

Abstract
Disulfide trapping studies have revealed that the pore-lining (P) segments of voltage-dependent sodium channels undergo sizable motions on a subsecond time scale. Such motions of the pore may be necessary for selective ion translocation. Although traditionally viewed as separable properties, gating and permeation are now known to interact extensively in various classes of channels. We have investigated the interaction of pore motions and voltage-dependent gating in μ1 sodium channels engineered to contain two cysteines within the P segments. Rates of catalyzed internal disulfide formation (kSS) were measured in K1237C+W1531C mutant channels expressed in oocytes. During repetitive voltage-clamp depolarizations, increasing the pulse duration had biphasic effects on the kSS, which first increased to a maximum at 200 msec and then decreased with longer depolarizations. This result suggested that occupancy of an intermediate inactivation state (IM) facilitates pore motions. Consistent with the known antagonism between alkali metals and a component of slow inactivation,kSS varied inversely with external [Na+]o. We examined the converse relationship, namely the effect of pore flexibility on gating, by measuring recovery from inactivation in Y401C+E758C (YC/EC) channels. Under oxidative conditions, recovery from inactivation was slower than in a reduced environment in which the spontaneous YC/EC cross-link is disrupted. The most prominent effects were slowing of a component with intermediate recovery kinetics, with diminution of its relative amplitude. We conclude that occupancy of an intermediate inactivation state facilitates motions of the P segments; conversely, flexibility of the P segments alters an intermediate component of inactivation.
Keywords: sodium channel, inactivation, permeation, cysteine mutagenesis, disulfide bond
Gating and permeation are conceptually distinct features of channels: gating processes determine whether the channel is open, whereas permeation focuses on the translocation of ions through the channel (Hodgkin and Huxley, 1952). The first insights into channel anatomy suggested a comfortable structural segregation to match the functional dichotomy. In the case of voltage-dependent K channels, we learned that fast inactivation was conferred by an N-terminal peptide whose removal did not alter channel conductance or selectivity, properties believed to reside far away in the S5–S6 linkers (pore-lining or “P” segments) (Hoshi et al., 1990; Zagotta and Aldrich, 1990). However, it soon became clear that residues that confer a slower form of inactivation (“C-type”) reside within the P segments; mutations of these residues altered not only gating but also permeation (Hoshi et al., 1991; Isacoff et al., 1991; McCormack et al., 1991; De Biasi et al., 1993). Numerous other examples have emerged to challenge the truism that gating and permeation are distinct and independent (Tomaselli et al., 1995; Balser et al., 1996; Chen et al., 1997; Townsend and Horn, 1997; Townsend et al., 1997; Yellen, 1997). Another long-held notion, which has recently been questioned, is that which presumes biological channel pores to be rigid structures, by analogy to the binding sites of ion-selective glass membranes (Eisenman and Krasne, 1975). The P segments of sodium channels are exceptionally mobile (Bénitah et al., 1997a;Tsushima et al., 1997), and evidence has been presented that such mobility enhances selectivity (Tsushima et al., 1997). Nevertheless, it is not yet clear whether P-segment mobility can influence channel gating or whether gating processes can influence the molecular dynamics of the pore.
To examine these questions, we expressed two previously characterized double-cysteine mutants of the μ1 sodium channel: K1237C+W1531C (KC/WC) and Y401C+E758C (YC/EC). Both channels contain two cysteines within the P segments for disulfide trapping experiments. KC/WC is representative of one class of such channels: it does not form an internal disulfide spontaneously, but it does so in the presence of a redox catalyst. We determined that either modifying the depolarizing pulse duration or altering the external permeant ion concentration ([Na+]o), maneuvers that alter the inactivated-state occupancy, changes the rates of catalyzed internal disulfide formation in KC/WC. To examine the converse issue, namely the effect of pore motions on gating, we studied YC/EC. This mutant spontaneously forms an internal disulfide bond between the 401 and 758 positions in the usual oxidized environment. In this cross-linked state, the pore is constrained and membrane current decreases; conductance increases when the channels are exposed to a reduced environment (Bénitah et al., 1996; Tsushima et al., 1997). The fact that conductance is measurable both before and after reduction makes YC/EC ideal for the comparison of gating transitions in cross-linked (constrained) and noncross-linked (freely mobile) channels.
We find that gating influences motions and vice versa. The direction of the effects is as follows. Greater flexibility decreases the occupancy of a particular inactivated state and slows its recovery kinetics; conversely, pulse protocols that favor the occupancy of this inactivated state facilitate KC/WC disulfide formation.
MATERIALS AND METHODS
Mutagenesis and channel expression. Site-directed mutagenesis of the α subunit of the μ1 Na channel cDNA was performed as described previously (Bénitah et al., 1997a). Briefly, mutations were introduced by recombinant PCR on the double-stranded cDNA using pfu DNA polymerase and verified by sequencing. Oocytes were removed through an abdominal incision from ovaries of adult female Xenopus laevis (XenopusI, Ann Arbor, MI, or Nasco, Ft. Atkinson, WI) under anesthesia and then treated for 1 hr with collagenase (Type 1A, Sigma, St. Louis, MO) in modified Barth’s solution containing (in mm): 88 NaCl, 1 KCl, 2.4 NaHCO3, 15 Tris base, 0.8 Ca(NO3)2–4 H2O, 0.41 CaCl2–6 H2O, 0.82 MgSO4–7 H2O, supplemented with 100 U/ml penicillin, 100 μg streptomycin, 250 ng fungizone, and 50 μg/ml gentamicin). Oocytes (stages V and VI) were coinjected with cRNA for the μ1 Na+ channel α subunit and the rat brain β1 subunit in an equimolar ratio (Bénitah et al., 1997a).
Electrophysiology and data analysis. Membrane currents were recorded 24–72 hr after injection using two microelectrodes, as described previously (Bénitah et al., 1997b), in frog Ringer’s solution containing (in mm): 96 NaCl, 2 KCl, 1 MgCl2, 5 HEPES, pH 7.6. Dithiothreitol (DTT; 1 mm) and Cu(II)(1, 10-phenanthroline)3[Cu(phe)3; 100 μm] were dissolved in the external solution as described previously (Bénitah et al., 1997a). Voltage protocols and data acquisition were managed by custom-written software as described previously (Bénitah et al., 1997b). Linear leak and capacitive currents were corrected using a P/4 protocol. Best data fits were performed using a nonlinear least-squares Marquardt–Levenberg algorithm (Origin, MicroCal, Northampton, MA). Average data are expressed as means ± SEM. Statistical significance levels were evaluated by ANOVA and paired ttest (Origin), as appropriate. A p value of < 0.05 was considered significant.
RESULTS
Cu(phe)3 catalyzes disulfide formation
We showed previously that multiple double-cysteine mutants of the μ1 Na channel can form internal cross-links, either spontaneously or in the presence of the redox catalyst Cu(phe)3. Because of the patterns and rates of the reactions, we concluded that the P segments that form the structural basis of the permeation pathway are flexible (Bénitah et al., 1997a). Figure1 shows an example of the occlusion of the pore by catalyzed internal disulfide formation. The panels show representative Na currents in response to consecutive 50 msec depolarizing test pulses (0.5 Hz) in wild-type (μ1 WT), in K1237C and W1531C single mutants, and in the paired KC/WC mutant channels. The wild-type and single-cysteine mutants were entirely insensitive to 100 μm Cu(phe)3 for ≥2 min. In contrast, exposure to the redox catalyst progressively inhibited the current through KC/WC channels. During 6 min of exposure to Cu(phe)3, we observed a progressive reduction of the current that could only be reversed by DTT or reduced glutathione (data not shown). The inset shows superimposed currents at a faster time scale; the decay kinetics of these currents did not change, suggesting that fast inactivation gating was not affected by the inhibition of the current. These findings are consistent with the idea that the cysteines at positions 1237 and 1531 form a disulfide bond in the presence of the redox catalyst, rendering the channels nonconducting. Single-cysteine mutants at each of these positions do not form a disulfide with endogenous cysteine residues that alter channel conductance. Only channels that are not yet cross-linked conduct current, such that the remaining current is simply scaled. Similar results were consistently observed for this double-cysteine mutant, as well as several other engineered cysteine pairs (Bénitah et al., 1997a). These observations imply the existence of motions in the pore; constraining such motions through disulfide bond formation eliminates or reduces ion flux.
Fig. 1.
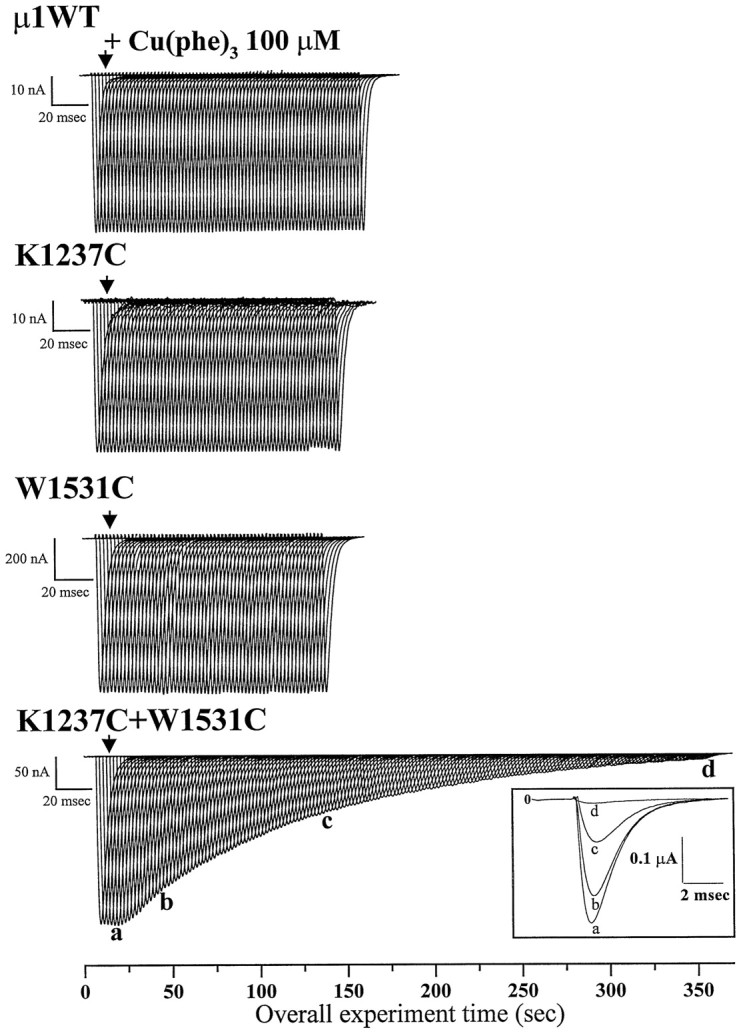
Effect of external application of Cu(phe)3 on wild-type and mutant μ1 Na current. Whole-cell Na currents were recorded from Xenopusoocytes coexpressing wild-type or mutant α subunits and the rat brain β1 subunit. The α subunits were (fromtop to bottom) μ1 wild-type, K1237C, W1531C, and the double-mutant K1237C+W1531C. Na currents were elicited by 50 msec pulses from −100 to −30 mV at 0.5 Hz. The calibration bar represents the time base at which the individual current records are displayed. The total experimental time in seconds is shown on the abscissa. The arrowhead indicates the time at which the redox catalyst Cu(phe)3 (100 μm) was applied. Currents from the wild-type and each of the single mutants were not modified by application of Cu(phe)3, whereas the current through the double-mutant K1237C+W1531C was progressively inhibited. Inset, Selected currents recorded from the double-mutant K1237C+W1531C during depolarizing voltage steps in the absence (a) and after 40 sec (b), 2 min (c), and 6 min (d) of exposure to 100 μmCu(phe)3.
To quantify the thermal backbone motions revealed by disulfide trapping methods, the oxidation catalysis reactions were analyzed according toCareaga and Falke (1992) as described previously (Bénitah et al., 1997a). Briefly, the oxidation chemistry of the Cu(phe)3catalysis yields two competing reactions that generate the intended disulfide bond product and undesired higher oxidation products of cysteine. Higher oxidation products may be formed in the wild-type and single-cysteine mutant channels but do not affect the current amplitude or gating (Fig. 1). Furthermore, both catalytic reactions are effectively irreversible under these oxidized experimental conditions. Application of Cu(phe)3 to the KC/WC mutant eliminates the current, presumably by disulfide bond formation, and is reversible under reducing conditions. Therefore the rate of formation of higher oxidation products is slowed compared with the rate of disulfide bond formation. The rate constants of the disulfide formation reaction (kss) were determined by fitting a single-component reaction scheme to the observed time course of the Cu(phe)3 inhibition of the current (see legend of Fig.2) (Bénitah et al., 1997a). Figure2 shows a representative disulfide reaction time course for the KC/WC channels, including a nonlinear fit illustrating good agreement between the single-component reaction scheme and the data.
Fig. 2.

Disulfide formation time course for the K1237C+W1531C channels. The normalized current amplitude of the superimposed current traces of the bottom panel of Figure 1 is plotted as function of the duration of redox catalyst Cu(phe)3exposure (plus symbols). A function describing the single-component reaction scheme (see Results) was fitted to the data: 1 − I/Imax = 1 − exp(−kss(t + {exp(−t/tp)} − 1). The fitted rate parameters kss for this reaction is 9.13 × 10−3 · sec−1 · molecule−1after correction for the rate of bath exchange (tp, 9.9 sec in this case) (Bénitah et al., 1997a).
Occupancy of a particular inactivated state facilitates disulfide formation
During depolarization, voltage-gated ion channels assume nonconducting inactivated states, which may be characterized as “fast” or “slow,” depending on the rate at which the channels recover from inactivation when subsequently repolarized (Rudy, 1978). Short depolarization favors fast inactivation, whereas slow-inactivated conformations develop with longer depolarizations. Although fast inactivation involves internal structures exposed to the cytoplasm [the N terminus in K channels (Hoshi et al., 1990) and the III–IV linker in Na channels (Stühmer et al., 1989)], slow inactivation seems to involve residues in the outer pore. In Shakerpotassium channels (Liu et al., 1996), a slow (C-type) inactivation process has been linked to dynamic rearrangement of residues in the P-segment. Furthermore, recent studies in μ1 Na+channels (Balser et al., 1996) have shown that cysteine substitution of a single P-segment tryptophan (W402C) reduces slow inactivation. Given the spatial proximity between W402 and both K1237 and W1531 (Bénitah et al., 1997a), we examined the relationship between slow inactivation and K1237C–W1531C disulfide bond formation. Our previous work (Bénitah et al., 1997a) showed that the effect of Cu(phe)3 on disulfide formation is not overtly voltage dependent; however, these experiments were not designed to discriminate among particular inactivated states. Figure3 illustrates that the time course of Cu(phe)3 inhibition depends on the duration of depolarization. Macroscopic currents from oocytes expressing KC/WC channels were monitored during a 0.05 Hz pulse train with depolarizations from −100 to −40 mV before and after addition of 100 μm Cu(phe)3 to the bath solution. Although with 50 or 5000 msec pulse duration complete inhibition requires >9 min, with 200 msec pulses the Cu(phe)3 effect is nearly complete within 5 min.
Fig. 3.

Effect of pulse duration on the rate of Cu(phe)3 catalysis. Representative examples are shown for Cu(phe)3 inhibition of the peak K1237C+W1531C Na current as a function of pulse duration. Whole-cell currents were elicited in 96 mm [Na+]o using 0.05 Hz pulse trains with individual depolarizations from −100 to −40 mV lasting 50, 200, or 5000 msec (from top tobottom). The total time of the experiment is shown on the x-axis; the time base of the individual currents is indicated by the calibration bar. The arrowheadindicates the time of application of the redox catalyst.
Figure 4 summarizes the rate constants of redox catalysis determined from a number of oocytes exposed to Cu(phe)3 and trains of depolarizing pulses ranging from 3 msec to 5 sec. In all cases, the pulse frequency (0.05 Hz) was slow enough to allow complete recovery from inactivation between depolarizations (data not shown). Disulfide bond formation hastened as the pulse lengthened, reaching a peak value at 200 msec (Figs. 3, 4). When even longer pulses were applied, kss slowed to a rate nearly the same as that in the briefest (3 msec) pulse. These results indicate that pulse durations of intermediate length enhance the rate of disulfide formation, suggesting that occupancy of an “intermediate” inactivated state may facilitate the catalytic effect of Cu(phe)3.
Fig. 4.

Cu(phe)3 catalyzed disulfide bond formation in the K1237C+W1531C double mutant has a biphasic dependence on pulse duration. The disulfide formation rates (kss) plotted as a function of the pulse duration for oocytes expressing the K1237C+W1531C double mutant. The Na currents were elicited by depolarizing pulses at a rate of 0.05 Hz and durations ranging from 3 msec to 5 sec. We detect no cumulative of inactivation at this slow stimulation frequency for even the longest (5 sec) depolarizations. Currents were measured before and after addition of 100 μm Cu(phe)3, and the reaction rate constants were determined as described for Figure 2. The plotted values (means ± SEM) represent measurements from at least four different oocytes for each data point. *, p < 0.02; **, p < 0.002.
When the α and β1 subunits of μ1 channels are coexpressed in Xenopus oocytes and subjected to 1 sec depolarizations, inactivation is characterized by a minimum of three readily distinguishable kinetic components (Nuss et al., 1995). These include an initial rapid component (τfast) with a recovery time constant of ∼1 msec, which exhibits marked sensitivity to mutations in the III–IV linker (West et al., 1992; Lawrence et al., 1996), and two slower components (τintermediate and τslow) with recovery time constants of ∼100 msec and 1 sec, which are sensitive to domain I, P-segment mutations (Balser et al., 1996; Kambouris et al., 1998). Although a number of gating schemes are consistent with these kinetic data, the following model provides a convenient framework for discussing the possible interaction between fast (IF), intermediate (IM), and slow-inactivated (IS) states and disulfide bond formation: Closed ↔ Open ↔ IF ↔IM ↔ IS.
To examine the hypothesis that the rate of disulfide formation is associated with entry into a particular inactivated state with intermediate kinetics (IM in the scheme above), we studied the rate of inactivation of the KC/WC mutant using a paired-pulse voltage-clamp protocol (Fig.5, top). Channels were depolarized for incremental periods, and fractional recovery was assessed after a 20 msec recovery interval (permitting recovery from the fast-inactivated state, IF). The rate of development of inactivation was well described by a biexponential function, with time constants of 66 msec and 5.2 sec (see legend to Fig. 5), consistent with time-dependent entry into two distinct inactivated states (IM +IS). Furthermore, the time constant of entry into the IM state is consistent with the pulse duration that induced the most rapid rate of disulfide formation (200 msec) (Fig. 4). Much shorter pulses would populate onlyIF, whereas longer pulses begin to recruit an appreciable fraction of IS. These findings suggest that disulfide formation in the outer pore is facilitated by occupancy of IM. Furthermore, the biphasic nature of kss suggests that channels occupying fast-inactivated states (IF) are less likely to form disulfides, as are the states favored by even longer depolarizations (IS).
Fig. 5.

Rate of development of intermediate and slow-inactivation for the double-mutant K1237C+W1531C. As shown in the voltage-clamp protocol (top), the duration of the depolarizing prepulse (−40 mV) was varied, and the extent of recovery after 20 msec at −100 mV was assessed using a 50 msec test pulse to −40 mV. The 20 msec recovery interval removed the most rapidly recovering inactivation component (IF) from consideration. Plotted is the fractional recovery from inactivation as a function of the prepulse duration. Data were collected from seven oocytes. The dotted line shows a nonlinear fit to the mean data using the function:y = A1exp(−t/τ1) + A2exp(−t/τ2). The least squares error was minimized when A1 = 0.2, τ1 = 66 msec, A2 = 0.8, and τ2= 5212 msec.
[Na+]o modulates the rate of disulfide formation
Although flux generally increases as a function of permeant ion concentration, an unanticipated inhibition of open probability by a reduction in external Na+ was described recently (Townsend et al., 1997). Both whole-cell and single-channel recordings of cardiac (hH1) Na channels revealed that raising [Na+]o inhibits the rate of slow inactivation at depolarized membrane potentials, as if binding of the alkali metal cation in the pore inhibits closure of the slow inactivation gate. We thus examined the time course of Cu(phe)3-catalyzed disulfide bond formation in oocytes bathed in different extracellular Na+concentrations. Figure6A plots the time course of current inhibition by Cu(phe)3 in representative oocytes bathed in 48, 96, or 140 mm Na+solutions. KC/WC current is inhibited slowly and completely within 10 min in 96 mm [Na+]o(open circles). Raising [Na+]o (open diamonds) slowed this time course, whereas decreasing [Na+]o (open triangles) accelerated the inhibitory effect of the Cu(phe)3. The time courses were analyzed as described previously to determine the rate constants of the redox reaction (Fig. 2) (Bénitah et al., 1997a). The rate constants of disulfide bond formation (kSS) changed notably as a function of the permeant ion concentration. Increasing [Na+]o (and therefore ion occupancy and relative conductance) slowed the kinetics of disulfide bond formation (Table 1). Although it remains possible that increasing Na+ occupancy in the pore had a direct and unanticipated inhibitory effect on disulfide formation, the decrease in kss with increasing ionic conductance is consistent with the inhibitory effect of [Na+]o on the rate of slow inactivation (Fig. 6B) (Townsend and Horn, 1997). Alteration in the rate of [Na+]o-induced occupancy of slow inactivated states is not unique to the KC/WC mutant. Entry into theIM state of the wild-type channel is accelerated in low [Na+]o (Fig.6B), suggesting that the KC/WC double mutant does not dramatically alter the outer pore structure. Thus, the observations of dependence on [Na+]o and on depolarizing pulse duration may share a common basis in the fractional occupancy of particular inactivated states (e.g.,IM).
Fig. 6.

External Na+ concentration modulates the rate of inactivation and disulfide bond formation.A, Representative examples of the time course of the inhibition of K1237C+W1531C by 100 mm Cu(phe)3obtained from oocytes bathed in either (▵) 48 mm[Na+]o, (○) 96 mm[Na+]o, or (⋄) 140 mm [Na+]o. Sorbitol was used as a substitute for Na+ to maintain constant osmolarity. The currents were elicited by repetitive 50 msec pulses from −100 to −35 mV (0.5 Hz). Plotted are the whole-cell Na current amplitudes normalized to the value measured before the application of Cu(phe)3 (100 μm). Summary data forkss as a function of extracellular [Na+]o are given in Table 1.B, Rates of the development of inactivation of wild-type μ1 channels coexpressed with the β1 subunit. The protocol is identical to that shown in Figure 5 for the KC/WC mutant. Reducing the [Na+]o enhances the rate of development of slow inactivation. In 96 mm[Na+]o (▪) there is little development of intermediate inactivation (< 3%). In contrast, in 10 mm [Na+]o (○) a 1 sec prepulse inactivates 35% of the current.
Table 1.
Effect of Na+ concentration on the rate of disulfide bond catalysis by Cu (Phe)3 on the double-mutant K1237C+W1531C (mean ± SD)
| [Na+]o = 48 mm | [Na+]o = 96 mm | [Na+]o = 140 mm | |
|---|---|---|---|
| kss(molecule−1 · sec−1) | 15.25 ± 6.61 (n = 5) | 7.58 ± 6.19 (n = 11) | 7.59 ± 1.94 (n = 6) |
Redox state modulates a slow-inactivated state transition
The experiments discussed above examine the influence of gating on pore mobility. To examine the converse issue, the influence of mobility on gating, we used another double-cysteine mutant, Y401C+E758C (YC/EC). YC/EC spontaneously forms a functionally evident disulfide bond (Bénitah et al., 1996) under oxidizing conditions, whereas a reducing environment breaks the covalent bond, reestablishing free motion in the outer pore. The unique characteristics of this double-cysteine mutation allowed us to examine the effect of a partially immobilized outer pore structure on the development of slow inactivation. Figure 7 shows the time dependence of recovery from inactivation of YC/EC. Oocytes were held at −100 mV, and a conditioning pulse was applied for either 50 msec (Fig.7A) or 1 sec (Fig. 7B). The conditioning pulse was followed by recovery at −100 mV for intervals varying from 10 msec to 1 sec, and the Na+ current was measured in response to a subsequent 50 msec test depolarization. Cumulative inactivation between experiments was excluded by using a long (30 sec) recovery interval at −100 mV after each test pulse, and by confirming that there was no variation in the Na current elicited by successive conditioning pulses. Oocytes were sequentially bathed in normal oxidized solution (left, solid symbols) and a reducing environment (right, open symbols). Exposure to DTT increased the magnitude of the Na current, presumably by breaking the disulfide bond (Bénitah et al., 1996). In addition, after the longer depolarizing prepulse (Fig. 7B), DTT significantly slowed the rate of recovery from inactivation. Figure 7C plots the fractional recovery of the peak Na+ current amplitude against the recovery interval at −100 mV. As shown previously for wild-type μ1 channels (Nuss et al., 1996), YC/EC recovery from inactivation exhibited three readily separable kinetic components. The recovery rates (τ) and the relative amplitudes of the three components as a function of the external environment and the conditioning pulse length are summarized in Table2. After the brief conditioning pulses (50 msec), <20% of the Na channels entered slow-inactivated states (Table 2), and the redox environment had no significant effect on the relative amplitudes or time constants of the three components of recovery. However, with longer conditioning pulses (1 sec), >60% of the channels entered slowly recovering inactivated states (IM, IS). Exposure to a reducing environment speeded recovery by decreasing the amplitude of the intermediate component (Table 2). The time constant of recovery from this intermediate component of inactivation was also increased (Table 2), although the extent to which this influenced the overall recovery profile was small. These data suggest that a reducing environment raised the energy barrier controlling entry to, and exit from, a slow-inactivated state with intermediate kinetics similar to that facilitating K1237C–W1531C disulfide bond formation (Figs. 3, 4). Our findings are consistent with the notion that “freezing” the outer pore with a disulfide bond facilitates both entry into and egress from an inactivated state that possesses intermediate recovery kinetics (IM).
Fig. 7.

Redox state modulates the rate of recovery from inactivation for the double mutant Y401C+E758C. A, B, A standard two-pulse protocol was used to compare recovery from inactivation of Y401C+E758C channels in oxidized control solution (left panels) and after addition of 1 mm DTT (right panels) in the same oocyte. Oocytes were held at −100 mV. After a 50 msec (A) or 1 sec (B) conditioning pulse to −35 mV (the peak of the IV relationship), a pulse to −100 mV ranging from 1 msec to 1000 msec was followed by a 50 msec test depolarization. C, Fractional recovery from inactivation as a function of time at −100 mV is plotted for the double mutant Y401C+E758C in oxidized control solution (solid symbols) and after paired addition of 1 mm DTT (open symbols). Data sets obtained using either a 50 msec (squares) or a 1 sec (circles) conditioning pulse are shown. A three-exponential function of the form y = A1exp(−t/τ1) + A2exp(−t/τ2) + A3exp(−t/τ3) was fitted to the individual data sets (fitted parameters are in Table 2). Fitted parameters to the mean data for a 50 msec prepulse (control vs DTT) were τ1 = 2.6 versus 2.4 msec, A1 = 0.84 versus 0.80; τ2 = 40.0 versus 40.9 msec, A2 = 0.11 versus 0.12; τ3 = 1565 versus 1315 msec, A3 = 0.05 versus 0.08. For the 1 sec prepulse, fitted parameters to the mean data (control vs DTT) were τ1 = 2.6 versus 3.3 msec, A1 = 0.51 versus 0.62; τ2 = 100.2 versus 49.8 msec, A2 = 0.23 versus 0.33; τ3 = 2660 versus 3449 msec, A3 = 0.16 versus 0.15.
Table 2.
The effect of redox environment on Y401C+E758C recovery from inactivation (mean ± SD)
| Prepulse 50 msec (n = 10) | Prepulse 1 sec (n = 7) | |||
|---|---|---|---|---|
| Control | DTT | Control | DTT | |
| τ1(msec) | 2.5 ± 0.9 | 2.3 ± 0.4 (NS) | 4.4 ± 1.1 | 4.9 ± 4.0 (NS) |
| A1 | 0.82 ± 0.13 | 0.79 ± 0.09 (NS) | 0.42 ± 0.18 | 0.45 ± 0.12 (NS) |
| τ2(msec) | 43.7 ± 24.1 | 36.2 ± 18.1 (NS) | 58.6 ± 37.8 | 123.1 ± 82.8 (p < 0.05) |
| A2 | 0.13 ± 0.10 | 0.13 ± 0.07 (NS) | 0.30 ± 0.09 | 0.22 ± 0.04 (p < 0.05) |
| τ3 (msec) | 728 ± 742 | 907 ± 640 (NS) | 4301 ± 2426 | 5852 ± 5930 (NS) |
| A3 | 0.05 ± 0.07 | 0.08 ± 0.09 (NS) | 0.28 ± 0.10 | 0.33 ± 0.12 (NS) |
DISCUSSION
Previous site-directed mutagenesis studies (Balser et al., 1996;Kambouris et al., 1998) suggest that slow kinetic components of Na+ channel inactivation involve conformational changes in the outer pore. W402C, a domain I P-segment substitution, markedly enhanced recovery by reducing the likelihood that depolarized μ1 channels would occupy slowly recovering inactivated states. Notably, μ1 channels are capable of entering two inactivated states with kinetics slower than the duration of theINa transient; these include both intermediate and slow kinetic components with recovery time constants at −100 mV on the order of ∼100 msec and 1 sec, respectively (Nuss et al., 1995,1996). Similar to the W402 P-segment mutations (Kambouris et al., 1998), the findings presented here suggest that disulfide formation in the outer pore of the KC/WC mutant is primarily sensitive to the inactivated state with intermediate kinetics (IM). It is possible that the mutations themselves alter channel conformation or movement in a way that is not representative of the wild-type μ1 channel. We observed no change in the gating of the W1531C component of the double mutant (Balser et al., 1996). The K1237C component alters both permeation (Favre et al., 1996) and the development of ultra-slow inactivation (Todt et al., 1997) but not the rate of occupancy of IM (data not shown). The biphasic dependence of the rate of disulfide formation (kss) (Figs. 3, 4) on pulse duration suggests that when either IF orIS is occupied, the rate of disulfide formation is slowed compared with that when IM is the predominant inactivated state. One possible interpretation, illustrated in Figure 8, is that the spatial relationship between the 1237 and 1531 cysteines is optimized by occupancy of the IM state. Consistent with this hypothesis, the rate of disulfide formation decreases when the rate of entry into slow-inactivated states is modified by elevating the external [Na+] (Fig. 6, Table 2) (Townsend et al., 1997). Admittedly, the sensitivity of particular slow inactivated states (e.g., IM vsIS) to alkali metal cations has not been characterized, and it remains possible that the inhibitory effect of external Na+ on disulfide formation results from a direct competition between the covalent reaction and Na+ binding in the pore. However, the converse experiment using a spontaneously forming disulfide (YC/EC) (Fig. 7) showed that freezing the pore with a disulfide selectively facilitated entry into, and recovery from, an inactivated state with intermediate gating kinetics (Table 2). Hence, our results suggest that greater flexibility decreases the occupancy of a particular inactivated state with intermediate kinetics and furthermore that occupancy of this inactivated state facilitates KC/WC disulfide formation. These findings are consistent with the general notion of microscopic reversibility: inactivation gating influences motion in the outer pore, and vice versa.
Fig. 8.

Occupancy of an intermediate inactivated state facilitates disulfide formation in the outer pore. The diagram illustrates sequential occupancy of three distinct inactivated states (IF,IM, andIS) as the length of the depolarization is extended. Time constants for entry intoIM and IS are based on the biexponential fit to the data in Figure 5, and the time constant for development of fast inactivation (IF) was estimated from the literature (Nuss et al., 1995). The scheme suggests that when theIM state is occupied, the 1237 and 1531 cysteines are spatially optimized in the outer pore for disulfide formation.
These data have several implications for the structure of the Na channel pore. In the recently published KcsA channel structure (Doyle et al., 1998), the tryptophan in the selectivity region makes important contacts with other aromatic residues facing away from the pore, forming an aromatic cuff around the selectivity filter. The structural data are consistent with the finding that some mutations at this tryptophan [e.g., W434F (Perozo et al., 1993)] eliminate ionic flux by “locking” the K channel in an inactivated state (Yang et al., 1997). This is distinct from the phenotype of the Na channel with mutations of a comparable ring of tryptophans (Tomaselli et al., 1995;Balser et al., 1996). The Na channel pore is not compromised by single mutations of the tryptophans in any of the domains. In domains I, III, and IV, mutagenesis data suggest that this side chain points into the pore, consistent with the findings of Lü and Miller (1995), who found that single tryptophan to cysteine mutations in the K channel pore were accessible to block by silver. In domain II the permeation phenotype of the Na channel mutant (W756C) is identical to that of the wild-type channel (Perez-Garcia et al., 1996). The tryptophans in the K channel are part of the pore helix on the amino terminal side of the selectivity filter. By sequence alignment with the K channel, this ring of tryptophans in the Na channel is predicted to be in the filter loop on the C-terminal side of the filter residues. It appears that the pore tryptophans do not have analogous roles in the Na and K channels.
The combination of paired cysteine mutagenesis and disulfide trapping is a powerful technique for indirect structural study of the channel pore and the role of motion of the external vestibule in channel gating. Several limitations of the technique should be enumerated. First, it is unclear whether the backbone or side chains of the substituted residues are exhibiting flexibility and permitting trapping. Structural models of the Na channel (Guy and Durell, 1995) and KcsA (Doyle et al., 1998) suggest that the residues (or analogous residues in the K channel) are sufficiently separated that simple side chain movement would not place the cysteinyls close enough to form a disulfide. Second, it is possible that disulfide trapping captures the channel in an infrequently occupied and unphysiological state. However, the enhancement of the rate of disulfide formation during occupancy ofIM, a state normally occupied by the channel, argues against trapping in a completely unnatural configuration. Finally, as noted previously, the mutations themselves may alter the flexibility and movement in the vestibule.
These data support the concept that analogous to C-type inactivation (Hoshi et al., 1991; Liu et al., 1996) in K channels, inactivation involving structural rearrangement in the outer pore occurs in the Na channel. It is possible that both intermediate inactivation (IM) (Kambouris et al., 1998) and ultraslow inactivation (IS) (Todt et al., 1997) involve structural changes in the outer pore, but only occupancy of IM influences the rate of disulfide bond formation between paired cysteine mutants examined in this study. Nonetheless, it is possible that other pairs of cysteine mutants will be more likely to disulfide bond in the IS state to support this hypothesis.
Footnotes
This research was supported by National Institutes of Health (P50 HL52307). Additional salary support was provided by the American Heart Association, Maryland Affiliate (J.-P.B.), and R01 HL52768 (E.M.), R01 GM56307 (J.R.B.), and R01 HL50411 (G.F.T.).
Correspondence should be addressed to Dr. Gordon F. Tomaselli, Section of Molecular and Cellular Cardiology, 844 Ross Building, Johns Hopkins University, Baltimore, MD 21205.
REFERENCES
- 1.Balser JR, Nuss HB, Chiamvimonvat N, Pérez-García MT, Marban E, Tomaselli GF. External pore residue mediates slow inactivation in μ1 rat skeletal muscle sodium channels. J Physiol (Lond) 1996;494.2:431–442. doi: 10.1113/jphysiol.1996.sp021503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bénitah J-P, Tomaselli GF, Marban E. Adjacent pore-lining residues within sodium channels identified by paired cysteine mutagenesis. Proc Natl Acad Sci USA. 1996;93:7392–7396. doi: 10.1073/pnas.93.14.7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bénitah J-P, Ranjan R, Yamagishi T, Janecki M, Tomaselli GF, Marban E. Molecular motions within the pore of voltage-dependent sodium channels. Biophys J. 1997a;73:603–613. doi: 10.1016/S0006-3495(97)78096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bénitah J-P, Balser JR, Marban E, Tomaselli GF. Proton inhibition of sodium channels: mechanism of gating shifts and reduced conductance. J Membr Biol. 1997b;155:121–131. doi: 10.1007/s002329900164. [DOI] [PubMed] [Google Scholar]
- 5.Careaga CL, Falke JJ. Thermal motions of surfacea-helces in the d-galactose chemosensory receptor: detection by disulfide trapping. J Mol Biol. 1992;226:1219–1235. doi: 10.1016/0022-2836(92)91063-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen FSP, Steele D, Fedida D. Allosteric effects of permeating cations on gating currents during K+ channel deactivation. J Gen Physiol. 1997;110:87–100. doi: 10.1085/jgp.110.2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Biasi M, Hartman HA, Drewe JA, Taglialatela M, Brown AM, Kirsch GE. Inactivation determined by a single site in K+ pore. Pflügers Arch. 1993;422:354–363. doi: 10.1007/BF00374291. [DOI] [PubMed] [Google Scholar]
- 8.Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 9.Eisenman G, Krasne S. The ion selectivity of carrier molecules, membranes and enzymes. In: Fox CF, editor. MTP international review of science, biochemistry series, Vol 2. Butterworths; London: 1975. pp. 27–59. [Google Scholar]
- 10.Favre I, Moczydlowski E, Schild L. On the structural basis for ionic selectivity among Na+, K+, and Ca2+ in the voltage-gated sodium channel. Biophys J. 1996;71:3110–3125. doi: 10.1016/S0006-3495(96)79505-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guy HR, Durell SR. Structural models of Na+, Ca2+ and K+ channels. Soc Gen Physiol Ser. 1995;50:1–16. [PubMed] [Google Scholar]
- 12.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol (Lond) 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 14.Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- 15.Isacoff EY, Jan YN, Jan LY. Putative receptor for the cytoplasmic inactivation gate in the Shaker K+ channel. Nature. 1991;353:86–90. doi: 10.1038/353086a0. [DOI] [PubMed] [Google Scholar]
- 16.Kambouris NG, Hastings L, Stepanovic S, Marban E, Tomaselli GF, Balser JR. Mechanistic link between local anaesthetic action and inactivation gating probed by outer pore mutations in the rat μ1 sodium channel. J Physiol (Lond) 1998;512:693–705. doi: 10.1111/j.1469-7793.1998.693bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawrence JH, Orias DW, Balser JR, Nuss HB, Tomaselli GF, Marban E. Single-channel analysis of inactivation-defective rat skeletal muscle sodium channels containing the F1304Q mutation. Biophys J. 1996;71:1285–1294. doi: 10.1016/S0006-3495(96)79329-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Jurman ME, Yellen G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- 19.Lü Q, Miller C. Silver as a probe of pore-forming residues in a potassium channel. Science. 1995;268:304–307. doi: 10.1126/science.7716526. [DOI] [PubMed] [Google Scholar]
- 20.McCormack K, Tanouye MA, Iverson LE, Lin J-W, Ramaswami M, McCormack T, Campanelli JT, Mathew MK, Rudy B. A role for hydrophobic residues in the voltage-dependent gating of Shaker K+ channels. Proc Natl Acad Sci USA. 1991;88:2931–2935. doi: 10.1073/pnas.88.7.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nuss HB, Chiamvimonvat N, Perez-Garcia MT, Tomaselli GF, Marban E. Functional association of the β1 subunit with human cardiac (hH1) and rat skeletal muscle (μ1) sodium channel α subunits expressed in Xenopus oocytes. J Gen Physiol. 1995;106:1171–1191. doi: 10.1085/jgp.106.6.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nuss HB, Balser JR, Orias DW, Lawrence JH, Tomaselli GF, Marban E. Coupling between fast and slow inactivation revealed by analysis of a point mutation (F1304Q) in μ1 rat skeletal muscle sodium channels. J Physiol (Lond) 1996;494.2:411–429. doi: 10.1113/jphysiol.1996.sp021502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perez-Garcia MT, Chiamvimonvat N, Marban E, Tomaselli GF. Structure of the sodium channel pore revealed by serial cysteine mutagenesis. Proc Natl Acad Sci USA. 1996;93:300–304. doi: 10.1073/pnas.93.1.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perozo E, MacKinnon R, Bezanilla F, Stefani E. Gating currents from a nonconducting mutant reveal open-closed conformations in Shaker K+ channels. Neuron. 1993;11:353–358. doi: 10.1016/0896-6273(93)90190-3. [DOI] [PubMed] [Google Scholar]
- 25.Rudy B. Slow inactivation of the sodium conductance in squid giant axons. Pronase resistance. J Physiol (Lond) 1978;238:1–21. doi: 10.1113/jphysiol.1978.sp012485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stühmer W, Conti F, Suzuki H, Wang X, Noda M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- 27.Todt H, Dudley S, Fozzard H. Ultra-slow inactivation in the skeletal muscle sodium channel is influenced by pore residues. Biophys J. 1997;72:A261. [Google Scholar]
- 28.Tomaselli GF, Chiamvimonvat N, Nuss HB, Balser JR, Pérez-García MT, Xu RH, Orias DW, Backx PH, Marban E. A mutation in the pore of the sodium channel alters gating. Biophys J. 1995;68:1814–1827. doi: 10.1016/S0006-3495(95)80358-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Townsend C, Horn R. Effect of alkali metal cations on slow inactivation of cardiac Na+ channels. J Gen Physiol. 1997;110:23–33. doi: 10.1085/jgp.110.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Townsend C, Hartmann H, Horn R. Anomalous effect of permeant ion concentration on peak open probability of cardiac Na+ channels. J Gen Physiol. 1997;110:11–21. doi: 10.1085/jgp.110.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsushima RG, Li RA, Backx PH. P-loop flexibility in Na+ channel pores revealed by single- and double-cysteine replacements. J Gen Physiol. 1997;110:59–72. doi: 10.1085/jgp.110.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.West J, Patton D, Scheuer T, Wang Y, Goldin A L, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc Natl Acad Sci USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Y, Yan Y, Sigworth FJ. How does the W434F mutation block current in Shaker potassium channels? J Gen Physiol. 1997;109:779–789. doi: 10.1085/jgp.109.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yellen G. Single channel seeks permeant ion for brief but intimate relationship. J Gen Physiol. 1997;110:83–85. doi: 10.1085/jgp.110.2.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zagotta WN, Aldrich RW. Voltage-dependent gating of Shaker A-type potassium channels in Drosophila muscle. J Gen Physiol. 1990;95:29–60. doi: 10.1085/jgp.95.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]