

Abstract
Activity-dependent facilitation was studied in bovine adrenal chromaffin cells. Stimulation with a train of depolarizations caused subsequent triggered exocytotic activity to be significantly enhanced. After the facilitating stimulus train, the readily releasable vesicle pool (RRP) size was estimated from capacitance jumps in response to paired depolarizations and found to be elevated for a period of at least 10 min. The time dependency of onset and degree of facilitation could be well fitted assuming protein kinase C (PKC)-dependent and independent Ca2+-mediated processes. Both processes increase the recruitment of vesicles from the reserve pool to the RRP, resulting in an greater number of releasable vesicles. The data suggest that cell activity can act as a trigger to increase cytosolic Ca2+ to a level sufficient to cause an increase in the number of readily releasable secretory vesicles, with the more persistent component of the evoked facilitation being mediated through activity-dependent activation of PKC.
Keywords: facilitation, exocytosis, vesicle pools, plasticity, membrane capacitance, chromaffin
Many excitatory cells undergo activity-dependent changes in their secretory response. Neuroendocrine cells, like many neurons, display secretory depression after intense stimulation (Thomas et al., 1993; Moser and Neher, 1997), augmentation after elevation of cytosolic calcium (Bittner and Holz, 1992; Thomas et al., 1993; von Rüden and Neher, 1993), long-lasting potentiation after pharmacological activation of protein kinases (Knight and Baker, 1983; Vitale et al., 1995; Gillis et al., 1996), and a block of potentiation after treatment with kinase inhibitors (Gillis et al., 1996; Renström et al., 1997; Kumar et al., 1998).
As first shown for the neuromuscular junction (Elmqvist and Quastel, 1965; Betz, 1970), secretory behavior of neuroendocrine cells can be simulated by sequential models that assume trafficking of exocytotic vesicles between a large reserve pool and a smaller readily releasable pool (RRP; Heinemann et al., 1993; Thomas and Waring, 1997; Neher, 1998). The amount of evoked transmitter release is determined by the number of vesicles inhabiting the RRP and the probability of each vesicle to undergo exocytosis in response to a stimulus. In bovine adrenal chromaffin cells, the RRP size has been shown to be increased by elevated [Ca2+]i in a protein kinase C (PKC)-independent manner and through the activation of PKC. PKC causes facilitation by increasing the delivery rate of vesicles from the reserve pool to the RRP (Smith et al., 1998). The goal of this study was to test whether the Ca2+-mediated activation of PKC was likely to play a physiological role in the modulation of stimulus-evoked secretion. It was asked whether heightened cell activity was indeed capable of raising [Ca2+]i to levels sufficient in magnitude and duration to activate PKC and result in a facilitation of the exocytotic process. To answer these questions, the RRP size was estimated using paired depolarizations (Gillis et al., 1996) before and at different times after conditioning trains of short depolarizations. Exocytosis was monitored by means of patch-clamp measurements of cell membrane capacitance (Cm, an index of vesicle-membrane fusion). Average [Ca2+]i was recorded using the calcium indicator fura-2. A computer simulation of the experimental protocol, based on a two-step model of secretion (Heinemann et al., 1993,1994) was used to determine the extent to which the increased RRP was responsible for the observed secretory augmentation, to examine the duration of the facilitation, and to track the activity of the PKC-enhanced recruitment of vesicles from a reserve population to the RRP.
Reported here are the results of a study in which secretory efficiency was measured during multiple trains of depolarizations. The data show that trains of stimuli in chromaffin cells evoke two forms of Ca2+-dependent facilitation previously described (Smith et al., 1998) and that the more persistent form of facilitation is sensitive to selective PKC blockers. Furthermore, facilitation is not likely caused by a shift in the Ca2+ sensitivity of the secretory process, rather to the increase in the total number of vesicles that inhabit a releasable state.
MATERIALS AND METHODS
Chromaffin cell culture. Adult bovine adrenal glands were acquired fresh from local slaughter houses, perfused with cooled sterile Locke’s Ringer’s solution, and transported to the laboratory. The glands were trimmed well of fat, and the cortex was cut around the circumference of the long axis of the gland. Glands were again perfused with Locke’s Ringer’s solution and placed in a 37°C shaking water bath for 5 min. Glands were then perfused with collagenase (type I; 1.0 mg/ml; Worthington, Freehold, NJ) that had been solublized in a DMEM growth medium (Life Technologies, Gaithersburg, MD). The DMEM growth medium was supplemented with penicillin (20 U/ml), streptomycin (20 μg/ml), and GMS-X (a defined serum substitute containing insulin, transferrin, and selenium; Life Technologies). The collagenase perfusion was performed twice, with each perfusion followed by a 15 min incubation in a 37°C shaking water bath. After the collagenase treatment, the cortex of the glands was manually separated from the medulla, and the medulla was minced into small cubes with a scalpel. The minced medulla was further manually dissociated with the blunt end of a sterile Pasteur pipette and filtered through nylon mesh (∼150 μm pore size). The filtrate was resuspended in 45 ml of Locke’s Ringer’s solution and spun at 1000 × g for 2 min. The pellet was then resuspended in Locke’s Ringer’s solution to a final volume of 14.7 ml combined with 15.3 ml Percoll (Pharmacia, Uppsala, Sweden) and 1.7 ml 10× Locke’s Ringer’s solution. The Percoll suspension was spun at ∼20,000 × g for 20 min at room temperature. Enriched chromaffin cells were recovered from the Percoll gradient and repelleted in 40 ml Locke’s Ringer’s solution by a 1000 × g spin for 2 min. The cells were resuspended in the DMEM growth medium (see above) and at an approximate concentration of 4.4 * 103 cells/mm2, plated onto coverslips and maintained at 37°C in 10% CO2. Experiments were performed 1–4 d after cell preparation.
Solutions. During recordings, cells were constantly superfused at a rate of ∼1 mm/min with a Ringer’s solution of the following composition (in mm): 150 NaCl, 10 HEPES-H, 10 Glucose, 2.5 CaCl2, 2.8 KCl, and 2 MgCl2, or as otherwise noted. The osmolarity was adjusted to 310 mOsm with mannitol, and pH was 7.2. The standard perforated-patch solution contained (in mm): 135 Cs glutamate, 10 HEPES-H, 9.5 NaCl, 0.5 TEA-Cl, and 0.53 amphotericin B, pH 7.2, and osmolarity 300 mOsm. Amphotericin B was prepared as described by Smith and Neher (1997). Pipettes were tip-dipped in amphotericin-free solution for 2–10 sec, and back-filled with freshly mixed amphotericin-containing solution. The liquid junction potential between the extracellular Ringer’s solution and the intracellular solution was measured to be ∼13 mV, and all potentials were adjusted accordingly. All chemicals were obtained from Sigma (St. Louis, MO), with the exception of CsOH (Aldrich, Milwaukee, WI) and amphotericin B (Calbiochem, La Jolla, CA), or as otherwise noted.
Electrophysiological measurements. Pipettes of ∼2–3 MΩ resistance were pulled from borosilicate glass, partially coated with a silicone compound (G.E. Silicones, Bergen Op Zoom, The Netherlands), and lightly fire-polished. For acquisition, an EPC-9 amplifier and Pulse software running on an Apple Macintosh were used. Cell capacitance (Cm) was estimated by the Lindau–Neher technique (for review, see Gillis, 1995) implemented as the “Sine + DC” feature of the Pulse lock-in module. A 700 Hz, 35 mV peak amplitude sinewave was applied to a holding potential of −83 mV, and the reversal potential of the lock-in module was set to 0 mV. For depolarization, the sinewave was interrupted only after a complete sine cycle and reinitiated at the start of a sine cycle. Data were acquired through a combination of the high time resolution Pulse software and the lower time resolution XChart plug-in module to the Pulse software. Briefly, membrane current was sampled at 10 kHz shortly (100 msec) before, during, and for 3.5 sec after the depolarizations and cell capacitance, cell conductance, and pipette access resistance were calculated at 700 Hz. Data acquired between high time resolution Pulse protocols were typically sampled at 12 Hz with the lower time resolution XChart plug-in. The higher time resolution Pulse data were then digitally filtered and merged into the lower time resolution XChart data off-line in IgorPro (Wavemetrics, Lake Oswego, OR) for display purposes.
Capacitance increases caused by depolarizations were determined from the high time resolution Cm traces as the difference between mean Cm measured in a 50 msec window starting 50 msec after the depolarization minus the mean prestimulus Cm, also measured over a 50 msec window. The first 50 msec of the postdepolarization capacitance was neglected to avoid influences of nonsecretory capacitance transients (Horrigan and Bookman, 1994). With the stimulus protocol used here, it was assumed that endocytosis was unlikely to be a contaminating factor in the estimation of exocytosis. Triggered endocytosis in response to Ca2+ influx of <90 pC, termed “compensatory endocytosis” (Smith and Neher, 1997), has been shown to take place with time constants of 6 sec or slower, or even to be totally absent after single secretion events totaling less than ∼50 fF (Engisch and Nowycky, 1998). Endocytosis on such a slow time scale would be unlikely to effect Cm measurements made 50 msec after a depolarization. Experiments were performed at 20–25°C. Data are presented as mean ± SE.
Cytosolic Ca2+ measurements. Cellular Ca2+ concentrations were measured in the perforated-patch configuration by preincubating the cells in growth medium containing a 1 μm concentration of the membrane-permeant acetyl methyl ester form of fura-2 (fura-2 AM; Molecular Probes, Eugene, OR; Grynkiewicz et al., 1985) for 5 min at 37°C. Cells were illuminated at either 360 or 390 nm light with a monochrometer (TILL Photonics, Planegg, Germany) attached to the fluorescence port of a Zeiss IM35 microscope (Zeiss, Jena, Germany). Excitation light was reflected through the objective (50× water immersion, NA = 1.0; Leitz, Wetzlar, Germany) by a 495 nm cutoff extended range dichroic mirror (TILL Photonics). Emitted light then was passed through an HQ535/50 nm bandpass filter (AHF, Tübingen, Germany) before sampling with a photo multiplier tube (model R928; Hamamatsu, Tokyo, Japan). The signal was passed into an analog-to-digital converter channel of the EPC-9 and processed with the fura extension of the Pulse Software. After the experimental protocol, the perforated patch was ruptured causing the intracellular fura-2 to dialyze out of the cell, allowing for the measurement of the autofluorescence of the cell. The autofluorescence values at 360 and 390 nm wavelength excitation were subtracted from values measured during the experiment, and the [Ca2+] was estimated after Grynkiewicz et al. (1985). No absolute estimate for the cytosolic fura concentration was made, but the emitted fluorescence in these experiments was roughly 0.2–0.5 times as bright as values measured with 100 μm K+-fura in the internal solution of whole-cell experiments. Also, after breaking into the whole-cell configuration to allow fura to dialyze out, it was found that the cell fluorescence dropped only by ∼50% (fura fluorescence was only as bright as autofluorescence). Taken together, these two points imply that the fura-AM loading protocol used in these experiments resulted in very low cytosolic fura concentrations, perhaps only a few tens of micromolar. These concentrations of fura would be very unlikely to significantly alter the cytosolic Ca2+-buffering and the time course of poststimulus Ca2+ clearance.
Computer simulation of measured RRP recovery. A computer simulation of the measured RRP characteristics was based on the “two-step model of secretion control” (Heinemann et al., 1993) with code written within IgorPro. A brief summary of the model and its current implementation is supplied here. The two-step scheme for secretion describes the transition of vesicle between three separate states. Pool A is considered to be a large reserve pool of vesicles that mature, in a Ca2+-dependent manner, into the release-ready vesicles of pool B. Vesicles in the B pool either revert and rejoin pool A, or undergo evoked Ca2+-dependent secretion, described as the transition to pool C.
| Equation 1 |
For the simulations, pool A was assumed to be equivalent to a number of vesicles totaling in capacitance to 5 pF (Heinemann et al., 1993). The forward rate constant k1between pools A and B is dependent on [Ca2+]i and is described by the Michaelis–Menten relationship in Equation 2:
| Equation 2 |
where a1 is the maximum possible value ofk1, and b1 has the meaning of a KD of a regulating site. In the simulations, the stimulus-evoked [Ca2+]i transient was approximated as monoexponential decay with a time constant of 5.5 sec (Smith et al., 1998), except after the last pulse of a train, where the [Ca2+]i decay was described by the time constants Caτ4 and Caτ5 (see Table1 and Fig. 4, legend, for description). The backward rate constant k−1 represents the spontaneous loss of vesicles from pool B to pool A. The rate of vesicle exocytosis, transition from pool B to C, is represented in the two-step model by the rate constant k2 and is defined by Equation 3:
| Equation 3 |
where a3 is calculated from the data ofHeinemann et al. (1994), according to the secretion study of Klingauf and Neher (1997) to be equal to 0.035 μm−3∗ sec−1 (see Smith et al., 1998 for a more detailed description of a3 calculation).
Table 1.
Summary of simulation parameters
| Train depolarization simulation control parameter set | Time constant | Magnitude |
|---|---|---|
| Basal[Ca2+]i | 153 nm | |
| Peak [Ca2+]i after initial depolarization in train | 871 nm | |
| Caτ1: Primary [Ca2+]i decay after pulses within train | 5.5 sec | |
| 1-aCaτ2: Fast Ca2+current rundown within train | 8.8 sec | 137 pA |
| 1-aCaτ3: Slow Ca2+current rundown within train | 116.9 sec | 253 pA |
| 1-aCaτ4: Fast [Ca2+]i decay after train | 22 sec | 250 nm |
| 1-aCaτ5: Slow [Ca2+]i decay after train | 77 sec | 250 nm |
| a3 value | 0.035 μm−3 sec−1 | |
| Pool A size | 5 pF | |
| a1 value (control) | 0.007 sec−1 | |
| a1value (activated) | 0.014 sec−1 | |
| k−1 value | 0.031 sec−1 | |
| b1 value | 1.9 μm |
All values either obtained from the literature or measured experimentally in this study.
See legend to figure 5 for description.
Fig. 4.
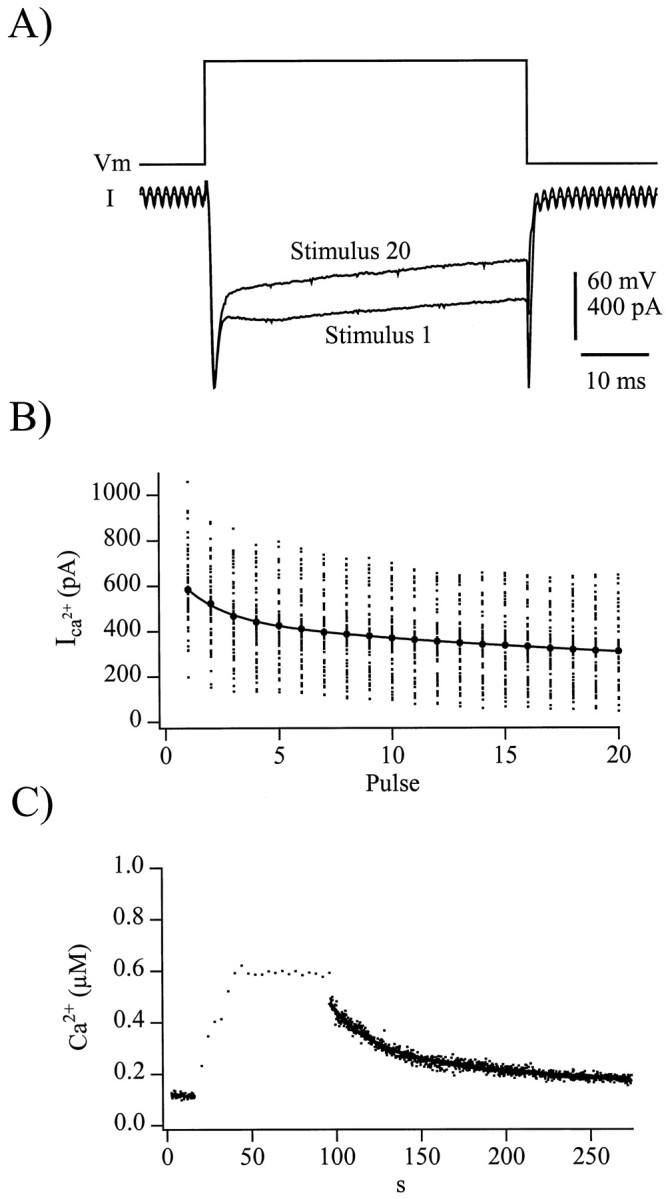
Ca2+ clearance is slowed after the train stimulation protocol. A, Voltage protocol (top trace) and representative evoked currents (bottom traces) from the first and last depolarization of a train are shown. B, Average evoked Ca2+ currents measured during each depolarization of a train stimuli from 38 individual cells is plotted against the pulse number (small dots). Only data from the first trains are included to avoid effects of Ca2+-dependent inhibition of calcium influx observed in the second trains of paired train stimulation protocols. For clarity, the current magnitudes from each pulse were averaged and overlay the individual points (large dots). The decay in current amplitude was fitted as a double exponential decay, resulting in time constants of 8.5 and 117.0 sec (Table 1, Caτ2 and Caτ3) with magnitude of 137 and 253 pA, respectively. C, [Ca+2]i measured in fura-AM ester-loaded chromaffin cells after train depolarizations shows a slowed clearance of Ca2+. A Ca2+record from a representative cell is displayed, and the post-train [Ca2+]i decay is fitted with a double exponential decay, with time constants of 22 and 77 sec (Table 1, Caτ4 and Caτ5), each of 250 nm in magnitude.
The [Ca2+]i-dependent steady state size of pool B is described by Equation 4:
| Equation 4 |
where A is the size of pool A in farads.
To estimate the poststimulus recovery of pool B, the differential equations derived from the model were solved using the Runge–Kutta integration method. The kinetic input parameters that matched closely the measured experimental data were determined by fitting both RRP recovery after depletion, as well as steady state size (Smith et al., 1998). The control kinetic input parameter set is summarized in Table1. For further description of the limitations and assumptions of the model, see Heinemann et al. (1993; 1994) and Smith et al. (1998).
RESULTS
Repetitive stimulation results in an increase in exocytotic efficiency
The efficacy with which trains of depolarizations evoked granule exocytosis was measured in single chromaffin cells in the perforated-patch configuration of the voltage-clamp technique (Zhou and Misler, 1995; Engisch and Nowycky, 1996; Engisch et al., 1997;Renström et al., 1997). For short experimental protocols, no quantifiable or significant difference in the kinetics of exocytosis can be detected between whole-cell and perforated-patch recordings (von Rüden and Neher, 1993; Smith et al., 1998). However, for the prolonged times required for the protocols in this study, experiments were performed in the perforated-patch configuration to avoid washout of vital cytosolic factors that support exocytosis (Zhou and Neher, 1993; Burgoyne, 1995; Engisch and Nowycky, 1996; Smith and Neher, 1997;Smith et al., 1998). Vesicle exocytosis was measured by monitoring the electrical capacitance of the cell, an index of cell surface area (Neher and Marty, 1982; Lindau and Neher, 1988). Total evoked exocytosis was measured as the difference in cell capacitance before and after the depolarizing stimuli, whereas stimulus intensity was quantified as the integrated evoked Ca2+ influx caused by the depolarization (Engisch and Nowycky, 1996; Seward and Nowycky, 1996; Engisch et al., 1997; Renström et al., 1997). Shifts in exocytotic efficiency would be reported as a steeper or shallower slope of the integrated evoked capacitance increases to the integrated Ca2+ influx, either caused by a change in the affinity of a Ca2+ or caused by a change in the number of readily releasable vesicles.
In neuroendocrine cells, studies designed to measure exocytotic efficiencies under conditions that approach the physiological behavior of cells have used depolarization trains as stimuli (Zhou and Misler, 1995; Engisch and Nowycky, 1996; Seward and Nowycky, 1996; Engisch et al., 1997; Renström et al., 1997). For this same reason, and so that results could be interpreted in the context of the existing literature, cells were also stimulated with depolarizing trains in this study. When stimulated with a train of depolarizations, chromaffin cells exhibit a heightened secretory efficiency during subsequent depolarization trains. Shown in Figure 1A is a cell challenged with two stimulation trains with the second train of stimuli delivered ∼3 min after the end of the first train. Evoked exocytosis was measured as the difference between cell capacitance before and shortly after depolarization. Evoked endocytosis, as seen between individual stimuli, took place on time scales too slow to play any significant role in the accurate estimation of triggered exocytosis (see Materials and Methods for a description of exocytosis estimation protocol). Total measured evoked capacitance increase is plotted against integrated Ca2+ influx in Figure1Aii. An increase in the slope of the secretion–stimulus relationship occurred after approximately the eighth depolarization of the train. Integrated evoked Cmincreases were divided by total integrated Ca2+influx for the calculation of “exocytotic efficiency”. This in effect gives the average efficiency within an entire train. The efficiency in many trains begins lower, but increases within the train, as seen in Figure 1Aii. This is likely caused by the buildup of [Ca2+]i during the train and therefore an increase in the rate of vesicle delivery to the RRP, and would therefore be limited to times of elevated [Ca2+]i. Efforts were not focused on quantification of the internal efficiency shifts within trains in this study, rather were limited to increases of efficiency between trains. Therefore, efficiencies are reported as mean values for all responses from a train. In Figure 1Aii, the mean exocytotic efficiency was 2.3 fF/pC, a result similar to the “standard curve” described by Engisch et al. (1997). The second train, after the first by ∼3 min, resulted in an average exocytotic efficiency of 3.1 fF/pC. The same protocol as described for Figure1A was repeated with a 500 nmconcentration of the membrane-permeant PKC inhibitor bisindolylmaleimide I (BIS; Calbiochem) in the bath (Fig.1B). Although the Ca2+ influx from the second stimulus train was smaller than that of the first, both trains resulted in ∼2.3 fF/pC exocytosis.
Fig. 1.
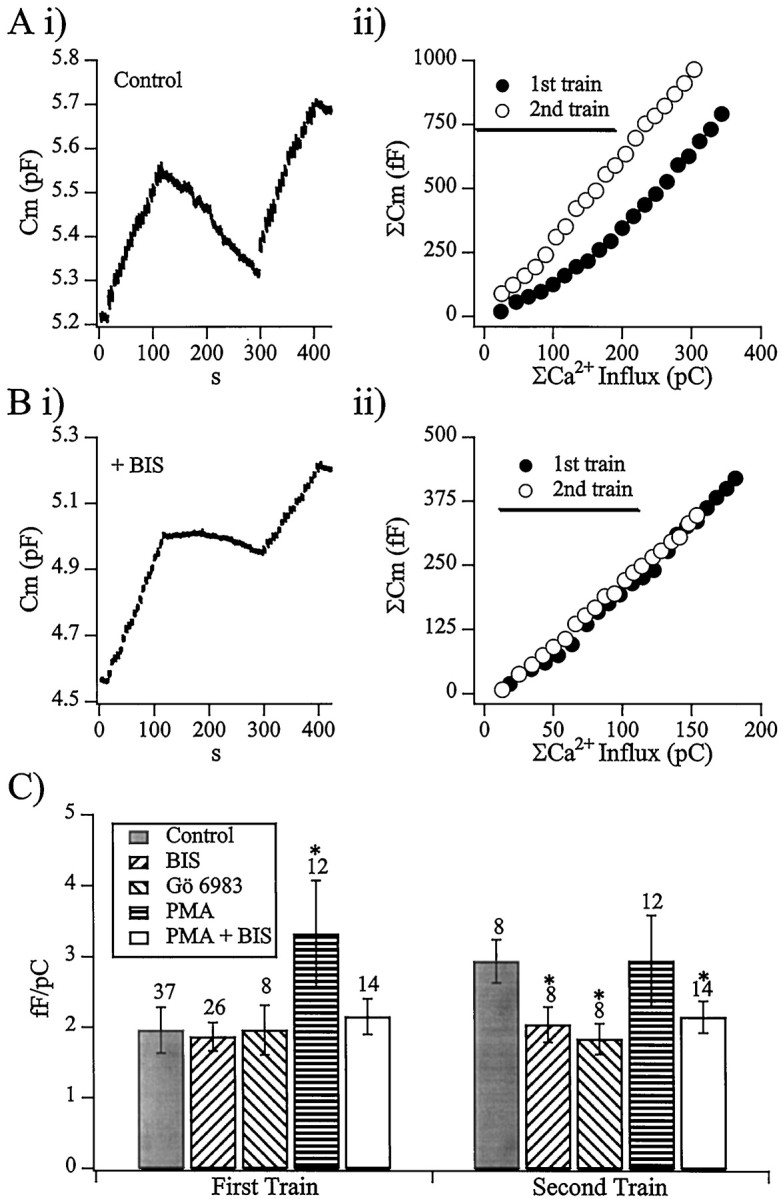
Exocytotic efficiency is increased by cell activity. Ai shows the capacitance record of a cell stimulated by two trains of depolarizations. Cells were held at a potential of −83 mV in the perforated-patch voltage-clamp technique. Each train consisted of 20 depolarizations to 7 mV, 50 msec long, separated by 5 sec intervals. In this experiment, the second train followed the first train by 189 sec. Aii, Evoked capacitance increases and Ca2+ influx were measured for each stimulus. The integrated capacitance response is plotted against the integrated Ca2+ influx for each train. The first train resulted in a total Cm increase of 792 fF caused by a Ca2+ influx of 343 pC, whereas the second train resulted in a total Cm increase of 966 fF in response to a Ca2+ influx of 304 pC. The same protocol as described in A was repeated with the specific membrane-permeant protein kinase C inhibitor BIS in the bath at a concentration of 500 nm. Bi, The cell capacitance recorded from the cell during the experiment is plotted.Bii, As in Aii, the integrated capacitance response is plotted against the integrated Ca2+ influx for each train. The first train resulted in a total Cm increase of 419 fF because of a Ca2+ influx of 181 pC, whereas the second train resulted in a total Cm increase of 347 fF in response to a Ca2+ influx of 154 pC. C, The same protocol and analysis as described in A andB was repeated on a cell treated with 100 nmGö 6983, 100 nm PMA, and a cocktail of 500 nm BIS and 100 nm PMA. The data for all tested conditions is summarized and displayed as measured femtofarads per picocoulombs from the first and second trains. Theasterisks denote values that are significantly different (p < 0.05; paired Student’st test) from the control value of that group.
To confirm that the BIS-sensitive facilitation was indeed caused by Ca2+-mediated activation of PKC, another potent inhibitor, Gö 6983 (100 nm, Calbiochem), was used, resulting again in a block of facilitation expected in the second train. Treatment of the cells with 100 nm PMA, a potent phorbol ester activator of PKC, resulted in an increase in secretory efficiency in both stimulus trains, obscuring the activity-dependent facilitation seen in control cells. Simultaneous treatment with 100 nm PMA and 500 nm BIS resulted in a block of the activity-evoked facilitation seen in control cells (results summarized in Fig. 1C). Taken together, these pharmacological perturbations point to Ca2+ activation of PKC as responsible for the activity-dependent facilitation observed in the control condition.
With intertrain delays shorter than the 180 sec demonstrated in Figure1, the second train showed a facilitated exocytotic efficiency in both the control and the BIS-treated cells, although the control cells always showed a greater increase than the BIS-treated cells (Fig.2). The facilitation observed in the BIS-treated cells with shorter intertrain delays most likely represents a residual Ca2+-evoked, but PKC-independent secretory facilitation (von Rüden and Neher, 1993; Thomas and Waring, 1997; Smith et al., 1998).
Fig. 2.

The increase in exocytotic efficiency is greater in cells with intact PKC activity. Exocytotic efficiencies for control (dark bars) and BIS-treated cells (light bars) was determined for first and second trains as total evoked increase in cell capacitance divided by total evoked Ca2+ influx. The time between the end of the first train and the onset of the second train was varied between ∼30 and 180 sec. The number of cells measured for each condition are reported above the data sets. Asterisks represent data sets (with and without BIS) that are significantly different as determined by a paired Student’s t test (p< 0.05).
An increase in the RRP size is responsible for the activity-dependent facilitation
Two possible mechanisms for the activity-evoked facilitation were considered. In the first scenario, heightened cell activity could act as a positive feedback mechanism for increasing the affinity of the Ca2+-sensitive trigger in the fusion process. In other words, activity would increase the likelihood that a given vesicle would undergo exocytosis in response to a stimulus. The result would be a greater exocytotic efficiency. The second scenario would be to maintain the probability of a single vesicle to undergo exocytosis but to supply more such vesicles, again effectively increasing the exocytotic efficiency of the cell. To distinguish between these two possibilities, a protocol was devised by which the total number of readily releasable vesicles was measured before and at varying times after a train of depolarizations (Fig.3). The RRP was measured through the use of a “dual-pulse paradigm” (Gillis et al., 1996; Moser and Neher, 1997; Smith et al., 1998). From the sum and the ratio of the capacitance increases (ΔCm) to two identical Ca2+-current injections given in rapid succession, an upper limit of the RRP, Bmax, is derived:
| Equation 5 |
where S represents the sum of the capacitance responses to the first (ΔCm1) and the second (ΔCm2) depolarizations, and R is defined as the ratio of ΔCm2:ΔCm1. A value <1 for R represents secretory depression, presumably caused by depletion of the RRP. In experimental analyses, all cells with an R value of >0.6 were discarded because accurate estimate of the RRP size is only possible when adequate vesicle depletion occurs (see Gillis et al., 1996 for a detailed description of the dual-pulse protocol and its limitations). This strict depression requirement eliminated 46% of the responses measured under the dual-pulse protocol from analysis.
Fig. 3.
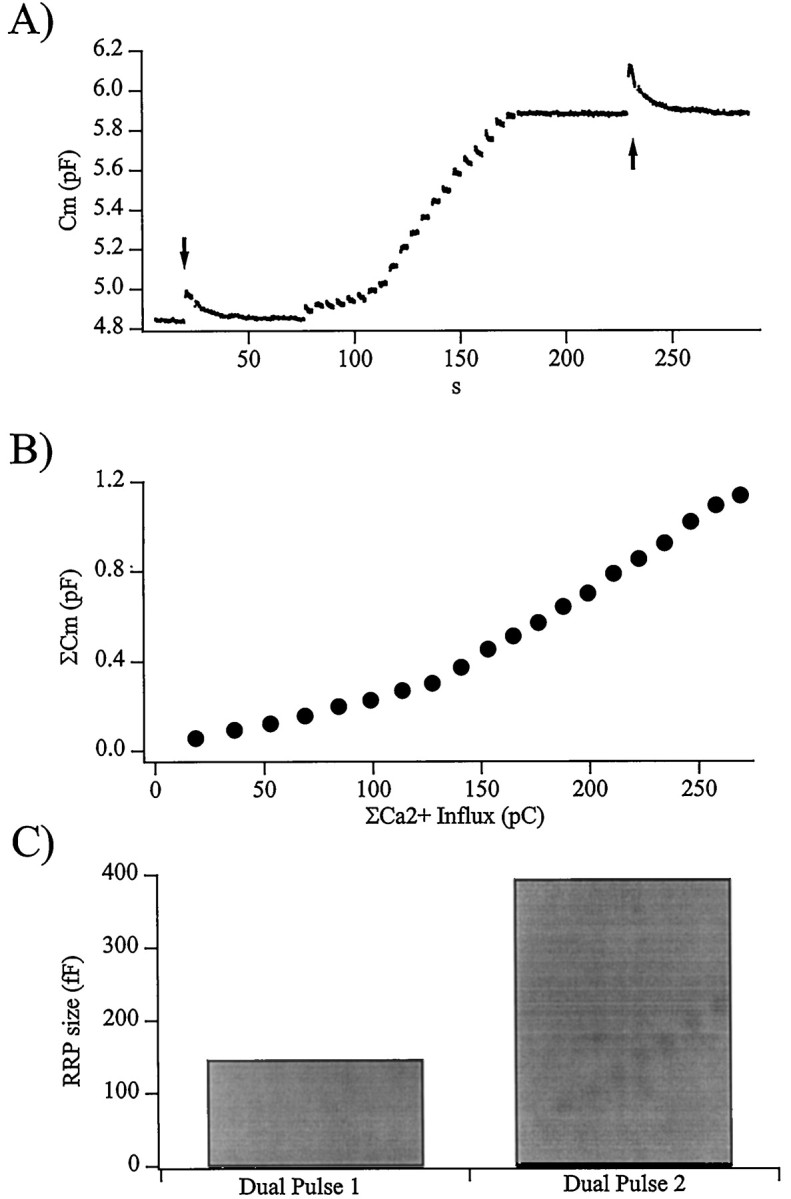
The activity-dependent facilitation is caused by a larger RRP of vesicles. A, Plotted is the membrane capacitance recorded from a cell stimulated by a pulse protocol designed to measure the activity-dependent changes in the number of vesicles in the readily releasable pool. The initial size of the RRP was determined through the use of a dual-pulse protocol (arrow). The RRP was then allowed to completely recover before the cell was stimulated by a single train of depolarizations as described in the legend to Figure 1. Again, after complete recovery to its steady state, the size of the RRP was measured by a dual-pulse stimulation protocol (arrow). The values R for the first and second dual-pulse stimuli in this cell were 0.28 and 0.57.B, As in Figures 1 and 2, the integrated evoked cell capacitance increase is plotted against total evoked Ca2+ influx, showing a shift in secretory efficiency during the train. The total evoked increase in Cm was 1136 fF in response to 269 pC total Ca2+ influx, an exceptionally high increase of exocytotic efficiency during the train. This cell was chosen for presentation because both dual-pulse stimuli resulted in an adequate amount of exocytotic depression for an accurate estimate of the RRP. C, In response to the train of depolarizations, the size of the RRP grew from a pretrain value of 146 fF to the increased value of 393 fF.
Figure 3 shows an example of an experiment in which the readily releasable pool size was probed before and 60 sec after a train of depolarizations (arrows). The RRP was a control value of 146 fF before the stimulus train but was found to have increased to 393 fF after the train. Note that the dual-pulse stimuli were followed by endocytosis returning Cm to prepulse baseline, whereas endocytosis was not as vigorous after depolarizations during the train (Fig. 1Ai,Bi). This observation is consistent with published studies that show endocytosis as dependent on Ca2+ influx, being slower and smaller in magnitude after stimuli that evoke less Ca2+ influx (Smith and Neher, 1997) and may not occur at all after stimuli that result in <50 fF increase in Cm (Engisch and Nowycky, 1998). Although the cell presented in Figure 3 represents a high degree of facilitation within the train, the train-evoked increase in RRP size was repeated in 38 different cells with variable time intervals between the train and RRP measurement (see Fig.6A). Therefore, it seems at least likely that a significant portion of the activity-dependent facilitation observed in the multiple-train excitation protocols is caused by an increase in the number of immediately releasable secretory vesicles. To identify the proportion of the facilitation that can be attributed to the increased RRP size, a computer simulation of the process was designed to imitate the experimental protocol shown in Figure 3. However, to accurately calibrate the simulation for a quantitative comparison to the measured data, a better understanding of the characteristics of Ca2+ clearance after a train of stimuli was first necessary, because many of the parameters in the kinetic secretion model are Ca2+-dependent.
Fig. 6.

The major component of evoked facilitation is caused by delayed PKC activation. A, The size of the readily releasable pool was measured in cells both before and at different times after a train of 20 depolarizations. The protocol was performed on both control and BIS-treated cells. The resulting RRP sizes are displayed for all time intervals and for pharmacological conditions (values accompanying each data point represent the number of cells). Also displayed are the corresponding results of the computer simulation of the experimental protocol, with the assumption that the facilitative effects of PKC action are not measured before a time delay of 120 sec, an assumption justified below. B, The results of an analysis designed to track the increase in RRP size caused by the effects of PKC activation is presented. The computer simulation (Fig. 5) was run in a repetitive analysis with Ca2+ activation of PKC assumed to occur immediately in the first repetition of the model and then delayed by 1 sec for each successive repetition (i.e., 45 sec delay on repetition number 46) for a total of 200 repetitions. The goodness-of-fit between the result of each simulation repetition was calculated as the χ2between the simulation and the data measured from cells (Bi represents trains of 20 depolarizations, andBii represents trains of 10 depolarizations). The minimum χ2 value was 0.23 and 0.42 (Bi andBii, respectively), occurring at a delay time of 178 and 172 sec (Bi and Bii, respectively) or ∼120 sec after the initial rise in [Ca2+]i from the first pulse.C, The activity-dependent facilitation observed persists undiminished for at least 10 min. Shown are the actual and simulated RRP magnitudes measured 600 sec after the end of a stimulus train. Theleft bars represent five experiments in which the cells were depolarized to 7 mV within the train, whereas thebars on the right represent control cells that were held constant at their clamped potential of −83 mV. The differences between the two conditions were judged as significant (paired Student’s t test).
Ca2+ clearance after trains of depolarizations is slow
A slow clearance of residual Ca2+ after tetanic stimuli has been strongly linked to a form of increased exocytotic activity in crayfish neuromuscular junction termed long-term facilitation (LTF; Tang and Zucker, 1997). A similar slow Ca2+ clearance can also be recognized in repetitively stimulated bovine chromaffin cells (Engisch et al., 1996, their Fig. 12). Any such slowed Ca2+ clearance would have dramatic effects on the Ca2+-dependent refilling of the RRP in chromaffin cells (von Rüden and Neher, 1993; Smith et al., 1998), resulting in a residual increase in RRP size after trains. Two major aspects to the cytosolic Ca2+ environment were considered for the calibration of the kinetic model, Ca2+ influx during the train and Ca2+ clearance after the train. Ca2+-modulated inhibition of Ca2+influx through voltage-gated calcium channels is well described in bovine chromaffin cells and would diminish the predicted rate of basal Ca2+ buildup in the cytosol of the cell or even cause the steady-state [Ca2+]i to slowly drop during a train of stimuli. As demonstrated in Figure 4, this was indeed the case. Data shown in Figure 4, A andB, demonstrate that during the train of 20 stimuli, the average magnitude of the Ca2+ currents decreased by 48% in a dual-exponential manner. The second attribute of the Ca2+ kinetic considered was the posttrain decay toward basal Ca2+. After less intense stimulation, [Ca2+]i recovers to baseline in a monoexponential manner with a time constant of ∼5–7 sec (Neher and Augustine, 1992; Smith et al., 1998). In response to the more prolonged Ca2+ influx evoked during the train stimulus, [Ca2+]i decayed much slower, perhaps because of a saturation of the faster Ca2+ clearance mechanisms seen after briefer stimuli (Xu et al., 1997) or because of a re-release of Ca2+ sequestered in cytosolic organelles (Tang and Zucker, 1997). The representative cell in Figure 4showed a posttrain [Ca2+]i decay described by a double exponential with time constant of 22 and 77 sec. The decaying magnitude of Ca2+ influx during the train and slow clearance after the train were then used to create a representative Ca2+ profile to serve as a template for the Ca2+-dependent processes of the secretion simulation (Fig. 5A). The Ca2+-dependent readily releasable pool behavior predicted from the simulation is also presented (Fig. 5A,dashed line). The simulated exocytosis efficiency of 2.16 fF/pC falls within the range measured for control cells (Fig. 2). Assuming that the elevated Ca2+ caused by the train of depolarizations, at some point, activates PKC, the steady-state RRP size should increase because of a heightened rate of vesicle recruitment. For the purposes of the simulation shown in Figure 4, the Ca2+-dependent effects of PKC action were delayed 120 sec (arrow), an assumption to be justified below. The size of the simulated RRP before and 60 sec after the train is plotted in Figure 4C. The predicted increased RRP size observed after the train is qualitatively similar to that experimentally measured in cells (Fig. 3). Assuming that sustained increased cell activity does indeed raise [Ca2+]i to levels sufficient to activate protein kinase C, then the facilitation observed seems to be at least mostly caused by an activity-dependent increase in the number of releasable vesicles.
Fig. 5.
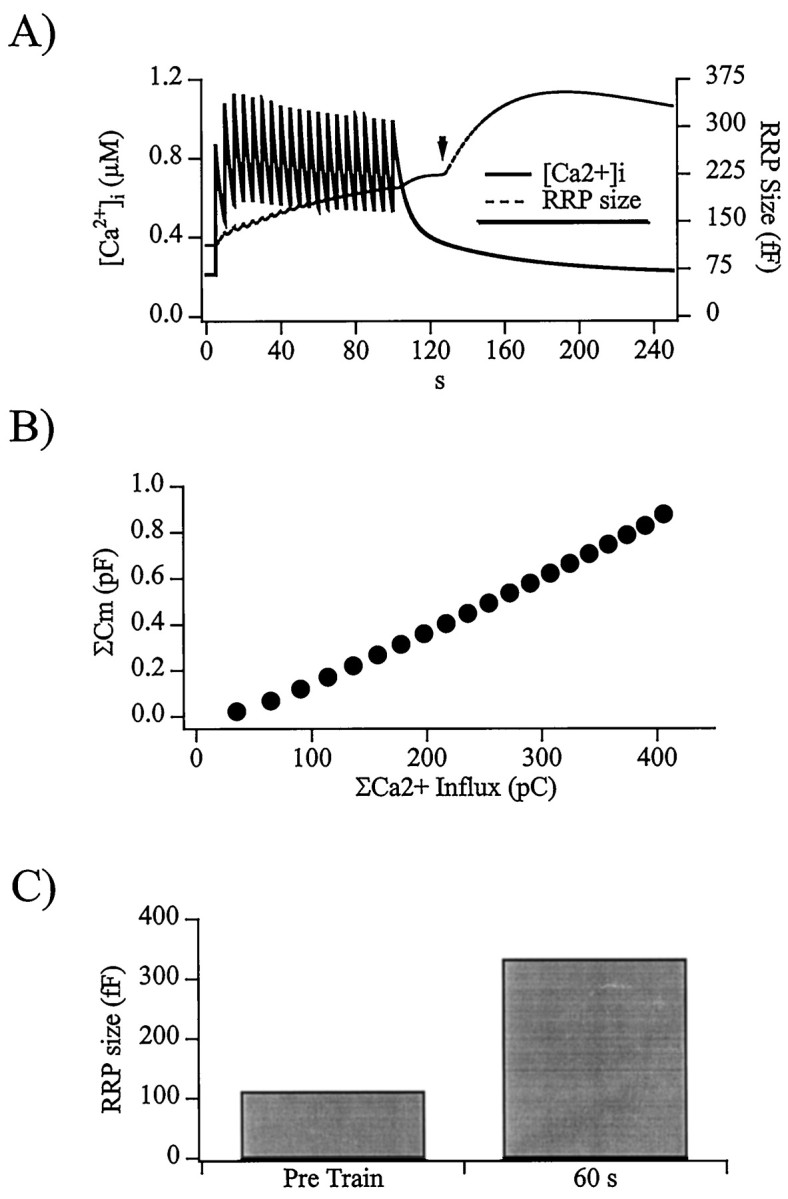
The computer secretion model reflects the activity-dependent increase in RRP size. A, The rundown in Ca2+ influx and post-train stimulus Ca2+ clearance characteristics measured from cells was incorporated into the two-step model for secretion control as modified by Smith et al. (1998). The Ca2+ profile before, during, and after a train of depolarizations was composed, and the resulting predicted RRP size was simulated. Activation of PKC through a sustained elevation in Ca2+ was simulated as occurring 125 sec into the protocol (arrow) or 120 sec after the initial increase in [Ca2+]i from the first 50 msec depolarization. B, The simulated plot of integrated, evoked capacitance increases versus integrated Ca2+influx, corresponding to Figures 1, B andD, and 4B, is displayed with an efficiency of 2.16 fF/pC. C, Shown are the steady-state RRP sizes predicted by the model both before and 60 sec after the end of the depolarization train, with magnitudes of 112 and 333 fF, respectively.
The activity-dependent increase in RRP size is delayed
To quantify the proportion of facilitation that could be attributed to the increased RRP size, measured and predicted train-evoked increases in RRP size were compared (Fig.6A). By definition, the measured and simulated RRP size before the facilitation train were the same, because it is under these conditions that the simulation is calibrated. Thirty seconds after a 20 pulse train, the measured RRP was found to increase by ∼100%, even in the BIS-treated cell, presumably because of residual effects of the slowly cleared cytosolic Ca2+. However, a further 30 sec later, a large difference in the RRP size was measured between the control and BIS-treated cells, with the control cells exhibiting a sudden and striking increase in the number of vesicles composing the readily releasable pool. The sudden increase in RRP size in control cells, with no such observation in the BIS-treated cells, indicates a delayed onset of the PKC-determined heightened recruitment of vesicles from the reserve pool A to the readily releasable pool B.
The large and abrupt BIS-sensitive increase in the RRP observed after stimulus trains implied a delayed but temporally synchronized onset of Ca2+-activated PKC action. To understand the time course of the activity-dependent facilitation better, a comparison of the experimentally measured data to a secretion simulation was made. The repetitive simulation described a delayed PKC activation, and subsequent increase in the maximal achievable rate constant (Eq. 2,a1; Fig. 5, arrow) that controls the recruitment of vesicles from the reserve pool A to the RRP B from 0.007 sec−1 to 0.014 sec−1 (see Smith et al., for a further discussion of the Ca2+-dependent increase in the kinetic constant a1). The first repetition contained an increase in a1 att = 0 sec, the second repetition incorporated the elevated value for a1 at t = 1 sec, and each successive repetition delayed the increase in the constant a1 by 1 additional second. The simulation was run 200 times. The predicted RRP size was compared with the measured data for each point (Fig. 6A), and the “goodness-of-fit” of the simulation result to the measured data was calculated as the χ2 between the two data sets. The resulting χ2 values for each of the 200 repetitions is plotted against the assumed PKC activity delay in Figure6Bi. Clearly, the time point at ∼180 sec (120 sec after the first pulse and subsequent increase in [Ca2+]i) fits the measured data best, implying that an increase in cytosolic Ca2+activates PKC but that the effects of the activation are not observed for ∼2 min.
The abrupt nature of the facilitation seen in Figure6A seemed suspiciously sharp, as if the train activated PKC but also had an inhibitory influence on the facilitation mechanism. Then after the train was over, the inhibition would be released and cause the observed abrupt increase in the readily releasable pool size. To determine whether this was actually the case, the same protocol used to produce the data in Figure6A was repeated, except that the trains were half the length, only 10 pulses delivered at 0.2 Hz. The RRP size was measured 60 sec before and 30, 60, and 180 sec seconds after the shorter train. The same χ2 analysis as in Figure 6Bwas then repeated on these data, with the expectation that if the BIS-sensitive facilitation was somehow masked during cell activity, only to appear after a period of rest, that the minimum χ2 value for the 10 pulse trains should occur earlier. This was not the finding, the minimum χ2 value for the 10 pulse trains was still only achieved 180 sec into the experimental protocol, 120 sec after the initial increase in [Ca2+]i (n = 9; Fig.6Bii). It is interesting to note that the χ2 of the 10 pulse trains failed to rise as strongly with longer PKC activation delays as that for the 20 pulse trains. This may indicate that the 10 pulse trains were not strong enough to fully activate the Ca2+-dependent PKC.
How long does the activity-triggered BIS-sensitive facilitation last? The RRP was measured 10 min after a 20 pulse protocol and found to be still as large as that predicted for maximal PKC action (Fig.6C). Therefore, it seems that once [Ca2+]i is elevated to a sufficient level for long enough, Ca2+-activation of the delivery of vesicle from the reserve to the readily releasable pool remains elevated even after [Ca2+]ireturns to baseline levels that would otherwise not activate the persistent facilitation. This may be caused by several mechanisms. Assuming that the BIS-sensitive, Ca2+-mediated facilitation does represent the activation of Ca2+-sensitive PKC, it would be expected that PKC would translocate to the plasma membrane (TerBush et al., 1988). It is not yet clear how long the activated form of PKC remains at the membrane. Alternately, the BIS- and Gö 6983-sensitive facilitation may represent the phosphorylation of a target peptide that experiences low basal phosphatase activity and therefore does not return to a control phosphorylation state within 10 min.
DISCUSSION
The study of exocytotic plasticity enjoys a long history in the field of synaptic as well as the neuroendocrine physiology. In cells in which the probability of exocytosis for a single vesicle is low, tetanic stimulation causes a build-up of residual Ca2+ and results in an increase in evoked secretion. Further stimuli begin to deplete vesicles, measured as secretory depression. In many excitable cells, a period of post-tetanic potentiation follows the stimulus train. Long-lasting modulation of synaptic strength in cells of the brain is thought to underlie the formation of memory and learning, both motor and cognitive (for review, see Malenka, 1994; Byrne and Kandel, 1996). Exocytotic plasticity in the neuroendocrine chromaffin cells exhibits many types of modulation similar to those in neurons. The modulation of secretion in chromaffin cells likely plays a role in an animal’s stress response, because they contribute to the “fight or flight” response of the sympathetic nervous system.
Evidence implies that activity-dependent secretory modulation may occur through mechanisms other than RRP modulation, relying rather on alteration of the fusogenic stimulus or the Ca2+sensitivity of the secretion process. The central synapse “calyx of Held” exhibits synaptic depression that is at least in part caused by activity-dependent rundown in presynaptic Ca2+influx (Forsythe et al., 1998). Also decreasing stimulus intensity, the autocrine action of presynaptically released ATP has been shown to lower evoked Ca2+ influx, thereby limiting transmitter exocytosis in crayfish neuromuscular junction (Lindgren and Smith, 1987) and chromaffin cells (Currie and Fox, 1996). In the giant synapse of the squid, decreased synaptic transmission occurs via adaptation of the secretory apparatus to elevated Ca2+ during periods of heightened activity (Hsu et al., 1996). Post-tetanic potentiation at the neuromuscular junction of crayfish, lasting minutes, is caused by the sequestration and slow re-release of Ca2+ from mitochondrial stores (Tang and Zucker, 1997). Other studies supply evidence that variable levels of cell activity mobilize entirely mechanistically, kinetically, or even morphologically different populations of vesicles (Vogel et al., 1996; Engisch et al., 1997; Takahashi et al., 1997; Kumar et al., 1998; Ma et al., 1998; Xu et al., 1998). Although activity-induced decreases in evoked Ca2+ influx were also observed in this study, stimulus normalization or the delivery of a saturating stimulus effectively allows for the quantification of the remaining exocytotic modulatory mechanisms.
Here it is reported that heightened cell activity is sufficient to trigger at least two forms of activity-induced facilitation in bovine adrenal chromaffin cells. The data show that a transient form is likely caused by the Ca2+-regulated increased recruitment of vesicles from the reserve to the readily releasable vesicle pool, as previously described in cells with chemically manipulated cytosolic [Ca2+]. This form of facilitation in seen within trains and seems likely to be very similar to the evoked facilitation seen with shorter and milder trains (Zhou and Misler, 1995; Engisch and Nowycky, 1996; Engisch et al., 1997). It may also be analogous to the activity-dependent facilitation described in neuronal systems such as the neuromuscular junction (for review, see Zucker, 1989, 1996; Fisher et al., 1997). The second form of stimulus-evoked facilitation seems consistent in magnitude and pharmacology to a facilitation previously shown under either treatment with phorbol ester or with prolonged elevations in buffered cytosolic Ca2+ and is sensitive to specific inhibitors of Ca2+-sensitive PKC (Gillis et al., 1996; Kumar et al., 1998; Smith et al., 1998). The BIS- and Gö 6983-sensitive form of facilitation has not been previously associated with heightened cell activity in the chromaffin system, perhaps because of a threshold Ca2+requirement that had not been reached in previous studies. Indeed, it seems that even strong stimuli such as the dual-pulse protocol used here are not sufficient to evoke the Ca2+-mediated activation of PKC. It is further shown that both forms of increased exocytosis are caused by an increase in the number of vesicles that make up the RRP and are likely not caused by a change in the affinity of the Ca2+-sensitive fusion step.
The ability for cells to regulate the number of releasable vesicles through the activity of kinases is not unique to chromaffin cells or even neuroendocrine cells. The observation that β-cells of the pancreas greatly increase their insulin secretion when treated with agents such as forskolin, which activates protein kinase A (PKA) through the cAMP pathways, is well known (Ämmälä et al., 1993, 1994). Inhibition of PKA blocks this facilitation (Renström et al., 1997). PKA, as well as PKC, also seems to play a role in facilitation measured and even a form of learning observed in the mollusk Aplysia (for review, see Byrne and Kandel, 1996). Phosphorylation of the synaptic vesicle-associated peptide Synapsin I by calcium–calmodulin-dependent protein kinase II is believed to make more vesicles available for release in central synapses such as neurons of the hippocampus (Greengard et al., 1993; Li et al., 1995) by releasing them from the cytoskeletal framework. A tetanus-induced facilitation lasting hours, termed LTF, has been demonstrated to be caused by a serotonin-evoked increase in the number of releasable vesicles (Wang and Zucker, 1998). Although no molecular pathway for LTF has yet been identified, serotonin has indeed been shown to be an effective activator of PKC activity in medullary respiratory neurons (Richter et al., 1997).
Facilitation evoked through stimulus trains observed by several groups in chromaffin cells (Zhou and Misler, 1995; Seward and Nowycky, 1996;Engisch et al., 1997) are accompanied by what initially seem to be differing interpretations. For example, a previous study (Engisch et al., 1997) concluded that different intensity trains cause secretory efficiency to deviate from a standard stimulus–exocytosis relationship, implying a shift in stimulus efficacy. The study concluded that the modulation was not caused by the decrease or increase in an RRP but rather to modulation through an unidentified second messenger pathway. After consideration, however, these results are not incompatible with the simpler two-step model shown in chromaffin cells (Heinemann et al., 1993; Smith et al., 1998), other neuroendocrine cells (Renström et al., 1997; Thomas and Waring, 1997), and from neuronal preparations (Elmqvist and Quastel, 1965; Betz, 1970; Stevens and Tujimoto, 1995). An examination of the shifts in Ca2+ efficacy reported by Engisch et al. (1997) show that under light stimulation, a given Ca2+ influx elicits more secretion than their control efficiency (standard curve) and that under heavier stimulation the secretion efficiency falls below the standard curve. If one assumes that after an initial decrease in RRP size, evident as an exocytotic burst (Neher and Zucker, 1993; Heinemann et al., 1994; Seward and Nowycky, 1996), that the rate-limiting step to ensuing exocytosis is the recruitment of vesicles from the reserve pool to the RRP, Ca2+ would then essentially begin to approach saturating concentrations for the release process, having a limited number of vesicles on which to act. The result would be that greater Ca2+ influx would on average be a less effective secretagogue, lowering secretory efficiency. If the control stimulus intensity of Engisch and Nowycky (1996) and Engisch et al. (1997) were such that the RRP were always in a semidepleted state, as indicated by the measured Ca2+ cooperativity of 1.5, [seeHeinemann et al. (1994) and Chow et al. (1994) for discussions of Ca2+ cooperativity as a function of stimulus intensity], one would expect exactly the reported facilitation and depression reported.
Finally, on what level does stimulus frequency really cause secretion and RPP modulation in chromaffin cells? Stimulus–secretion coupling in chromaffin cells does not appear to be as tight as in neurons (Chow et al., 1996; Klingauf and Neher, 1997). One could draw the conclusion that single action potentials in chromaffin cells are designed to elicit exocytosis, as occurs in neurons. However, it also seems possible that, not single action potentials, rather their frequency may play a larger role in triggering chromaffin cell secretion. But why would a secretory cell chose to lose the stimulus–response fidelity observed in many neurons? The answer may be twofold. (1) They don’t need it. The secreted products from chromaffin cells are mainly released into the bloodstream for remote action, not requiring the very high time fidelity observed in neurons. (2) It may not be such a great sacrifice. As outlined in an elegant review by Zador and Dobrunz (1997), recent studies have implicated a form of synaptic modulation that does not depend on frequency of single action potentials, rather on the change in their frequency. In this model the changes in firing frequency result in slight changes in the number of vesicles composing the RRP, perhaps by changing the basal [Ca2+]i and therefore the vesicle delivery rate to the RRP. This results in an increase or decrease in stimulus–secretion efficacy. A similar model may apply to chromaffin cells in which not single action potentials, but rather changes in firing frequency, modulate secretion efficiency as already considered by Zhou and Misler (1995).
Footnotes
This work was supported by a grant from the Human Science Frontiers Program (RG-4/95B). I thank Dr. E. Neher for valuable discussion of this manuscript and for both material and intellectual support throughout the project. I also thank F. Friedlein, I. Herfort, and M. Pilot for expert technical assistance.
Correspondence should be addressed to C. Smith, Medical College of Georgia, Department of Physiology and Endocrinology, School of Medicine, Augusta, GA 30912-3000.
REFERENCES
- 1.Ämmälä C, Ashcroft FM, Rorsman P. Calcium-independent potentiation of insulin release by cyclic AMP in single b cells. Nature. 1993;363:356–358. doi: 10.1038/363356a0. [DOI] [PubMed] [Google Scholar]
- 2.Ämmälä C, Eliasson L, Bokvist K, Berggren P-O, Honkanen RE, Sjöholm Å, Rorsman P. Activation of protein kinases and inhibition of protein phosphatases play a central role in the regulation of exocytosis in mouse pancreatic b-cells. Proc Natl Acad Sci USA. 1994;91:4343–4347. doi: 10.1073/pnas.91.10.4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Betz WJ. Depression of transmitter release at the neuromuscular junction of the frog. J Physiol (Lond) 1970;206:629–644. doi: 10.1113/jphysiol.1970.sp009034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bittner MA, Holz RW. Kinetic analysis of secretion from permeabilized adrenal chromaffin cells reveals distinct components. J Biol Chem. 1992;267:16219–16225. [PubMed] [Google Scholar]
- 5.Burgoyne RD. Fast exocytosis and endocytosis triggered by depolarization in single adrenal chromaffin cells before rapid Ca2+ current run-down. Pflügers Arch. 1995;430:213–219. doi: 10.1007/BF00374652. [DOI] [PubMed] [Google Scholar]
- 6.Byrne JH, Kandel ER. Presynaptic facilitation revisited: state and time dependence. J Neurosci. 1996;16:425–435. doi: 10.1523/JNEUROSCI.16-02-00425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chow RH, Klingauf J, Neher E. Time course of Ca2+ concentration triggering exocytosis in neuroendocrine cells. Proc Natl Acad Sci USA. 1994;91:12765–12769. doi: 10.1073/pnas.91.26.12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chow RH, Klingauf J, Heinemann C, Zucker RS, Neher E. Mechanisms determining the time course of secretion in neuroendocrine cells. Neuron. 1996;16:369–76. doi: 10.1016/s0896-6273(00)80054-9. [DOI] [PubMed] [Google Scholar]
- 9.Currie KPM, Fox AP. ATP serves as a negative feedback inhibitor of voltage-gated Ca2+ channel currents in cultured bovine adrenal chromaffin cells. Neuron. 1996;16:1027–1036. doi: 10.1016/s0896-6273(00)80126-9. [DOI] [PubMed] [Google Scholar]
- 10.Elmqvist D, Quastel DMJ. A quantitative study of end-plate potentials in isolated human muscle. J Physiol (Lond) 1965;178:505–529. doi: 10.1113/jphysiol.1965.sp007639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engisch KL, Nowycky MC. Calcium dependence of large dense-cored vesicle exocytosis evoked by calcium influx in bovine adrenal chromaffin cells. J Neurosci. 1996;16:1359–1369. doi: 10.1523/JNEUROSCI.16-04-01359.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engisch KL, Nowycky MC. Compensatory and excess retrieval: two types of endocytosis following single step depolarizations in bovine adrenal chromaffin cells. J Physiol (Lond) 1998;506:591–608. doi: 10.1111/j.1469-7793.1998.591bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engisch KL, Chernevskaya NI, Nowycky MC. Short-term changes in the Ca2+-exocytosis relationship during repetitive pulse protocols in bovine adrenal chromaffin cells. J Neurosci. 1997;17:9010–9025. doi: 10.1523/JNEUROSCI.17-23-09010.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisher SA, Fischer TM, Carew TJ. Multiple overlapping processes underlying short-term synaptic enhancement. Trends Neurosci. 1997;20:170–177. doi: 10.1016/s0166-2236(96)01001-6. [DOI] [PubMed] [Google Scholar]
- 15.Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- 16.Gillis KD. Chapter 7. Techniques for membrane capacitance measurements. In: Sakmann B, Neher E, editors. Single-channel recording, Ed 2. Plenum; New York: 1995. pp. 155–198. [Google Scholar]
- 17.Gillis KD, Möβner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- 18.Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- 19.Grynkiewicz G, Poenie M, Tsien R. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 20.Heinemann C, von Rüden L, Chow RH, Neher E. A two-step model of secretion control in neuroendocrine cells. Pflügers Arch. 1993;424:105–122. doi: 10.1007/BF00374600. [DOI] [PubMed] [Google Scholar]
- 21.Heinemann C, Chow R, Neher E, Zucker R. Kinetics of the secretory response in bovine chromaffin cells following flash photolysis of caged Ca++. Biophys J. 1994;67:2546–2557. doi: 10.1016/S0006-3495(94)80744-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horrigan FT, Bookman RJ. Releasable pools and the kinetics of exocytosis in adrenal chromaffin cells. Neuron. 1994;13:1119–1129. doi: 10.1016/0896-6273(94)90050-7. [DOI] [PubMed] [Google Scholar]
- 23.Hsu S-F, Augustine GJ, Jackson MB. Adaptation of Ca2+-triggered exocytosis in presynaptic terminals. Neuron. 1996;17:501–512. doi: 10.1016/s0896-6273(00)80182-8. [DOI] [PubMed] [Google Scholar]
- 24.Klingauf J, Neher E. Modeling buffered Ca2+ diffusion near the membrane: implications for secretion in neuroendocrine cells. Biophys J. 1997;72:674–690. doi: 10.1016/s0006-3495(97)78704-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knight D, Baker P. Stimulus-secretion coupling in isolated bovine adrenal medullary cells. Q J Exp Physiol. 1983;68:123–143. doi: 10.1113/expphysiol.1983.sp002691. [DOI] [PubMed] [Google Scholar]
- 26.Kumar GK, Overholt JL, Bright GR, Kwong YH, Hongwen L, Gratzl M, Prabhakar NR. Release of dopamine and norepinephrine by hypoxia from PC-12 cells. Am J Physiol. 1998;274:C1592–C1600. doi: 10.1152/ajpcell.1998.274.6.C1592. [DOI] [PubMed] [Google Scholar]
- 27.Li L, Chin LS, Shupliakov O, Brodin L, Sihra TS, Hvalby O, Jensen V, Zheng D, McNamara JO, Greengard P. Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc Natl Acad Sci USA. 1995;92:9235–9239. doi: 10.1073/pnas.92.20.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindau M, Neher E. Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflügers Arch. 1988;411:137–146. doi: 10.1007/BF00582306. [DOI] [PubMed] [Google Scholar]
- 29.Lindgren CA, Smith DO. Extracellular ATP modulates calcium uptake and transmitter release at the neuromuscular junction. J Neurosci. 1987;7:1567–1573. doi: 10.1523/JNEUROSCI.07-05-01567.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma XM, Lightman SL. The arginine vasopressin and corticotropin-releasing hormone gene transcription responses to varied frequencies of repeated stress in rats. J Physiol (Lond) 1998;510:605–614. doi: 10.1111/j.1469-7793.1998.605bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malenka RC. Synaptic plasticity in the hippocampus: LTP and LTD. Cell. 1994;78:535–8. doi: 10.1016/0092-8674(94)90517-7. [DOI] [PubMed] [Google Scholar]
- 32.Moser T, Neher E. Rapid exocytosis in single chromaffin cells recorded from mouse adrenal slices. J Neurosci. 1997;17:2314–2323. doi: 10.1523/JNEUROSCI.17-07-02314.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 34.Neher E, Augustine GJ. Calcium gradients and buffers in bovine chromaffin cells. J Physiol (Lond) 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neher E, Marty A. Discrete changes in cell membrane capacitance observed under conditions of enhanced secretion in bovine adrenal chromaffin cells. Proc Natl Acad Sci USA. 1982;79:6712–6716. doi: 10.1073/pnas.79.21.6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neher E, Zucker RS. Multiple calcium-dependent processes related to secretion in bovine chromaffin cells. Neuron. 1993;10:21–30. doi: 10.1016/0896-6273(93)90238-m. [DOI] [PubMed] [Google Scholar]
- 37.Renström E, Eliasson L, Rorsman P. Protein kinase A-dependent and -independent stimulation of exocytosis by cAMP in mouse pancreatic B-cells. J Physiol (Lond) 1997;502:105–118. doi: 10.1111/j.1469-7793.1997.105bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richter DW, Lalley PM, Pierrefichte O, Haji A, Bischoff AM, Wilken B, Hanefeld F. Intracellular signal pathways controlling respiratory neurons. Respir Physiol. 1997;110:113–123. doi: 10.1016/s0034-5687(97)00077-7. [DOI] [PubMed] [Google Scholar]
- 39.Seward EP, Nowycky MC. Kinetics of stimulus-coupled secretion in dialyzed bovine chromaffin cells in response to trains of depolarizing pulses. J Neurosci. 1996;16:553–562. doi: 10.1523/JNEUROSCI.16-02-00553.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith C, Neher E. Multiple forms of endocytosis in bovine adrenal chromaffin cells. J Cell Biol. 1997;139:885–894. doi: 10.1083/jcb.139.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith C, Moser T, Xu T, Neher E. Cytosolic Ca2+ acts by two separate pathways to modulate the supply of release-competent vesicles in chromaffin cells. Neuron. 1998;20:1243–1253. doi: 10.1016/s0896-6273(00)80504-8. [DOI] [PubMed] [Google Scholar]
- 42.Stevens CF, Tsujimoto T. Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proc Natl Acad Sci USA. 1995;92:846–849. doi: 10.1073/pnas.92.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takahashi N, Kadowaki T, Yazaki Y, Miyashita Y, Kasai H. Multiple exocytotic pathways in pancreatic beta cells. J Cell Biol. 1997;138:55–64. doi: 10.1083/jcb.138.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang Y, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18:483–491. doi: 10.1016/s0896-6273(00)81248-9. [DOI] [PubMed] [Google Scholar]
- 45.TerBush DR, Bittner MA, Holz RW. Ca2+ influx causes rapid translocation of protein kinase C to membranes. Studies of the effects of secretagogues in adrenal chromaffin cells. J Biol Chem. 1988;263:18873–18879. [PubMed] [Google Scholar]
- 46.Thomas P, Waring DW. Modulation of stimulus-secretion coupling in single rat gonadotrophs. J Physiol (Lond) 1997;504:705–719. doi: 10.1111/j.1469-7793.1997.705bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas P, Wong JG, Lee AK, Almers W. A low affinity Ca2+ receptor controls the final steps in peptide secretion from pituitary melanotrophs. Neuron. 1993;11:93–104. doi: 10.1016/0896-6273(93)90274-u. [DOI] [PubMed] [Google Scholar]
- 48.Vitale ML, Seward EP, Trifaró J-M. Chromaffin cell cortical actin network dynamics control the size of the release-ready vesicle pool and the initial rate of exocytosis. Neuron. 1995;14:353–363. doi: 10.1016/0896-6273(95)90291-0. [DOI] [PubMed] [Google Scholar]
- 49.Vogel SS, Blank PS, Zimmerberg J. Poisson-distributed active fusion complexes underlie the control of the rate and extent of exocytosis by calcium. J Cell Biol. 1996;134:329–338. doi: 10.1083/jcb.134.2.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.von Rüden L, Neher E. A Ca-dependent early step in the release of catecholamines from adrenal chromaffin cells. Science. 1993;262:1061–1065. doi: 10.1126/science.8235626. [DOI] [PubMed] [Google Scholar]
- 51.Wang C, Zucker RS. Regulation of synaptic vesicle recycling by calcium and serotonin. Neuron. 1998;21:155–167. doi: 10.1016/s0896-6273(00)80523-1. [DOI] [PubMed] [Google Scholar]
- 52.Xu T, Binz T, Niemann H, Neher E. Multiple kinetic components of exocytosis distinguished by neurotoxin sensitivity. Nat Neurosci. 1998;1:192–200. doi: 10.1038/642. [DOI] [PubMed] [Google Scholar]
- 53.Xu T, Naraghi M, Kang H, Neher E. Kinetic studies of Ca2+ binding and Ca2+ clearance in the cytosol of adrenal chromaffin cells. Biophys J. 1997;73:532–545. doi: 10.1016/S0006-3495(97)78091-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zador AM, Dobrunz LE. Dynamic synapses in the cortex. Neuron. 1997;19:1–4. doi: 10.1016/s0896-6273(00)80341-4. [DOI] [PubMed] [Google Scholar]
- 55.Zhou Z, Misler S. Action potential-induced quantal secretion of catecholamines from rat adrenal chromaffin cells. J Biol Chem. 1995;270:3498–3505. [PubMed] [Google Scholar]
- 56.Zhou Z, Neher E. Mobile and immobile calcium buffers in bovine adrenal chromaffin cells. J Physiol (Lond) 1993;469:245–273. doi: 10.1113/jphysiol.1993.sp019813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zucker R. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]
- 58.Zucker RS. Exocytosis: a molecular and physiological perspective. Neuron. 1996;17:1049–1055. doi: 10.1016/s0896-6273(00)80238-x. [DOI] [PubMed] [Google Scholar]