

Abstract
The developing cerebral cortex undergoes a period of substantial cell death. The present studies examine the role of the suicide receptor Fas/Apo[apoptosis]-1 in cerebral cortical development. Fas mRNA and protein are transiently expressed in subsets of cells within the developing rat cerebral cortex during the peak period of apoptosis. Fas-immunoreactive cells were localized in close proximity to Fas ligand (FasL)-expressing cells. The Fas-associated signaling protein receptor interacting protein (RIP) was expressed by some Fas-expressing cells, whereas Fas-associated death domain (FADD) was undetectable in the early postnatal cerebral cortex. FLICE-inhibitory protein (FLIP), an inhibitor of Fas activation, was also expressed in the postnatal cerebral cortex. Fas expression was more ubiquitous in embryonic cortical neuroblasts in dissociated culture compared to in situ within the developing brain, suggesting that the environmental milieu partly suppresses Fas expression at this developmental stage. Furthermore, FADD, RIP, and FLIP were also expressed by subsets of dissociated cortical neuroblasts in culture. Fas activation by ligand (FasL) or anti-Fas antibody induced caspase-dependent cell death in primary embryonic cortical neuroblast cultures. The activation of Fas was also accompanied by a rapid downregulation of Fas receptor expression, non-cell cycle-related incorporation of nucleic acids and nuclear translocation of the RelA/p65 subunit of the transcription factor NF-κB. Together, these data suggest that adult cortical cell number may be established, in part, by an active process of receptor-mediated cell suicide, initiatedin situ by killer (FasL-expressing) cells and that Fas may have functions in addition to suicide in the developing brain.
Keywords: CNS, rat, interleukin-1β-converting enzyme, ICE protease inhibitors, FasL, FADD, FLIP, RIP, PCNA, BrdU
The developing cortical neuroepithelium generates more neurons than are retained in the adult. Neuronal attrition occurs developmentally, by apoptotic cell suicide. A prominent hypothesis is that cell death is partly a consequence of competition for limited target-derived growth factors (Oppenheim, 1985). However, cell death in other organ systems also results from receptor-mediated cell suicide, triggered by specific intercellular interactions (Nagata, 1997). The present study examined the neural expression and activation of the suicide receptor Fas/Apo [apoptosis]-1/CD95.
Fas, a member of the tumor necrosis factor receptor (TNFr) family (Yonehara et al., 1989; Itoh et al., 1991; Cleveland and Ihle, 1995), first detected in the immune system and, subsequently in several non-neural tissues (Watanabe-Fukunaga et al., 1992; Oishi et al., 1994;Esser et al., 1995), plays a critical role in limiting cell proliferation by apoptosis (Nagata and Goldstein, 1995). One transmembrane, cell surface ligand [Fas ligand (FasL)] has been identified (Suda et al., 1993), suggesting that intercellular contacts are important for Fas induction of cell suicide. Mutations in Fas (lpr) and FasL (gld) loci lead to lymphadenopathy and autoimmune disease (Rieux-Laucat et al., 1995;Watanabe et al., 1995). Although previous studies reported FasL expression in the developing brain (French et al., 1996), expression of Fas, until now, has only been associated with tumorogenesis and adult neurological diseases. For example, Fas is expressed in neural-derived tumors (Tachibana et al., 1995) and is associated with apoptosis in neuroblastomas (Weller et al., 1998). Recent reports have identified Fas in adult postmortem brains of patients with neurodegenerative disorders, including Alzheimer’s disease (Nishimura et al., 1995) and multiple sclerosis (D’Souza et al., 1996; Bonetti and Raine, 1997). Fas mRNA is also expressed in periventricular and perivascular cells in postischemic, adult mouse brains (Matsuyama et al., 1995). Adult brain expression may be caused by direct injury to neural cells or invasion of immune cells into the nervous system.
Activated Fas associates with several signal cascades including Fas-associated death domain (FADD)/caspase-8, receptor-interacting protein (RIP) or RIP-associated ICH-1/CED-3 homologous protein (RAIDD)/caspase-2 complexes or Fas death domain-associated protein (DAXX) (Boldin et al., 1995; Chinnaiyan et al., 1995, 1996; Kischkel et al., 1995; Stanger et al., 1995; Muzio et al., 1996; Ahmad et al., 1997; Duan and Dixit, 1997) to induce interleukin-1β-converting enzyme/cysteine-aspartate protease (ICE/caspase-1) and CPP-32/caspase-3 protease cascades (Thornberry et al., 1992; Enari et al., 1995, 1996;Los et al., 1995; Milligan et al., 1995) or Jun kinase (JNK) activation (Xiaolu et al., 1997). In contrast, recently described negative regulators of Fas, such as FLICE-inhibitory protein (FLIP), prevent Fas activation of caspase cascades (Irmler et al., 1997). Although Fas interactions with caspases lead to cell death (Boldin et al., 1995,1996; Chinnaiyan et al., 1995), Fas–RIP interactions can additionally activate the transcription factor NF-κB (Oishi et al., 1994). RIP overexpression induces both NF-κB activation (Hsu et al., 1996) and apoptosis (Stanger et al., 1995). However, NF-κB activation generally appears to promote cell survival (Baeuerle and Baltimore, 1997), suggesting that Fas may activate competitive cell death and survival pathways.
Although Fas has a crucial role in immune system function, little is known about its expression or role during normal brain development. We hypothesized that cell death during cerebral cortical development may be induced by receptor-mediated mechanisms involving, in part, Fas–FasL interactions. The present studies examined the expression of Fas/Apo-1 in the developing cerebral cortex and its role in cell death induction.
MATERIALS AND METHODS
Animals
Timed pregnant rats (Sprague Dawley) were purchased from Harlan. Embryonic day zero (E0) was defined as the day on which dams were sperm-positive, and postnatal day one (P1) was the day of birth. A total of 114 rat pups (E11.5, E13.5, E15, E18, E19, P1–11, P15, P18, and P22) were used for in vivo analysis, and 93 rat pups were used for in vitro experiments. In all cases, care was taken to minimize any pain and discomfort to the animals. Pregnant dams were anesthetized with phenobarbital for prenatal experiments. Pups younger than P10 were anesthetized with ice, whereas older pups were anesthetized with phenobarbital.
In situ hybridization
To determine the temporal expression of Fas mRNA and its association with the protein RIP, single (nonisotopic) and double (isotopic with nonisotopic) in situ hybridization of cryostat-sectioned rat brains were performed according to our published protocols (Miranda and Toran-Allerand, 1992; Miranda et al., 1993a,b,1994; Sohrabji et al., 1994b; Donovan et al., 1995). Embryonic brain tissue was fixed by immersion in buffered paraformaldehyde with 2.5% dimethylsulfoxide while postnatal brain tissue was obtained from perfusion-fixed animals. Brains were cryoprotected in 15% sucrose in PBS and frozen in M1 mounting media (Lipshaw). Briefly, 20 μm cryostat-sectioned brain slices from the septohippocampal region were thaw-mounted onto treated slides. Brain sections from six different ages were combined onto one slide, and slides containing different age series were processed simultaneously to limit experimental variability. A specific 45 base oligonucleotide sequence complimentary to Fas mRNA (5′GTG TGC AAG GCT CAA GGA TGT CTT CAA GTC CAC ACG AGG TGC AGT3′) or RIP mRNA (5′TTC TCC GTG TTT GCA TTG ATG TCA TTC AGG TGT TGT TCG GGT GCC A3′) and sense controls were 3′ end-labeled with digoxigenin-11-dUTP or γ-[33P]dATP, respectively. After hybridization with Fas or RIP probes, sections were washed in 0.1× SSC (15 mm sodium chloride and 1.5 mm sodium citrate, 2 hr, 50°C) then in 1× SSC (0.15 m sodium chloride and 0.015 m sodium citrate, overnight, 50°C). Slides being processed for combined in situ hybridization were then incubated with the second oligonucleotide probe or sense control. Sections were then incubated with an alkaline-phosphatase-linked anti-digoxigenin antibody, (Boehringer Mannheim, Indianapolis, IN) and processed for alkaline phosphatase-linked histochemistry. Sections processed for combined in situ hybridization were dehydrated briefly through an ethanol gradient containing 0.3 m sodium acetate. Slides were subsequently coated with autoradiographic emulsion (Ilford, K5) and processed for the detection of the33P-labeled oligonucleotide probe. Hybridization with33P-labeled probes was considered specific when the accumulation of exposed silver grains exceeded 5 times background (Arnold, 1980). Sense controls did not exhibit any hybridization.
RT-PCR
RT-PCR was used to confirm the expression of Fas mRNA in the developing rat cortex, according to previously published protocols (Sohrabji et al., 1994a; Donovan et al., 1995; Miranda et al., 1996). Forward: 5′AAG AGG CAA CCT GGT GAC CC3′ and reverse: 5′GGG TCA CCA GGT TGC CTC TT3′ rat-specific primers were designed to cross exon–intron boundaries and have a melting temperature difference of 0.2°C. RT-PCR was performed on DNase-treated, total RNA (P6 cortex) using a kit (Gene-Amp; Perkin-Elmer, Emeryville, CA) and manufacturer’s instructions. cDNA was synthesized using reverse transcriptase and reverse primers specific to Fas and cyclophillin (a control) (Sohrabji et al., 1995). After heat denaturation of reverse transcriptase, the cDNA product was amplified for Fas or cyclophillin mRNA. The PCR program (MJ Research PTC200 thermal cycler), 94°C for 1 min, 45°C for 30 sec, and 72°C for 1 min, was cycled 30 times. The PCR product was size-fractionated on a 2% agarose gel. A control transcript (cyclophillin) was also reverse-transcribed and amplified. No bands were observed for RNA samples not exposed to reverse transcriptase but incubated with Fas primers and processed for PCR. An AvaII restriction digest of the PCR product yielded two fragments of the expected size (202 and 248 bp, respectively).
Western analysis
The expression of Fas, its associated proteins FADD and RIP, the negative regulator FLIP, and the ligand FasL was verified by Western immunoblot analysis of P6 cerebral cortex, according to our previously published protocols (Donovan et al., 1995; Miranda et al., 1996). The polyclonal FLIP antibody was generated in New Zealand white rabbits against a 26 amino acid (SAEVIHQVEEALDTDEKEMLLFLCRD) sequence spanning amino acids 2–27 at the N terminus of FLIP using multiple antigen technology (Francis et al., 1991). This sequence is common among all of the reported isoforms of human FLIP (Hu et al., 1997; Irmler et al., 1997; Srinivasula et al., 1997) and highly homologous to the corresponding sequence of murine FLIP (Irmler et al., 1997). The IgG fraction of the antiserum was isolated using an E-Z-SEP kit (Pharmacia, Piscataway, NJ) and affinity-purified on the corresponding peptides coupled to Affi-Gel 15 gel (Bio-Rad, Hercules, CA).
To determine whether Fas lead to changes in cell cycle, Western blot analysis was also performed for proliferating cell nuclear antigen (PCNA) expression. PCNA is a G1-S phase cell cycle marker whose expression is rapidly upregulated during DNA replication (Celis and Celis, 1985; Kurki et al., 1986).
Detergent (1% SDS)-soluble protein was isolated using the Trizol reagent (Life Technologies, Gaithersburg, MD). Protein samples were size-fractionated on an 8% SDS-polyacrylamide gel and blotted onto supported nitrocellulose (Hibond-C-super; Amersham, Arlington Heights, IL). Blots were blocked [5% milk, TBS (1.4 mNaCl, 0.2 m Tris)-0.1% Tween 20], exposed to primary antibody [mouse monoclonal anti-Fas antibody (Transduction Laboratories, Lexington, KY; 1:500); mouse monoclonal RIP antibody (Transduction Laboratories, 1:500); mouse monoclonal anti-FADD antibody (Transduction Laboratories, 1:500); affinity-purified rabbit polyclonal anti-FLIP antibody (1:500); anti-FasL antibody (Transduction Laboratories; 1:500); mouse monoclonal anti-PCNA antibody (Calbiochem, La Jolla, CA; 1:66)], washed and exposed to horse anti-mouse secondary antibody (Vector Laboratories, Burlingame, CA; 1:1000) or donkey anti-rabbit (Jackson ImmunoResearch, West Grove, PA; 1:5000), washed again and exposed to streptavidin horseradish-peroxidase conjugate (Amersham). Immunoreactive bands were detected using enzyme-linked chemiluminescence (NEN).
Appropriate-sized bands (Fas, 45 kDa; RIP, 74 kDa; FADD, 24 kDa; FasL, 37 kDa) were also observed in a size-fractionated positive control (Jurkat cell total protein lysate). To test the specificity of anti-FLIP antibody, protein obtained from COS7 cells transfected with the coding sequence of FLIP in an expression vector were used as controls. The antibody recognizes FLIP (at ∼60 kDa) in transfected COS7 cells (see Fig. 6D), human endothelial cells (M. Sata and K. Walsh, unpublished observations), and murine C2C12 skeletal myoblast cell lines (T. Mano and K. Walsh, unpublished observations). Western blot analysis for PCNA yielded a band of expected size (37 kDa).
Fig. 6.

The expression of the Fas-associated proteins RIP (A), FADD (B), FLIP (C), and FasL (D) was examined by Western analysis of 50 μg of detergent-soluble protein obtained from P6 cerebral cortex obtained from four separate animals (lanes 1-4). Bands corresponding to RIP (74 kDa) and FasL (37 kDa) were detected in 10 μg of Jurkat cell lysates (a positive control) and in P6 cortex. FLIP expression in P6 cortex was confirmed by the appearance of a similar band just below the 66 kDa molecular weight marker, corresponding to the expected size of ∼60 kDa, in protein extract from COS7 cell transfected with the full-length FLIP plasmid (positive control). In contrast, we did not detect any bands corresponding to the expected size for FADD in the cerebral cortex, although a band of the approximate size (24 kDa) was observed in Jurkat cell-derived protein (a positive control).Mkr, Biotinylated marker.
Immunohistochemistry and immunofluorescence
To determine the temporal expression of the Fas receptor, the expression of the associated protein RIP and the transcription factor NF-κB, immunohistochemistry and immunofluorescence were performed on fixed 20-μm-thick thaw-mounted sections (Fas, RIP) or dissociated cortical cultures (NF-κB), according to previously published protocols (Toran-Allerand et al., 1991; Donovan et al., 1995; Miranda et al., 1996). Binding of the primary antibody [mouse monoclonal antibody to Fas (Transduction Laboratories, 1:500); mouse monoclonal antibody to RIP (Transduction Laboratories, 1:500); rabbit polyclonal antibody to p65/RelA subunit of NF-κB (Santa Cruz Biotechnology, Santa Cruz, CA; 1:250)] was detected using the appropriate secondary antibody conjugated to biotin [horse anti-mouse (Vector Laboratories, 1:1000) or donkey anti-rabbit (Jackson ImmunoResearch, 1:500)], either linked to horseradish peroxidase via an avidin–biotin complex (Vector ABC elite) and diaminobenzidine as a chromogenic substrate for immunohistochemistry or conjugated to streptavidin-FITC or rhodamine-avidin for immunofluorescence. Cultures immunostained for the p65/RelA subunit of NF-κB were also counterstained with a nuclear stain (Hoechst dye #33342).
For combined immunohistochemistry with Fas and neuronal markers or Fas with either FasL, FADD, or FLIP, after immunohistochemistry for Fas [mouse monoclonal anti-Fas antibody (Transduction Laboratories) or rabbit polyclonal anti-Fas antibody (Santa Cruz; secondary donkey anti-rabbit antibody from Jackson ImmunoResearch), 1:500], tissue sections or cultures were exposed to either a cocktail of antibodies [rabbit polyclonal anti-microtubule associated protein (MAP) antibody (selective for MAP2, Sigma), rabbit polyclonal anti-neurofilament 150 kDa antibody, and rabbit polyclonal anti-neurofilament 200 kDa antibody (Genosys), all at 1:500 dilution] or rabbit polyclonal anti-FasL antibody (Santa Cruz, 1:500), mouse monoclonal anti-FADD antibody (Transduction Laboratories, 1:250), and rabbit polyclonal anti-FLIP antibody (1:500). Binding of the second, primary antibody was visualized by the appropriate secondary antibody, either a biotinylated donkey anti-rabbit antibody (Jackson ImmunoResearch, 1:500) or horse anti-mouse (Vector Laboratories, 1:1000) coupled to either streptavidin-FITC or rhodamine-avidin. A cocktail of antibodies to neuron-specific markers was used to maximize the labeling of both processes and the soma of neurons at various stages of differentiation. In controls, no staining was observed in sections exposed to preimmune serum in place of the primary antibody.
Detection of apoptotic profiles
To determine the normal temporal expression of apoptosis in the developing cerebral cortex, apoptotic profiles were detected by nonisotopic terminal deoxynucleotidyl transferase-mediated digoxigenin-dUTP nick end labeling (TUNEL) of nucleosomal fragments generated by endonuclease cleavage. Tissue sections (10 μm) were extensively dilipidated, then rehydrated again. Slides were treated with proteinase K (20 μg/ml Sigma, 15 min at room temperature), washed extensively and exposed for 30 min, to equilibration buffer [1× TDT buffer (Life Technologies) and BSA]. Each slide was then incubated for 3 hr, at 37°C, with a solution containing 1× TDT buffer, BSA (1 mg/ml), 37 U TDT, and 1 pmol digoxigenin-11-dUTP. Slides were washed in Tris-buffered saline and processed for immunohistochemistry with an alkaline phosphatase-conjugated, anti-digoxigenin antibody.
The presence of apoptotic profiles was also confirmed by staining with Hoechst dye #33342. Hoechst dye #33342 binds specifically to A-T base regions in DNA and emits blue immunofluorescence at 350 nm. The dye was administered to fixed cells at 10 μg/ml for 30 min. Apoptotic profiles exhibit a highly condensed nuclear fluorescence, whereas viable cells exhibit a diffuse nuclear fluorescence.
Dissociated cell culture
Fas induction of cell death. To examine whether activated Fas could induce cell death, cultures of dissociated cortical neuroblasts were obtained from E15 rat brains. Neuroblasts were dispersed by trituration in 0.5% trypsin and 6.84 mm EDTA (Sigma) and were plated on mouse-laminin-coated (0.1 mg/ml) 96-well plates. Experimental groups were exposed to a soluble Fas Ligand (sFasL; 5 ng/ml, Alexis Corporation) with enhancer (1 μg/ml, Alexis Corporation), the monoclonal anti-Fas antibody alone [at 1:500 dilution in media, Transduction Laboratories (the antibody was specific to an 18.2 kDa peptide corresponding to amino acids 1–163 of the extracellular domain)], or either sFasL or the anti-Fas antibody together with one of three ICE-like protease/caspase inhibitors [ICE inhibitor I (acetyl-YVAD-aldehyde), ICE inhibitor II (acetyl-YVAD-chloromethylketone), or ICE inhibitor III (acetyl-YVAD-acyloxymethylketone)], all at 100 μg/ml of medium, Calbiochem). Control wells were exposed to either culture medium (89% DMEM, 10% gelding serum, and 1% penicillin–streptomycin) alone or culture medium containing one of three ICE inhibitors (100 μg/ml of medium). Inhibitors were chosen for their ability to nonspecifically block the caspase-mediated cell death pathway downstream of Fas activation in immune tissue (Enari et al., 1996). Cultures were maintained at 37°C for 48 hr after treatment and then fixed and stained with hemotoxillin and eosin.
5-Bromodeoxyuridine labeling assay. To determine whether loss of E15 cortical neuroblast cultures after Fas activation was, in part, a result of changes in cell proliferation, the incorporation of a DNA replication marker, 5-bromodeoxyuridine (BrdU), was determined by a cell proliferation ELISA assay (Boehringer Mannheim). Cultures were established and exposed to Fas activators on E15 according to the protocol described above. Cultures were fixed 8.5 hr after administration of a 10 μm BrdU pulse and assayed for BrdU incorporation according to the kit manufacturer’s instructions (Boehringer Mannheim). Briefly, cells were exposed to 20 μl of fixative for 30 min at room temperature and then incubated with 100 μl anti-BrdU conjugated to peroxidase for 120 min. Culture wells were subsequently washed and exposed to color substrate solution (tetramethylbenzidine). Spectrophotometric analysis was performed using a standard microtiter plate reader (ELX808; Bio-Tek Instruments) with absorbance measured at 340 nm with a reference wavelength at 490 nm. Furthermore, to determine whether BrdU incorporation is caused by scheduled versus unscheduled DNA synthesis (DNA repair), we examined the Fas-related expression of the cell cycle protein PCNA using Western immunoblot analysis (see above) and the proportion of cells in cell cycle using fluorescence-assisted cell sorting (FACS) analysis (see below).
FACS analysis of cell cycle. Flow cytometric analysis was used to measure DNA content and the proportion of neuroblasts in cell cycle by the fluorescence intensity of propidium iodide intercalation. Dissociated E15 cortical neuroblasts were cultured on laminin substrate and administered control medium or sFasL (5 ng/ml, Alexis Corporation) for 8 hr. The cultures were exposed to 0.5% trypsin and 6.84 mm EDTA (Sigma) to bring the cells into solution. The cells were washed twice with cold PBS and centrifuged at 350 ×g, before the pellet was resuspended in ice-cold 70% ethanol and fixed overnight at 4°C. The cells were stained in PBS, 0.1% Triton X-100 (Sigma), 0.5 mmol/l EDTA, 0.05 mg/ml RNase A (Life Technologies), and 50 μg/ml propidium iodide (Boehringer Mannheim) at 4°C for 4 hr. DNA content was analyzed from 104cells using an FACS flow cytometer (Becton Dickinson) with excitation at 488 nm (argon laser) and detection at 620–700 nm. To determine whether there was a decrease in cell cycle, the number of cells with greater than diploid DNA content (G2 + S) was ratioed to the number of cells with normal diploid DNA content (G0 + G1).
Fas activation of the p65/RelA subunit of NF-κB. To determine the role of Fas in NF-κB activation, cultures of cortical neuroblasts were plated on glass laminin-coated slides compartmentalized with a silicon grid (Flexiperm; Harheaus Instruments). Experimental groups were exposed to sFasL (5 ng/ml) with enhancer (1 μg/ml) or sFasL with ICE inhibitor III (at 100 μg/ml of medium). Control wells were exposed to culture medium alone. Cultures were maintained at 37°C for either 1, 2, 4, 8, or 24 hr, then fixed. NF-κB localization was determined by immunofluorescence (as described above) and nuclei were counterstained with Hoechst dye #33342.
Data analysis
The density of apoptotic and Fas mRNA-positive cells were determined using a standard image analysis package (Bioquant). The density of TUNEL-positive cells in periseptal isocortex were averaged over counts of 10 random fields (200 × 260 μm) to generate a single sample value for each animal. For dissociated cultures, the total pixel area occupied by cell colonies was counted over a 1.6 × 2.6 mm area. Densitometric analysis of Western immunoblots was performed using standard analytical software (Molecular Analyst; Bio-Rad). After FACS analysis, the ratio of cells in cell cycle was calculated by the formula (G2 + S)/(G0 + G1). Statistical differences were calculated using ANOVAs followed by post hoc tests (Student-Newman-Keuls, p < 0.05).
RESULTS
Expression of Fas mRNA in the telencephalon
Fas mRNA expression was detected by nonisotopic in situhybridization in embryonic and postnatal rat forebrain (Fig.1) and confirmed by RT-PCR (Fig.2A). The cerebral cortex and hippocampus (data not shown) were the principle sites of Fas mRNA expression.
Fig. 1.
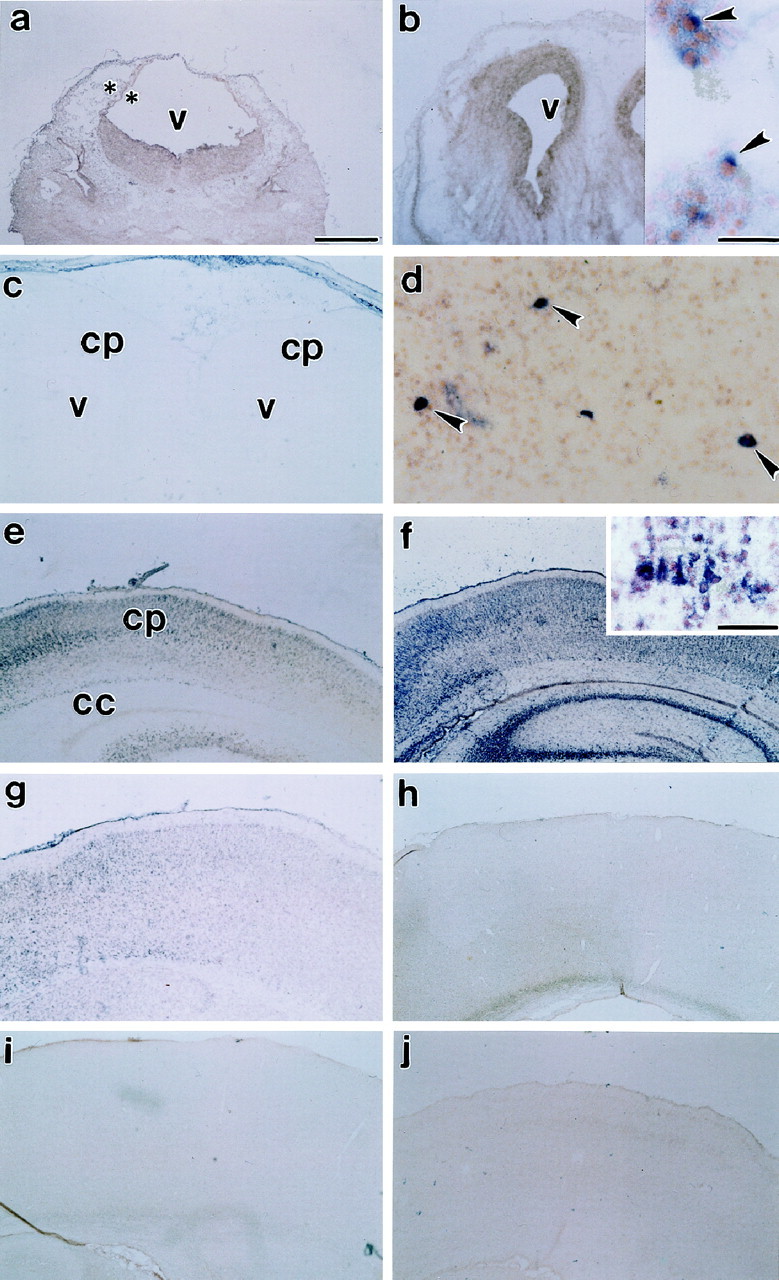
Age-related alterations in cortical Fas mRNA expression. Fas mRNA expression was not observed at E13 (a, asterisks indicate boundaries of cortical neuroepithelium). Expression was observed in a few cells at E15 (b, inset,arrowheads), then drops to virtually undetectable levels in cingulate and isocortex just before birth, E19 (c). Fas mRNA is re-expressed postnatally. Expression is observed in only a few cells on P1 (d,arrows). Expression increases from P3 (e) to the end of the first postnatal week, P6 (f, inset shows higher magnification of hybridization to cortical cells), and declines thereafter from P9 (g) to P11 (h) to undetectable levels by P22 (i). Sense controls [P6 cortex (j)] showed no hybridization. v, Ventricle; cp, cortical plate; cc, corpus callosum. Scale bars: a–c,e–j, 570 μm; b,inset, d, 30 μm; f,inset, 60 μm.
Fig. 2.

A, Expression of Fas mRNA in the developing cerebral cortex (P6) was confirmed by RT-PCR. Size-fractionation of the product revealed a single band corresponding to the expected cDNA length (450 bp). RT-PCR of cyclophillin mRNA and RNA amplified with the Fas primer in the absence of reverse transcriptase were included as controls. B, The expression of Fas-like immunoreactivity was confirmed by Western analysis of 100 μg of detergent-soluble protein obtained from P6 cerebral cortex. A protein band at 45 kDa was Fas-immunoreactive, corresponding to the expected size for Fas. Size fractionation of 20 μg of total protein obtained from jurkat cells (a positive control) revealed a band of similar size.
Fas mRNA was not observed at E13 (Fig. 1a). Fas mRNA was initially expressed in the developing cerebral cortex at E15 (Fig.1b, inset). There were a few mRNA-expressing cells in the differentiating cortical primordial plexiform and neuroepithelial layers. At E19, Fas mRNA-expressing cells were observed within the basal forebrain and piriform cortex but not in isocortical or other allocortical regions or in the ventricular zone (Fig.1c). At P1 and P3, a few cells within the cortical plate (Fig. 1d,e), subplate, and ventricular zone (data not shown) expressed Fas mRNA. Expression was also observed in cells within the interhemispheric fissure, at the base of the corpus callosum (data not shown). Expression in these areas increased during the first postnatal week and peaked by P6 (Fig. 1f), declining thereafter. At P6, there was increased expression in multiple regions of the cerebral cortex, including isocortex (Fig.1f), piriform, and cingulate cortices, as well as the residual ventricular epithelium. A few cells among the afferent and efferent cortical pathways also expressed Fas mRNA. Most notable among these were the morphologically distinguishable magnocellular neurons in the corpus callosum. By P9 (Fig. 1g), there was a decline in Fas mRNA expression until, by P11–P22, expression (Fig. 1h,i) was similar to the sense controls (Fig.1j).
A few Fas mRNA-expressing cells were observed in other regions of the developing forebrain as well. For example, Fas mRNA was expressed in the postnatal hippocampus. At P4, Fas mRNA was expressed in all of the hippocampus. By P6, Fas mRNA was expressed in CA 2 and 3 but no expression was observed in either CA1 or the dentate gyrus of the hippocampal formation. Toward the end of the first postnatal week, Fas mRNA expression was also observed in other telencephalic structures such as the amygdaloid complex and the basal forebrain. Some expression was also observed in the thalamus and the hypothalamus (the arcuate nucleus, ventromedial hypothalamus, and lateral hypothalamus). However, at the peak period of Fas mRNA expression, the greatest expression was observed in the cerebral cortex and hippocampus.
Developmental expression of Fas immunoreactivity
An immunoreactive protein band at ∼45 kDa, corresponding to the expected size for Fas (Oehm et al., 1992) (Fig. 2B) was observed in detergent-soluble protein from P6 cortex as well as protein isolated from Jurkat cells. Jurkat cells were used as a positive control because they have been previously shown to express Fas (Marimoto et al., 1994; Weis et al., 1995). At E11.5 (data not shown) and E13.5 (Fig. 3a), no Fas immunoreactivity was observed in the developing brain. By E15, however, Fas-immunoreactive cells were observed in the developing cerebral isocortex (Fig. 3b–d, arrowheads). Some Fas-like immunoreactivity was also observed in the diencephalon, spinal cord, and dorsal root ganglia. Between E18 and E20, no Fas-like immunoreactivity was observed in isocortical areas (Fig.3e). However, Fas-like immunoreactivity was observed within the soma of cells in the basal forebrain, the piriform cortex, and neuroepithelium (ventricular zone, data not shown).
Fig. 3.

Immunohistochemical analysis of Fas expression in the developing cerebral cortex. Fas immunoreactivity was undetectable at E13 (a, asterisks indicate boundaries of the cortical neuroepithelium). Expression was observed in a few cells at E15 (b–d,arrowheads) then drops to undetectable levels in cingulate and isocortex by E19 (e). Fas immunoreactivity is re-expressed postnatally. Expression is observed in only a few cells on P1 (f, g,arrowheads) and increases to the end of the first postnatal week, P6 (h) then declines thereafter from P8 (i) to P11 (j,arrowheads indicate staining in the corpus callosum) to undetectable levels by P18 (k) and P22 (l). Control sections lacking the antibody showed no staining: P6 (m). v, Ventricle. Scale bars: a, b,e, f, i–m, 114 μm; c, d, g, 30 μm; e, 300 μm; h, 60 μm.
At P1, Fas immunoreactivity was re-expressed within the perikaryon of a few immature cells in the isocortex (Fig. 3f,g,arrowheads) as well as within the soma of cingulate cortical cells and ventricular zone neuroblasts. Between P1 and P6, fewer perikaryon expressed Fas immunoreactivity. Instead, immunoreactive product was increasingly limited to intracortical fibers [Figs.3h, 4A, arrowheads show localization primarily to apical dendrites of cortical pyramidal neurons (Fas immunofluorescence at P6)] and to fibers of passage within the corpus callosum and anterior commisure (data not shown). Fas-like immunoreactivity was initially observed only in the most medial portion of the corpus callosum. Immunoreactivity decreased during the second postnatal week (Fig.3i,j, arrowheads) and was undetectable by P18 (Fig. 3k,l). From P18 (Fig. 3k,l) onward, staining was comparable to preimmune serum controls (Fig. 3m). At the peak period of Fas expression, the greatest expression was observed in the cerebral cortex, in corticofugal pathways, and in structures that form part of the hippocampal formation, the alveus, and fornix (data not shown).
Fig. 4.
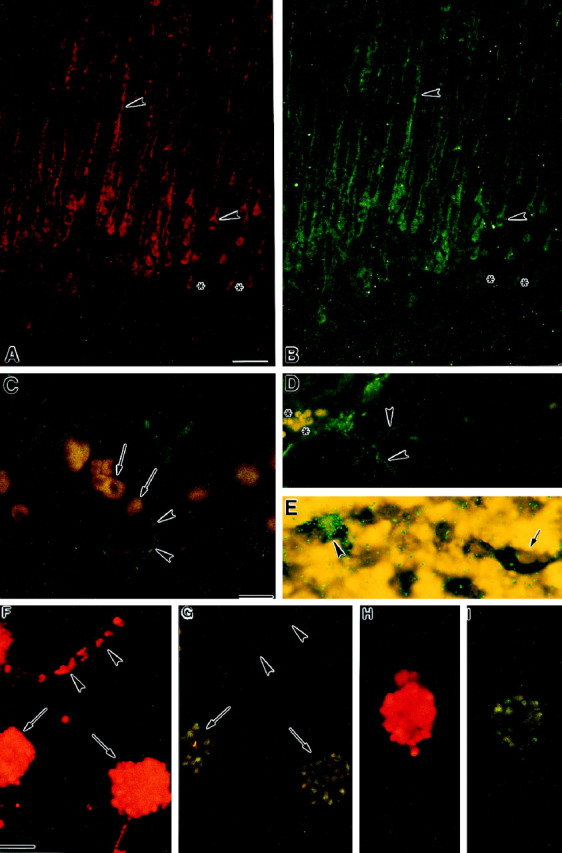
A, B, Colocalization of Fas-like immunofluorescence (A, rhodamine) to (B, FITC) immunofluorescence for a cocktail of neuronal markers (MAP2, NF150, NF200) in P6 cerebral cortex suggests that Fas is localized to neurons. Arrowheads indicate overlapping immunofluorescence for Fas and neuronal markers in the cell soma and fibers. Asterisks indicate cells that express Fas but do not show immunofluorescence for the neuronal markers. C, Combined immunohistochemistry for Fas [(FITC) greenimmunofluorescence] and FasL [(rhodamine) orangeimmunofluorescence] in P2 superficial cortical plate. Fas-immunoreactive fibers (arrowheads) are observed in the proximity of FasL-expressing cells (arrows).D, Immunohistochemical localization of RIP [(FITC)green immunofluorescence] in P6 cortical plate. RIP-immunoreactive fibers (arrowheads) are observed in the superficial cortex. Asterisks correspond to nonspecific fluorescence of red blood cells. E, Combined isotopic and nonisotopic in situ hybridization for RIP and Fas mRNA, respectively, in P6 cortical plate. RIP mRNA (green grains) is expressed in a subset of Fas mRNA-expressing cells (blue reaction product).Arrowhead indicates cell coexpressing Fas and RIP mRNA, whereas arrow indicates cell expressing only Fas mRNA.F, G, Combined immunofluorescence for Fas (F, rhodamine) and FADD (G, FITC) in embryonic dissociated cortical neuroblast cultures.Arrows indicate subsets of Fas-expressing cells in colonies that also express FADD. Arrowheads indicate that FADD is not expressed by individual, isolated Fas-expressing cells. H, I, Colocalization of Fas-like immunofluorescence (H, rhodamine) to immunofluorescence for FLIP (I, FITC) in a subset of Fas-expressing E15 cortical neuroblasts. Scale bars: A, B,F–I, 29 μm;C–E, 30 μm.
Fas is expressed in developing neurons
We examined whether Fas expression in the developing cerebral cortex was localized to neurons. Combined immunohistochemistry for Fas and a cocktail of neuronal markers indicated that Fas was expressed in a subset of neurons (Fig.4A,B; see arrowheads vs asterisks) of the developing cerebral cortex. Fas expression appeared particularly strong in the apical dendrites of these neurons and is comparable to the pattern of immunostaining at P6 depicted in Figure 3h.
Localization of Fas-associated signaling components, FasL, RIP, FADD, and FLIP in relationship to Fas expression
Fas activation by its cognate ligand, FasL, in the immune system leads, in turn, to the activation of several Fas-associated proteins, including FADD and RIP. To determine whether the developing brain expresses similar signaling mechanisms, the expression of FasL, FADD, RIP, and the inhibitor FLIP, was examined in the cortical plate. Western immunoblot analysis for FasL indicated the presence of an immunoreactive band at 37 kDa in P6 cortex and in Jurkat cells (a positive control, see Fig. 6D). Combined immunohistochemistry of Fas and FasL confirmed that the ligand is localized to regions of Fas expression. In the developing ventricular zone, in particular, FasL was expressed in cells neighboring those expressing Fas (Fig.5A,B). Within the cortical plate, Fas-immunoreactive fibers were present proximate to FasL-immunoreactive cells (Fig. 4C).
Fig. 5.
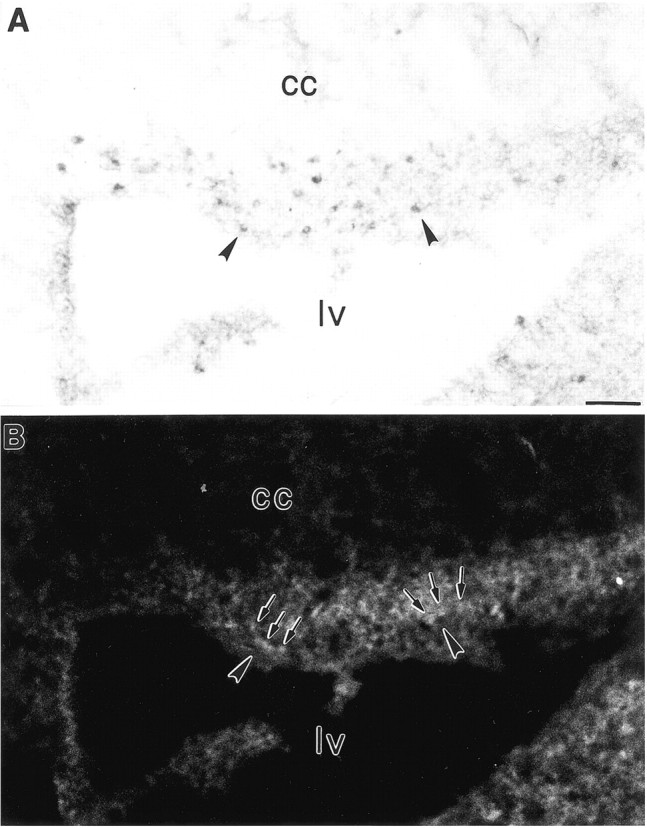
A, B, Combined immunohistochemistry for Fas (A) and immunofluorescence for FasL (B) in the ventricular zone of the developing cerebral cortex indicate that FasL is expressed in cells (arrows) neighboring Fas-expressing cells (arrowheads). lv, Lateral ventricle; cc, corpus callosum. Scale bars, 60 μm.
Combined isotopic and nonisotopic in situ hybridization for RIP and Fas, respectively, at P6 indicates that RIP mRNA (green grains) are expressed in a subset of Fas mRNA-expressing cells (blue--black color product) in the developing cerebral cortex (Fig. 4E). Immunohistochemical analysis indicates that RIP localizes to neurites in the cortical plate (Fig. 4C). Western blot analysis of detergent-soluble protein obtained from P6 cortex indicated immunoreactivity for a protein band at ∼74 kDa, corresponding to the expected size for RIP (Stanger et al., 1995) (Fig.6A). A similar sized band was observed in total protein isolated from Jurkat cells, a positive control.
In contrast, we were unable to detect the expression of FADD in the postnatal rat cortex by Western immunoblot analysis, although a band corresponding to the expected size (24 kDa) was expressed in the positive control, Jurkat cell lysate (Fig.6B). Similarly, FADD protein and mRNA were not detected by immunohistochemistry and in situhybridization, respectively, within the developing cerebral cortex (data not shown).
Western immunoblot analysis indicated that FLIP, the negative regulator of Fas activation was also expressed by the postnatal rat brain. A band just below the 66 kDa molecular weight marker corresponding to the expected size of ∼60 kDa was detected in soluble protein obtained from P6 cortex and corresponded to a similar-sized band in a protein sample obtained from COS7 cells transfected with the FLIP plasmid in an expression vector. The lower molecular weight band in transfected COS7 cells (Fig. 6D) may represent a cleavage product (Irmler et al., 1997).
We also examined the expression of Fas-associated proteins in cultured E15 cortical neuroblasts. RIP (data not shown), FADD, and FLIP were expressed in E15 cortical neuroblasts cultures. Subsets of Fas-expressing cells also expressed FADD and FLIP (Fig.4F–I). Furthermore, FADD localized to Fas-expressing cells that were clustered together in colonies (Fig.4F,G, arrows) but not to individual, isolated, Fas-expressing cells (Fig. 4F,G,arrowheads).
Apoptosis in the developing cerebral cortex
Because Fas activation induces apoptosis in non-neural tissues, we examined the temporal regulation of apoptosis in the developing cerebral cortex. Apoptotic profiles were visualized by end-labeling of nucleosomal fragments (Fig.7a,b,f). There was a significant, age-related decline in the density of apoptotic profiles from the first to the second postnatal weeks (Fig.7c). Apoptotic profiles were observed throughout all laminae of the cortical plate as well as within the cortical subplate and ventricular zone (regions that also expressed Fas mRNA and immunoreactivity). The highest density of apoptotic cells was observed in the cingulate cortex.
Fig. 7.

Apoptotic profiles were detected in the developing cerebral cortex by TUNEL labeling of free 3′ ends of fragmented nuclear DNA. There was a significant age-related decline in apoptotic particles (c) from P4 (a) to P15 (b). This pattern overlaps closely the temporal expression of Fas mRNA and protein (Figs. 1-3). The spatial expression pattern of Fas mRNA (d) and Fas-like immunoreactivity (e) overlaps the expression of apoptotic profiles (f) in the P1 ventricular zone. Fas mRNA and protein are both localized to the cytoplasm of neuroblasts (d and e,arrowheads, respectively), whereas labeling of fragmented DNA is localized to the nuclei (f,arrowhead). mz, Marginal zone;cp, cortical plate. Scale bars: a, 60 μm; b, 120 μm; c–e, 12 μm.
Fas activation induces neural cell death by a caspase-dependent pathway, and downregulates the expression of the Fas receptor
We examined whether Fas activation leads to the death of dissociated embryonic cortical neuroblasts in primary cultures. Dissociated embryonic cortical neuroblasts (Fig.8A, left panel) proliferate in vitro to form neuroblast colonies (Fig. 8A, right panel) and ubiquitously express Fas immunoreactivity (Fig.8B, left panel; Fig.4F,H) as compared with preimmune serum controls (Fig. 8B, right panel). The addition of sFasL (Fig.8C1,C2) to these cortical cultures led to a significant (p < 0.05) 22.6-fold decrease in the density of these neuroblast colonies. Similarly, the addition of an activating anti-Fas antibody (Fig.8D1,D2) led to a significant (p < 0.05) 4.3-fold decrease in neuroblast density. Fas activation by its ligand was tested in dissociated cell cultures to decrease cell–cell interactions and to minimize the actions of endogenous FasL. Concurrent addition of caspase inhibitors ICEI, ICEII, or ICEIII, significantly attenuated (p < 0.05) sFasL-induced cell loss (Fig.8C1,C2). However, only ICEIIIsignificantly attenuated (p < 0.05) anti-Fas antibody-induced cell loss (Fig.8D1,D2).
Fig. 8.

A, Phase-contrast photomicrograph of living dissociated E15 cortical neuroblasts (left panel) indicate that these cells proliferate to form neuroblast colonies in vitro within 24 hr (right panel). B, Extensive Fas immunoreactivity was observed in dissociated E15 cortical neuroblasts (left panel). Preimmune serum immunohistochemical controls showed no staining (right panel).C1, D1, Low magnification photomicrographs of fixed neuroblast colonies stained with hemotoxylin and eosin indicate that cultures administered either sFasL or an anti-Fas antibody exhibit significant loss of cell colonies, relative to controls, whereas the caspase inhibitor ICEIIIattenuates Fas-induced cell loss. C2, D2, Graphs represent quantification of changes in cell density after Fas activation. Caspase inhibitors I (ICEI), II (ICEII), or III (ICEIII) attenuated sFasL-induced cell loss (C2). However, only ICEIII attenuated anti-Fas antibody-induced cell loss (D2). Asterisks (C2,D2) indicate that cell loss in the respective treatment group was significantly (p < 0.05) different from controls (n = 5). E, Hoechst-stained (dye #33258) neuroblasts administered either sFasL or anti-Fas antibody exhibited nuclear condensation. In contrast, control cells appeared normal. Scale bars: A, 5 μm;B, 60 μm; C1, D1, 190 μm; E, 15 μm.
In a second set of experiments, cortical neuroblasts were stained with the Hoechst dye #33258 (a nuclear stain specific for double stranded DNA). After Fas activation by FasL, nuclei of cells became condensed as compared with control cultures (Fig. 8E), suggesting that Fas induces apoptotic cell death.
Western immunoblot analysis indicates that exposure of E15 cortical neuroblast cultures to FasL leads to a significant (p < 0.05) decrease in the ratio of Fas to total soluble protein within 8 hr, suggesting that Fas activation leads to a loss of the Fas receptor (Fig. 9). Fas-mediated receptor downregulation was partially attenuated by concurrent exposure to the caspase inhibitor ICEIII, because the expression of Fas after FasL + ICEIII treatment was not different from controls. ICEIII by itself had no significant effect on Fas expression.
Fig. 9.

Western immunoblot blot analysis of 50 μg of soluble protein from E15 cortical neuroblasts showed that Fas activation by soluble FasL leads to reduction of Fas protein (lane 2; p < 0.05) as compared with controls. In contrast, no reduction was seen in cultures administered sFasL + ICEIII (caspase inhibitor) or ICEIIIalone (lanes 3, 4). The top panel represents a sample from a single immunoblot, whereas thebottom panel represents densitometric analysis of the data. Error bars indicate SE for each treatment (n= 4).
Fas activation does not inhibit cell cycle activity
To determine whether Fas activation decreases cell proliferation rather than inducing cell death alone, we examined the effect of Fas activation on the incorporation of BrdU into nuclear DNA. We hypothesized that Fas may decrease BrdU incorporation. However, 8.5 hr after sFasL or anti-Fas antibody addition, there was a significant (p < 0.05) increase in BrdU incorporation into dissociated primary cultures of cortical neuroblasts (Fig.10). BrdU incorporation was attenuated by concurrent exposure to ICEIII. However, ICEIII treatment by itself had no effect on BrdU incorporation.
Fig. 10.

BrdU incorporation in E15 cortical neuroblasts, assayed at 8.5 hr, was significantly higher (p < 0.05) after Fas activation with anti-Fas antibody and soluble FasL than controls. The administration of a caspase inhibitor (ICEIII) alone or in conjunction with Fas activators lead to BrdU incorporation similar to controls. Error bars indicate SE for each treatment (n = 5).
To determine whether BrdU incorporation was indicative of DNA synthesis, we examined the Fas-mediated regulation of PCNA, a cofactor for DNA polymerase δ (Celis and Celis, 1985; Kurki et al., 1986) that shows increased expression at the G1 to S phase transition in the cell cycle. sFasL or anti-Fas antibody did not lead to a significant change in PCNA (Fig.11A1,A2) at 8.5 hr, suggesting that the BrdU incorporation was not caused by DNA replication. Furthermore, we examined the cell cycle-specific uptake of the intercalative DNA-binding dye propidium iodide. Using FACS analysis, we found that sFasL treatment for 8 hr did not lead to an increase in the proportion of cells in cell cycle [hyper-diploid cells (G2 + S) to diploid cells (G0 + G1)] relative to controls (Fig.11B1,B2).
Fig. 11.
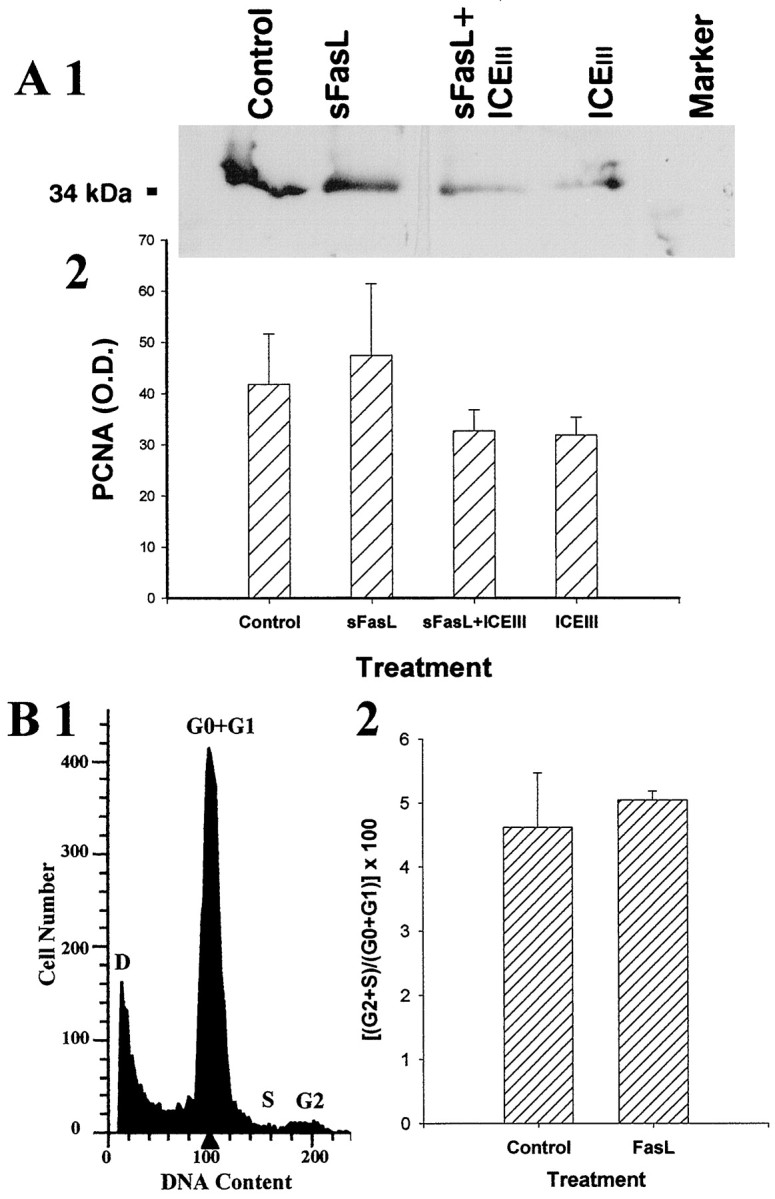
A, Western analysis for PCNA in E15 cortical neuroblasts administered either soluble FasL alone (lane 2), FasL with a caspase inhibitor (ICEIII), or ICEIII alone (lanes 3, 4) indicated that there is no change in PCNA expression associated with Fas activation. The top panel (A1) represents a sample immunoblot, whereas the bottom panel (A2) represents densitometric analysis of the data. Error bars indicate SE for each treatment (n = 5). B, FACS analysis in E15 cortical neuroblasts administered soluble FasL confirm that there is no change in cell cycle associated with Fas activation. Theleft panel shows a sample FACS analysis (B1) containing cell cycle peaks G0/G1, S, and G2/M.D represents debris. The right panel(B2) shows that administration of FasL did not increase the ratio of cells in cell cycle (G2 + S) to diploid (G0 + G1) cell number (n = 8).
Fas activation induces nuclear localization of NF-κB
The Fas-associated protein RIP has been implicated in the activation of the nuclear transcription factor NF-κB (Hsu et al., 1996). Furthermore, Fas can activate the transcription factor NF-κB in fibroblasts (Rensing-Ehl et al., 1995). Control and FasL or FasL + ICEIII-treated cultures were immunostained for the RelA/p65 subunit of NF-κB (Fig. 12, arrowheads) and counterstained with a nuclear dye (Hoechst dye #33342; Fig.12, arrows). In control cultures, immunoreactivity for the RelA/p65 subunit of NF-κB was associated primarily with the cytoplasm and proximal processes (Fig.12a,b). Virtually no immunofluorescence was observed in the nucleus. Both of the Fas activators, sFasL (Fig. 12c,d) or anti-Fas antibody (data not shown), led to a nuclear localization of immunofluorescence. Furthermore, within 2 hr after Fas activation, immunoreactive cells appeared to take on a condensed and irregular morphology relative to controls. Counterstaining with the Hoechst dye #33342 indicated that even in cells with fragmented nuclei, the highest density of NF-κB immunoreactivity was associated with nuclear fragments (Fig. 12d1,d2, inset,arrowheads). Concurrent administration of the caspase inhibitor ICEIII (Fig.12e,f) prevented the condensation of nuclei but did not prevent the localization of RelA to the nucleus since the highest fluorescence intensity continued to be localized over the nucleus.
Fig. 12.

Dissociated embryonic cortical cells were administered sFasL. Cultures were processed for immunofluorescence analysis of p65/RelA (an NF-κB subunit) expression (a1, b1, c1,d1, e1,f1) and counterstained with Hoechst dye #33342 to visualize nuclear DNA (a2, b2,c2, d2, e2,f2). NF-κB in control cultures was limited to the cytoplasm (a1, b1,arrowheads). Counterstained nuclei were intact (a2, b2, arrows). Within 2 hr after treatment with sFasL, immunofluorescence for p65/RelA was observed throughout the cell, which now appeared condensed and irregular (c1, arrowhead;d1). Furthermore, nuclei of activated cells exhibited a condensed and irregular morphology characteristic of apoptosis (c2, d2, arrows). In panels d1 and d2, a single apoptotic cell (arrows) is magnified (5×) as an inset. The inset in d1 shows a condensed cell with a dense “V”-shaped NF-κB immunofluorescence at the lateral margins of the cell (arrowheads). Theinset in d2 (arrowhead) shows that the dense NF-κB immunofluorescence overlies two condensed nuclear profiles within that cell. ICEIII partially attenuated nuclear condensation but not nuclear localization of p65/RelA (e1, f1,arrowheads). Scale bar, 29 μm.
DISCUSSION
Substantial cell suicide occurs during cortical development (Ferrer et al., 1990, 1992; Blaschke et al., 1996). Previous reports indicated that Fas expression in neural tissue is associated primarily with tumorogenesis and with adult neurodegenerative diseases. Fas-mediated apoptosis in the nervous system may reflect neural cell injury or invasion of immune cells into the brain. In contrast, the data from the current study indicate that Fas/Apo-1 is transiently expressed by neurons and is a candidate cell suicide regulator in the normal, developing brain.
Within the telencephalon, Fas mRNA and protein were primarily expressed in developing cerebral cortical and hippocampal neurons. Fas is also expressed in transient neural structures, including the ventricular epithelium, subplate, and marginal zone (Luskin and Shatz, 1985;Al-Ghoul and Miller, 1989; Shatz et al., 1990). Laminar and cellular compartmentalization of Fas expression coincides with expression of cell death signal transduction elements, including caspase-3 and JNK (Krajewski et al., 1997; Migheli et al., 1997; Chen et al., 1998). FasL was also expressed in ventricular epithelium and cortical plate. However, expression of Fas and FasL in vivo may be permissive but not sufficient for cell suicide induction. Contact between Fas and FasL-expressing cells is requisite for cell suicide induction. Availability of key Fas-associated proteins or the presence of inhibitors may also limit Fas-induced suicide. We were unable to localize the caspase-8 adapter, FADD, to the postnatal, developing brain. In contrast, others have observed prenatal FADD expression in the developing mouse brain (Yeh et al., 1998), and we observed FADD in embryonic cortical cultures, suggesting that there may be temporal regulation of FADD expression. Furthermore, other Fas-associated adapter proteins, including CRADD/RAIDD (Ahmad et al., 1997; Duan and Dixit, 1997) and DAXX (Xiaolu et al., 1997), may substitute for FADD in the neonatal cerebral cortex and signal through either redundant (caspase-2) or alternate (JNK) pathways. CRADD/RAIDD may heterodimerize with caspase-2 and RIP (Ahmad et al., 1997) to transduce Fas-death signals. Although RIP was expressed in the postnatal brain, RIP immunoreactivity was observed in a few fibers, and RIP mRNA colocalized to only a subset of Fas mRNA-expressing cells. These observations do not eliminate possible Fas associations with other unique signaling mechanisms (including nonsuicide mechanisms) in the brain. Finally, inhibitors of Fas activation such as FLIP have recently been described (Irmler et al., 1997; Srinivasula et al., 1997). Our data indicate that FLIP is expressed in the developing postnatal cerebral cortex and in subsets of cultured embryonic Fas-positive cells. Such negative regulators could have important, as yet undetermined, developmental roles in shaping cellular specificity of Fas activation and suggest that not all Fas-positive cells may undergo cell suicide normally, during cortical development.
Overall, the spatiotemporal expression of Fas patterns correlates well with apoptosis. Our data indicate distinct prenatal and postnatal peaks of Fas expression in the cerebral cortex. An embryonic peak, at E15, corresponds to the tail of cortical preplate genesis (Bayer and Altman, 1991). Postnatal expression corresponds to the peak apoptotic period in the developing cerebral cortex, suggesting that Fas may be a candidate regulator of developmentally specific cell suicide. Although apoptotic nuclei were observed in all Fas-expressing regions of the cerebral cortex, we were unable to colocalize Fas immunoreactivity to apoptotic cells. One possible explanation is that DNA fragmentation may be accompanied by a loss of cellular proteins and cell surface receptors including Fas. The observed reduction of Fas expression in cortical neuroblasts after activation supports this hypothesis.
Induction of Fas-mediated neural cell death
Our data indicate that soluble FasL and activating anti-Fas antibodies induced death in embryonic cerebral cortical progenitors. Concurrent administration of nonselective caspase inhibitors, particularly ICEIII, attenuated Fas-mediated loss of neuroblasts. These data are consistent with the notion that Fas induces cell death by activation of caspase mechanisms (Nagata, 1997). Only ICEIII attenuated anti-Fas antibody-induced cell loss, whereas all inhibitors attenuated sFasL-induced cell loss. This differential effect of ICE inhibitors may be related to their relative efficacies and bioavailabilities. ICEI (an aldehyde-linked peptide) is a reversible inhibitor, whereas ICEIII (an acyloxymethylketone derivative) is more stable (Thornberry et al., 1992; Walker et al., 1994) and irreversibly inhibits caspases (Thornberry et al., 1994). The anti-Fas antibody, although less efficacious than sFasL, may have exhibited a prolonged bioactivity, attenuatable only by an irreversible and more stable ICE inhibitor (ICEIII), although all inhibitors attenuated the actions of the soluble ligand.
Although normal embryonic cerebral cortex has low levels of Fas expression, dissociated E15 cortical cultures expressed Fas and RIP ubiquitously. Furthermore, subsets of Fas-positive cultured cortical neuroblasts also expressed FADD and FLIP. This suggests that disruption of local developmental environments may induce suicide receptor mechanisms. Such context-dependent expression is consistent with data from other laboratories, indicating that Fas is expressed in neural tumors (Tachibana et al., 1995; Weller et al., 1998) that have presumably escaped their normal developmental controls.
FADD expression was limited to neuroblast colonies, whereas Fas was expressed in both single, isolated cells and neuroblast colonies. Specific localization of FADD to neuroblast colonies that presumably were the product of cell proliferation suggests that Fas-associated caspase-8 activation may play some role in limiting cell proliferation, similar to that described in the immune system. However, Fas-induced cell loss is not caused by reduction in cell proliferation per se. To the contrary, activated cells exhibit caspase-dependent incorporation of BrdU (a DNA replication marker) before death. However, Fas activation does not lead to a simultaneous change in expression of the G1-S phase marker PCNA or the proportion of cells in G2 and S phase relative to G0 and G1. This suggests that BrdU incorporation is not related to DNA replication but rather to other mechanisms such as DNA repair. BrdU incorporation has also been associated with viral induction of suicide (Belyavskyi et al., 1998) and suggests that DNA repair mechanisms may be initiated by neural cells after receipt of death signals.
Although we and others have shown that Fas can be activated in normal (our data) and tumorigenic (Weller et al., 1998) tissues of neural origin, Fas (lpr) and FasL (gld) mutations have not been reported to express neural phenotypes. However, recent findings indicate that caspase-3-deficient mice have ectopic cerebral cortical cell masses, suggesting that neural viability is increased by blocking downstream elements of the Fas pathway (Kuida et al., 1996). Several factors may explain the apparent lack of neural phenotype inlpr and gld animals. First, to our knowledge, no published reports have included a thorough analysis of neural tissue from these mutations. Second, lpr appears to be a leaky mutation (Nagata, 1996), suggesting an incomplete block of Fas function or an effect on only a subset of signal transduction pathways induced by Fas activation. Furthermore, some FasL mRNA expression has been observed in gld mice (Chu et al., 1995), while activated T-cells from lpr mice can reportedly induce Fas-mediated target cell death (Ramsdell et al., 1994). There may also be limited redundancy in cell suicide control mechanisms. Thus, TNFα induces T-cell death (Zheng et al., 1995), similar to Fas, and peripheral T-cell deletion does occur in lpr and gld mice (Zheng et al., 1995), suggesting that other suicide receptors may assume Fas-related functions. Finally, there may also be tissue-selective responses to deletion of Fas family members. For example, deletion of p75NTR gene (another member of the TNFr/Fas family) leads to increased survival of forebrain cholinergic neurons (Van der Zee et al., 1996). However, such enhanced survival has not been observed in other p75NTR-expressing regions of the developing nervous system. Thus, lpr and gld mice may retain cell suicide activity because of limited Fas functionality or through functionally similar receptors.
Fas-mediated signal transduction appears not to be limited to caspase cascades. For example, in fibroblasts, leukemic T-lymphocytes and epithelioid carcinomas, Fas activation also leads to induction of NF-κB (Rensing-Ehl et al., 1995; Ponton et al., 1996). RIP appears to mediate activation of NF-κB (Hsu et al., 1996). Our results indicate that Fas activation can also lead to nuclear translocation of the p65 subunit of NF-κB in neuroblasts. However, in contrast to the Fas-mediated protease cascade, NF-κB has been implicated in cell death prevention. For example, NF-κB inhibition enhances sensitivity to TNF-induced death (Beg and Baltimore, 1996; Liu et al., 1996; Van Antwerp et al., 1996; Wang et al., 1996). Recent evidence also indicates that NF-κB activation prevents apoptosis in neurons after both oxidative stress (Mattson et al., 1997) and exposure to mutated presenilin-1 (Guo et al., 1998). Our preliminary data also suggest that embryonic cortical neuroblasts die after inhibition of NF-κB activation (Z. Cheema and R. Miranda, unpublished observations). Thus, responses of neural cells to Fas activation may be complex, involving initiation of opposing cell death and survival mechanisms. The temporal predominance of any given mechanism may ultimately determine cell fate.
Western analysis indicates that Fas activation leads to rapid downregulation of the Fas receptor. The mechanism for Fas autoregulation is unclear. However, our data indicating that Fas activates nuclear translocation of NF-κB and the presence of NF-κB consensus elements in the Fas promoter (Behrmann et al., 1994) suggest that Fas may downregulate its own expression in part, viaNF-κB activation. Precedence for such a repressor function for NF-κB comes from observations that NF-κB downregulates glucocorticoid receptor-dependent transcription (Caldenhoven et al., 1995). Downregulation of Fas may serve to render cells refractory to additional death signals.
Immunohistochemical analysis indicates that FasL is expressed in cortical cells adjacent to those expressing Fas, and Fas-immunoreactive fibers appear to make contact with FasL-expressing cells. Data from other laboratories also indicate that FasL localizes to regions in the brain (French et al., 1996) that constitute target substrates for Fas-expressing neurons in the cerebral cortex. In contrast to immune cells, where FasL and Fas may be coexpressed, a lack of coexpression in the cerebral cortex may confer selective survival advantages to FasL-expressing cells. The proximity of Fas-immunoreactive fibers to FasL-expressing neural cells suggests that FasL-positive cells may kill Fas-positive cells that make contact. Cellular segregation of Fas and FasL suggests intercellular interactions may be important for cell suicide induction during cerebral cortical development.
Footnotes
This work was supported by grants from National Institutes of Health (MH55724) and Texas A & M University (FMG96–26) to R.C.M. We thank Scott Weiters, Li-Yu Huang, Chuck Kim, and Robert McAlhany for technical assistance and Drs. Lori Bernstein, Wei-Jung Chen, and Julian Leibowitz (Texas A & M University) for critical review of this manuscript.
Correspondence should be addressed to Rajesh C. Miranda, Department of Human Anatomy and Medical Neurobiology, Texas A & M Health Science Center, 228 Reynolds Medical Building, College Station, TX 77843.
REFERENCES
- 1.Ahmad M, Srinivasula SM, Wang L, Talanian RV, Litwack G, Fernandes-Alnemri T, Alnemri ES. CRADD, a novel human apoptotic adaptor molecule for caspase-2, and FasL/tumor necrosis factor receptor-interacting protein RIP. Cancer Res. 1997;57:615–619. [PubMed] [Google Scholar]
- 2.Al-Ghoul WM, Miller MW. Transient expression of Alz-50 immunoreactivity in developing rat neocortex: a marker for naturally occurring cell death? Brain Res. 1989;481:361–367. doi: 10.1016/0006-8993(89)90815-9. [DOI] [PubMed] [Google Scholar]
- 3.Arnold AP. Quantitative differences in hormone accumulation in the zebra finch brain: methodological and theoretical issues. J Comp Neurol. 1980;189:421–436. doi: 10.1002/cne.901890302. [DOI] [PubMed] [Google Scholar]
- 4.Baeuerle PA, Baltimore D. NF-kappaB: ten years after. Cell. 1997;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 5.Bayer SA, Altman J. Neocortical development. Raven; New York: 1991. [Google Scholar]
- 6.Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 7.Behrmann I, Walczak H, Krammer PH. Structure of the human Apo-1 gene. Eur J Immunol. 1994;24:3057–3062. doi: 10.1002/eji.1830241221. [DOI] [PubMed] [Google Scholar]
- 8.Belyavskyi M, Levy GA, Leibowitz JL. The pattern of induction of apoptosis during infection with MHV-3 correlates with strain variation in resistance and susceptibility to lethal hepatitis. In: Enjuanes L, Siddell S, Spaan W, editors. Coronaviruses and arteriviruses. Plenum; New York: 1998. pp. 619–625. [DOI] [PubMed] [Google Scholar]
- 9.Blaschke AJ, Staley K, Chun J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development. 1996;122:1165–1174. doi: 10.1242/dev.122.4.1165. [DOI] [PubMed] [Google Scholar]
- 10.Boldin MP, Varfolomeev EE, Pancer Z, Mett IL, Camonis JH, Wallach D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J Biol Chem. 1995;270:7795–7798. doi: 10.1074/jbc.270.14.7795. [DOI] [PubMed] [Google Scholar]
- 11.Boldin MP, Goncharov TM, Golstev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease in FAS/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 12.Bonetti B, Raine CS. Multiple sclerosis: oligodendrocytes display cell death-related molecules in situ but do not undergo apoptosis. Ann Neurol. 1997;42:74–84. doi: 10.1002/ana.410420113. [DOI] [PubMed] [Google Scholar]
- 13.Caldenhoven E, Liden J, Wissink S, Van de Stolpe A, Raaijmakers J, Koenderman L, Okret S, Gustafsson JA, Van der Saag PT. Negative cross-talk between RelA and the glucocorticoid receptor: a possible mechanism for the antiinflammatory action of glucocorticoids. Mol Endicronol. 1995;9:401–412. doi: 10.1210/mend.9.4.7659084. [DOI] [PubMed] [Google Scholar]
- 14.Celis JE, Celis A. Cell cycle-dependent variations in the distribution of the nuclear protein cyclin proliferating cell nuclear antigen in cultured cells: subdivision of S phase. Proc Natl Acad Sci USA. 1985;82:3262–3266. doi: 10.1073/pnas.82.10.3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, Nagayama T, Jin K, Stetler A, Zhu RL, Graham SH, Simon RP. Induction of Caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 17.Chinnaiyan AM, Tepper CG, Seldin MF, O’Rourke K, Krischel FC, Hellbardt S, Krammer PH, Dixit VM. FADD/MORT is a common mediator of CD95 (Fas/Apo-1) and TNF-receptor-induced apoptosis. J Biol Chem. 1996;271:4961–4965. doi: 10.1074/jbc.271.9.4961. [DOI] [PubMed] [Google Scholar]
- 18.Chu JL, Ramos P, Rosendorff A, Nikolic-Zugic J, Lacy E, Matsuzuwa A, Elkon KB. Massive upregulation of the Fas ligand in lpr and gld mice: implications for Fas regulation and the graft-versus-host disease-like wasting syndrome. J Exp Med. 1995;181:393–398. doi: 10.1084/jem.181.1.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cleveland J, Ihle J. Contenders in FasL/TNF death signaling. Cell. 1995;81:479–482. doi: 10.1016/0092-8674(95)90068-3. [DOI] [PubMed] [Google Scholar]
- 20.D’Souza SD, Bonetti B, Balasingam V, Cashman NR, Barker PA, Troutt AB, Raine CS, Antel JP. Multiple sclerosis: Fas signaling in oligodendrocyte cell death. J Exp Med. 1996;184:2361–2370. doi: 10.1084/jem.184.6.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donovan M, Miranda R, Kraemer R, McCaffrey T, Tessarollo L, Mahadeo D, Kaplan D, Tsoulfas P, Parada L, Toran-Allerand D, Hajjar D, Hempstead B. Neurotrophin and neurotrophin receptors in vascular smooth muscle cells: regulation of expression in response to injury. Am J Pathol. 1995;147:309–324. [PMC free article] [PubMed] [Google Scholar]
- 22.Duan H, Dixit VM. RAIDD is a new “death” adaptor molecule. Nature. 1997;385:86–89. doi: 10.1038/385086a0. [DOI] [PubMed] [Google Scholar]
- 23.Enari M, Hug H, Nagata S. Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature. 1995;375:78–81. doi: 10.1038/375078a0. [DOI] [PubMed] [Google Scholar]
- 24.Enari M, Talanian RV, Wong WW, Nagata S. Sequential activation of ICE-like and CPP-32-like proteases during Fas-mediated apoptosis. Nature. 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- 25.Esser P, Heimann K, Abts H, Fontana A, Weller M. CD95 (Fas/Apo-1) antibody mediated apoptosis in human retinal pigmented epithelium cells. Biochem Biophys Res Commun. 1995;213:1026–1035. doi: 10.1006/bbrc.1995.2231. [DOI] [PubMed] [Google Scholar]
- 26.Ferrer I, Bernet E, Soriano E, Del Rio T, Fonseca M. Naturally occurring cell death in the cerebral cortex of the rat and removal of dead cells by transitory phagocytes. Neuroscience. 1990;39:451–458. doi: 10.1016/0306-4522(90)90281-8. [DOI] [PubMed] [Google Scholar]
- 27.Ferrer I, Soriano E, DelRio J, Alcantara S, Auladell C. Cell death and removal in the cerebral cortex during development. Prog Neurobiol. 1992;39:1–43. doi: 10.1016/0301-0082(92)90029-e. [DOI] [PubMed] [Google Scholar]
- 28.Francis MJ, Hastings GZ, Brown F, McDermed J, Lu YA, Tam JP. Immunological evaluation of the multiple antigen peptide (MAP) system using the major immunogenic site of foot-and-mouth disease virus. Immunology. 1991;73:249–254. [PMC free article] [PubMed] [Google Scholar]
- 29.French LE, Hahne M, Viard I, Radlgruber G, Zanone R, Becker K, Muller C, Tschopp J. Fas and Fas ligand in embryos and adult mice: ligand expression in several immune-privileged tissues and coexpression in adult tissues characterized by apoptotic cell turnover. J Cell Biol. 1996;133:335–343. doi: 10.1083/jcb.133.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo Q, Robinson N, Mattson MP. Secreted beta-amyloid precursor protein counteracts the proapoptotic action of mutant presenilin-1 by activation of NF-kappaB and stabilization of calcium homeostasis. J Biol Chem. 1998;273:12341–12351. doi: 10.1074/jbc.273.20.12341. [DOI] [PubMed] [Google Scholar]
- 31.Hsu H, Huang J, Shu H-B, Bauchwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4:387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- 32.Hu S, Vincenz C, Ni J, Gentz R, Dixit VM. I-FLICE, a novel inhibitor of tumor necrosis factor receptor-1 and CD-95-induced apoptosis. J Biol Chem. 1997;272:17255–17257. doi: 10.1074/jbc.272.28.17255. [DOI] [PubMed] [Google Scholar]
- 33.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer J-L, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 34.Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 1991;66:233–243. doi: 10.1016/0092-8674(91)90614-5. [DOI] [PubMed] [Google Scholar]
- 35.Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins (CAP) form a death-inducing signalling complex (DISC) with the receptor. EMBO J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krajewski M, Wang H, Krajewski S, Zapata JM, Shabaik A, Gascoyne R, Reed JC. Immunohistochemical analysis of in vivo patterns of expression of CPP32 (Caspase-3), a cell death protease. Cancer Res. 1997;57:1605–1613. [PubMed] [Google Scholar]
- 37.Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell R. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 38.Kurki P, Vanderlaan M, Dolbeare F, Gray J, Tan EM. Expression of proliferating cell nuclear antigen (PCNA)/cyclin during the cell cycle. Exp Cell Res. 1986;166:209–219. doi: 10.1016/0014-4827(86)90520-3. [DOI] [PubMed] [Google Scholar]
- 39.Liu Z-G, Hsu H, Goeddel D, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 40.Los M, Van de Craen M, Penning L, Schenk H, Westendorp M, Baeuerle P, Droge W, Krammer P, Fiers W, Schulze-Osthoff K. Requirement of an ICE/CED-3 protease for FAS/APO-1-mediated apoptosis. Nature. 1995;375:81–83. doi: 10.1038/375081a0. [DOI] [PubMed] [Google Scholar]
- 41.Luskin MB, Shatz CJ. Studies of the earliest generated cells of the cat’s visual cortex: cogeneration of subplate and marginal zones. J Neurosci. 1985;5:1062–1075. doi: 10.1523/JNEUROSCI.05-04-01062.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marimoto C, Lord CI, Zhang C, Duke-Cohen JS, Letvin NL, Schlossman SF. Role of CD26/dipeptidyl peptidase IV in human immunodeficiency virus type 1 infection and apoptosis. Proc Natl Acad Sci USA. 1994;91:9960–9964. doi: 10.1073/pnas.91.21.9960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsuyama T, Hata R, Yamamoto Y, Tagaya M, Akita H, Uno H, Wanaka A, Furuyama J, Sugita M. Localization of Fas antigen mRNA induces in postischemic murine forebrain by in situ hybridization. Mol Brain Res. 1995;34:166–172. doi: 10.1016/0169-328x(95)00162-l. [DOI] [PubMed] [Google Scholar]
- 44.Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K. Activation of NF-kappaB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res. 1997;49:681–697. doi: 10.1002/(SICI)1097-4547(19970915)49:6<681::AID-JNR3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 45.Migheli A, Piva R, Atzori C, Troost D, Schiffer D. c-Jun, JNK/SAPK kinases and transcription factors NF-kappaB are selectively activated in astrocytes, but not motor neurons, in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1997;56:1314–1322. doi: 10.1097/00005072-199712000-00006. [DOI] [PubMed] [Google Scholar]
- 46.Milligan CE, Prevette D, Yaginuma H, Homma S, Cardwell C, Fritz LC, Tomaselli KJ, Oppenheim RW, Schwartz LM. Peptide inhibitors of the ICE protease family arrest programmed cell death of motoneurons in vivo and in vitro. Neuron. 1995;15:385–393. doi: 10.1016/0896-6273(95)90042-x. [DOI] [PubMed] [Google Scholar]
- 47.Miranda R, Toran-Allerand CD. Developmental expression of estrogen receptor mRNA in the rat cerebral cortex: a nonisotopic in situ hybridization histochemistry study. Cereb Cortex. 1992;2:1–15. doi: 10.1093/cercor/2.1.1. [DOI] [PubMed] [Google Scholar]
- 48.Miranda R, Sohrabji F, Toran-Allerand CD. Neuronal co-localization of mRNAs for neurotrophins and their receptors in the developing central nervous system suggests a potential for autocrine interactions. Proc Natl Acad Sci USA. 1993a;90:6439–6443. doi: 10.1073/pnas.90.14.6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miranda R, Sohrabji F, Toran-Allerand CD. Presumptive estrogen target neurons express mRNAs for both the neurotrophins and neurotrophin receptors: a basis for potential developmental interactions of estrogen with the neurotrophins. Mol Cell Neurosci. 1993b;4:510–525. doi: 10.1006/mcne.1993.1063. [DOI] [PubMed] [Google Scholar]
- 50.Miranda R, Sohrabji F, Toran-Allerand CD. Interactions of estrogen with the neurotrophins and their receptors during neural development. Horm Behav. 1994;28:367–375. doi: 10.1006/hbeh.1994.1033. [DOI] [PubMed] [Google Scholar]
- 51.Miranda R, Sohrabji F, Singh M, Toran-Allerand CD. Nerve growth factor (NGF) regulation of estrogen receptors in explant cultures of the developing forebrain. J Neurobiol. 1996;31:77–87. doi: 10.1002/(SICI)1097-4695(199609)31:1<77::AID-NEU7>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 52.Muzio M, Chinnaiyan AM, Kischel FC, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bertz JD, Zhang M, Gentz R, Mann M, Krammer PH, Peter ME, Dixit VM. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (FAS/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 53.Nagata S. Apoptosis mediated by the Fas system. Prog Mol Subcell Biol. 1996;16:87–103. doi: 10.1007/978-3-642-79850-4_6. [DOI] [PubMed] [Google Scholar]
- 54.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 55.Nagata S, Goldstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 56.Nishimura T, Akiyama H, Yonehara S, Kondo H, Ikeda K, Kato M, Iseki E, Kenji K. Fas antigen expression in brains of patients with Alzheimer’s-type dementia. Brain Res. 1995;695:137–145. doi: 10.1016/0006-8993(95)00699-q. [DOI] [PubMed] [Google Scholar]
- 57.Oehm A, Behrmann I, Falk W, Pawlita M, Maier G, Klas C, Li-Weber M, Richards S, Dhein J, Trauth B, Ponstingl H, Krammer P. Purification and molecular cloning of the APO-1 cell surface antigen, a member of the tumor necrosis factor/nerve growth factor receptor family. J Biol Chem. 1992;15:10709–10715. [PubMed] [Google Scholar]
- 58.Oishi M, Maeda K, Sugiyama S. Distribution of apoptosis-mediating Fas antigen in human skin and effects of anti-Fas monoclonal antibody on human epidermal keratinocytes and squamous cell carcinoma cell lines. Arch Dermatol Res. 1994;286:396–407. doi: 10.1007/BF00371800. [DOI] [PubMed] [Google Scholar]
- 59.Oppenheim R. Naturally occurring cell death during neural development. Trends Neurosci. 1985;8:487–493. [Google Scholar]
- 60.Ponton A, Clement M-V, Stamenkovic I. The CD95 (Apo-1/Fas) receptor activates NF-kappaB independently of its cytotoxic function. J Biol Chem. 1996;271:8991–8995. doi: 10.1074/jbc.271.15.8991. [DOI] [PubMed] [Google Scholar]
- 61.Ramsdell F, Seaman MS, Miller RE, Tough TW, Alderson MR, Lynch DH. gld/gld mice are unable to express a functional ligand for Fas. Eur J Immunol. 1994;24:928–933. doi: 10.1002/eji.1830240422. [DOI] [PubMed] [Google Scholar]
- 62.Rensing-Ehl A, Hess S, Ziegler-Heitbrock HW, Riethmuller G, Engelmann H. Fas/Apo-1 activates nuclear factor kappa B and induces interleukin-6 production. J Inflamm. 1995;45:161–174. [PubMed] [Google Scholar]
- 63.Rieux-Laucat F, Le Deist F, Hivroz C, Roberts I, Debatin K, Fischer A, de Villartay J. Mutations in Fas associated with lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–1349. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- 64.Shatz CJ, Ghosh A, McConnell SK, Allendoerfer KL, Friauf E, Antonini A. Pioneer neurons and target selection in cerebral cortical development. Cold Spring Harb Symp Quant Biol. 1990;55:469–480. doi: 10.1101/sqb.1990.055.01.046. [DOI] [PubMed] [Google Scholar]
- 65.Sohrabji F, Greene L, Miranda R, Toran-Allerand CD. Reciprocal regulation of estrogen and NGF receptors by their ligands in PC12 cells. J Neurobiol. 1994a;25:974–988. doi: 10.1002/neu.480250807. [DOI] [PubMed] [Google Scholar]
- 66.Sohrabji F, Miranda R, Toran-Allerand CD. Estrogen differentially regulates estrogen and nerve growth factor receptor mRNAs in adult sensory neurons. J Neurosci. 1994b;14:459–471. doi: 10.1523/JNEUROSCI.14-02-00459.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sohrabji F, Miranda R, Toran-Allerand D. Identification of a putative estrogen response element in the gene encoding brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:11110–11114. doi: 10.1073/pnas.92.24.11110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Srinivasula SM, Ahmad M, Ottilie S, Bullrich F, Banks S, Wang Y, Fernandes-Alnemri T, Croce C, Litwack G, Tomaselli KJ, Armstrong RC, Alnemri ES. FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. J Biol Chem. 1997;272:18542–18545. doi: 10.1074/jbc.272.30.18542. [DOI] [PubMed] [Google Scholar]
- 69.Stanger BZ, Leder P, Lee T-H, Kim E, Seed B. RIP: a novel protein containing a death domain that interacts with Fas/APO1 (CD95) in yeast and causes cell death. Cell. 1995;81:513–523. doi: 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- 70.Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75:1169–1178. doi: 10.1016/0092-8674(93)90326-l. [DOI] [PubMed] [Google Scholar]
- 71.Tachibana O, Nakazawa H, Lampe J, Watanabe K, Kleihues P, Ohgaki H. Expression of Fas/Apo-1 during the progression of astrocytomas. Cancer Res. 1995;55:5528–5530. [PubMed] [Google Scholar]
- 72.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, Ellington KO, Ayala JM, Casano FJ, Chin J, Ding GJ-F, Egger LA, Gaffney EP, Limjuco G, Palyha OC, Raju SM, Rolando AM, Salley JP, Yamin T-T, Lee TD, Shively JE, MacCross M, Mumford RA, Schmidt JA, Tocci MJ. A novel heterodymeric cysteine protease is required for interleukin-1β processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 73.Thornberry NA, Peterson EP, Zhao JJ, Howard AD, Griffin PR, Chapman KT. Inactivation of interleukin-1β converting enzyme by peptide (acyloxy) methyl ketones. Biochemistry. 1994;33:3934–3940. doi: 10.1021/bi00179a020. [DOI] [PubMed] [Google Scholar]
- 74.Toran-Allerand CD, Bentham W, Miranda R, Anderson JP. Insulin influences astroglial morphology and glial fibrillary acidic protein (GFAP) expression in organotypic cultures. Brain Res Dev Brain Res. 1991;558:296–304. doi: 10.1016/0006-8993(91)90781-p. [DOI] [PubMed] [Google Scholar]
- 75.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 76.Van der Zee CEEM, Ross GM, Riopelle RJ, Hagg T. Survival of cholinergic forebrain neurons in developing p75NGFR-deficient mice. Science. 1996;274:1729–1732. doi: 10.1126/science.274.5293.1729. [DOI] [PubMed] [Google Scholar]
- 77.Walker NPC, Talanian RV, Brady KD, Dang LC, Bump NJ, Ferenz CR, Franklin S, Ghayur T, Hackett MC, Hammill LD, Herzog L, Hugunin M, Houy W, Mankovich JA, McGuiness L, Orlewicz E, Paskind M, Pratt CA, Reiss P, Summani A, Terranova M, Welch JP, Xiong L, Moller A, Tracey DE, Kamen R, Wong WW. Crystal structure of the cysteine protease interleukin-1β-converting enzyme: a (p20/p10)2 homodimer. Cell. 1994;78:343–352. doi: 10.1016/0092-8674(94)90303-4. [DOI] [PubMed] [Google Scholar]
- 78.Wang C-Y, Mayo MW, Balwin AS., Jr TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 79.Watanabe D, Suda T, Hashimoto H, Nagata S. Constitutive acti- vation of the Fas ligand gene in mouse lymphoproliferative disorders. EMBO J. 1995;14:12–18. doi: 10.1002/j.1460-2075.1995.tb06970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Watanabe-Fukunaga R, Brannan CI, Itoh N, Yonehara S, Copeland NG, Jenkins NA, Nagata S. THe cDNA structure, expression and chromosomal assignment of the mouse Fas antigen. J Immunol. 1992;148:1274–1279. [PubMed] [Google Scholar]
- 81.Weis M, Sclegel J, Kass GE, Holmstrom TH, Peters I, Eriksson J, Orrenius S, Chow SC. Cellular events in Fas/Apo-1-mediated apoptosis in Jurkat T-lymphocytes. Exp Cell Res. 1995;219:699–708. doi: 10.1006/excr.1995.1281. [DOI] [PubMed] [Google Scholar]
- 82.Weller M, Kleihues P, Dichgans J, Ohgaki H. CD95 ligand - lethal weapon against malignant gliomas. Brain Pathol. 1998;8:285–293. doi: 10.1111/j.1750-3639.1998.tb00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xiaolu Y, Khosravi-Far R, Chang HY, Baltimore D. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell. 1997;89:1067–1076. doi: 10.1016/s0092-8674(00)80294-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yeh W-C, de la Pompa JL, McCurrach ME, Shu H-B, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, El-Deiry WS, Lowe SW, Goeddel DV, Mak TW. FADD: Essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998;279:1954–1958. doi: 10.1126/science.279.5358.1954. [DOI] [PubMed] [Google Scholar]
- 85.Yonehara S, Ishii A, Yonehara M. A cell-killing monoclonal antibody (anti-Fas) to a cell surface antigen co-downregulated with the receptor of tumor necrosis factor. J Exp Med. 1989;169:1747–1756. doi: 10.1084/jem.169.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature. 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]