

Abstract
Nigrostriatal dopaminergic neurons undergo sprouting around the margins of a striatal wound. The mechanism of this periwound sprouting has been unclear. In this study, we have examined the role played by the macrophage and microglial response that follows striatal injury. Macrophages and activated microglia quickly accumulate after injury and reach their greatest numbers in the first week. Subsequently, the number of both cell types declines rapidly in the first month and thereafter more slowly. Macrophage numbers eventually cease to decline, and a sizable group of these cells remains at the wound site and forms a long-term, highly activated resident population. This population of macrophages expresses increasing amounts of glial cell line-derived neurotrophic factor mRNA with time. Brain-derived neurotrophic factor mRNA is also expressed in and around the wound site. Production of this factor is by both activated microglia and, to a lesser extent, macrophages. The production of these potent dopaminergic neurotrophic factors occurs in a similar spatial distribution to sprouting dopaminergic fibers. Moreover, dopamine transporter-positive dopaminergic neurites can be seen growing toward and embracing hemosiderin-filled wound macrophages. The dopaminergic sprouting that accompanies striatal injury thus appears to result from neurotrophic factor secretion by activated macrophages and microglia at the wound site.
Keywords: sprouting, BDNF, GDNF, macrophage, microglia, dopamine, striatal injury
Collateral sprouting is a process whereby neurons form additional axonal branches. In the CNS, sprouting occurs at the margins of traumatic wounds and in the terminal fields of incompletely lesioned pathways.
In the nigrostriatal system, dopaminergic neurons undergo sprouting at the margins of striatal wounds. Such periwound sprouting was initially observed around the margins of adrenal medullary grafts implanted into the striatum (Bohn et al., 1987; Fiandaca et al., 1988; Bankiewicz et al., 1994). Subsequent experiments have demonstrated that nigrostriatal dopaminergic neurons will also form a halo of sprouting fibers around a striatal wound when other tissue types are implanted (Bankiewicz et al., 1988, 1991) or even when the striatum is simply wounded and no tissue is implanted (Plunkett et al., 1990; Liberatore et al., 1996).
The exact mechanism by which neurons are induced to sprout is unknown, but it is believed that neurotrophic factors (NTFs) are involved (Gallo and Letourneau, 1998). Basic fibroblast growth factor (bFGF) and ciliary neurotrophic factor (CNTF) have some effect on enhancing the survival of dopaminergic neurons in vitro (Hagg and Varon, 1993; Mayer et al., 1993) and are produced by striatal reactive astrocytes (Asada et al., 1995; Ho and Blum, 1997). Astroglial proliferation is stimulated by interleukin-1 (IL-1) (Giulian et al., 1988), and striatal implants of IL-1 stimulate both periwound astrocytosis and dopaminergic sprouting (Wang et al., 1994). On this basis, it has been hypothesized that reactive astrocytosis may be primarily responsible for inducing periwound dopaminergic sprouting. Activated macrophages and microglia have been hypothesized to play an indirect role in this process by secreting IL-1 (Bankiewicz et al., 1988; Wang et al., 1991, 1994).
The two NTFs, however, which have demonstrated the greatest capacity to stimulate dopaminergic neurons to undergo sprouting, are brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF). In vitro, both factors stimulate profuse neurite extension from dopaminergic neurons (Hyman et al., 1991; Lin et al., 1993). In vivo models of Parkinson’s disease [1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-lesioned primates and 6-OHDA lesioned rats] demonstrate that GDNF dramatically restores the function of dopaminergic neurons and ameliorates neurological deficits (Gash et al., 1996), whereas BDNF also protects nigrostriatal neurons from MPTP toxicity (Beck, 1992). With regards to dopaminergic sprouting, tyrosine hydroxylase-positive (TH+ve) fibers can be seen to sprout and extend neurites into striatal grafts of fibroblasts genetically modified to produce BDNF (Lucidi-Phillipi et al., 1995), as well as encapsulated cells producing GDNF (Linder et al., 1995). In addition, the injection of BDNF into the striatum induces a halo of dopaminergic fibers to sprout around the injection site (Shults et al., 1995), whereas TH+ve fibers have also been shown to grow into the site of nigral injections of GDNF (Bowenkamp et al., 1995). With regards to the potential involvement of these two factors in periwound dopaminergic sprouting, our own data demonstrate that the synthesis of both BDNF (Wong et al., 1997) and GDNF (Liberatore et al., 1997) substantially increases around the site of striatal injury.
The aims of this study were twofold. The first was to determine which cells around the striatal wound synthesize BDNF and GDNF. The second was to determine whether sprouting dopaminergic fibers grow toward and are associated with these cells, thus implicating their involvement in generating the sprouting response.
MATERIALS AND METHODS
Surgical procedure. Male C-57 Black/6J mice (17–25 gm, aged 6–8 weeks) were anesthetized (Nembutal 70 mg/kg and atropine sulfate 0.5 mg/kg, i.p.) and placed in a stereotactic frame. A Scouten wire knife (model 120, blade 121-A; Kopf Instruments) was inserted into the striatum via a burr hole (anteroposterior, 0.4 mm; lateral, 2.0 mm; dorsoventral, 2.5 mm relative to bregma and the dural surface), and once in position, the blade was extended to give a cut diameter of ∼0.5 mm. Rotating the blade housing 10 times about its vertical axis and then twice reducing the cannula depth by 0.25 mm and rotating the blade an additional 10 times at each position created a cylinder of damaged striatum ∼0.5 mm in diameter and 0.5 mm deep. The scalp was then closed.
Experimental groups. A total of 200 mice were used: 122 underwent surgery and 78 were kept as unlesioned controls. For the macrophage and microglial time course experiment, there were 28 mice in both the surgical and control groups. Cell counts of activated macrophages and microglia were performed on four lesioned animals and four control animals at 0, 1, 3, 7, 14, 30, and 120 d after injury. In the astrocyte time course experiment, 20 mice underwent surgery and 20 were kept as controls, and counts of glial fibrillary acidic protein (GFAP)-positive reactive astrocytes were made in four lesioned animals and four control animals at 0, 1, 3, 7, and 30 d after injury.
For GDNF quantitation, 28 mice underwent surgery and were killed 0 (n = 4), 1 (n = 7), 7 (n = 5), 14 (n = 4), 30 (n = 5), and 120 (n = 3) d after injury. Fourteen mice were kept as uninjured controls and were killed at 0 (n = 3), 1 (n = 3), 7 (n = 3), and 30 (n = 5) d. Sixteen injured and 16 control mice were used for BDNF quantitation, and four from each group were killed after 0, 7, 30, and 120 d.
An additional 30 injured mice were used in the GDNF (n= 12) and BDNF (n = 12) colocalization studies and dopamine transporter (DAT) immunohistochemistry (n = 6).
Tissue preparation. To prepare fresh frozen tissue, mice were killed by Nembutal overdose (600 mg/kg, i.p.), and the brains were snap frozen in dry ice-cooled isopentane before storage at −80°C. A series of 20 μm coronal sections spanning the damage site were then cut from each brain using a cryostat (Bright Instruments) maintained at −20°C, thaw-mounted onto 3-aminopropyltriethoxysilane-coated glass slides, desiccated, and stored at −80°C.
Perfusion-fixed tissue was prepared by deeply anesthetizing and intracardially perfusing mice with 30 ml of cold 0.1 m PBS, pH 7.4, followed by 80 ml of cold 4% paraformaldehyde in 0.1m PBS, pH 7.4. Animals were then decapitated, and brains were fixed for a further 12 hr in the same solution, followed by immersion overnight at 4°C in 20% sucrose. The brains were then frozen in dry ice-cooled isopentane and stored at −80°C before cutting 40 μm coronal sections on a cryostat.
Histochemistry. For nonspecific esterase (NSE) histochemistry, fresh frozen 20 μm sections were first post-fixed in a 0.1 m buffered potassium phosphate solution, pH 6.6, containing 25% formaldehyde and 45% acetone for 8 min. NSE was then identified using the method described by Koski et al. (1976).
The iron component of hemosiderin was demonstrated using Perls prussian blue reaction for ferric irons. Fresh frozen 20 μm sections were first stained for NSE as described above. Sections were then washed and incubated for 30 min in a solution containing 0.3 mhydrochloric acid and 2.4 mm potassium ferrocyanide. Slides were subsequently washed before being dehydrated and coverslipped.
Anti-MAC-1 is a rat monoclonal antibody that labels mouse macrophages and microglia. For MAC-1 immunohistochemistry, fresh frozen tissue was first post-fixed as described for NSE cytochemistry. Slide-mounted sections were then processed as detailed below, except that the methanol and serum blocking steps were omitted. For DAT and GFAP immunohistochemistry, free-floating sections were washed in 0.1m PBS, pH 7.4, (and subsequently washed three times for 5 min each in the same buffer between each step) and then incubated for 30 min in 0.01% H2O2 in methanol, followed by 10% normal horse serum for 20 min. Sections were incubated at 4°C in primary antibody [overnight with rat monoclonal MAC-1 (Serotec, Indianapolis, IN) at a dilution of 1:50; 2 weeks with rat monoclonal anti-DAT (Chemicon, Temecula, CA) at a dilution of 1:1000; overnight with rabbit anti-GFAP polyclonal (Sigma, St. Louis, MO) at a dilution of 1:1000] containing 2% normal horse serum and 0.3% Triton X-100, before being incubated for 60 min at room temperature with biotinylated secondary anti-rat IgG (anti-rabbit IgG for GFAP) diluted 1:200 (Vector Laboratories, Burlingame, CA). Tissue was then incubated for 60 min in avidin–biotin–peroxidase complex (Vectastain Elite kit, 1:200; Vector Laboratories), before finally being incubated for 10 min with 0.05% 3,3′diaminobenzidine tetrahydrochloride (DAB) containing 4.2 mm cobalt chloride and 2.5 mm ammonium nickel sulfate. Hydrogen peroxide was added to a final concentration of 0.001%, and sections were reacted for 30 min. All sections were subsequently washed in PBS, mounted on 0.1% gelatin-coated slides, dried, cleared in xylene, and coverslipped.
Macrophages and activated microglia were counted in five successive sections (one section through the middle of the wound and two sections on either side), and the figures were summed. Group averages were then calculated to give a mean number of cells (per five representative sections) at each time point. Macrophages were defined as MAC-1- or NSE-positive cells (>7.5 μm in diameter) without any significant cytoplasmic processes. These criteria are similar to those described previously (Riva-Depaty et al., 1994). On the basis of observations made in uninjured animals, MAC-1-positive resting microglia were defined as cells with a relatively small cell body (<7.5 μm in diameter) and long processes. After injury, activated microglia were identified as more intensely stained cells with a larger cell body and relatively short processes. Using a combination of morphological criteria and a cell body diameter cutoff of 7.5 μm, microglia were classified as either resting or activated. An estimate of average macrophage cell size was made by determining the diameter (using a 2.5 μm/division grid with a 40× objective) of 100 MAC-1- and NSE-positive wound macrophages (25 from each of four animals) at each time point. Only clearly defined, positively stained cells within the wound were sized.
Astrocytes were counted using the method described by Miyake et al. (1987) from a minimum of three sections from within the body of the striatal wound (one section through the middle of the wound and a section on either side). The number of astrocytes was then averaged to give a mean figure per section at each time point.
In situ hybridization. GDNF and BDNF mRNA were detected using our previously described in situ hybridization methods (Liberatore et al., 1997; Wong et al., 1997). Briefly, for GDNF mRNA detection, two 50-mer antisense oligonucleotides were used. For BDNF mRNA detection, a single 50-mer antisense oligonucleotide probe was used. These probes were dissolved in dH2O to give a stock concentration of ∼3 μg/ml, end-labeled by a standard kinase protocol using [γ-33P]ATP (Amersham International, Cardiff, UK) and T4 polynucleotide kinase (New England Biolabs, Beverly, MA), and then purified using S200 Microspin columns (Pharmacia, Piscataway, NJ). Hybridization was performed in a drop of 20 mm sodium phosphate buffer, pH 7.0 (containing 600 mm NaCl, 60 mm sodium citrate, 0.02% Ficoll, 0.02% bovine serum albumin, 0.02% poly(vinylpyrrolidone), 10% dextran sulfate, 0.1% degraded herring sperm DNA, 1 mmdithiothreitol, and 50% de-ionized formamide), placed on the section, and incubated at 42°C for 18 hr in a humidified container. Sections were then washed four times for 15 min each in 1× SSC at 55°C before dehydration in ethanol and drying at room temperature.
Detection of GDNF and BDNF hybridized mRNA was first performed by exposing the slides (together with laboratory-prepared 33P standards) to Hyperfilm (Amersham International). Exposure times were 4 weeks for GDNF and 1 week for BDNF. Quantitation of dry film autoradiograms was performed using a microcomputer imaging device (Imaging Research Inc., Brock University, St. Catharines, Ontario, Canada). For GDNF, densitometry was performed on the entire striatum visible in each hemisphere after counter-staining the sections. For BDNF, densitometry was performed over a 1-mm-diameter region that encompassed the wound site, as well as a corresponding area in the contralateral hemisphere. Standardization was achieved by comparing binding densities with a standard curve created from the33P standards exposed with each film.
Controls were provided by incubating sections with complementary sense probes, by using excess unlabeled oligonucleotides to competitively abolish probe binding and by preincubation with RNase-A degrade mRNA (Liberatore et al., 1997; Wong et al., 1997). No signal above background was detected with these controls for either BDNF or GDNFin situ hybridization.
For colocalization studies, cytochemistry or immunohistochemisry was performed in autoclaved water as detailed above (DAB was left unintensified). Tissue was then digested with proteinase K (20 μg/ml PBS, 10 min) before being post-fixed for 10 min in 0.4% paraformaldehyde. In situ hybridization was then performed as described above. Slides were dipped in LM-1 Hypercoat nucleic emulsion (Amersham International) at 43°C and exposed for 1 (BDNF) or 8 (GDNF) weeks. After development, emulsion-dipped sections were dehydrated and coverslipped and then photographed under bright-field illumination on an Olympus BX60 microscope (Olympus Optical, Tokyo, Japan).
Statistical analysis. All values are expressed as mean ± SEM. Comparisons between contralateral and ipsilateral striatum within animals and differences between groups of animals were analyzed using one-way ANOVA with Tukey honestly significant differencepost hoc error correction. In all analyses, the null hypothesis (that there was no difference between means) was rejected atp ≤ 0.05, p ≤ 0.005, orp ≤ 0.0005 as stated. The Mann–Whitney Utest was used to ascertain whether the number of NSE- and MAC-1-positive macrophages detected 4 months after injury were identical. Statistical analysis was performed with Simstat for Windows (version 7; Provalis Research, Montreal, Canada) on a Pentium computer.
RESULTS
Appearance of activated macrophages, microglia, and reactive astrocytes
MAC-1 immunostained resting microglia had a small cell body, long processes, and relatively faint immunoreactivity (Fig.1A). In contrast, activated microglia had a larger cell body, short retracted processes, and intense immunoreactivity (Fig. 1B). Activated microglia were first visible in and around the wound 1 d after lesion. Peak numbers of activated microglia occurred 1 week after striatal injury and then declined rapidly in the subsequent 3 weeks, with a slow (but not statistically significant) decline thereafter (Fig. 2).
Fig. 1.
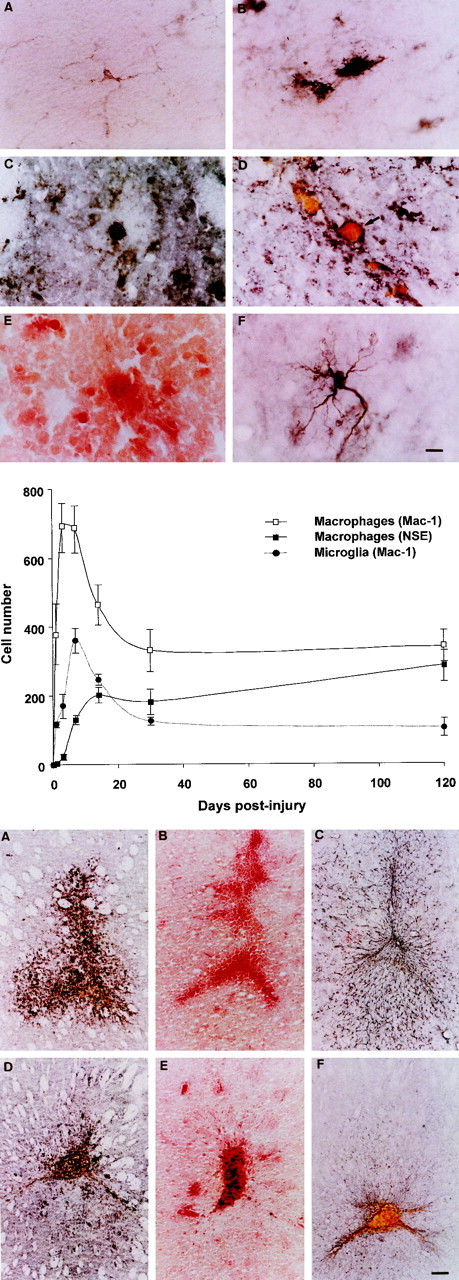
Top. Photomicrographs of periwound reactive glia. A, MAC-1 immunostained resting microglial cell. Note the small cell body, long processes, and relatively faint immunoreactivity. B, MAC-1 immunostained activated microglial cell. Note the relatively large cell body, short, retracted processes, and intense immunoreactivity. C, MAC-1 immunostained wound macrophage at day 1. D, MAC-1 immunostained wound macrophage at 1 month. Note, the diameter has significantly increased, and the cell is filled with hemosiderin.E, NSE-positive macrophage (30 d). F, GFAP-positive periwound reactive astrocyte. Scale bar, 10 μm.
Fig. 2.

Middle. Line graphs showing the changing number of MAC-1-positive macrophages, NSE-positive macrophages, and MAC-1-positive microglia in groups of four mice at 0, 1, 3, 7, 14, 30, and 120 d after striatal injury. All values are the mean number of cells in five representative sections through the wound (1 section through the middle of the wound and 2 sections either side). Error bars indicate SEM.
The MAC-1 antibody identifies the membrane-bound complement 3 receptor (Beller et al., 1982). Hence, within the wound area, MAC-1 immunolabeled macrophages appeared as large circumferentially stained cells without any significant cytoplasmic processes (Fig.1C,D). NSE+vemacrophages had a similar morphology (Fig.1E), but in this case, staining was cytoplasmic. At higher magnification, the interior of these cells could be seen to be filled with a conglomeration of intensely red, fine intracytoplasmic granules.
MAC-1 immunostained macrophages (Fig.1C,D) were first visible in the striatal wound 1 d after injury. The number of such macrophages peaked at 3–7 d after lesion and then progressively declined until only half the peak number were present 1 month after lesion. Thereafter, macrophage numbers remained constant, with a resident population of cells continuing to be present 4 months after injury. This resident population contained approximately half the number of macrophages present at 3 d after lesion (Fig. 2).
In contrast to MAC-1+ve macrophages, NSE+ve macrophages (Fig. 1E) were not visible in significant numbers until 1 week after injury (Fig. 2). Thereafter, they continued to slowly increase in both number and staining intensity until 4 months after injury, when the numbers of NSE+ve (286 ± 47, mean ± SEM) and MAC-1+ve (340 ± 47) macrophages were identical (Mann–Whitney U test; p = 0.49).
At 1 week or thereafter, many macrophages contained large cytoplasmic deposits of a granular yellow-brown pigment (Fig.1D). This pigment was identified as iron-rich hemosiderin by its intense blue coloration with Perls stain. It was also observed that the MAC-1 or NSE+ve macrophages present at 1 week or later (Fig.1D,E) were considerably larger than MAC-1 immunostained macrophages present at day 1 (Fig. 1C). MAC-1+ve/NSE negative (NSE−ve) macrophages at day 1 had an average cell body diameter of 8.3 ± 0.1 μm, and at day 3, their average diameter had increased to 14.0 ± 0.1 μm. MAC-1+ve or NSE+ve macrophages present at 1 week and later had average cell body diameters of 15.8 ± 0.3 and 15.0 ± 0.3 (1 week), 17.5 ± 0.4 and 17.3 ± 0.3 (2 weeks), 18.6 ± 0.9 and 18.8 ± 0.5 (1 month), and 19.1 ± 0.6 and 19.1 ± 0.7 (4 months) μm, respectively.
Periwound GFAP+ve astrocytes (Fig.1F) were not visible immediately after injury but were detected in small numbers (54 ± 13 per 4 mm2) on the first day after injury. The number of reactive astrocytes increased at day 3 (131 ± 16 per 4 mm2) and peaked at 1 week (656 ± 88 per 4 mm2)(Fig.3C) after striatal trauma. Subsequently, numbers of reactive astrocytes fell and by 1 month after injury had decreased significantly to 171 ± 31 per 4 mm2.
Fig. 3.
Bottom. Photomicrographs demonstrating the changing number and distribution of reactive glia. A, MAC-1 immunostained section 1 week after injury demonstrating numerous periwound-activated microglia. B, NSE stained section 7 d after injury. NSE staining is relatively faint at this time point, and few cells are labeled. C, Section demonstrating the relatively widespread periwound distribution of GFAP-positive reactive astrocytes 7 d after injury.D, MAC-1 immunostained section 30 d after injury. At this time point, far fewer activated microglia are present. Note the large number of hemosiderin macrophages in the body of the wound.E, NSE stained section 30 d after injury. Macrophages at this time point appear as large intensely stained cells within the body of the wound. F, Section stained for GFAP 30 d after injury. At this time, reactive astrocytes are restricted to the immediate area of the wound. Scale bar, 100 μm.
One week after injury, MAC-1 immunoreactive cells were present both within the wound and in a clearly defined band of tissue around the wound (Fig. 3A). These cells comprised a population of MAC-1 immunoreactive macrophages centered within the wire knife cuts and numerous periwound-activated microglia. At this time, relatively few NSE stained macrophages were present, but these too were centered on the wire knife cuts. In contrast, a much larger number of GFAP immunoreactive astrocytes could be seen throughout the wounded striatum and beyond into many other structures in the wounded hemisphere (Fig.3C). One month after injury, a relatively small number of large MAC-1 immunoreactive microglia were present around the wound (Fig. 3D). Within the wound, both microglia (Fig.3D) and a large number of hemosiderin-filled macrophages were clearly seen (Fig.3D,F), with the distribution of the later closely matching the pattern of intense NSE staining (Fig.3E). One month after injury, GFAP immunoreactivity had contracted to a scattering of faintly positive cells around the wound and a narrow band of still intensely positive cells at the margins of the wound (Fig. 3F).
Expression of BDNF mRNA by activated microglia and macrophages
The silver grains of the BDNF in situ signal were located over cells within and in the neighborhood of the striatal wound on unstained emulsion-dipped sections. Colocalization studies combining BDNF in situ hybridization with MAC-1 immunolabeling revealed that the silver grains of the in situ signal were deposited in foci over the brown DAB deposits of activated microglia (Fig. 4A–C). The largest and most intense silver grain deposits were located over the biggest and morphologically most highly activated microglia. Silver grains were also present over wound macrophages, but their density was considerably less than the deposits over activated microglia. No silver grain deposits could be seen over GFAP+veastrocytes.
Fig. 4.
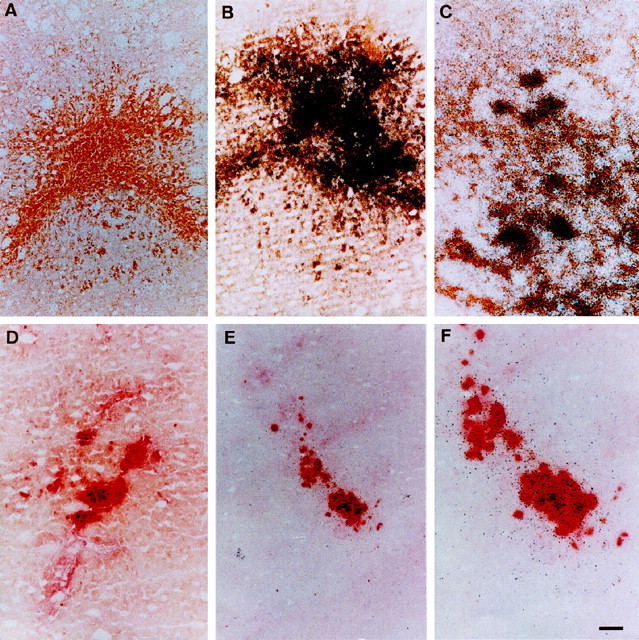
Colocalization of neurotrophins to activated microglia and macrophages. A, MAC-1 immunostained section 7 d after injury. DAB is unintensified, and activated microglia and macrophages are stained yellow-brown.B, MAC-1 immunostained section 7 d after injury in which BDNF in situ hybridization has also been performed. Note the intense silver grain deposits over MAC-1 immunostained cells. C, High-power photomicrograph showing the colocalization of the BDNF in situ signal (black silver grain deposits) with MAC-1 immunolabeled activated microglia. D, Section stained for NSE 30 d after injury. Macrophages appear as large red cells within the wound area. E, Section stained for NSE 30 d after injury in which GDNF in situhybridization has also been performed. The silver grains of thein situ signal are colocalized with the NSE-positive macrophages. F, High-power photomicrograph ofE. Scale bar: A, B,D, E, 100 μm; C,F, 25 μm.
Quantitation of the BDNF signal by dry film autoradiography at each time point (Fig. 5) revealed that there was a peak in BDNF expression 1 week after surgery. At this time, BDNF expression had increased 2½-fold over baseline (p ≤ 0.0005). Subsequently, BDNF expression decreased. A relatively rapid decline in expression occurred in the following 3 weeks, and there was no statistically significant change thereafter. The temporal pattern of BDNF expression closely matched the temporal pattern of microglial activation (r2 = 0.96; p = 0.018) (Fig.6).
Fig. 5.
Top, left. Autoradiograms and line graph showing the changing expression of BDNF after striatal injury. The line graph shows striatal BDNF mRNA expression (counts per minute/square millimeter) 0, 7, 30, and 120 d after injury in ipsilateral and contralateral striatum (n = 4 at each time point). All values are mean ± SEM. The autoradiographs are representative sections at key time points showing the changing expression of BDNF. Note that the expression of BDNF at 7 d is relatively widespread, whereas at 30 d it is restricted to the immediate periwound area. Increasing signal intensity is represented by color change fromblue to red. *p ≤ 0.0005; $p ≤ 0.05.
Fig. 6.
Bottom, left. Line graphs showing the temporal correlation between numbers of activated microglia (mean number of cells in 5 representative sections through the wound) and the quantitative expression of BDNF (counts above baseline in counts per minute/square millimeter). Linear regression analysis is shown as aninset.
The BDNF dry film autoradiography images showed that BDNF mRNA production at 1 week was relatively widespread within the striatum, whereas at 1 month, production was restricted to a thin band of tissue immediately adjacent to the wound. This spatial change in BDNF expression was very similar to the change in activated microglia distribution (Fig. 5).
BDNF in situ hybridization controls showed no signal above background on sections pretreated with RNase-A, after competition with 100-fold excess of unlabeled probe or when using the corresponding sense probes.
Expression of GDNF mRNA by activated macrophages and microglia
The silver grains of the GDNF in situ signal were located over large yellow-brown deposits of hemosiderin in unstained emulsion-dipped sections. As mentioned, hemosiderin deposits were located within the cytoplasm of MAC-1 and NSE+vemacrophages, and thus the synthesis of GDNF by these cells could be inferred. Direct evidence that macrophages express GDNF mRNA was obtained by combining NSE histochemistry with GDNF in situhybridization. This combined procedure demonstrated that the silver grains of the GDNF in situ signal were exquisitely colocalized with red NSE+ve macrophages (Fig.4D–F). Occasional clusters of silver grains could seen adjacent to these cells, and these were most likely NSE−ve activated microglia, as detailed below. After 1 week, few wound macrophages could be seen that did not also exhibit GDNF expression.
GDNF in situ hybridization was also combined with MAC-1 immunohistochemistry. In this case, the silver grains of the GDNFin situ signal were primarily colocalized with MAC-1+ve macrophages. Some activated microglia around the wound site were also faintly GDNF+ve.
Quantitation of the GDNF signal at each time point revealed that there was a progressive increase in GDNF expression (Fig.7). One week after surgery, GDNF expression was increased twofold (p ≤ 0.0005), whereas 4 months after surgery, expression was increased fourfold (p ≤ 0.0005). The pattern of this increase almost exactly matched the progressive increase in NSE+ve macrophage numbers (r2 = 0.85; p ≤ 0.005) (Fig.8). No correlation was evident between GDNF production and the numbers of reactive astrocytes or activated microglia.
Fig. 7.
Top, right. Autoradiograms and line graph showing the changing expression of GDNF after striatal injury. The line graph shows striatal GDNF mRNA expression (counts per minute/square millimeter) 0, 1, 3, 7, 14, 30, and 120 d after injury in ipsilateral and contralateral striatum (n = 4, 7, 10, 5, 4, 5, and 3 at each time point, respectively). All values are mean ± SEM. The autoradiographs are representative sections at key time points showing the changing expression of GDNF. Note that the expression of GDNF is restricted to the location of the wound at all time points. Increasing signal intensity is represented by color change from blue to red. *p≤ 0.0005; #p ≤ 0.005.
Fig. 8.
Bottom, right. Line graphs showing the temporal correlation between numbers of NSE-positive activated macrophages (mean number of cells in 5 representative sections through the wound) and the quantitative expression of GDNF (counts above baseline in counts per minute/square millimeter). Linear regression analysis is shown as an inset.
GDNF in situ hybridization controls showed no signal above background on sections pretreated with RNase-A after competition with 100-fold excess of unlabeled probe or when using the corresponding sense probes.
Relationship of sprouting dopaminergic fibers to macrophages
In the uninjured striatum, DAT immunohistochemisry enabled clear visualization of fine dopaminergic fibers and terminals. These processes were of a relatively narrow diameter, and at high magnification, terminal boutons could easily be seen.
DAT immunohistochemistry performed 2 weeks after injury demonstrated that within and in the immediate neighborhood of the wound there were many bundles of intensely stained, large diameter, sprouting dopaminergic fibers (Fig. 9). These fibers were orientated toward the wound and could be seen growing toward groups of hemosiderin-containing macrophages that filled the wound (Fig. 9B). At high magnification, these sprouting fibers could be seen curling around and embracing individual macrophages (Fig. 9C–E).
Fig. 9.
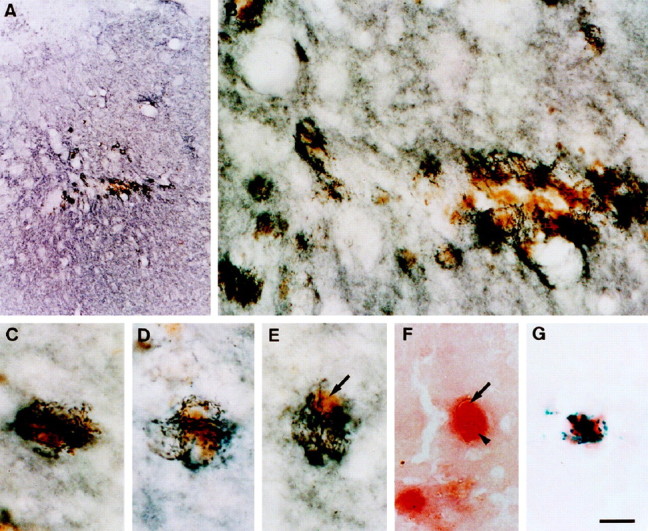
Photomicrographs showing DAT-positive sprouting dopaminergic fibers growing toward and around hemosiderin-filled wound macrophages. A, Low-magnification image showing bundles of dark DAT-positive fibers sprouting in the periwound margin. B, High-magnification image showing sprouting fibers approaching and growing around hemosiderin-containing macrophages. C–E, Higher magnification images showing the intimate association of sprouting dopaminergic fibers to a number of individual hemosiderin-filled (arrow) wound macrophages. F, Photomicrograph demonstrating that hemosiderin (arrow) is located within macrophage cytoplasm (arrowhead) (NSE stain). G, NSE-positive macrophage containing prussian blue deposits of iron-containing hemosiderin. Scale bar:A, 375 μm; B, 25 μm;C–G, 10 μm.
DISCUSSION
The appearance of activated macrophages and microglia
Macrophages become visible within CNS wounds 1–3 d after injury (Giulian et al., 1989; Riva-Depaty et al., 1994), with the majority originating from circulating monocytes (Del Cerro and Monjan, 1979). We found MAC-1+ve macrophages as early as 1 d after injury but did not detect significant numbers of NSE-stained macrophages until 1 week after injury (Fig. 2). The MAC-1 antibody identifies the complement 3 receptor (Beller et al., 1982), which is constitutively expressed by both microglia and macrophages, whereas NSE histochemistry identifies a lysosomal esterase (Koski et al., 1976) expressed in macrophages but not microglia (Ulvestad et al., 1994). With activation, macrophages increase in size, lysosomal content, and synthetic capabilities (Kumar et al., 1992). Li et al. (1996) found that in early multiple sclerosis plaques, many immunolabeled macrophages are visible, but few are NSE+ve, whereas in chronic lesions, the majority of macrophages are NSE+ve. This finding is analogous to our data, with many MAC-1+ve, but few NSE+ve, macrophages present in early lesions, whereas after 4 months, the number of NSE+ve and MAC-1+vemacrophages are equal. Therefore, whereas MAC-1 immunostaining identifies both resting and activated macrophages, NSE histochemistry appears to identify macrophages in an activated state. This concept is further supported by the observation that MAC-1+ve/NSE−ve macrophages present at day 1 are considerably smaller (and hence less activated) than NSE+ve/MAC-1+vemacrophages at 1 week or later.
The number of MAC-1+ve macrophages (Fig. 2) peaked 3–7 d after injury. By 1 month, their numbers had fallen by half, leaving a stable population of macrophages that was still present 4 months after injury. In contrast, the number and staining intensity of NSE+ve macrophages increased steadily and achieved parity with the number of MAC-1+ve macrophages 4 months after injury. It appears, therefore, that after loss of half of the initial rapid influx of macrophages the remainder form a resident population that become more activated with time (Fig.3B,E). The precise function of these two subpopulations of macrophages is unclear. Although the transient population may fulfill the traditional role for macrophages, phagocytosis, and debris removal (Giulian et al., 1989), we argue below that one role of the resident population of highly activated macrophages may be the induction of neuritic sprouting and tissue repair.
The rapid rise and fall in macrophage numbers was consistent with previous studies (Bunge et al., 1994; Riva-Depaty et al., 1994). However, although others have also noted the presence of macrophages for many weeks after injury (Hirsch et al., 1990; Kordower et al., 1991; Bunge et al., 1994), the size of this population has not been considered significant. It is now clear that large numbers of highly activated macrophages can persist in the striatum for many months after injury.
Expression of GDNF and BDNF mRNA by macrophages and microglia
In situ hybridization colocalized the majority of GDNF expression to activated macrophages (Fig.4D–F) and, to a much lesser extent, highly activated microglia. The close correlation between the amount of GDNF mRNA expressed and the number of NSE+ve macrophages (but not MAC-1+ve macrophages) gave strong additional evidence that GDNF was expressed by activated macrophages that persist after injury (Fig. 8).
BDNF mRNA expression was localized over large activated microglia (Fig.4A–C) and, to a lesser extent, wound macrophages. The predominantly microglial localization was also reflected in the close correlation between BDNF expression and appearance and disappearance of microglia (Figs. 5, 6).
Although the time course of BDNF expression also, to some extent, matches that of GFAP reactive astrocytosis, its spatial distribution does not. Reactive astrocytosis occurs throughout and even beyond the striatum (Fig. 3C), whereas BDNF mRNA production and reactive microgliosis only occur in a smaller periwound area (Figs.3A,D, 4A–C). Furthermore, colocalization studies found no association between BDNF expression and GFAP+ve astrocytes.
The data therefore suggest that the initial peak in BDNF mRNA expression after injury is caused by synthesis by the large but transient population of periwound-activated microglia, whereas ongoing production is by a relatively small number of activated microglia and, to a lesser extent, macrophages, which remain close to the margins of the wound.
Therefore, after striatal injury, GDNF is produced predominantly by activated macrophages and, to a lesser extent, by activated microglia, whereas the converse is true for BDNF.
Microglia, macrophages, and dopaminergic sprouting
Wounding of the striatum results in dopaminergic sprouting. Increased TH immunoreactivity is seen around grafts of dopaminergic and nondopaminergic tissue in humans, nonhuman primates, and rodents (Bohn et al., 1987; Bankiewicz et al., 1988, 1991, 1994; Fiandaca et al., 1988; Hirsch et al., 1990; Plunkett et al., 1990; Date et al., 1991;Kordower et al., 1991). It is important to note that sprouting occurs despite poor or absent graft survival (Bankiewicz et al., 1988, 1991;Fiandaca et al., 1988; Hirsch et al., 1990; Plunkett et al., 1990; Date et al., 1991; Kordower et al., 1991). Indeed, localized striatal injury alone is sufficient to upregulate markers of dopaminergic sprouting (Howells et al., 1993, 1996; Liberatore et al., 1996).
The mechanism by which sprouting is induced is unclear, but as outlined in the introductory remarks, the available evidence suggests that neurotrophic factors, in particular BDNF and GDNF, play an important role.
For these factors to be involved in periwound dopaminergic sprouting, they must be produced at the injury site. Such increased production does occur, with both BDNF (Plunkett et al., 1997; Wong et al., 1997) and GDNF (Liberatore et al., 1997) mRNA being expressed in and around the site of striatal trauma. In addition, fluid extracted from injured striatum promotes neurite growth and contains a significant BDNF-like component (Asada et al., 1996).
It appears that the macrophage and microglial response accompanying striatal injury is responsible for periwound expression of BDNF and GDNF mRNA. The dopaminergic sprouting accompanying striatal injury may thus result from the secretion of potent dopaminergic NTFs by activated macrophages and microglia at the wound site. Dramatic illustration of this concept is given by the images of DAT-positive fibers sprouting in the immediate periwound area, where production of these factors takes place. Fibers extend toward the wound and at high magnification can be seen to be growing toward and embracing hemosiderin-filled macrophages at the injury site (Fig. 9).
Although sprouting occurs after a simple wound to the striatum, the literature suggests that a greater degree of sprouting takes place when a tissue is grafted into the striatum (Bohn et al., 1987), despite limited or no graft survival. Indeed, Bresjanac et al. (1997) recently reported that it is adrenal graft rejection, rather than survival, that leads to increased TH immunoreactivity. The concept that it is the reactive microglia and macrophages within the wound, which induce neurons to sprout, offers a potential explanation for this, with increased numbers of reactive macrophages being present in grafted subjects, particularly in which grafts degenerate. This idea is given additional support by the observation that the implantation of activated leukocytes or microglia in rat models of parkinsonism improves rotational behavior and increases TH immunoreactivity (Wang et al., 1991; Ewing et al., 1992). Furthermore, autopsy studies performed on patients 4–30 months after adrenal medullary implantation have noted that, although there are few or no surviving adrenal chromaffin cells, there is often a substantial dopaminergic sprouting response in the periwound area and numerous macrophages present within the graft site (Hirsch et al., 1990; Kordower et al., 1991).
In addition to secreting the neurotrophic factors necessary for sprouting to occur, reactive macrophages and microglia also produce other factors likely to be involved in the sprouting process. IL-1 is produced by these cells when activated (Giulian et al., 1986) and induces periwound astrocytosis (Giulian et al., 1988). Reactive astrocytes are capable of forming an attractive substrate for axonal growth via the production of cell surface and extracellular matrix (ECM) molecules that facilitate axon elongation, as well as various NTFs (Ridet et al., 1997). Striatal reactive astrocytes have been found to produce bFGF (Ho and Blum, 1997) and CNTF (Asada et al., 1995), both of which have some effect on enhancing the survival of dopaminergic neurons in vitro (Hagg and Varon, 1993; Mayer et al., 1993). Thus, in the striatum, reactive astrocytes may provide additional trophic support to sprouting dopaminergic fibers, as well as an ECM framework over which to grow. The stimulation of reactive astrocytosis and dopaminergic sprouting by intrastriatal implants of IL-1 possibly occurs through this mechanism (Wang et al., 1994). Activated macrophages and microglia therefore appear to stimulate dopaminergic sprouting both directly, by the secretion of NTFs, and indirectly, by the secretion of IL-1 and the stimulation of reactive astrocytosis.
Axonal sprouting also takes place in other brain regions after local trauma. After spinal cord injury, Beattie et al. (1997) found that the density of axonal sprouting was proportional to the degree of local tissue damage and that sprouting neurites entered lesion cavities containing large numbers of macrophages. In another injury model, that of fimbria–fornix transection, both cholinergic and catecholaminergic sprouting occurs in the lateral septal area, which borders the lesion site (Gage et al., 1986). Local neuronal sprouting in this region could also be stimulated by activated macrophages and microglia.
These observations lead to intriguing questions. Do these cells secrete a broad range of NTFs and cytokines in which GDNF and BDNF form part of a generic repair mechanism that all neurons respond to regardless of neurochemical specificity? Alternatively, do macrophages and microglia secrete factors specific for the type of neuronal sprouting occurring at each injury site?
Finally, the ability to induce robust dopaminergic sprouting in the striatum would offer a potential therapy for patients afflicted with Parkinson’s disease. Appropriately activated macrophages and microglia attracted to or infused into the striatum may be well suited, by virtue of their ability to secrete a variety of NTFs and cytokines, to providing a rich environment promoting the formation of a dense network of dopaminergic fibers.
In conclusion, our data suggest that NTF secretion (in particular BDNF and GDNF) by activated macrophages and microglia at the wound site induces the dopaminergic sprouting that accompanies striatal injury. This concept offers a novel explanation to the mechanism of dopaminergic sprouting and would potentially unify much of the data regarding the occurrence of sprouting after injury to the nigrostriatal pathway and after grafting in human Parkinson’s disease.
Footnotes
This work was supported by the National Health and Medical Research Council of Australia, the Austin Hospital Medical Research Foundation, and Parkinson’s Victoria.
Correspondence should be addressed to Dr. David W. Howells, Department of Neurology, Austin and Repatriation Medical Centre, Heidelberg, Victoria 3084, Australia.
REFERENCES
- 1.Asada H, Ip NY, Pan L, Razack N, Parfitt MM, Plunkett RJ. Time course of ciliary neurotrophic factor mRNA expression is coincident with the presence of protoplasmic astrocytes in traumatized rat striatum. J Neurosci Res. 1995;40:22–30. doi: 10.1002/jnr.490400104. [DOI] [PubMed] [Google Scholar]
- 2.Asada H, Kaseloo PA, Lis A, Petti DM, Plunkett RJ. Traumatized rat striatum produces neurite-promoting and neurotrophic activities in vitro. Exp Neurol. 1996;139:173–187. doi: 10.1006/exnr.1996.0092. [DOI] [PubMed] [Google Scholar]
- 3.Bankiewicz K, Palmatie M, Plunkett RJ, Cummins A, Oldfield EH. Reversal of hemiparkinsonian syndrome in nonhuman primates by amnion implantation into caudate nucleus. J Neurosurg. 1994;81:869–876. doi: 10.3171/jns.1994.81.6.0869. [DOI] [PubMed] [Google Scholar]
- 4.Bankiewicz KS, Plunkett RJ, Kopin IJ, Jacobowitz DM, London WT, Oldfield EH. Transient behavioral recovery in hemiparkinsonian primates after adrenal medullary allografts. Prog Brain Res. 1988;78:543–549. doi: 10.1016/s0079-6123(08)60329-5. [DOI] [PubMed] [Google Scholar]
- 5.Bankiewicz KS, Plunkett RJ, Jacobowitz DM, Kopin IJ, Oldfield EH. Fetal nondopaminergic neural implants in parkinsonian primates. Histochemical and behavioral studies. J Neurosurg. 1991;74:97–104. doi: 10.3171/jns.1991.74.1.0097. [DOI] [PubMed] [Google Scholar]
- 6.Beattie MS, Bresnahan JC, Komon J, Tovar CA, Van Meter M, Anderson DK, Faden AI, Hsu CY, Noble LJ, Salzman S, Young W. Endogenous repair after spinal cord contusion injuries in the rat. Exp Neurol. 1997;148:453–463. doi: 10.1006/exnr.1997.6695. [DOI] [PubMed] [Google Scholar]
- 7.Beck KD. Pretreatment of dopaminergic neurons in culture with brain-derived neurotrophic factor attenuates toxicity of MPP+. Neurodegeneration. 1992;1:27–36. [Google Scholar]
- 8.Beller DI, Springer TA, Schreiber RD. Anti-Mac-1 selectively inhibits the mouse and human type three complement receptor. J Exp Med. 1982;156:1000–1009. doi: 10.1084/jem.156.4.1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bohn MC, Cupit L, Marciano F, Gash DM. Adrenal medulla grafts enhance recovery of striatal dopaminergic fibers. Science. 1987;237:913–916. doi: 10.1126/science.2887034. [DOI] [PubMed] [Google Scholar]
- 10.Bowenkamp KE, Hoffman AF, Gerhardt GA, Henry MA, Biddle PT, Hoffer BJ, Granholm AC. Glial cell line-derived neurotrophic factor supports survival of injured midbrain dopaminergic neurons. J Comp Neurol. 1995;355:479–489. doi: 10.1002/cne.903550402. [DOI] [PubMed] [Google Scholar]
- 11.Bresjanac M, Sagen J, Seigel G, Paino CL, Kordower J, Gash DM. Xenogeneic adrenal medulla graft rejection rather than survival leads to increased rat striatal tyrosine hydroxylase immunoreactivity. J Neuropathol Exp Neurol. 1997;56:490–498. doi: 10.1097/00005072-199705000-00005. [DOI] [PubMed] [Google Scholar]
- 12.Bunge MB, Holets VR, Bates ML, Clarke TS, Watson BD. Characterization of photochemically induced spinal cord injury in the rat by light and electron microscopy. Exp Neurol. 1994;127:76–93. doi: 10.1006/exnr.1994.1082. [DOI] [PubMed] [Google Scholar]
- 13.Date I, Felten SY, Felten DL. The nigrostriatal dopaminergic system in MPTP-treated mice shows more prominent recovery by syngeneic adrenal medullary graft than by allogeneic or xenogeneic graft. Brain Res. 1991;545:191–198. doi: 10.1016/0006-8993(91)91286-a. [DOI] [PubMed] [Google Scholar]
- 14.Del Cerro M, Monjan AA. Unequivocal demonstration of the hematogenous origin of brain macrophages in a stab wound by a double-label technique. Neuroscience. 1979;4:1399–1404. doi: 10.1016/0306-4522(79)90167-2. [DOI] [PubMed] [Google Scholar]
- 15.Ewing SE, Weber RJ, Zauner A, Plunkett RJ. Recovery in hemiparkinsonian rats following intrastriatal implantation of activated leukocytes. Brain Res. 1992;576:42–48. doi: 10.1016/0006-8993(92)90607-b. [DOI] [PubMed] [Google Scholar]
- 16.Fiandaca MS, Kordower JH, Hansen JT, Jiao SS, Gash DM. Adrenal medullary autografts into the basal ganglia of Cebus monkeys: injury-induced regeneration. Exp Neurol. 1988;102:76–91. doi: 10.1016/0014-4886(88)90080-5. [DOI] [PubMed] [Google Scholar]
- 17.Gage FH, Wictorin K, Fischer W, Williams LR, Varon S, Bjorklund A. Retrograde cell changes in medial septum and diagonal band following fimbria-fornix transection: quantitative temporal analysis. Neuroscience. 1986;19:241–255. doi: 10.1016/0306-4522(86)90018-7. [DOI] [PubMed] [Google Scholar]
- 18.Gallo G, Letourneau PC. Localized sources of neurotrophins initiate axon collateral sprouting. J Neurosci. 1998;18:5403–5414. doi: 10.1523/JNEUROSCI.18-14-05403.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gash DM, Zhang Z, Ovadia A, Cass WA, Yi A, Simmerman L, Russell D, Martin D, Lapchak PA, Collins F, Hoffer BJ, Gerhardt GA. Functional recovery in parkinsonian monkeys treated with GDNF. Nature. 1996;380:252–255. doi: 10.1038/380252a0. [DOI] [PubMed] [Google Scholar]
- 20.Giulian D, Baker TJ, Shih LC, Lachman LB. Interleukin 1 of the central nervous system is produced by ameboid microglia. J Exp Med. 1986;164:594–604. doi: 10.1084/jem.164.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giulian D, Woodward J, Young DG, Krebs JF, Lachman LB. Interleukin-1 injected into mammalian brain stimulates astrogliosis and neovascularization. J Neurosci. 1988;8:2485–2490. doi: 10.1523/JNEUROSCI.08-07-02485.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giulian D, Chen J, Ingeman JE, George JK, Noponen M. The role of mononuclear phagocytes in wound healing after traumatic injury to adult mammalian brain. J Neurosci. 1989;9:4416–4429. doi: 10.1523/JNEUROSCI.09-12-04416.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagg T, Varon S. Ciliary neurotrophic factor prevents degeneration of adult rat substantia nigra dopaminergic neurons in vivo. Proc Natl Acad Sci USA. 1993;90:6315–6319. doi: 10.1073/pnas.90.13.6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirsch EC, Duyckaerts C, Javoy-Agid F, Hauw JJ, Agid Y. Does adrenal graft enhance recovery of dopaminergic neurons in Parkinson’s disease? Ann Neurol. 1990;27:676–682. doi: 10.1002/ana.410270615. [DOI] [PubMed] [Google Scholar]
- 25.Ho A, Blum M. Regulation of astroglial-derived dopaminergic neurotrophic factors by interleukin-1 beta in the striatum of young and middle-aged mice. Exp Neurol. 1997;148:348–359. doi: 10.1006/exnr.1997.6659. [DOI] [PubMed] [Google Scholar]
- 26.Howells DW, Donnan GA, Wong JY, Kaczmarczyk SJ, Chilcho PJ, Fabinyi GC, Mendelsohn FA. Surgical damage stimulates proliferation of dopamine uptake sites in normal mouse brain. Brain Res. 1993;622:285–288. doi: 10.1016/0006-8993(93)90830-g. [DOI] [PubMed] [Google Scholar]
- 27.Howells DW, Liberatore GT, Wong JY, Donnan GA. Dopaminergic responses to striatal damage. J Neurol Sci. 1996;139:125–130. [PubMed] [Google Scholar]
- 28.Hyman C, Hofer M, Barde YA, Juhasz M, Yancopoulos GD, Squinto SP, Lindsay RM. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature. 1991;350:230–232. doi: 10.1038/350230a0. [DOI] [PubMed] [Google Scholar]
- 29.Kordower JH, Cochran E, Penn RD, Goetz CG. Putative chromaffin cell survival and enhanced host-derived TH-fiber innervation following a functional adrenal medulla autograft for Parkinson’s disease. Ann Neurol. 1991;29:405–412. doi: 10.1002/ana.410290411. [DOI] [PubMed] [Google Scholar]
- 30.Koski IR, Poplack DG, Blaese RM. A nonspecific esterase stain for the identification of monocytes and macrophages. In: Bloom BR, David IR, editors. In vitro methods in cell mediated and tumour immunity. Academic; New York: 1976. pp. 359–362. [Google Scholar]
- 31.Kumar V, Cotran RS, Robins SL. Basic pathology, pp 28–46. Saunders; London: 1992. Acute and chronic inflammation. [Google Scholar]
- 32.Li H, Cuzner ML, Newcombe J. Microglia-derived macrophages in early multiple sclerosis plaques. Neuropathol Appl Neurobiol. 1996;22:207–215. [PubMed] [Google Scholar]
- 33.Liberatore GT, Finkelstein DI, Wong JY, Horne MK, Donnan GA, Howells DW. Increased dopaminergic activity caused by striatal injury is associated with axonal sprouting. Soc Neurosci Abstr. 1996;22:592.18. [Google Scholar]
- 34.Liberatore GT, Wong JY, Porritt MJ, Donnan GA, Howells DW. Expression of glial cell line-derived neurotrophic factor (GDNF) mRNA following mechanical injury to mouse striatum. NeuroReport. 1997;8:3097–3101. doi: 10.1097/00001756-199709290-00018. [DOI] [PubMed] [Google Scholar]
- 35.Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- 36.Lindner MD, Winn SR, Baetge EE, Hammang JP, Gentile FT, Doherty E, McDermott PE, Frydel B, Ullman MD, Schallert T. Implantation of encapsulated catecholamine and GDNF-producing cells in rats with unilateral dopamine depletions and parkinsonian symptoms. Exp Neurol. 1995;132:62–76. doi: 10.1016/0014-4886(95)90059-4. [DOI] [PubMed] [Google Scholar]
- 37.Lucidi-Phillipi CA, Gage FH, Shults CW, Jones KR, Reichardt LF, Kang UJ. Brain-derived neurotrophic factor-transduced fibroblasts: production of BDNF and effects of grafting to the adult rat brain. J Comp Neurol. 1995;354:361–376. doi: 10.1002/cne.903540306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayer E, Dunnett SB, Pellitteri R, Fawcett JW. Basic fibroblast growth factor promotes the survival of embryonic ventral mesencephalic dopaminergic neurons. I. Effects in vitro. Neuroscience. 1993;56:379–388. doi: 10.1016/0306-4522(93)90339-h. [DOI] [PubMed] [Google Scholar]
- 39.Miyake T, Hattori T, Fukuda M, Kitamura T, Fujita S. Quantitative studies on proliferative changes of reactive astrocytes in mouse cerebral cortex. Brain Res. 1987;451:133–138. doi: 10.1016/0006-8993(88)90757-3. [DOI] [PubMed] [Google Scholar]
- 40.Plunkett RJ, Bankiewicz KS, Cummins AC, Miletich RS, Schwartz JP, Oldfield EH. Long-term evaluation of hemiparkinsonian monkeys after adrenal autografting or cavitation alone. J Neurosurg. 1990;73:918–926. doi: 10.3171/jns.1990.73.6.0918. [DOI] [PubMed] [Google Scholar]
- 41.Plunkett RJ, Ip NY, Asada H, Friedman B, Pan L, Kaseloo PA, Parfitt MM. Trauma-induced striatal CNTF and BDNF mRNA in hemiparkinsonian rats. NeuroReport. 1997;8:507–511. doi: 10.1097/00001756-199701200-00026. [DOI] [PubMed] [Google Scholar]
- 42.Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- 43.Riva-Depaty I, Fardeau C, Mariani J, Bouchaud C, Delhaye-Bouchaud N. Contribution of peripheral macrophages and microglia to the cellular reaction after mechanical or neurotoxin-induced lesions of the rat brain. Exp Neurol. 1994;128:77–87. doi: 10.1006/exnr.1994.1114. [DOI] [PubMed] [Google Scholar]
- 44.Shults CW, Kimber T, Altar CA. BDNF attenuates the effects of intrastriatal injection of 6-hydroxydopamine. NeuroReport. 1995;6:1109–1112. doi: 10.1097/00001756-199505300-00009. [DOI] [PubMed] [Google Scholar]
- 45.Ulvestad E, Williams K, Mork S, Antel J, Nyland H. Phenotypic differences between human monocytes/macrophages and microglial cells studied in situ and in vitro. J Neuropathol Exp Neurol. 1994;53:492–501. doi: 10.1097/00005072-199409000-00008. [DOI] [PubMed] [Google Scholar]
- 46.Wang J, Bankiewicz KS, Plunkett RJ, Sheng J, Jacobowitz DM. Transplantation of microglia reduces experimental parkinsonism in rats. In: Lindvall O, Bjorklund A, Winder H, editors. Intracerebral transplantation in movement disorders. Elsevier; Amsterdam: 1991. pp. 313–323. [Google Scholar]
- 47.Wang J, Bankiewicz KS, Plunkett RJ, Oldfield EH. Intrastriatal implantation of interleukin-1. Reduction of parkinsonism in rats by enhancing neuronal sprouting from residual dopaminergic neurons in the ventral tegmental area of the midbrain. J Neurosurg. 1994;80:484–490. doi: 10.3171/jns.1994.80.3.0484. [DOI] [PubMed] [Google Scholar]
- 48.Wong JY, Liberatore GT, Donnan GA, Howells DW. Expression of brain-derived neurotrophic factor and TrkB neurotrophin receptors after striatal injury in the mouse. Exp Neurol. 1997;148:83–91. doi: 10.1006/exnr.1997.6670. [DOI] [PubMed] [Google Scholar]