

Abstract
In a slice preparation of rat visual cortex, we discovered that paired-pulse stimulation (PPS) elicits a form of homosynaptic long-term depression (LTD) in the superficial layers when carbachol (CCh) or norepinephrine (NE) is applied concurrently. PPS by itself, or CCh and NE in the absence of synaptic stimulation, produced no lasting change. The LTD induced by PPS in the presence of NE or CCh is of comparable magnitude with that obtained with prolonged low-frequency stimulation (LFS) but requires far fewer stimulation pulses (40 vs 900). The cholinergic facilitation of LTD was blocked by atropine and pirenzepine, suggesting involvement of M1 receptors. The noradrenergic facilitation of LTD was blocked by urapidil and was mimicked by methoxamine, suggesting involvement of α1 receptors. β receptor agonists and antagonists were without effect. Induction of LTD by PPS was inhibited by NMDA receptor blockers (completely in the case of NE; partially in the case of CCh), suggesting that one action of the modulators is to control the gain of NMDA receptor-dependent homosynaptic LTD in visual cortex. We propose that this is a mechanism by which cholinergic and noradrenergic inputs to the neocortex modulate naturally occurring receptive field plasticity.
Keywords: visual cortex, development, synaptic plasticity, long-term potentiation, long-term depression, acetylcholine, norepinephrine
It is well established that synapses in sensory neocortex can be modified by experience. For example, brief deprivation of normal vision during early postnatal development can lead to a depression of synaptic transmission that renders visual cortical neurons unresponsive to retinal stimulation. This type of synaptic plasticity obviously depends on information of retinal origin. However, there is evidence that experience-dependent plasticity also requires that animals be awake, alert, and paying attention to sensory stimuli (for review, see Singer, 1995). Thus, issues of great interest are the mechanisms of experience-dependent synaptic plasticity and their modulation by behavioral state.
Extrathalamic inputs convey information to the cortex about behavioral state. Attention has focused mainly on the inputs arising from the locus coeruleus and the basal telencephalon that use norepinephrine (NE) and acetylcholine (ACh), respectively, as neurotransmitters. In one early study, it was shown that partial destruction of the noradrenergic or the cholinergic inputs alone did not disrupt deprivation-induced synaptic depression in visual cortex. However, their combined destruction produced a large deficit in this form of experience-dependent plasticity (Bear and Singer, 1986). The results of this experiment suggested that both of these inputs to visual cortex facilitate synaptic plasticity. Furthermore, because the simultaneous loss of both noradrenergic and cholinergic inputs was required to produce the defect in plasticity, the suggestion was made that these two modulators may substitute for one another and act via a common molecular mechanism.
The notion that ACh and NE modulate naturally occurring cortical plasticity has received ample support (Kasamatsu and Pettigrew, 1979;Gordon et al., 1990; Juliano et al., 1991; Gu and Singer, 1993;Osterheld-Haas et al., 1994; Bakin and Weinberger, 1996; Baskerville et al., 1997; Kilgard and Merzenich, 1998; Sachdev et al., 1998; Zhu and Waite, 1998). The important question that remains, of course, is precisely how these neurotransmitters affect the cortical synapses that carry detailed information about sensory experience.
Over the past several years, slice preparations of visual neocortex have been used in an effort to clarify the elementary mechanisms of activity-dependent synaptic plasticity. One hypothesis that derived from this work is that modulation of inhibition in the cortex might be a way in which ACh and NE control plasticity (Kirkwood and Bear, 1994a). Because the state of functional inhibition in the cortex can be assayed by recording layer III responses to paired-pulse stimulation (PPS) of the white matter (Luhmann and Prince, 1991; Metherate and Ashe, 1994), we set out to examine the effects of ACh and NE on responses to PPS. In the course of this investigation we made the unexpected discovery that PPS in the presence of ACh or NE triggers a form of long-term synaptic depression (LTD). Here we report that ACh, acting via M1 receptors, and NE, acting via α1 receptors, dramatically facilitate NMDA receptor-dependent homosynaptic LTD in visual cortex. We suggest that this reflects a mechanism whereby these modulators facilitate experience-dependent synaptic plasticity in sensory neocortex.
MATERIALS AND METHODS
The experiments described in this paper were performed on transverse slices prepared from the visual cortex of 3- to 5-week-old Long–Evans rats. Each animal was deeply anesthetized by exposure to methoxyflurane vapors and was decapitated soon after the disappearance of any corneal reflexes. The brain was rapidly removed and immersed in ice-cold dissection buffer containing (in mm): sucrose, 212.7; KCl, 5; NaH2PO4, 1.25; MgSO4, 3; CaCl2, 1; NaHCO3, 26; dextrose, 10; and kynurenate, 10. A block of visual cortex was removed and sectioned in the coronal plane into 0.4-mm-thick slices using a microslicer (DTK 1000; Ted Pella, Redding, CA). The slices were gently transferred to an interface storage chamber containing artificial CSF (ACSF) and maintained at room temperature for at least an hour before recording. The ACSF was saturated with 95% O2/5% CO2 and contained (in mm): NaCl, 124; KCl, 5; NaH2PO4, 1.25; MgCl2, 1; CaCl2, 2; NaHCO3, 26; and dextrose, 10. The experiments were performed on submerged slices continuously perfused at a rate of 2 ml/min with 30°C ACSF saturated with 95% O2/5% CO2. Microelectrodes were filled with ACSF (1–2 MΩ) for extracellular recording or 3m potassium acetate (80–120 MΩ) for intracellular recording. Only cells with resting membrane potentials more negative than −70 mV and input resistances >20 MΩ were studied. A site in the middle of the cortical thickness, confirmed histologically to correspond to layer IV and upper layer V, was stimulated to evoke field potentials (FPs) in layer III, as described previously (Kirkwood and Bear, 1994a,b). The amplitude of the maximum negative FP in layer III was used as a measure of the evoked population excitatory synaptic current. Changes in the amplitude of the maximum negative FP reflect changes in the magnitude of a synaptic current sink (Mitzdorf, 1985; Aizenman et al., 1996) and correlate with changes in the initial slope of EPSPs recorded intracellularly in layer III neurons (Kirkwood and Bear, 1994a,b). Baseline responses were obtained every 15 sec with a stimulation intensity that yielded a half-maximal response. PPS was used throughout the experiment unless stated otherwise. At least 10 min of stable baseline recordings were made before drugs were applied. When NE was applied, 40 μm sodium ascorbate was included in the ACSF to prevent oxidation of the drug. Sodium ascorbate was also included with the application of noradrenergic agonists and antagonists. Carbachol and atropine were purchased from Sigma (St. Louis, MO); all other drugs were purchased from Research Biochemicals (Natick, MA).
Only data from slices with stable recordings (<3% change over the baseline period) were included in the analysis. The data were analyzed as follows: (1) the maximum negative FP amplitude data for each experiment were expressed as percentages of the preconditioning baseline average; (2) the time scale in each experiment was converted to time from the onset of conditioning, and the four responses recorded in each minute were averaged; and (3) then, the time-matched, normalized data were averaged across experiments and expressed as the means (± SEM). Within each group, the statistical significance of a change produced by conditioning stimulation was assessed with a pairedt test, comparing values immediately before the application of neuromodulators with those 30 min after the end of the application. Statistical significance across groups was assessed with an unpairedt test.
RESULTS
Visual cortical slices were prepared from 3- to 5-week-old rats. Initially, layer III synaptic responses were evoked with stimulation applied to the underlying white matter. Our original goal was to study the effects of cholinergic and noradrenergic stimulation on paired-pulse suppression, i.e., the attenuation of the synaptic responses when two pulses are given in rapid succession. In these experiments, the stimulation consisted of two pulses (40 msec apart) delivered repetitively every 15 sec throughout the experiment. To activate cholinergic receptors, we bath applied 50 μmcarbachol (CCh), and to activate noradrenergic receptors, we bath applied 40 μm NE in ascorbate (40 μm). When we realized that the PPS in the presence of these modulators caused LTD, we shifted the stimulating electrode to layer IV to activate a less complex circuit. All the data presented below were obtained with this stimulation-recording configuration.
Paired-pulse stimulation in carbachol induces a long-lasting depression of the synaptic responses
Figure 1 illustrates the effects of a 10 min application of 50 μm CCh on the FP responses to paired pulses. Under control conditions, the response amplitude to the second pulse is usually somewhat smaller than the response amplitude to the first pulse. In the experiments shown in Figure 1, the ratio of the second response to the first response is 0.59 ± 0.08 (n = 11). Exposure to CCh strongly reduced the response to the first pulse (64 ± 4% of control measured at 10 min of CCh), but it had a smaller effect on the response to the second pulse. Thus, paired-pulse suppression was virtually eliminated in the presence of CCh; the ratio of the second to the first response was 0.99 ± 0.14 at the end of the 10 min of CCh perfusion.
Fig. 1.

The cholinergic agonist CCh induces a lasting depression of the layer III synaptic responses to layer IV stimulation. A, B, Effects of a brief (10 min) bath application of CCh (50 μm;open horizontal bar in B) on the field responses evoked with paired-pulse stimulation [interstimulus interval (ISI) = 40 msec] are shown. A, FPs are from an experiment in which the CCh application resulted in a clear depression of the response to the first pulse but had virtually no effect on the response to the second pulse. Thetraces were recorded immediately before (control) and during the application of CCh and 30 min after washout of the drug. B, Time course of the average of 11 experiments is shown. Solid circles are responses to the first pulse; open circles are responses to the second pulse. C, Prolonged paired-pulse stimulation alone has no effect on the FPs (n = 9). D,E, The effects of CCh on FPs correlate with changes in simultaneously recorded intracellularEPSPs. D, Traces are averages of four consecutive intracellular (top) and extracellular (bottom) responses recorded before (control) and 30 min after washout of CCh.E, Average time course of seven similar experiments is shown. Top, Changes in FP amplitude (open circles) and initial slope of the EPSP(filled triangles) are presented.Bottom, Changes in the membrane potential are shown.
These acute effects of CCh—that is, the transient and reversible reduction of the synaptic responses and the reduction in paired-pulse suppression—have been reported previously (Vaknin and Teyler, 1991;Murakoshi, 1995). These effects are likely to reflect a reduced probability of glutamate release (Markram and Tsodyks, 1996; Gil et al., 1997), probably because of the action of CCh on cholinergic receptors located on glutamatergic presynaptic terminals (Valentino and Dingledine, 1981; Segal, 1982; Dodt et al., 1991; Vaknin and Teyler, 1991). An unexpected result, however, was that after removal of CCh the responses to the first stimulation pulse never returned to the original level. After an initial partial recovery, the response magnitude reached a stable level 20 min after washing out the CCh and remained depressed (80 ± 6% of control) even after an additional 30 min of washout. The LTD was a specific consequence of the CCh, because prolonged PPS by itself had no effect on the FPs (Fig.1C).
To confirm that the changes in the FP reflect changes in excitatory synaptic transmission, we repeated the experiment using intracellular recording. Simultaneous recordings were made of the evoked FP in layer III and the EPSP in a nearby layer III neuron. PPS during CCh resulted in a reduction of FP amplitude (70 ± 6%) that was paralleled by a comparable reduction in the initial slope of the EPSPs (73 ± 8%; n = 7), indicating that a reduction of the synaptic responses of layer II/III cells is reflected in the reduction of the field responses. The application of CCh also produced a modest depolarization that was rapidly reversed after removal of the drug. Thus the LTD of FP responses is not caused by a tonic depolarization of postsynaptic neurons. To determine whether the slight depolarization of the neuron by CCh during PPS was the cause of LTD, we injected current intracellularly that yielded similar levels of depolarization (10 mV for 12 min; n = 3). However, in no case was LTD observed, suggesting that the depolarization alone (at least in the cell soma) is not sufficient to cause expression of the LTD.
The LTD induced by PPS in 50 μm CCh was very reliable, so this procedure was used to characterize the phenomenon. However, we did examine the CCh dose–response relationship. By the use of a 20 min application during PPS, LTD of increasing magnitude (measured 30 min after washout) was observed with 0.1 μm (94 ± 2% of baseline; n = 7), 1 μm (81 ± 3%; n = 7), 10 μm (84 ± 3%;n = 7), 50 μm (72 ± 3%;n = 8), and 100 μm CCh (68 ± 4%;n = 7).
Synaptic depression induced in the presence of carbachol requires synaptic stimulation and is input specific
An important issue to be resolved concerned the role of synaptic activation in the depression induced with CCh. Thus we asked whether CCh applied alone, without concurrent synaptic activation, would be sufficient to produce LTD. Figure 2 shows that in the absence of stimulation, CCh application has minor effects on the synaptic responses (97 ± 4% of baseline; pairedt test, p > 0.2; n = 5). However, subsequent administration of CCh in the same slices during PPS did result in substantial depression (78 ± 4%; p< 0.02). These results indicate that CCh permits or facilitates an activity-dependent form of synaptic depression. These findings also eliminate the possibility that the lasting depression is caused by tonic activation of ACh receptors (i.e., incomplete washout of CCh).
Fig. 2.

Synaptic depression induced withCCh requires synaptic stimulation. A,Left, In the absence of stimulation, CChfailed to produce any significant change in the FP responses.Right, Subsequently, depression was reliably produced when CCh was applied with PPS. In bothpanels the responses were normalized with respect to the average response during the initial baseline period beforeCCh application. The time that elapsed between thelast data point on the left and thefirst data point on the right varied but was not >30 min. B, The depression induced byCCh is input specific. Right, The stimulation-recording configuration used to assess input specificity is shown. Layer IV was stimulated on either side of a radial cut that extended from the white matter (WM) through layer IV. Left, The graph shows thatCCh application induces depression only on the side that was stimulated. In all experiments designed to test for input specificity, responses from the two inputs showed summation and were of similar amplitudes.
Next we wished to determine whether this form of depression is input specific, that is, whether the reduction in synaptic efficacy was confined to the stimulated inputs only. To address this question, separate inputs to layer III were isolated by making a radial cut in the slice that extended from the white matter to layer IV, and baseline stimulation was applied in layer IV on each side of the cut. During CCh, PPS was applied to only one input, the other serving as an unstimulated control. The results, summarized in Figure2B, showed that only inputs receiving stimulation during CCh show the LTD (stimulated path, 82 ± 6% of baseline; control path, 98 ± 1%; n = 5). Thus, cholinergic stimulation promotes LTD only in active inputs.
The results described above indicate that CCh in conjunction with paired-pulse stimulation induces an activity-dependent and homosynaptic form of LTD. We were interested to know whether the CCh effect is specific to inputs receiving PPS or whether CCh exerts a more general facilitation of homosynaptic LTD mechanisms. In hippocampal and visual cortical slices, a homosynaptic form of LTD can also be reliably induced with low-frequency stimulation (LFS; typically 1 Hz), but only if this stimulation is prolonged [usually 10–15 min (Dudek and Bear, 1992; Kirkwood et al., 1993)]. To see whether CCh can facilitate LFS-induced LTD, we combined application of the drug with brief epochs of LFS (1 Hz for 5 min; Fig.3). When the brief LFS was applied alone, little or no LTD was induced (94 ± 3%; n = 11). However, when brief LFS was applied in conjunction with CCh, substantial LTD was induced (81 ± 3; n = 17;p < 0.005; Fig. 3C). These results indicate that CCh application facilitates the induction of homosynaptic LTD with LFS, as well as with PPS.
Fig. 3.

CCh promotes the induction of LTD with a LFS. A, Results from an experiment in which a brief epoch of LFS (300 pulses at 1 Hz; solid horizontal bar) failed to induce LTD under control conditions but did produce LTD when it was subsequently applied in the presence ofCCh. The baseline was collected with single-pulse stimulation delivered every 15 sec. B, Changes in the FP amplitude induced by LFS alone (open circles;n = 11) and by LFS in the presence of 50 μmCCh (filled circles; n = 17). C, Cumulative probability distribution of the changes induced by LFS in the absence (dashed line) and in the presence (solid line) of CCh. The percentage change over the baseline was measured at 30 min after conditioning. Data are from the same experiments shown in B.
Involvement of NMDA receptors and M1 receptors in the induction of synaptic depression in the presence of carbachol
The induction of one prominent form of homosynaptic LTD in visual cortex requires the activation of NMDA receptors (Kirkwood and Bear, 1994b). To test whether this is also the case for the synaptic depression promoted by CCh, we attempted to block its induction with the NMDA receptor antagonist 2-amino-5-phosphonovaleric acid (AP5). Figure 4, A–C, illustrates the effects of bath-applied 100 μm AP5 on the induction of depression in CCh. In three cases, one of them shown in Figure 4A, AP5 completely blocked LTD induced with the aid of CCh, and this blockade was relieved after removal of AP5. On average, LTD induced by PPS in the presence of CCh was significantly reduced but not eliminated by AP5 (AP5, 91 ± 9%;n = 12; control, 77 ± 12; n = 10;p < 0.01; Fig. 4C). The data indicate that cholinergic activation promotes the induction of an NMDA receptor-dependent form of LTD. However, the fact that the inhibition of LTD with AP5 is incomplete suggests that an NMDA receptor-independent form of LTD may also be promoted by CCh. In this regard, it is worth mentioning that in the CA1 region it has been demonstrated that two forms of LTD can be induced at the same synapses (Oliet et al., 1997).
Fig. 4.

Synaptic depression induced by PPS inCCh is partially dependent on NMDA receptor activation.A, Results are from one of the three experiments in which application of 100 μmAP5 reversibly blocked the depression induced with CCh.B, On average, application of 100 μmAP5 reduced the depression induced withCCh. The graph shows the effects of CChon the first response in the presence (filled circles; n = 10) and in the absence (open circles; n = 12) of 100 μmAP5. C, Cumulative probability distribution of the changes in the field potential amplitude measured 30 min after washout of CCh is shown. Experiments were done in the presence of AP5(solid line) and in the absence of AP5(dashed line). Data are from the same experiments shown in B.
In adults, cholinergic terminals and muscarinic receptors are distributed throughout the depth of the visual cortex. Nicotinic receptors, in contrast, are confined to the middle layers. It seemed reasonable to assume, therefore, that the effects of CCh on layer III synaptic responses depend on the activation of muscarinic receptors. To test this assumption, we studied the effects of the muscarinic antagonist atropine on the synaptic depression induced with CCh. In the presence of 5 μm atropine, the application of CCh failed to produce either the transient or the lasting depression of the field responses (98 ± 3.4% of baseline; n = 6), whereas substantial depression was induced in the interleaved controls (87 ± 2; n = 7; Fig.5A). A major postsynaptic muscarinic receptor type is the M1 receptor (Mrzljak et al., 1993; Wang and McCormick, 1993), and this receptor has been implicated in the regulation of visual cortical plasticity (Gu and Singer, 1993). As shown in Figure 5B, in the presence of the selective M1 antagonist pirenzepine (50 μm), application of CCh failed to induce LTD [98 ± 3% of baseline (n = 10) as compared with 80 ± 4% (n = 10) in the interleaved controls]. In contrast, the M2 receptor antagonist gallamine (50 μm) had no effect (79 ± 3% of baseline 30 min after CCh;n = 7; data not shown). Thus, facilitation of LTD by CCh depends, at least in part, on M1 receptor activation coincident with synaptic activation of NMDA receptors.
Fig. 5.

Synaptic depression induced by CChis dependent on the activation of M1 muscarinic receptors.A, Time course of the effects of CChapplied in the absence (open circles;n = 6) and presence (filled circles; n = 7) of the muscarinic antagonist atropine (5 μm). B, Time course of the effects of CCh applied in the absence (open circles; n = 10) and presence (filled circles; n = 10) of the specific M1 muscarinic antagonist pirenzepine (50 μm).
Norepinephrine induces a long-lasting depression of the synaptic responses
The suggestion has been made from lesion studies in vivo that ACh and NE might have similar modulatory effects on synaptic plasticity in visual cortex (Bear and Singer, 1986). Therefore, we investigated whether NE can also promote synaptic plasticity of layer III synaptic responses in visual cortical slices. In these experiments we used the same stimulation paradigms used in the CCh experiments. Figure 6, Aand B, shows that application of 40 μm NE for 10 min results in an acute reduction of the responses to PPS. The ratio of the second to the first response, which was 0.90 ± 0.05 (n = 8) at the beginning of the experiments, grew to 0.98 ± 0.06 during NE application and remained at that value (1.00 ± 0.05) after the washout of NE. As was the case for CCh, when NE was removed the response to the first pulse remained depressed for as long as the response was recorded (82 ± 2% of control at 60 min after NE). Simultaneous intracellular and extracellular recordings revealed that NE reduced the EPSP (90 ± 2%;n = 11) and the field potential (88 ± 3%) to a similar extent and caused a slight and transient depolarization (Fig.6C,D) that disappeared after washout of the drug. Thus, the lasting depression of the field responses induced with the aid of NE reflects, at least in part, changes in synaptic efficacy.
Fig. 6.

PPS in the presence of NE induces a lasting depression of the layer III synaptic responses to layer IV stimulation. A, B, Effects of a brief (10 min) bath application of NE (40 μm) on the field responses evoked with paired-pulse stimulation (ISI = 40 msec) are shown. A, FPs were recorded immediately before (control) and during the application of NE and 30 min after washout of the drug. B, Time course of the average of nine experiments is shown. Solid circles are responses to the first pulse; open circles are responses to the second pulse. C, D, The effects ofNE on FPs correlate with changes in simultaneously recorded intracellular EPSPs. C,Traces are averages of four consecutive intracellular (top) and extracellular (bottom) responses recorded before and 30 min after washout ofNE. D, Average time course of 11 similar experiments is shown. Top, Changes in FP amplitude (open circles) and initial slope of theEPSP (filled triangles) are presented. Bottom, Changes in the membrane potential are shown.
Norepinephrine promotes an activity- and NMDA receptor-dependent form of LTD
The striking similarity of the long-term effects of CCh and NE led us to examine further whether LTD induced with the aid of NE also requires synaptic activity. In these experiments we stimulated and recorded simultaneously from two independent sites in the same slice (see Fig. 7). Stimulation at one site was suspended during the application of NE and resumed 5 min after washout of the drug; the other site was stimulated throughout the experiment and served as a control. As shown in Figure 7, very little depression developed in the nonstimulated site (99 ± 3%; n = 5), whereas robust depression was observed in the control site (83 ± 6%).
Fig. 7.

Synaptic depression induced with NErequires synaptic activity. Left, Time course of the changes induced by NE on the amplitude of the first response in two sites recorded simultaneously in the same slices (n = 5). One site was stimulated throughout the experiment (solid circles), whereas in the other site stimulation was suspended during NE application and resumed 5 min after washout (open circles).Right, Stimulus-recording arrangement.
Next we investigated the involvement of NMDA receptors in the induction of LTD by PPS in the presence of NE, using the NMDA receptor antagonist AP5. As shown in Figure 8, in the presence of 100 μm AP5, PPS in NE failed to induce LTD (103 ± 3%; n = 6), but it did induce substantial LTD after washout of the antagonist (84 ± 3%; p< 0.005). Taken together, these results support the conclusion that noradrenergic activation, like cholinergic activation, facilitates the induction of homosynaptic LTD in visual cortex.
Fig. 8.

Synaptic depression induced with NEis reversibly blocked by the NMDA receptor antagonistAP5. Left, In the presence ofAP5 (100 μm), NE failed to produce any change in the FP response to test stimulation.Right, After washout of the drug, however, depression was reliably produced. In both panels the responses were normalized with respect to the average response during the initial baseline period before NE application. The time that elapsed between the last data point on theleft and the first data point on theright varied but was not >30 min.
α1 receptors are involved in the noradrenergic facilitation of LTD
NE can activate a variety of adrenergic receptors in the superficial layers of visual cortex, including α1-, α2-, and β-adrenergic receptors. To determine the receptor subtype involved in the facilitation of LTD, we investigated the effect of a battery of receptor-specific antagonists and agonists.
Figure 9, A and B, shows that β-adrenergic receptors are unlikely to be involved in the facilitation of LTD. PPS in the presence of the β-adrenergic receptor agonist isoproterenol (40 μm) produced no LTD (97 ± 4%; n = 6), and application of the β receptor antagonist propranolol (40 μm) had no effect on the induction of LTD in the presence of NE (74 ± 4%;n = 5). These results contrast with the effects of α1 receptor agonists and antagonists (Fig.9C,D). PPS during application of the α1 receptor agonist methoxamine (40 μm) caused a sustained depression of the synaptic responses (84 ± 3;n = 8) that mimicked the LTD induced with NE (Fig.9C). Moreover, the α1 antagonist urapidil (40–80 μm) effectively blocked the effects of NE (98 ± 3%; n = 12) as compared with interleaved controls (85 ± 2%; n = 12; Fig. 9D). Together, these data indicate that α1, but not β, receptors are involved in the facilitation of LTD by NE.
Fig. 9.

Noradrenergic facilitation of LTD is mediated by the activation of α1 receptors. A, The β-adrenergic receptor agonist isoproterenol (40 μm;ISO) fails to facilitate LTD. B, Application of the β-adrenergic receptor antagonist propranolol (40 μm) does not block the facilitation of LTD byNE. C, The α1-adrenergic agonist methoxamine (40 μm; METHOX) mimics NE. D, The α1-adrenergic antagonist urapidil (40 μm) blocks the effects ofNE (open symbols, NE + urapidil;filled symbols, NE controls). E, Bath application of the α2-adrenergic agonist clonidine (40 μm) transiently potentiates the response to synaptic stimulation. F, Application of the α2-adrenergic antagonist yohimbine (40 μm) depresses the synaptic responses.
The pharmacological manipulation of α2 receptors produced some perplexing results. The α2 agonist clonidine had the opposite effect of NE; PPS in clonidine produced a large, but transient, potentiation of the synaptic responses (130 ± 8% at 20 min of clonidine application). After clonidine was removed, the responses slowly came back to control levels (103 ± 3% at 50 min of washout;n = 4; Fig. 9E). The α2 antagonist yohimbine (40 μm) mimicked the effects of NE and profoundly reduced the field responses. When the drug was removed, the responses recovered only partially, reaching a stably depressed level (70 ± 4%; n = 7; Fig. 9F). The paradoxical results obtained with yohimbine and clonidine might be attributable, at least in part, to their effects on presynaptic α2 inhibitory autoreceptors. By blocking the autoreceptors, yohimbine might enhance the release of endogenous NE and facilitate the induction of LTD.
Differential effects of neuromodulators in visual cortex and hippocampus
Previous studies indicate that synaptic plasticity evoked in visual cortical layer III and synaptic plasticity evoked in the CA1 region of the hippocampus share crucial similarities in their mechanism of induction (Kirkwood et al., 1993). Therefore, it was of interest to investigate the effect of the neuromodulators on the CA1 synaptic responses. In these experiments, the responses to PPS were recorded simultaneously in visual cortex and hippocampus using slices that contained both structures (Fig.10A). During the CCh application, the CA1 responses were the most affected (Fig.10B). After removal of the drug, however, the responses in both visual cortex and hippocampus stabilized at a similar, depressed level (82 ± 7% in visual cortex; 89 ± 4% in CA1; n = 8). On the other hand, whereas during the application of NE both CA1 and visual cortex were depressed to the same level (Fig. 10C), only the visual cortical responses remained depressed after the removal of the drug (82 ± 3% in visual cortex; 103 ± 6% in CA1; n = 10).
Fig. 10.
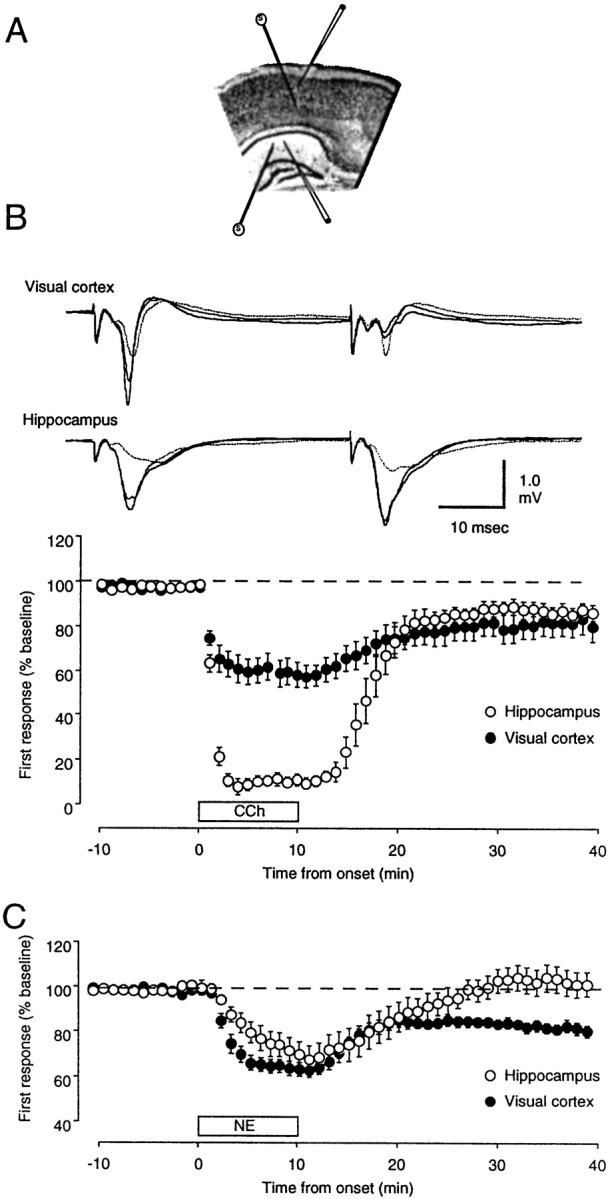
Differential effects of CCh andNE in hippocampus and visual cortex. A, The drawing depicts the electrode configuration used for simultaneous recording in CA1 and visual cortex. B, Application ofCCh yields comparable LTD in CA1 and visual cortex.Top, The traces are averages of four consecutive recordings taken before (thick line), during (dotted line), and 30 min after the application ofCCh (thin line). Bottom, The graph shows the time course of the changes induced in the first response recorded simultaneously in the CA1 region of the hippocampus (initial slope of the field potential; open circles) and in the visual cortex (field potential amplitude; filled circles). C, The time course of the changes induced by bath application of NE is shown. PPS in the presence of NE failed to produce LTD in the hippocampus.
DISCUSSION
The principal finding of our study is that cholinergic or noradrenergic receptor activation dramatically facilitates the induction of homosynaptic LTD in layer III of visual cortex. In fact, when PPS is used, the neuromodulators enable induction and expression of synaptic plasticity that otherwise would not be observed. Thus, our results provide strong support for the view that ACh and NE can serve as “enabling factors” for activity-dependent cortical plasticity (Singer, 1979, 1995; Dykes et al., 1990; Dykes, 1997). Because this robust modulation of plasticity can be observed under controlled conditions in vitro, the paradigm we describe here may be useful for the further dissection of the molecular mechanism of experience-dependent cortical plasticity and its modulation by behavioral state.
Properties of LTD induced in the presence of ACh and NE
In recent years, it has become clear that many synapses in the CNS can undergo activity-dependent LTD. In the hippocampus and neocortex, LTD can be induced reliably with prolonged (e.g., 15 min), low-frequency (e.g., 1 Hz) synaptic stimulation. LTD induced in this way occurs only at the stimulated synapses and thus is said to be homosynaptic. In the CA1 region of hippocampus and in layer III of visual cortex, homosynaptic LTD evoked with LFS requires NMDA receptor activation under most experimental conditions (e.g., Dudek and Bear, 1992; Kirkwood et al., 1993).
The properties of LTD in visual cortex induced by PPS in the presence of NE suggest that it uses a similar induction mechanism. In particular, the LTD is completely blocked by application of AP5, an NMDA receptor antagonist. Thus, NE may act as a sort of gain-control mechanism for NMDA receptor-dependent homosynaptic LTD. The situation for ACh, however, may be more complicated. Although the LTD induced by PPS in the presence of CCh was input specific, it was only partially blocked by AP5. Thus, in addition to facilitating NMDA receptor-dependent homosynaptic LTD, ACh may also promote other mechanisms of LTD induction. In this context, it should be noted that NMDA receptor-independent forms of homosynaptic LTD have been described, both in visual cortex (Artola et al., 1990) and in hippocampus, including area CA1 (Stanton and Sejnowski, 1989; Bolshakov and Siegelbaum, 1994; Oliet et al., 1997).
With PPS, a brief application of NE or CCh resulted in LTD of magnitude comparable with that obtained with prolonged LFS, the standard method to induce LTD in many laboratories. However, PPS induced LTD with far fewer stimulation pulses (40 vs 900). PPS may be a particularly efficacious stimulation regime, in part because it facilitates NMDA receptor-mediated responses (Metherate and Ashe, 1994). However, the facilitation of LTD by the neuromodulators is not unique to PPS, because we find that LFS-induced LTD is also enhanced. In this context it should be noted that Kojic et al. (1997) reported that another neuromodulator, serotonin, could also greatly facilitate LFS-induced LTD in visual cortex.
The LTD induced by PPS in the presence of neuromodulators was associated with an increase in the paired-pulse response ratio. This effect was more pronounced for ACh than it was for NE. These findings could be taken as support for a presynaptic expression mechanism for LTD in the neocortex (i.e., reduced probability of glutamate release in response to the first pulse). However, interpretation of a change in paired-pulse responses in our preparation is not straightforward. The paired-pulse ratio depends importantly on the magnitude of the response to the first pulse and on the state of inhibition (Luhmann and Prince, 1991; Metherate and Ashe, 1994; Frank et al., 1995). Thus, elucidation of the expression mechanism(s) for LTD in the neocortex will require additional experiments, using intracellular recordings of responses to minimal stimulation of a small number of synaptic inputs.
Mechanism of LTD facilitation in visual cortex
Our data suggest that the effect of CCh on LTD is mediated via muscarinic receptors of the M1 subtype and that the effect of NE is mediated via α receptors of the α1 type. Both of these receptor types are highly expressed in the superficial layers of visual cortex (Shaw et al., 1986; Schliebs et al., 1989), and interestingly, both are coupled to phospholipase C.
Although the exact site(s) of modulation and the precise mechanisms by which the modulation occurs remain to be determined, there are some obvious possibilities. The LTD triggered by PPS depends, at least in part, on NMDA receptor activation. Because the NMDA receptor is voltage dependent, cholinergic and noradrenergic modulation of LTD could occur indirectly by regulating the excitability of the postsynaptic neuron or by altering the properties of inhibition. In addition, the NMDA receptor itself is a phosphoprotein that is subject to regulation (Roche et al., 1994). Finally, regulation of LTD could occur downstream of NMDA receptor activation, for example, via release of Ca2+ from intracellular stores or by altering the activity of the protein kinases and phosphatases that control synaptic efficacy.
Indeed, studies in neocortex and hippocampus have shown that activation of M1 (Valentino and Dingledine, 1981; Markram and Segal, 1990, 1992; Behrends and Buggencate, 1993; Murakoshi, 1995; Kimura and Baughman, 1997) and α1 receptors (Madison and Nicoll, 1988) reduces evoked GABA release by inhibitory interneurons. In addition, M1 receptor activation increases the excitability of pyramidal neurons (McCormick and Prince, 1986; Dutar and Nicoll, 1988) and directly enhances NMDA receptor function (Segal, 1992; Aramakis et al., 1997). Any or all of these effects could contribute to the modulation of LTD in visual cortex, and it will be of interest to investigate these possible mechanisms in future studies. It should be noted, however, that there is already evidence that decreasing inhibition can facilitate homosynaptic LTD in CA1 (Wagner and Alger, 1995).
Neuromodulators and bidirectional synaptic plasticity
To our knowledge, our study is the first to show facilitation of LTD by ACh and NE. However, there have been many reports on the effects of ACh and NE on long-term potentiation (LTP). As is the case for LTD, there are multiple forms of LTP with different induction and expression mechanisms. One form, which is highly expressed in CA1 and in the superficial layers of neocortex (e.g., Kirkwood et al., 1993), depends on NMDA receptor activation for its induction. This type of LTP is of particular interest because it is the functional inverse of NMDA receptor-dependent LTD (for review, see Bear and Kirkwood, 1996).
As we have shown for LTD, NMDA receptor-dependent LTP is enhanced by muscarinic receptor activation (Tanaka et al., 1989; Blitzer et al., 1990; Burgard and Sarvey, 1990; Brocher et al., 1992; Huerta and Lisman, 1993; Sokolov and Kleschevnikov, 1995). In fact, Auerbach and Segal (1994, 1996) report that low concentrations of CCh (0.25–0.75 μm) can directly trigger LTP in CA1. We did not observe a similar effect in visual cortex, however.
Similarly, NMDA receptor-dependent LTP is enhanced by NE. However, this facilitatory effect is mediated by β receptors and not α receptors as is the case for LTD (Stanton and Sarvey, 1987; Brocher et al., 1992;Kato, 1993; Thomas et al., 1996; Katsuki et al., 1997). Moreover, in CA1 10 μm NE not only facilitates LTP but also inhibits induction of LTD with LFS (Blitzer et al., 1995; Thomas et al., 1996). The apparent discrepancy between the effects of NE on LTD in visual cortex and hippocampus could be explained by differences in the balance of α and β receptor activation by NE in the two preparations. Indeed, our direct comparison of hippocampus and visual cortex revealed that synaptic transmission in the two structures responds differently to bath-applied NE. Unlike in the visual cortex, there was no LTD induced in CA1 by PPS in the presence of NE.
Relevance to experience-dependent cortical plasticity
The stimulus selectivity of cortical neurons is subject to experience-dependent modification, and such changes are likely to reflect the storage of information by cortical synapses. Theoretical studies have shown that synaptic modifications with the properties of LTP and LTD are well suited to account for experience-dependent shifts in selectivity (Bienenstock et al., 1982; Bear et al., 1987; Bear, 1996). The striking facilitation by NE and ACh of both LTP and LTD suggests that these modulators could function generally as a gain-control mechanism for the synaptic plasticity that underlies receptive field plasticity and learning. Indeed, the notion that cortical synaptic plasticity is somehow “gated” by ACh and NE has received abundant support from studies performed in vivo on visual, auditory, and somatosensory cortex (Kasamatsu and Pettigrew, 1979; Bear and Singer, 1986; Gordon et al., 1990; Juliano et al., 1991;Kasamatsu, 1991; Gu and Singer, 1993; Edeline et al., 1994; Baskerville et al., 1997; Dykes, 1997; Kilgard and Merzenich, 1998; Zhu and Waite, 1998).
Although plasticity is an important feature of adult cortical organization, it is clearly most robust during early postnatal development. A classic example of developmental plasticity in the visual cortex is the loss of responsiveness to stimulation of an eye that has been briefly deprived of vision. Like other forms of cortical plasticity, the deprivation-induced synaptic depression is disrupted by lesions that interrupt cholinergic and noradrenergic inputs to cortex (Bear and Singer, 1986; Gordon et al., 1990). Interestingly, although partial destruction of each input alone was insufficient to produce a detectable loss of deprivation-induced synaptic depression, their combined destruction did disrupt this form of plasticity. This finding led to the suggestion that the two inputs might be able to substitute for one another in the control of cortical plasticity. Our discovery that ACh and NE produce a qualitatively similar facilitation of LTD is entirely consistent with this idea. Thus, the study of LTD and its modulation may provide new insights into the mechanisms of experience-dependent synaptic plasticity in the neocortex.
Footnotes
This work was partly supported by grants from the National Eye Institute, the National Science Foundation, and the Charles A. Dana Foundation. We thank Dr. Kim Huber for helpful comments.
Correspondence should be addressed to Dr. Mark Bear, Howard Hughes Medical Institute and Department of Neuroscience, Brown University, Providence, RI 02912.
REFERENCES
- 1.Aizenman C, Kirkwood A, Bear M. Current source density analysis of evoked responses in visual cortex in vitro: implications for the regulation of long-term potentiation. Cereb Cortex. 1996;6:751–758. doi: 10.1093/cercor/6.6.751. [DOI] [PubMed] [Google Scholar]
- 2.Aramakis VB, Bandrowski AE, Ashe JH. Activation of muscarinic receptors modulates NMDA receptor-mediated responses in auditory cortex. Exp Brain Res. 1997;113:484–496. doi: 10.1007/pl00005601. [DOI] [PubMed] [Google Scholar]
- 3.Artola A, Brocher S, Singer W. Different voltage-dependent thresholds for inducing long-term depression and long-term potentiation in slices of rat visual cortex. Nature. 1990;347:69–72. doi: 10.1038/347069a0. [DOI] [PubMed] [Google Scholar]
- 4.Auerbach JM, Segal M. A novel cholinergic induction of long-term potentiation in rat hippocampus. J Neurophysiol. 1994;72:2034–2040. doi: 10.1152/jn.1994.72.4.2034. [DOI] [PubMed] [Google Scholar]
- 5.Auerbach JM, Segal M. Muscarinic receptors mediating depression and long-term potentiation in rat hippocampus. J Physiol (Lond) 1996;492:479–493. doi: 10.1113/jphysiol.1996.sp021323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bakin JS, Weinberger NM. Induction of a physiological memory in the cerebral cortex by stimulation of the nucleus basalis. Proc Natl Acad Sci USA. 1996;93:11219–11224. doi: 10.1073/pnas.93.20.11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baskerville KA, Schweitzer JB, Herron P. Effects of cholinergic depletion on experience-dependent plasticity in the cortex of the rat. Neuroscience. 1997;80:1159–1169. doi: 10.1016/s0306-4522(97)00064-x. [DOI] [PubMed] [Google Scholar]
- 8.Bear MF. A synaptic basis for memory storage in the cerebral cortex. Proc Natl Acad Sci USA. 1996;93:13453–13459. doi: 10.1073/pnas.93.24.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bear MF, Kirkwood A. Bidirectional plasticity of cortical synapses. In: Fazeli MS, Collingridge GL, editors. Cortical plasticity: LTP and LTD. Bios Scientific; Oxford: 1996. pp. 191–205. [Google Scholar]
- 10.Bear MF, Singer W. Modulation of visual cortical plasticity by acetylcholine and noradrenaline. Nature. 1986;320:172–176. doi: 10.1038/320172a0. [DOI] [PubMed] [Google Scholar]
- 11.Bear MF, Cooper LN, Ebner FF. A physiological basis for a theory of synaptic modification. Science. 1987;237:42–48. doi: 10.1126/science.3037696. [DOI] [PubMed] [Google Scholar]
- 12.Behrends JC, Buggencate GT. Cholinergic modulation of synaptic inhibition in the guinea pig hippocampus in vitro: excitation of GABAergic interneurons and inhibition of GABA release. J Neurophysiol. 1993;69:626–629. doi: 10.1152/jn.1993.69.2.626. [DOI] [PubMed] [Google Scholar]
- 13.Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blitzer RD, Gil O, Landau EM. Cholinergic stimulation enhances long-term potentiation in the CA1 region of the hippocampus. Neurosci Lett. 1990;119:207–210. doi: 10.1016/0304-3940(90)90835-w. [DOI] [PubMed] [Google Scholar]
- 15.Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- 16.Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- 17.Brocher S, Artola A, Singer W. Agonists of cholinergic and noradrenergic receptors facilitate synergistically the induction of long-term potentiation in slices of rat visual cortex. Brain Res. 1992;573:27–36. doi: 10.1016/0006-8993(92)90110-u. [DOI] [PubMed] [Google Scholar]
- 18.Burgard EC, Sarvey JM. Muscarinic receptor activation facilitates the induction of long-term potentiation (LTP) in the rat dentate gyrus. Neurosci Lett. 1990;116:34–39. doi: 10.1016/0304-3940(90)90382-j. [DOI] [PubMed] [Google Scholar]
- 19.Dodt H-U, Pawelzik P, Zieglgänsberger W. Actions of noradrenaline on neocortical neurons in vitro. Brain Res. 1991;545:307–331. doi: 10.1016/0006-8993(91)91303-i. [DOI] [PubMed] [Google Scholar]
- 20.Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc Natl Acad Sci USA. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dutar P, Nicoll RA. Classification of muscarinic responses in hippocampus in terms of receptor subtypes and second-messenger systems: electrophysiological studies in vitro. J Neurosci. 1988;8:4214–4224. doi: 10.1523/JNEUROSCI.08-11-04214.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dykes RW. Mechanisms controlling neuronal plasticity in somatosensory cortex. Can J Physiol Pharmacol. 1997;75:535–545. [PubMed] [Google Scholar]
- 23.Dykes RW, Tremblay N, Warren RA, Bear MF. Cholinergic modulation of synaptic plasticity in sensory neocortex. In: Richardson RT, editor. Activation to acquisition: functional aspects of the basal forebrain cholinergic system. Birkhäuser; Boston: 1990. pp. 325–346. [Google Scholar]
- 24.Edeline JM, Hars B, Maho C, Hennevin E. Transient and prolonged facilitation of tone-evoked responses induced by basal forebrain stimulations in the rat auditory cortex. Exp Brain Res. 1994;97:373–386. doi: 10.1007/BF00241531. [DOI] [PubMed] [Google Scholar]
- 25.Frank H, Kirkwood A, Paradiso MA, Bear MF. Probing the “plasticity gate” in visual cortex using paired-pulse stimulation. Soc Neurosci Abstr. 1995;21:2024. [Google Scholar]
- 26.Gil Z, Connors BW, Amitai Y. Differential regulation of neocortical synapses by neuromodulators and activity. Neuron. 1997;19:679–686. doi: 10.1016/s0896-6273(00)80380-3. [DOI] [PubMed] [Google Scholar]
- 27.Gordon B, Mitchell B, Mohatadi K, Roth E, Tseng Y, Turk F. Lesions of non-visual inputs affect plasticity, norepinephrine content and acetylcholine content of visual cortex. J Neurophysiol. 1990;64:1851–1860. doi: 10.1152/jn.1990.64.6.1851. [DOI] [PubMed] [Google Scholar]
- 28.Gu Q, Singer W. Effects of intracortical infusion of anticholinergic drugs on neuronal plasticity in kitten striate cortex. Eur J Neurosci. 1993;5:475–485. doi: 10.1111/j.1460-9568.1993.tb00514.x. [DOI] [PubMed] [Google Scholar]
- 29.Huerta PT, Lisman JE. Heightened synaptic plasticity of hippocampal CA1 neurons during a cholinergically induced rhythmic state. Nature. 1993;364:723–725. doi: 10.1038/364723a0. [DOI] [PubMed] [Google Scholar]
- 30.Juliano SL, Ma W, Eslin D. Cholinergic depletion prevents expansion of topographic maps in somatosensory cortex. Proc Natl Acad Sci USA. 1991;88:780–784. doi: 10.1073/pnas.88.3.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kasamatsu T. Adrenergic regulation of visuocortical plasticity: a role of the locus coeruleus system. Prog Brain Res. 1991;88:599–616. doi: 10.1016/s0079-6123(08)63837-6. [DOI] [PubMed] [Google Scholar]
- 32.Kasamatsu T, Pettigrew JD. Restoration of visual cortical plasticity by local microperfusion of norepinephrine. J Comp Neurol. 1979;185:163–182. doi: 10.1002/cne.901850110. [DOI] [PubMed] [Google Scholar]
- 33.Kato N. Mechanisms of beta-adrenergic facilitation in rat visual cortex. NeuroReport. 1993;4:1087–1090. [PubMed] [Google Scholar]
- 34.Katsuki H, Izumu Y, Zorumski CF. Noradrenergic regulation of synaptic plasticity in the hippocampal CA1 region. J Neurophysiol. 1997;77:3013–3020. doi: 10.1152/jn.1997.77.6.3013. [DOI] [PubMed] [Google Scholar]
- 35.Kilgard MP, Merzenich MM. Cortical map reorganization enabled by nucleus basalis activity. Science. 1998;279:1714–1718. doi: 10.1126/science.279.5357.1714. [DOI] [PubMed] [Google Scholar]
- 36.Kimura F, Baughman RW. Distinct muscarinic receptor subtypes suppress excitatory and inhibitory synaptic responses in cortical neurons. J Neurophysiol. 1997;77:709–716. doi: 10.1152/jn.1997.77.2.709. [DOI] [PubMed] [Google Scholar]
- 37.Kirkwood A, Bear MF. Hebbian synapses in visual cortex. J Neurosci. 1994a;14:1634–1645. doi: 10.1523/JNEUROSCI.14-03-01634.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirkwood A, Bear MF. Homosynaptic long-term depression in the visual cortex. J Neurosci. 1994b;14:3404–3412. doi: 10.1523/JNEUROSCI.14-05-03404.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kirkwood A, Dudek SM, Gold JT, Aizenman CD, Bear MF. Common forms of synaptic plasticity in the hippocampus and neocortex in vitro. Science. 1993;260:1518–1521. doi: 10.1126/science.8502997. [DOI] [PubMed] [Google Scholar]
- 40.Kojic L, Gu Q, Douglas RM, Cynader MS. Serotonin facilitates synaptic plasticity in kitten visual cortex: an in vitro study. Dev Brain Res. 1997;101:299–304. doi: 10.1016/s0165-3806(97)00083-7. [DOI] [PubMed] [Google Scholar]
- 41.Luhmann HJ, Prince DA. Postnatal maturation of the GABAergic system in rat neocortex. J Neurophysiol. 1991;65:247–263. doi: 10.1152/jn.1991.65.2.247. [DOI] [PubMed] [Google Scholar]
- 42.Madison DV, Nicoll RA. Norepinephrine decreases synaptic inhibition in the rat hippocampus. Brain Res. 1988;442:131–138. doi: 10.1016/0006-8993(88)91440-0. [DOI] [PubMed] [Google Scholar]
- 43.Markram H, Segal M. Acetylcholine potentiates responses to N-methyl-d-aspartate in the rat hippocampus. Neurosci Lett. 1990;113:62–65. doi: 10.1016/0304-3940(90)90495-u. [DOI] [PubMed] [Google Scholar]
- 44.Markram H, Segal M. The inositol 1,4,5-triphosphate pathway mediates cholinergic potentiation of rat hippocampal neuronal responses to NMDA. J Physiol (Lond) 1992;447:513–533. doi: 10.1113/jphysiol.1992.sp019015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Markram H, Tsodyks M. Redistribution of synaptic efficacy between neocortical pyramidal neurons. Nature. 1996;382:807–810. doi: 10.1038/382807a0. [DOI] [PubMed] [Google Scholar]
- 46.McCormick DA, Prince DA. Mechanisms of action of acetylcholine in the guinea-pig cerebral cortex in vitro. J Physiol (Lond) 1986;375:169–194. doi: 10.1113/jphysiol.1986.sp016112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Metherate R, Ashe JH. Facilitation of an NMDA receptor-mediated EPSP by paired-pulse stimulation in rat neocortex via depression of GABAergic IPSPs. J Physiol (Lond) 1994;481:331–348. doi: 10.1113/jphysiol.1994.sp020443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mitzdorf U. Current source-density method and application in cat cerebral cortex: investigation of evoked potentials and EEG phenomena. Physiol Rev. 1985;65:37–100. doi: 10.1152/physrev.1985.65.1.37. [DOI] [PubMed] [Google Scholar]
- 49.Mrzljak L, Levey A, Goldman-Rakic PS. Association of m1 and m2 muscarinic receptor proteins with asymmetric synapses in the primate cerebral cortex: morphological evidence for cholinergic modulation of excitatory neurotransmission. Proc Natl Acad Sci USA. 1993;90:5194–5198. doi: 10.1073/pnas.90.11.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murakoshi T. Cholinergic modulation of synaptic transmission in the rat visual cortex in vitro. Vision Res. 1995;35:25–35. doi: 10.1016/0042-6989(94)e0056-q. [DOI] [PubMed] [Google Scholar]
- 51.Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- 52.Osterheld-Haas MC, Van der Loos H, Hornung JP. Monoaminergic afferents to cortex modulate structural plasticity in the barrelfield of the mouse. Dev Brain Res. 1994;77:189–202. doi: 10.1016/0165-3806(94)90196-1. [DOI] [PubMed] [Google Scholar]
- 53.Roche K, Tingley W, Huganir R. Glutamate receptor phosphorylation and synaptic plasticity. Curr Opin Neurobiol. 1994;4:383–388. doi: 10.1016/0959-4388(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 54.Sachdev R, Lu S, Wiley R, Ebner F. Role of the basal forebrain cholinergic projection in somatosensory cortical plasticity. J Neurophysiol. 1998;79:3216–3228. doi: 10.1152/jn.1998.79.6.3216. [DOI] [PubMed] [Google Scholar]
- 55.Schliebs R, Walch C, Stewart MG. Laminar pattern of cholinergic and adrenergic receptors in rat visual cortex using quantitative receptor autoradiography. J Hirnforsch. 1989;30:303–311. [PubMed] [Google Scholar]
- 56.Segal M. Multiple actions of acetylcholine at a muscarinic receptor studied in the hippocampal slice. Brain Res. 1982;246:77–87. doi: 10.1016/0006-8993(82)90144-5. [DOI] [PubMed] [Google Scholar]
- 57.Segal M. Acetylcholine enhances NMDA-evoked calcium rise in hippocampal neurons. Brain Res. 1992;587:83–87. doi: 10.1016/0006-8993(92)91430-m. [DOI] [PubMed] [Google Scholar]
- 58.Shaw C, Wilkinson M, Cynader M, Needler MC, Aoki C, Hall SE. The laminar distributions and postnatal development of neurotransmitter and neuromodulator receptors in cat visual cortex. Brain Res Bull. 1986;16:661–671. doi: 10.1016/0361-9230(86)90137-1. [DOI] [PubMed] [Google Scholar]
- 59.Singer W. Central-core control of visual cortex functions. In: Schmitt FO, Worden FG, editors. The neurosciences fourth study program. MIT; Cambridge, MA: 1979. pp. 1093–1109. [Google Scholar]
- 60.Singer W. Development and plasticity of cortical processing architectures. Science. 1995;270:758–759. doi: 10.1126/science.270.5237.758. [DOI] [PubMed] [Google Scholar]
- 61.Sokolov M, Kleschevnikov A. Atropine suppresses associative LTP in CA1 region of rat hippocampal slices. Brain Res. 1995;672:281–284. doi: 10.1016/0006-8993(94)01376-s. [DOI] [PubMed] [Google Scholar]
- 62.Stanton PK, Sarvey JM. Norepinephrine regulates long-term potentiation of both the population spike and dendritic EPSP in hippocampal dentate gyrus. Brain Res Bull. 1987;18:115–119. doi: 10.1016/0361-9230(87)90039-6. [DOI] [PubMed] [Google Scholar]
- 63.Stanton PK, Sejnowski TJ. Associative long-term depression in the hippocampus induced by Hebbian covariance. Nature. 1989;229:215–218. doi: 10.1038/339215a0. [DOI] [PubMed] [Google Scholar]
- 64.Tanaka Y, Sakurai M, Hayashi S. Effect of scopolamine and HP 029, a cholinesterase inhibitor, on long-term potentiation in hippocampal slices of the guinea pig. Neurosci Lett. 1989;98:179–193. doi: 10.1016/0304-3940(89)90506-5. [DOI] [PubMed] [Google Scholar]
- 65.Thomas MJ, Moody TD, Makhinson M, O’Dell TJ. Activity-dependent β-adrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron. 1996;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- 66.Vaknin G, Teyler TJ. Ontogenesis of the depressant activity of carbachol on synaptic activity in rat visual cortex. Brain Res Bull. 1991;26:211–214. doi: 10.1016/0361-9230(91)90228-c. [DOI] [PubMed] [Google Scholar]
- 67.Valentino RJ, Dingledine R. Presynaptic inhibitory effect of acetylcholine in the hippocampus. J Neurosci. 1981;1:784–792. doi: 10.1523/JNEUROSCI.01-07-00784.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wagner JJ, Alger BE. GABAergic and developmental influences on homosynaptic LTD and depotentiation in rat hippocampus. J Neurosci. 1995;15:1577–1586. doi: 10.1523/JNEUROSCI.15-02-01577.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Z, McCormick DA. Control of firing mode of corticotectal and corticopontine layer V burst-generating neurons by norepinephrine, acetylcholine, and 1S,3R-ACPD. J Neurosci. 1993;13:2199–2216. doi: 10.1523/JNEUROSCI.13-05-02199.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu XO, Waite PME. Cholinergic depletion reduces plasticity of barrel field cortex. Cereb Cortex. 1998;8:63–72. doi: 10.1093/cercor/8.1.63. [DOI] [PubMed] [Google Scholar]