, Ca2+ influx into the cytoplasm alters the physical state of intravesicular contents in preparation for release.
Keywords: GFP, peptide hormone, exocytosis, alkalinization, secretory vesicle, Ca2+
Neurons and endocrine cells store peptides in aggregate or even crystalline forms inside secretory vesicles (Palade, 1975; Arrandale and Dannies, 1994). Peptide condensation is promoted by the acidic environment inside secretory vesicles. For example, chromogranin A (CgA) and chromogranin B (CgB), two of the major proteins inside secretory vesicles, aggregate because of pH- and Ca2+-dependent conformational changes (Yoo and Lewis, 1996). Similar conditions, an acidic pH environment and millimolar concentrations of Ca2+, are required to obtain optimal aggregation of the peptide hormone atrial natriuretic factor (ANF) (Canaff et al., 1996). Under these same conditions, granule content proteins from pituitary and chromaffin cells spontaneously aggregate (Colomer et al., 1996). Moreover, insulin is believed to exist as an insoluble Zn2+-bound hexamer inside acidic secretory vesicles of β cells, and release is promoted by alkalinization (see below) (Aspinwall et al., 1997). Thus, under normal resting conditions, a low intravesicular pH is important in maintaining peptides in an aggregate or solid state.
Although such a physical state inside secretory vesicles is advantageous for packaging and storage, it may hinder peptides from readily escaping the vesicles during exocytosis. For highly condensed hormones to be released, it is apparent that they must decondense and dissolve. For neurotransmitters, solubilization has been proposed to occur after vesicle fusion with the plasma membrane as a consequence of exposure to extracellular medium (for review, see Rahamimoff and Fernandez, 1997). In contrast, studies in peptidergic systems have revealed that drug-induced alkalinization of secretory vesicles promotes peptide solubilization before exocytosis. For example, vesicle matrix proteins (including CgA and CgB) bind to the vesicle membrane at resting intravesicular pH but become freed from vesicle membrane when intravesicular pH was raised toward physiological pH (Yoo, 1993). More recently, amperometric detection of insulin secretion showed that chemically alkalinizing vesicles speeds the kinetics of individual quantal responses and induces the appearance of amperometric “feet” (Aspinwall et al., 1997). This latter effect is usually interpreted as flux through the fusion pore (for review, see Artalejo et al., 1998). These results suggest that a physiological alkalinization of secretory vesicles before exocytosis could promote peptide decondensation and thus alter the kinetics of peptidergic neurotransmission.
To date, it has not been possible to monitor whether such an alkalinization occurs specifically in peptidergic secretory vesicles in live cells. However, the fluorescence from green fluorescent protein (GFP)-tagged peptides inside secretory vesicles can be assayed in live neuroendocrine cells (Burke et al., 1997; Lang et al., 1997). For example, we have engineered a cDNA construct using a human codon-optimized enhanced GFP variant (EGFP) and rat proANF. The expressed fusion protein is localized to secretory vesicles and undergoes regulated release after expression in nerve growth factor (NGF)-treated rat PC12 pheochromocytoma cells (Burke et al., 1997). Here, we use this live cell system to demonstrate that Ca2+ influx into the cytoplasm rapidly alkalinizes the contents of peptidergic secretory vesicles. This finding suggests for the first time that the physical state of neuropeptides is changed in preparation for release.
MATERIALS AND METHODS
Plasmids, cell culture, and transfection. The proANF-Sapphire construct was made by subcloning theAgeI–NotI fragment of the PCR product of pGFPsph-b[R] (Packard Instrument Company, Meriden, CT) into the corresponding site of proANF-EGFP construct (Burke et al., 1997). The following primers were used to introduce the AgeI andNotI sites: TCCACCGGTCGCCACCATGGTGAGCAAG on the 5′ side and CGGGCGGCCGCCCCGACTCTAGTCGA on the 3′ side. PC12 cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) at 37°C in a 5% CO2 incubator. Cells were transfected 1 d after plating on poly-lysine-coated coverslips with the above vectors using Tfx-50 (Promega, Madison, WI) in serum-free DMEM. Two hours later, the medium was replaced with 10% FBS-supplemented DMEM plus NGF (2.5 S NGF, 50 ng/ml; Life Technologies, Gaithersburg, MD). All experiments were performed 2–5 d after transfection.
When inhibition of exocytosis was required, cells were pretreated with 0.2 mmN-ethylmaleimide (NEM) on ice for 15 min, rinsed, and then incubated with 1 mm dithiothreitol (DTT) on ice for 15 min to quench residual NEM (Chavez et al., 1996). For altering the pH inside vesicles, cells were fixed and permeabilized with PBS containing 4% paraformaldehyde and 0.1% Triton X-100 for 15 min at 4°C. The above solution was then removed and replaced with buffered solutions.
Microfluorimetry and electrophysiological recording.Microfluorimetry was performed on an Olympus Opticals (Tokyo, Japan) inverted microscope using either a 40× (NA 1.3) or 100× (NA 1.3) oil immersion objective. Cells were bathed in normal saline [containing (in mm): 140 NaCl, 5.4 KCl, 0.8 MgCl2, 5 CaCl2, 10 Na-HEPES, and 10 glucose, pH 7.4] and illuminated with light from an attenuated mercury arc lamp passing through a standard wide band FITC optic cube. Emitted light was collected by a photomultiplier tube powered by a dual channel ratio fluorimeter (Biomedical Instrumentation Group, University of Pennsylvania, Philadelphia, PA). The signal was then displayed and stored using the X-chart module of the PULSE program (HEKA Electronik, Lambrecht/Pfalz, Germany) on a Power Macintosh computer. Standard whole-cell voltage-clamp recording (Hamill et al., 1981) was performed with an EPC-9 patch-clamp amplifier (HEKA Electronik) using the PULSE program (HEKA Electronik) on the same computer. Patch pipettes were filled with a solution containing (in mm): 140 KCl, 1 MgCl2, 10 Na-HEPES, 10 EGTA, and 3 MgATP, pH 7.4. Normal saline was used as bath solution. For K+-induced depolarization, 100 mm NaCl in the normal saline was substituted with 100 mm KCl. For Ba2+ experiments, Ba2+ was used as a substitute for Ca2+ in 100 mmK+ saline.
Spectral measurements. Coverslips with the fixed and permeabilized cells were illuminated with a 75 W xenon bulb-based monochromator (Applied Scientific Instrumentation, Eugene, OR) at 300–512 nm by 2 nm increments was passed through a quartz light guide and neutral density filters, which attenuated the light by >99%. A 515 nm dichroic mirror reflected light onto the cells through a 40× (NA 1.3) oil immersion objective. Emitted light was passed through a bandpass filter (535 ± 12.5 nm) and projected onto an intensified CCD camera (CCD 72 STX camera fitted with a Gen II Sys image intensifier; Dage-MTI, Michigan City, IN). Mean intensity levels of regions of interest were obtained on-line using SIMCA software (Compix Inc., Cranberry, PA). Cells were initially bathed in normal saline at pH 7.4 and subsequently switched to normal saline at various pH levels.
Data analysis. Data are presented as mean ± SEM. Only one cell or neurite was assayed per coverslip.n refers to number of cells or neurite endings measured. Statistical comparison was done first with ANOVA, followed by the F test. In cases with only two conditions, the Student’s t test was used.
RESULTS
It has been shown previously that EGFP-tagged proANF is targeted to large secretory vesicles and undergoes regulated release (Burke et al., 1997). Furthermore, the release of the fusion protein can be monitored in real time by measuring peptide fluorescence. It was expected that fluorescence would begin to decrease immediately after membrane depolarization (Huang et al., 1995). However, the apparent onset of fluorescent secretory responses by growth cones when cells were depolarized with high K+ saline appeared to be preceded by a delay or sometimes by a fluorescence increase (Fig.1A,B). This increase occurred within seconds when cells were directly depolarized via a patch-clamp electrode (Fig. 1C). Because more labeled peptides could not have been synthesized and packaged into secretory vesicles on this time scale, an increase in the intrinsic fluorescence of the fusion protein must have occurred before release.
Fig. 1.

Depolarization-induced proANF-EGFP fusion protein release is accompanied by an increase in fluorescence. InA and B, cells were superfused first in normal saline and then switched to 100 mmK+ saline (arrows). The fluorescence decrease, indicative of peptide release, was preceded by either a delay (A) or an increase (B) in fluorescence. C, A double-pulse depolarization stimulus (S, arrow), from a holding potential of −80 to 10 mV for 500 msec with a 5 sec interpulse, was delivered to the cell via a patch pipette in whole-cell configuration. Note the the increase in fluorescence was evident immediately after the double pulse. D, A similar fluorescence increase was observed in response to the double-pulse stimulus (S,arrow), without a net fluorescence decrease.
The rapid increase of peptide fluorescence could be caused by its exposure to extracellular medium at a postfusion stage (i.e., after formation of the fusion pore). However, the size of the increase was not proportional to the size of the secretory response. We even observed an increase in peptide fluorescence that was not followed by a net decrease in fluorescence (Fig. 1D). Furthermore, if the fluorescence increase occurred at a postfusion stage, then blocking exocytosis should abolish the response. NEM has been shown to inhibit secretion in Chinese hamster ovary and AtT20 cells (Chavez et al., 1996). To verify that NEM pretreatment produces a similar effect in PC12 cells, ANF release from PC12 cells was measured by radioimmunoassay (Burke et al., 1997). NEM was found to reduce K+-induced peptide release by sixfold (data not shown). Thus, NEM allowed us to test whether the increase in peptide fluorescence would be attenuated by inhibiting exocytosis. Contrary to the postfusion model, optical measurements after exposure to NEM revealed an uncontaminated increase in fluorescence (Fig.2A). Thus, the increase in the peptide signal occurs independently of fusion. Furthermore, as expected from the data in Figure 1, this effect was much faster (t½ = 38 ± 5 sec;n = 5) than typical release responses (Burke et al., 1997) (Fig. 1; see also Fig. 6). These kinetics, coupled with the lack of involvement of exocytosis, imply that the physical state of peptides inside secretory vesicles changes before fusion.
Fig. 2.

The depolarization-induced fluorescence change does not depend on exocytosis but requires extracellular Ca2+. Cells were pretreated with 0.2 mmNEM to block peptide release. A, Cells were superfused with normal saline and then with 100 mmK+ saline (bar). Note the apparent and uncontaminated increase in fluorescence. B, Cells were superfused with Ca2+-free normal saline, then with Ca2+-free 100 mmK+ saline, and finally with 100 mmK+ saline containing 5 mmCa2+. The fluorescence increase induced by 100 mm K+ saline occurred only when Ca2+ was present. C, Cd2+ prevented fluorescence increase caused by Ca2+. Cells were superfused with normal saline containing 0.2 mm Cd2+ and then with 100 mm K+ saline containing 0.2 mm Cd2+. D, Ba2+ mimicked Ca2+ in causing the fluorescence increase. Cells were initially superfused with normal saline and then with Ca2+-free Ba2+ containing 100 mmK+ saline. E, Quantification of divalent dependence of the effect of depolarization after NEM treatment. n = 4, 4, 6, and 4 for 0 Ca2+, 5 mm Ca2+, 5 mm Ca2+ plus 0.2 mmCd2+, and 5 mm Ba2+, respectively.
Fig. 6.

The apparent delay after application of 100 mm K+ saline is no longer observed when using proANF-Sapphire. A, A cell was initially superfused with normal saline and then with 100 mmK+ saline. Note that fluorescence decrease occurred almost immediately after 100 mm K+application. Also, no increase in fluorescence was observed with proANF-Sapphire. B, Comparison of delay from the application of 100 mm K+ saline to the onset of fluorescence decrease in cells transfected with proANF-EGFP (EGFP) and those transfected with proANF-Sapphire (Sph). n = 11 and 12 for proANF-EGFP and proANF-Sapphire, respectively. *p < 0.01 versus EGFP.
This finding suggests that a specific signal regulates the environment inside secretory vesicles. A potential candidate for a mediator of the effect of depolarization is the rise in cytoplasmic [Ca2+] that occurs after activation of voltage-gated Ca2+ channels. Consistent with this hypothesis, depolarization failed to alter peptide fluorescence in the absence of extracellular Ca2+. Rather, addition of bath Ca2+ to predepolarized cells produced the effect (Fig. 2B). Furthermore, blocking Ca2+ channels with 0.2 mmCd2+ prevented depolarization from causing the fluorescence brightening response (Fig. 2C,E). Ba2+, which is permeable through membrane Ca2+ channels, mimicked the effect of Ca2+ in causing the fluorescence increase (Fig.2D,E). Therefore, it is likely that Ca2+ enters the cytoplasm via voltage-gated channels and acts on secretory vesicles to alter peptide fluorescence.
How could cytoplasmic Ca2+ change the fluorescence of peptides inside secretory vesicles? It is known that the lumen of secretory vesicles is acidic, with a pH of ∼5.5. Furthermore, the fluorescence of wild-type GFP and EGFP has been shown to be sensitive to pH (Terry et al., 1995; Patterson et al., 1997). We found that the fluorescence of the fusion protein also varies with pH. Figure3A shows the excitation spectra of proANF-EGFP at two different pH values. An increase in fluorescence without any spectral shift was observed when the pH was increased from 5.5 to 7.4. The pH response was rapid and reversible (Fig. 3B), with an apparent pK of ∼5.7 (Fig.3C). This is similar to the value obtained with pure EGFP protein in solution (Patterson et al., 1997). Therefore, it seemed possible that the fluorescence increase was caused by Ca2+-induced alkalinization of secretory vesicles in stimulated cells.
Fig. 3.

Fluorescence of EGFP-tagged peptide hormone is sensitive to pH. A, Fluorescence excitation spectra of proANF-EGFP fusion protein at two different pH values.B, Microfluorimetric recordings of the responses of proANF-EGFP fusion protein expressed in cells to solutions at various pH levels. The response of the fluorescence of proANF-EGFP to changing pH was quick and reversible. C, Titration curve for the relative fluorescence of proANF-EGFP.
If alkalinization of secretory vesicles is responsible for the fluorescence increase, collapsing the pH gradient across the vesicle membrane should first increase peptide fluorescence and then prevent any further fluorescence increase by depolarization. Three membrane-permeant pH-collapsing agents, monensin (1 μm), nigericin (1 μm), and carbonyl cyanidep-(tri-fluoromethoxy) phenylhydrazone (FCCP) (1 μm), each increased vesicle fluorescence. Furthermore, after treatment with any of these agents, 100 mmK+ saline-induced depolarization failed to cause a further brightening of peptide fluorescence (Fig.4A–C). Moreover, these agents eliminated the apparent delay in secretory responses (Fig.4D). Thus, these pH-collapsing agents mimic and occlude the brightening effect of depolarization-induced Ca2+ influx.
Fig. 4.

The depolarization-induced increase in intravesicular fluorescence requires a pH gradient. Cells were initially superfused with normal saline containing a pH-collapsing agent [1 μm monensin (A), 1 μm nigericin (B), or 1 μm FCCP (C)] and then with 100 mm K+ saline containing the same pH-collapsing agent. Note that fluorescence decrease occurred almost immediately after 100 mmK+ application. D, Comparison of delay from the application of 100 mm K+to the onset of fluorescence decrease in cells without pretreatment (C) and those pretreated with monensin (M), nigericin (N), or FCCP (F). n = 11, 8, 5, and 5 for control, monensin, nigericin, and FCCP, respectively. *p < 0.01 versus control.
The hypothesis that the Ca2+-dependent fluorescence increase is caused by intravesicular alkalinization would also predict that the apparent delay or the fluorescence increase should no longer be observed if a pH-insensitive GFP variant is used in place of EGFP. The proANF-Sapphire fusion protein has a similar spectrum with Sapphire GFP (data not shown). However, in contrast to proANF-EGFP, proANF-Sapphire fluorescence is not sensitive to pH changes (Figs.5A–C). In live cells, proANF-Sapphire fluorescence did not increase when NEM-pretreated cells were subjected to monensin (see below) or 100 mm K+saline (percent of change after 100 mmK+, −0.001 ± 0.004; n = 7). Furthermore, the apparent delay after high K+application was no longer evident when proANF-Sapphire was used in place of proANF-EGFP (Fig. 6). These results indicate that the fluorescence increase of proANF-EGFP was reporting a Ca2+-induced alkalinization of the inside of secretory vesicles.
Fig. 5.
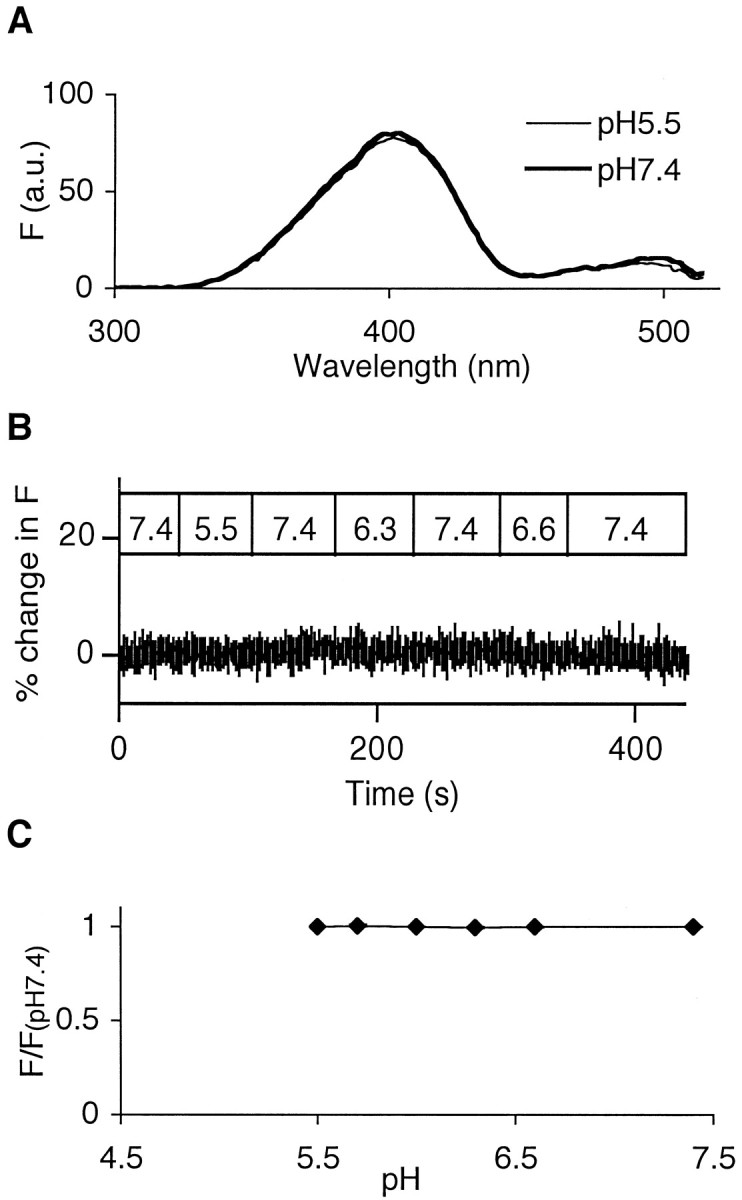
Fluorescence of proANF-Sapphire fusion protein is insensitive to pH. A, Fluorescence excitation spectra of proANF-Sapphire GFP fusion protein at two different pH values.B, Microfluorimetric recordings of the responses of proANF-Sapphire fusion protein to bath superfusion of normal saline at various pH levels. C, Titration curve for the relative fluorescence of Sapphire-tagged proANF.
NEM is known to block vesicular proton pumps and thus is expected to raise intravesicular pH (Flatmark et al., 1982). A rise of intravesicular pH should then cause a brightening of proANF-EGFP. Consistent with this prediction, NEM caused a gradual increase in proANF-EGFP fluorescence (Fig. 7A). However, NEM did not appear to neutralize intravesicular pH after 15 min of treatment (i.e., the same amount of time used in exocytosis inhibition procedures), because addition of monensin caused a much quicker and stronger increase in proANF-EGFP fluorescence (Fig. 7A). When similar treatments were used on cells that were transfected with pH-insensitive proANF-Sapphire, no increase in fluorescence was observed (Fig.7B,C). Therefore, a brief treatment with NEM causes only limited intravesicular alkalinization. This suggests that secretory vesicle pH is highly buffered and/or that resting H+-flux is low in the absence of elevated cytoplasmic Ca2+ levels.
Fig. 7.

NEM increases fluorescence in cells that are transfected with proANF-EGFP but not those with proANF-Sapphire.A, A cell expressing proANF-EGFP was initially superfused with normal saline, then with normal saline containing 0.2 mm NEM, and finally with normal saline containing 1 μm monensin. Note that fluorescence increased gradually after the start of NEM superfusion and that fluorescence increased very quickly when superfusion was switched to monensin (M). B, A cell expressing proANF-Sapphire was initially superfused with normal saline and then with normal saline containing 0.2 mm NEM. NEM did not have any effect on fluorescence of proANF-Sapphire. C, A proANF-Sapphire-expressing cell was pretreated with NEM and DTT. After initial superfusion with normal saline, the solution was switched to normal saline containing 1 μm monensin.
DISCUSSION
In the present report, we describe for the first time a depolarization-induced physical change of peptides inside secretory vesicles. It was found that intravesicular peptides are deprotonated before exocytosis after depolarization-induced Ca2+-influx. Ca2+-induced deprotonation caused by a rise in the pH inside vesicles may facilitate solubilization of peptides and perhaps their release, as well. Therefore, these changes may represent a novel role of Ca2+ in regulating exocytosis.
Possible mechanisms for Ca2+-dependent vesicular alkalinization
Our findings indicate that there must be a mechanism by which cytoplasmic Ca2+ induces secretory vesicle alkalinization. Because patch-clamp experiments revealed that this effect is very rapid and because secretory vesicle pH is thought to be highly buffered by ATP, slow proton transport mechanisms are not likely to be involved. Therefore, it is unlikely that cytoplasmic Ca2+ acted on synporters or antiporters. An action on the proton pump is also excluded by the persistence of the effect after treatment with NEM, a known inhibitor of the vesicular proton pump (Flatmark et al., 1982). Rather, it is more likely that a channel was activated that either changed vesicle membrane potential to indirectly promote proton flux or directly supported proton movement. In fact, secretory vesicles contain a variety of ion channels (Woodbury, 1995). Perhaps the most relevant of these to this study is a 130 pS channel that is present in peptidergic neurohypophysial granules (Lee et al., 1992). Interestingly, this channel is regulated by Ca2+ and is permeable to both divalent and monovalent cations. Therefore, it is possible that Ca2+ influx through plasma membrane channels opens these cation channels in the vesicles and causes a quick H+ loss from the vesicles that produces the increase of intravesicular pH.
Intravesicular alkalinization and peptide solubilization
As described in the introductory remarks, vesicle alkalinization promotes peptide decondensation and release. Such an effect could significantly affect the kinetics of peptidergic neurotransmission and release with “kiss and run” events (Artalejo et al., 1998). Our results show for the first time that vesicle alkalinization occurs as part of the normal response to cytoplasmic Ca2+influx. One potential concern is that the observed fluorescence change in this study is small, representing ∼0.2 U of pH change based on the proANF-EGFP titration curve of pH of ∼5.5. It should be noted that secretory peptide aggregation steeply changes between pH 5 and 6 (Colomer et al., 1996). Therefore, this effect could be significant. Furthermore, several lines of evidence suggest that our measurements could underestimate individual vesicle pH changes. First, in live cells, the fluorescence increase caused by intravesicular alkalinization is opposed by a fluorescence decrease caused by peptide release. Thus, release may have minimized the apparent change. Second, NEM raises intravesicular pH somewhat by inhibiting v-type ATPase in the vesicles. Finally, the fluorescence change measured in whole growth cones is an average from all of the secretory vesicles. However, because this response is dependent on Ca2+, vesicles near the membrane in which [Ca2+]i is high may have a bigger increase of pH. This would be optimal, because peptides would become decondensed only in vesicles that are likely to undergo exocytosis. Therefore, certain individual vesicles may have a pH increase great enough to initiate decondensation or dissolution of intravesicular contents.
Finally, it should be kept in mind that pH refers to proton concentration in bulk aqueous solutions. However, packaged peptides are normally in a solid or precipitated state that is only indirectly exposed to the bulk solution. Furthermore, it is possible that the pH requirements for peptide solubilization in real vesicles that contain a “smart gel” matrix (Rahamimoff and Fernandez, 1997) is quite different from that found in laboratory test tubes. Thus, even the low degree of peptide deprotonation detected on average may still have a significant influence in the unusual environment inside secretory vesicles. It is also interesting to note that a sustained alkalinization of the vesicle will promote loss of catecholamines that are packaged with peptides in large dense core vesicles in PC12 and some other cells (Sulzer and Rayport, 1990). Therefore, a rapid and moderate alkalinization may be optimal for initiating peptide decondensation, without major loss of catecholamines.
Footnotes
This study was supported by National Institutes of Health Grant NS32385 and an Established Investigator Award from the American Heart Association to E.S.L. We thank Dr. Nancy A. Burke for her help with radioimmunoassay experiments.
Correspondence should be addressed to Dr. Edwin S. Levitan, Department of Pharmacology, E1351 Biomedical Science Tower, University of Pittsburgh, Pittsburgh, PA 15261.
REFERENCES
- 1.Arrandale JM, Dannies PS. Inhibition of rat prolactin (PRL) storage by coexpression of human PRL. Mol Endocrinol. 1994;8:1083–1089. doi: 10.1210/mend.8.8.7997234. [DOI] [PubMed] [Google Scholar]
- 2.Artalejo CR, Elhamdani A, Palfrey HC. Secretion: dense-core granules can kiss-and-run too. Curr Biol. 1998;8:R62–R65. doi: 10.1016/s0960-9822(98)70036-3. [DOI] [PubMed] [Google Scholar]
- 3.Aspinwall CA, Brooks SA, Kennedy RT, Lakey JRT. Effects of intravesicular H+ and extracellular H+ and Zn2+ on insulin secretion in pancreatic beta cells. J Biol Chem. 1997;272:31308–31314. doi: 10.1074/jbc.272.50.31308. [DOI] [PubMed] [Google Scholar]
- 4.Burke NV, Han W, Li D, Takimoto K, Watkins SC, Levitan ES. Neuronal peptide release is limited by secretory granule mobility. Neuron. 1997;19:1095–1102. doi: 10.1016/s0896-6273(00)80400-6. [DOI] [PubMed] [Google Scholar]
- 5.Canaff L, Brechler V, Reudelhuber TL, Thibault G. Secretory vesicle targeting of atrial natriuretic peptide correlates with its calcium-mediated aggregation. Proc Natl Acad Sci USA. 1996;93:9483–9487. doi: 10.1073/pnas.93.18.9483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chavez RA, Miller SG, Moore H-P H. A biosynthetic regulated secretory pathway in constitutive secretory cells. J Cell Biol. 1996;133:1177–1191. doi: 10.1083/jcb.133.6.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colomer V, Kicska GA, Rindler MJ. Secretory granule content proteins and the luminal domains of granule membrane proteins aggregate in vitro at mildly acidic pH. J Biol Chem. 1996;271:48–55. doi: 10.1074/jbc.271.1.48. [DOI] [PubMed] [Google Scholar]
- 8.Flatmark T, Gronberg M, Husebye E, Jr, Berge SV. Inhibition of N-ethylmaleimide of the MgATP-driven proton pump of the chromaffin granules. FEBS Lett. 1982;149:71–74. doi: 10.1016/0014-5793(82)81074-0. [DOI] [PubMed] [Google Scholar]
- 9.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth F. Improved patch-clamp techniques for high resolution current recordings from cells and cell free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 10.Huang L, Shen H, Atkinson MA, Kennedy RT. Detection of exocytosis at individual pancreatic β cells by amperometry at a chemically modified microelectrode. Proc Natl Acad Sci USA. 1995;92:9608–9612. doi: 10.1073/pnas.92.21.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lang T, Wacker I, Steyer J, Kaether C, Wunderlich I, Soldati T, Gerdes HH, Almers W. Ca2+-triggered peptide secretion in single cells imaged with green fluorescent protein and evanescent-wave microscopy. Neuron. 1997;18:857–863. doi: 10.1016/s0896-6273(00)80325-6. [DOI] [PubMed] [Google Scholar]
- 12.Lee CJ, Dayanithi G, Nordmann JJ, Lemos JR. Possible role during exocytosis of a Ca2+-activated channel in neurohypophysial granules. Neuron. 1992;8:335–342. doi: 10.1016/0896-6273(92)90299-s. [DOI] [PubMed] [Google Scholar]
- 13.Palade G. Intracellular aspects of the process of protein synthesis. Science. 1975;189:347–358. doi: 10.1126/science.1096303. [DOI] [PubMed] [Google Scholar]
- 14.Patterson GH, Knobel SM, Sharif WD, Kain SR, Piston DW. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys J. 1997;73:2782–2790. doi: 10.1016/S0006-3495(97)78307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rahamimoff R, Fernandez JM. Pre- and postfusion regulation of transmitter release. Neuron. 1997;18:17–27. doi: 10.1016/s0896-6273(01)80043-x. [DOI] [PubMed] [Google Scholar]
- 16.Sulzer D, Rayport S. Amphetamine and other psychostimulants reduce pH gradients in midbrain dopaminergic neurons and chromaffin granules: a mechanism of action. Neuron. 1990;5:797–808. doi: 10.1016/0896-6273(90)90339-h. [DOI] [PubMed] [Google Scholar]
- 17.Terry BR, Matthews EK, Haseloff J. Molecular characterization of recombinant green fluorescent protein by fluorescence correlation microscopy. Biochem Biophys Res Commun. 1995;217:21–27. doi: 10.1006/bbrc.1995.2740. [DOI] [PubMed] [Google Scholar]
- 18.Woodbury DJ. Evaluation of the evidence for ion channels in synaptic vesicles. Mol Membr Biol. 1995;12:165–171. doi: 10.3109/09687689509027504. [DOI] [PubMed] [Google Scholar]
- 19.Yoo SH. pH-dependent binding of chromogranin B and secretory vesicle matrix proteins to the vesicle membrane. Biochim Biophys Acta. 1993;1179:239–246. doi: 10.1016/0167-4889(93)90078-4. [DOI] [PubMed] [Google Scholar]
- 20.Yoo SH, Lewis MS. Effects of pH and Ca2+ on heterodimer and heterotetramer formation by chromogranin A and chromogranin B. J Biol Chem. 1996;271:17041–17046. doi: 10.1074/jbc.271.29.17041. [DOI] [PubMed] [Google Scholar]