

Abstract
The antimitotic nucleoside cytosine arabinoside (araC) causes apoptosis in postmitotic neurons for which two mechanisms have been suggested: (1) araC directly inhibits a trophic factor-maintained signaling pathway required for survival, effectively mimicking trophic factor withdrawal; and (2) araC induces apoptosis by a p53-dependent mechanism distinct from trophic factor withdrawal. In rat sympathetic neurons, we found that araC treatment for 12 hr induced ∼25% apoptosis without affecting NGF-maintained signaling; there was neither reduction in the activity of mitogen actived protein kinase/extracellular signal-regulated kinase (MAPK/ERK) or protein kinase B/Akt, a kinase implicated in NGF-mediated survival, nor was there c-Jun N-terminal kinase (JNK) activation or c-Jun N-terminal phosphorylation, events implicated in apoptosis induced by NGF withdrawal. However, araC treatment, but not NGF-withdrawal, elevated expression of p53 protein before and during apoptosis. Additionally, araC-induced apoptosis was suppressed in sympathetic neurons from p53 null mice. Although MAPK/ERK activity is not necessary for NGF-induced survival, it protected against toxicity by araC, because inhibition of the MAPK pathway by PD98059 resulted in a significant increase in the rate of apoptosis induced by araC in the presence of NGF. Consistent with this finding, ciliary neurotrophic factor, which does not cause sustained activation of MAPK/ERK, did not protect against araC toxicity. Our data show that, in contrast to NGF deprivation, araC induces apoptosis via a p53-dependent, JNK-independent mechanism, against which MAPK/ERK plays a substantial protective role. Thus, NGF can suppress apoptotic mechanisms in addition to those caused by its own deprivation.
Keywords: superior cervical ganglion neurons, MAPK/ERK, PKB/Akt, JNK, c-Jun phosphorylation, p53, CNTF, PD98059, signal transduction, DNA damage
The nucleoside analog cytosine arabinoside (araC) is a potent antineoplastic agent for hematopoietic malignancies (for review, see Grant, 1998), but its use is complicated by toxicity to the nervous system (for review, see Baker et al., 1991). AraC also induces apoptosis in several types of postmitotic neuronsin vitro (Deckwerth and Johnson, 1993; Tomkins et al., 1994;Dessi et al., 1995; Sanz-Rodriguez et al., 1997). In mitotic cells, araC is thought to cause cell death primarily by disrupting DNA replication (Grant, 1998), but this would not be expected to affect postmitotic neurons.
Two mechanisms have been proposed for the neurotoxicity of araC. The first postulates that araC directly inhibits a crucial aspect of growth factor signaling, effectively mimicking trophic factor withdrawal and so causing death (Wallace and Johnson, 1989; Martin et al., 1990;Sanz-Rodriguez et al., 1997). Martin et al. (1990) reported that although araC was toxic to sympathetic neurons, other arabinose nucleosides, as well as other inhibitors of DNA synthesis, were not. They also found that 2′-deoxycytidine, the natural analog of araC, but not other deoxynucleosides, could inhibit araC toxicity. It was therefore suggested that araC was interfering with a 2′-deoxycytidine-dependent pathway other than the one involved in DNA synthesis, which is required for trophic factors to cause neuronal survival.
The second mechanism proposes that araC causes neuronal apoptosis by a p53-dependent pathway, independent of signaling by a survival stimulus. This was suggested because cerebellar granule neurons from p53 null mice were more resistant to araC compared with p53+/+ or p53+/− neurons, whereas apoptosis caused by removal of the depolarizing survival stimulus was independent of the p53 status of the neurons (Enokido et al., 1996a). This observation fitted with our previous finding that the rate of apoptosis of sympathetic neurons in the absence of NGF was accelerated by treatment with araC (Tomkins et al., 1994), suggesting that araC induces apoptosis by mechanisms other than inhibition of NGF survival signaling. Additionally, Park et al. (1998) reported that the caspase inhibitor zVAD-fluoromethylketone could inhibit apoptosis caused by NGF withdrawal but not that caused by araC, camptothecin, or UV irradiation (DNA-damaging agents).
In sympathetic neurons, NGF causes the sustained activation of several signaling pathways, including extracellular signal-regulated kinases 1 and 2 (ERK1/2) or mitogen activated protein kinases (MAPKs) (Virdee and Tolkovsky, 1995) and protein kinase B/Akt (Akt), Akt being implicated in mediating at least part of NGF-induced survival (Crowder and Freeman, 1998; K. Virdee and A. M. Tolkovsky, unpublished observations). On the other hand, NGF also suppresses c-Jun N-terminal kinase (JNK) activity and c-Jun N-terminal phosphorylation, which have been implicated in the induction of neuronal apoptosis after withdrawal of survival stimulus (Virdee et al., 1997; Eilers et al., 1998; Watson et al., 1998).
In this study, we have investigated the effect of araC on NGF-maintained signaling pathways, in particular those pathways thought to be involved in the survival and death of sympathetic neurons. We have also examined the involvement of p53 in apoptosis caused by araC.
MATERIALS AND METHODS
Preparation of neurons. Single-cell suspensions of superior cervical ganglion (SCG) neurons were prepared from <1-d-old Wistar rat pups as described previously (Buckmaster et al., 1991;Virdee and Tolkovsky, 1995). Briefly, ganglia were digested in 0.1% trypsin for ∼30 min at 37°C and then triturated through a narrow bore flame-polished Pasteur pipette. The resulting cell suspension was then preplated twice on collagen in L15–CO2 medium (Hawrot and Patterson, 1979) containing 5% rat serum in a humidified atmosphere of 5% CO2–95% air at 37°C. The nonadhering cells were collected by centrifugation and kept in L15 plating medium at 4°C until use in experiments.
Culture conditions. Neurons were cultured on poly-l-lysine–laminin-coated wells in growth medium (L15–CO2 containing 3% dialyzed rat serum) and additives as indicated. Rat serum (5 ml) was dialyzed twice against 1 l lots of 10 mm HEPES, pH 7.4, at 4°C. A 50 mm stock of 2′-amino-3′-methoxyflavone (PD98059) (Calbiochem-Novabiochem, Nottingham, UK) in DMSO was diluted into culture medium immediately before use, and its supply was replenished between 6 and 7 hr of treatment (at least 80% of the medium in all conditions was replaced). NGF (2.5S) was prepared from male mouse submaxillary glands (Mobley et al., 1976) and was used at 20–100 ng/ml. Rat ciliary neurotrophic factor (CNTF) (Peprotech EC, London, UK) was used at 100 ng/ml.
Cell counting and scoring of apoptosis. Neurons were fixed at the indicated times by the addition of an equal volume of fixing medium (3:1 mix of methanol/acetic acid) to the culture medium. The fixed cells were usually stored at 4°C before scoring for apoptosis. Nuclei were visualized using Hoechst 33342 (1.7 μg/ml, final concentration; Sigma, Poole, UK), and the fixing medium and dye was replaced by PBS before counting. A minimum of 500 neurons in a 600-μm-wide strip extending through the center of the well were scored for apoptosis using a Leica (Wetzlar, Germany) DMIL microscope, with only those neurons which had clearly segmented and condensed chromatin being counted as apoptotic.
Preparation of cell extracts and immunoblotting. Neurons were collected at the indicated times, pelleted, and washed with cold PBS, and then ice-cold lysis buffer was added (20 mmTris–acetate, pH 7.5, 0.27 m sucrose, 1 mmEDTA, 1 mm EGTA, 10 mmsodium-β-glycerophosphate, 50 mm sodium fluoride, 5 mm sodium pyrophosphate, 1% (v/v) Triton X-100, 1 mm sodium orthovanadate, 0.1% (v/v) β-mercaptoethanol, 0.2 mm PMSF, and 1 mm benzamidine). Lysis was achieved on ice over a period of 20–30 min with occasional vortexing, after which gel loading buffer was added, and lysates were heated at 100°C for 10 min and then stored at −20°C before processing. In all experiments, parallel cultures were set up to determine the amount of apoptosis at the time of cell collection.
Cell extracts were resolved on 10% polyacrylamide-SDS gels before electroblotting onto nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany). Blots were blocked for 1 hr at room temperature or overnight at 4°C in Tris-buffered saline–Tween 20 (TBST) (100 mm NaCl, 10 mm Tris, pH 7.5, and 0.1% (v/v) Tween 20) containing 5% or 2% skimmed milk powder (when using polyclonal or monoclonal antibodies, respectively). Blots were probed either overnight at 4°C or room temperature for 1 hr, with the following antibodies: PhosphoPlus JunII (Ser 63), PhosphoPlus Akt (Ser 473), PhosphoPlus SAPK/JNK (Thr 183/Tyr 185; New England Biolabs, Beverley, MA), anti-active MAPK (Promega, Madison, WI), anti-p53 (FL-393; Santa Cruz, Santa Cruz, CA); and anti-MAPK (MK12; Transduction Laboratories, Lexington, KY). After incubation with the appropriate horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA), immunoreactive bands were visualized by chemiluminescence and exposure to Kodak X-OMAT film (Eastman Kodak, Rochester, NY). In some cases, blots were stripped (in buffer containing 62.5 mm Tris, 2% SDS, and 0.7% (v/v) β-mercaptoethanol, pH 6.7) for 1 hr at room temperature, followed by extensive washing in TBST before reblocking and reprobing. Intensity of bands was analyzed by optical densitometry using the Leica Q500 image analysis system. To average the data from several experiments, the intensity of bands for the proteins being investigated was divided by the intensity of the bands of total ERK1/2. An arbitrary value of 1 was assigned to the ratio obtained from cultures treated with NGF alone. For a quantitative analysis of signaling events, a time point of 12 hr was chosen, which spanned a range of apoptotic values up to ∼40% depending on the conditions of treatment. When apoptosis exceeded 40% (or the incubation time was much longer than 12 hr) there was often too much protein loss, which precluded accurate normalization.
p53+/+, p53+/−, and p53−/− mice.Founder mice deficient in the p53 gene (p53+/− and p53−/−) were a kind gift from Dr. Alan Clarke (University of Edinburgh, Scotland), and their generation has been described previously (Clarke et al., 1993). SCG used in experiments were taken from pups obtained from breeding p53 heterozygote adults. Litters were taken at 3–4 d postnatally, and the pair of ganglia from each animal was removed and processed separately. After digestion for 30 min in 0.1% trypsin and trituration with a p200 pipette tip, the cells were preplated on tissue culture plastic for 4–5 hr in preplating medium before being plated in growth medium on poly-l-lysine–laminin-coated wells for experiments. NGF was used at 100 ng/ml and araC at 100 μm. Cells were fixed to determine the extent of apoptosis as described above, and a minimum of 200 cells were counted per well. The genotype of each animal was determined by PCR only after counting had been completed. The DNA for PCR was obtained by heating tail tissue at 100°C in 100 mm NaOH for at least 5 min. PCR was initiated using the primers described by Clarke et al. (1993) (product for wild-type allele, 642 bp; product for neomycin allele, 510 bp) by hot start (after 2 min at 94°C), followed by 35 cycles (45 sec at 94°C, 1 min at 62°C, 2 min 20 sec at 72°C) and then 10 min at 7°C. The genotype of adult mice was determined by PCR on DNA obtained from hair roots using the DNAce Clinipure system (Bioline, London, UK).
Statistical analysis. Data for experiments from rat SCG were analyzed by ANOVA and the Student–Newman–Keuls post hoc test or by an unpaired two-tailed Student’st test. Data from experiments on mice SCG were analyzed by Student’s t test for each pair of conditions obtained with and without araC. The value of n signifies the number of independent experiments.
RESULTS
Matching rates of apoptosis caused by withdrawal of NGF or araC treatment in the presence of NGF
To address the question of whether araC inhibits NGF signaling, thereby mimicking NGF withdrawal, we compared changes in signaling processes that occur on NGF deprivation with those induced by araC in the presence of NGF. To draw direct comparisons between these death-inducing conditions, a similar rate of apoptosis is required in both models. We achieved this requirement in newly isolated rat sympathetic neurons by using 100 μm araC and dialyzed serum (apoptosis at 14 hr: no NGF, 32.4% ± 3.9; NGF and araC, 31.2% ± 7.8; mean ± SD; n = 3) (Fig.1). Nondialyzed rat serum was found to reduce cellular uptake of araC and therefore reduce the rate at which araC kills the neurons (data not shown). It should also be noted from Figure 1 that apoptosis occurred at an accelerated rate when araC was added in the absence of NGF compared with the rate with no NGF or araC added, suggesting that araC can induce apoptosis by mechanisms other than by inhibiting NGF signaling as suggested previously (Tomkins et al., 1994).
Fig. 1.

Matching rates of apoptosis caused by NGF deprivation or araC treatment in the presence of NGF. Newly isolated neurons were plated into medium without NGF or with NGF (100 ng/ml) or araC (100 μm) for the indicated times before being fixed, stained, and counted. The results shown are the mean ± SD of three independent experiments. See Figure 8 for photomicrographs of stained nuclei under these conditions.
AraC does not affect NGF-induced phosphorylation of MAPK/ERK or Akt
To determine whether araC interferes with NGF-induced intracellular signaling in the neurons, we first examined whether araC inhibits the activation of three protein kinases whose sustained activity is dependent on NGF, namely the MAPKs p44-ERK1 and p42-ERK2 (ERK1/2) and Akt (Virdee and Tolkovsky, 1995; Virdee and Tolkovsky, unpublished observations). In sympathetic neurons, NGF causes the activation of ERK1/2, as long as NGF is present. If the MAPK pathway is inhibited, however, neuronal survival in the presence of NGF is unaffected (Creedon et al., 1996; Virdee and Tolkovsky, 1996). An NGF-induced pathway that is reported to be involved in the survival of sympathetic (and other) neurons is the phosphatidylinositol 3-kinase (PI 3-kinase) pathway with its downstream target Akt (Dudek et al., 1997; Crowder and Freeman, 1998; Virdee and Tolkovsky, unpublished observations). The MAPKs ERK1/2 and Akt need to be phosphorylated on key residues for their activation (Ray and Sturgill, 1988; Gotoh et al., 1990; Alessi et al., 1996;Andjelkovic et al., 1996). We therefore investigated whether araC affected these activation steps using antibodies specific for the appropriately phosphorylated forms of these enzymes. We have confirmed that the extent of activation of these kinases measured using in-gel kinase assays (ERKs) or immunocomplex kinase assays (Akt) is matched by their phosphorylation, which can be detected by the anti-phospho-kinase-specific antibodies used in this study (K. Virdee and A. M. Tolkovsky, unpublished observations). It can be seen in Figure 2 that during the period of active cell death, when 24.8 ± 2.5 and 25.3 ± 4.5% apoptosis (at 12 hr: mean ± SEM; n = 6 had occurred in the presence of araC and NGF or with no additions, respectively, the phosphorylation of neither Akt nor that of ERK1/2 was affected (fold-phosphorylation compared with a value of 1 arbitrarily assigned to the result obtained from cultures treated with NGF alone: Akt, 1.2 ± 0.2; mean ± range; n = 2; ERK1/2, 1.1 ± 0.3; mean ± SD; n = 3). During NGF deprivation, in contrast, dephosphorylation of all these kinases occurred (Akt, 0.14 ± 0.1; n = 2; ERK1/2, 0.1 ± 0.05; n = 3). The time point of 12 hr was chosen to enable accurate comparison with experiments shown below in which >40% of apoptosis was obtained (see Materials and Methods). However, it should be noted that araC did not affect levels of ERK phosphorylation after 14 hr, when apoptosis in the presence of NGF and araC reached ∼30% (data not shown).
Fig. 2.

NGF-induced phosphorylation of p44-ERK1, p42-ERK2, and Akt is not inhibited by araC. SCG neurons were treated for 12 hr with NGF alone (20 ng/ml) (lane 1), NGF and 100 μm araC (lane 2), and no NGF or araC (lane 3), and the protein extracts were prepared and probed as described in Materials and Methods. A,Top panel shows phospho-Akt probed for phosphorylation at serine 473 and phospho-ERK1/2 probed with anti-active MAPK antibody;bottom panel shows total ERKs as an indication of protein loading. B, Fold-phosphorylation as determined by optical densitometry (see Materials and Methods). Top panel, Phospho-Akt; bottom panel, phospho-ERKs; both normalized to total ERKs. Data for Akt and ERKs are the mean ± range and mean ± SD for two and three independent experiments, respectively. Numbers below the lanesgive the mean ± SEM of percentage of apoptosis for six independent experiments.
AraC does not cause activation of JNK or phosphorylation of c-Jun at serine 63
To investigate further the effects of araC with respect to possible inhibition of NGF signaling, we examined whether araC activates similar signaling processes to those which occur after the withdrawal of NGF and during the subsequent period of apoptosis. The JNKs are activated within a few hours of withdrawal of NGF and remain active throughout the period of apoptosis (Virdee et al., 1997; Eilers et al., 1998). JNK activity has also been implicated in the induction of neuronal apoptosis (Xia et al., 1995; Eilers et al., 1998; Maroney et al., 1998). We therefore examined the effect of araC treatment on the phosphorylation step required for JNK1 activation using a polyclonal phospho-specific antibody. Figure3A shows that araC did not cause any measurable increase in c-Jun N-terminal phosphorylation in the presence of NGF at the time when apoptosis was occurring (12 hr) (see above and Fig. 2 for percentages of apoptosis), although, as expected, JNK was strongly phosphorylated (∼10-fold) at its activation sites because of NGF withdrawal.
Fig. 3.
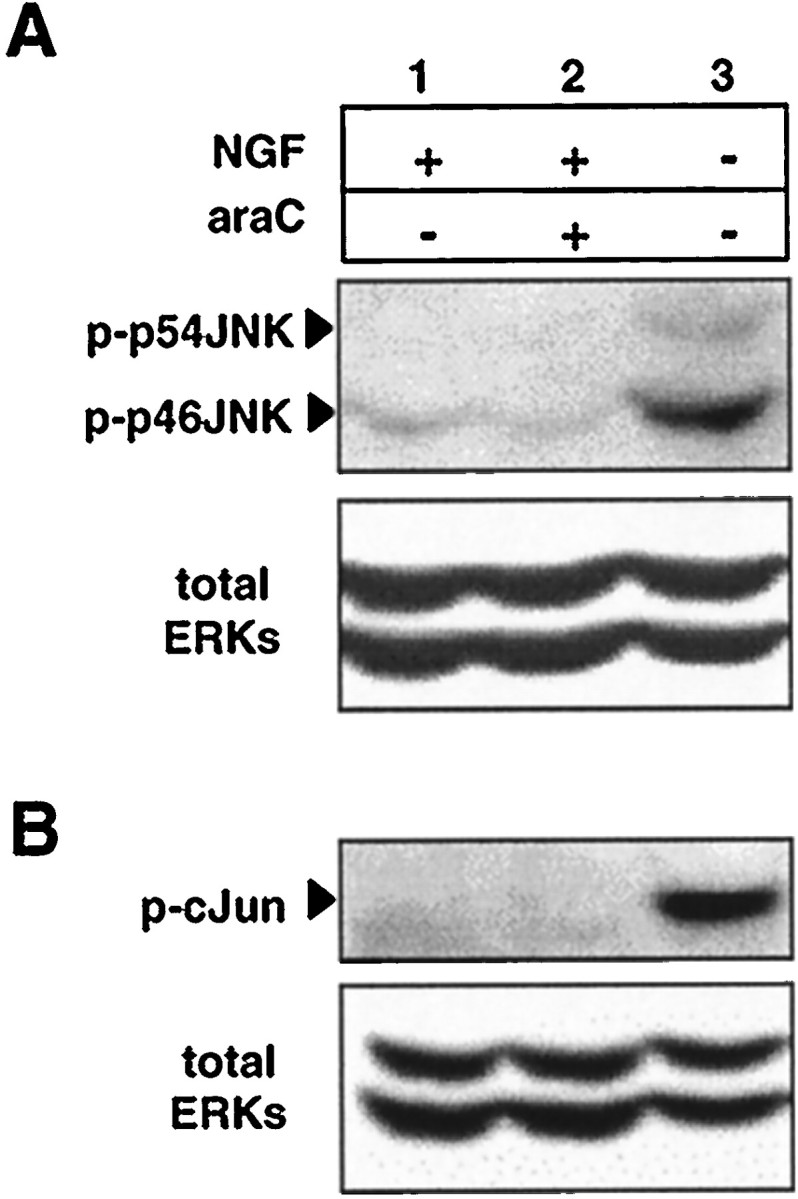
JNK activation and c-Jun phosphorylation at serine 63 are not required for araC-induced apoptosis. SCG neurons were treated for 12 hr with NGF alone (lane 1), NGF and 100 μm araC (lane 2), and no NGF or araC (lane 3), and the protein extracts were prepared and processed as in Materials and Methods. A, The blot was probed first for JNK phosphorylated at residues threonine 183 and tyrosine 185 (top panel) and then for total ERKs as an indication of protein loading (bottom panel). For these experiments, NGF was used at 100 ng/ml. The result shown is representative of three independent experiments. B, In two other independent experiments, the amount of N-terminal phosphorylation of c-Jun was determined by probing with an antibody specific for c-Jun phosphorylated at residue serine 63 (top panel). Immunoblotting for total ERKs is shown (bottom panel) as an indication of protein loading. NGF was used at a concentration of 20 ng/ml. See Figure 2 for percentage of apoptosis for each condition.
Because the transcription factor c-Jun is reported to be required for neuronal apoptosis (Estus et al., 1994; Ham et al., 1995) and particularly its N-terminal phosphorylation at serine 63 (Eilers et al., 1998; Watson et al., 1998), we investigated whether c-Jun phosphorylation at serine 63 was elevated in neurons undergoing apoptosis after araC addition. Figure 3B shows that despite ongoing apoptosis, phosphorylation of c-Jun at serine 63 was not stimulated by the addition of araC in the presence of NGF, consistent with the results showing lack of activation of JNK, whereas intense phosphorylation of c-Jun at serine 63 (∼5- to 10-fold) occurred in the absence of NGF, consistent with previous reports (Virdee et al., 1997; Eilers et al., 1998). The lack of significant JNK or c-Jun phosphorylation observed in some experiments in the presence of NGF precluded obtaining a reliable quantitative measurement of the change in activity. However, similar results showing lack of c-Jun phosphorylation were also obtained after 14 hr of araC treatment (data not shown). These data indicate that araC does not induce or require JNK activation and/or c-Jun N-terminal phosphorylation to cause apoptosis and further suggest that the suppression of JNK activity and c-Jun phosphorylation by NGF is not affected by araC treatment.
p53 is required in araC-induced apoptosis
It has been reported that cerebellar granule neurons from p53−/− animals were resistant to several antimitotic agents that also cause apoptosis in neurons, including araC, in contrast to neurons from p53+/− or p53+/+ mice (Enokido et al., 1996a,b). We therefore examined whether apoptosis induced by NGF withdrawal or araC treatment in sympathetic neurons was dependent on the genotype of p53 by comparing littermates from p53+/− parents. Figure4 shows that at 16 hr, no significant apoptosis occurred because of araC treatment in p53−/− neurons in the presence or absence of NGF (apoptosis at 16 hr: NGF, 14 ± 6%; NGF with araC, 16 ± 3%; no NGF, 68 ± 10%; no NGF with araC, 63 ± 14%; mean ± SD; n = 6). When one copy of the p53 gene was present, however, a significant increase in apoptosis was observed in response to araC treatment (NGF: no araC, 15 ± 5%; with araC, 29 ± 6%; no NGF: no araC, 68 ± 10%; with araC, 80 ± 7%; mean ± SD; n = 13; p < 0.001 for both comparisons). Similar differences were found in neurons from p53+/+, mice although the difference in percent apoptosis between neurons cultured with no NGF, without or with araC, did not quite achieve significance (0.1 > p > 0.05), most probably because of the inherent lack of accuracy of scoring apoptotic neurons when >70% apoptosis has occurred. Figure 4 also shows that the response of sympathetic neurons to NGF withdrawal was not dependent on the genotype of p53, consistent with previous reports (Davies and Rosenthal, 1994; Vogel and Parada, 1998).
Fig. 4.

SCG neurons from p53−/− mice are resistant to apoptosis induced by araC, but not NGF, withdrawal. SCG neurons were prepared from individual mice from litters born to p53+/− parents and cultured for 16 hr with or without NGF (100 ng/ml) and with or without araC (100 μm) before being fixed, stained, and counted as described in Materials and Methods. The p53 status of each animal tested was determined after counting had been completed. The results shown are the mean ± SEM for 6 (p53+/+), 13 (p53+/−), and 6 (p53−/−) animals, respectively. Student’st tests gave the significance values as indicated (***p < 0.001; †0.1 > p> 0.05). Inset, PCR results showing the three genotypes. Top band (642 bp) represents the presence of wild-type p53 gene; bottom band (510 bp) represents the absence of the p53 gene.
Because it appeared that there was a selective requirement for p53 in araC-induced apoptosis but not in NGF withdrawal-induced apoptosis, we determined whether araC caused any increase in the cellular levels of p53 protein in rat sympathetic neurons before and during induction of apoptosis, as would be expected if p53 played a causal role in araC-mediated apoptosis (for review, see Levine, 1997). Figure5A shows that araC treatment caused an increase in p53 protein as early as 4 hr after its addition (approximately threefold), a time point at which there were no significant differences in the extents of apoptosis between any of the experimental conditions (Fig. 1). This elevation in the level of p53 protein in araC-treated neurons was maintained for up to 14 hr (when significant apoptosis was occurring) and occurred irrespective of the presence of NGF (Fig. 5B). In NGF-maintained cultures, we observed detectable but low levels of p53 protein, which were not elevated after NGF withdrawal (Figs. 5,7B), in keeping with the results of Sadoul et al. (1996). Thus, there is an induction of and requirement for p53 in araC-induced apoptosis but not that caused by NGF-withdrawal.
Fig. 5.

AraC causes an early and sustained elevation of p53 protein levels. Rat SCG neurons were treated for the indicated times before being collected, resolved on SDS-PAGE, and electroblotted as described in Materials and Methods. NGF was used at 100 ng/ml, and araC was used at 100 μm. A, Lanes 1–4, araC; lanes 1–5, NGF. Top panel shows the results of probing for p53; bottom panel shows results for total ERKs as an indication of protein loading. The results shown are representative of two independent experiments. B, Cultures were treated for 10 or 14 hr before collection, processing, and probing as in A.Lanes 1, 2, 5,6, NGF; lanes 2, 4,6, araC. The results shown are representative of three independent experiments.
Fig. 7.

Suppression of ERK activity by PD98059 renders neurons more susceptible to araC: a comparison of signaling pathways.A, Neurons were cultured in the presence or absence of NGF (20 ng/ml), araC (100 μm), or PD98059 (50 μm) for 14 hr in the indicated conditions before being fixed, stained, and counted. All media contained 0.2% DMSO. Results shown are the mean ± SEM of three to five independent experiments (**p < 0.002; Student’s t test). See Figure 8 for photomicrographs of apoptotic nuclei.B, Neurons were treated with 20 ng/ml NGF (lanes 1, 2, 4, 5), 100 μm araC (lanes 2, 5), and 50 μm PD98059 (lanes 4, 5) for 12 hr and then processed for immunoblotting as in Materials and Methods. The panels show the results of probing with the following antibodies (top to bottom): anti-phospho-Akt (serine 473), anti-phospho-ERK1/2, anti-phospho-c-Jun (serine 63), anti-p53, and anti-ERK1/2. Results shown are representative of three independent experiments, except for anti-phospho-Akt and anti-phospho-c-Jun, which are representative of two independent experiments. The numbers below the lanes indicate the percentage of apoptosis and are the mean ± SD of three independent experiments.C, Fold-phosphorylation as determined by optical densitometry (see Materials and Methods): top panel, phospho-Akt (n = 2); bottom panel, phospho-ERKs (n = 3); both normalized to total ERK. Error bars represent the SD and range for ERK and Akt, respectively.
NGF protects against araC toxicity via the MAPK/ERK pathway
The survival of newly isolated sympathetic neurons can be sustained by CNTF for several days when laminin is used as substrate (Nobes and Tolkovsky, 1995; Virdee and Tolkovsky, 1995). Figure 6 shows that when we used CNTF to support the neurons, the rate of apoptosis induced by araC was significantly greater than that caused by araC in the presence of NGF (apoptosis at 14 hr: NGF and araC, 33.1 ± 6.9%; CNTF and araC, 69.6 ± 4.5%; mean ± SD;n = 3; p < 0.001). In addition, the extent of apoptosis induced by araC in the presence of CNTF at 14 hr was not significantly different from that obtained when neurons were treated with araC in the absence of any survival factor (77.3 ± 4.8%; mean ± SD; n = 3) (Fig.8, corresponding micrographs). These results suggest that NGF, but not CNTF, stimulates one or more signaling pathways that protect against araC-induced apoptosis.
Fig. 6.

AraC-induced apoptosis is delayed by NGF but not by CNTF. Neurons were cultured with 100 ng/ml CNTF, 20 ng/ml NGF, or with no growth factor additions (none) in the absence (black bars) or presence (hatched bars) of 100 μm araC as indicated. The cells were fixed at 14 hr before staining and counting. Results are the mean ± SD of three independent experiments. A Student–Newman–Keuls multiple comparisons test gave significant differences (***p< 0.001) for all comparisons, except between NGF versus CNTF (bars 1, 3), NGF with araC versus no growth factor additions (bars 2, 5), and CNTF with araC versus no growth factors with araC (bars 4, 6).
Fig. 8.

Nuclear morphologies of SCG neurons under various conditions as visualized with Hoechst 33342. Neurons were treated with 100 ng/ml NGF (a, d, g), 100 ng/ml CNTF (b, e, h), or without growth factors (-) (c,f). AraC was used at 100 μm(d–f), and PD98059 was used at 50 μm (g, h). Hoechst 33342 was added to the cultures at 14 hr, and photographs were taken before fixing and counting. Scale bar, 10 μm.
One difference between NGF and CNTF signaling is that CNTF only transiently activates the MAPK pathway, with ERK1/2 activity returning to nonstimulated values by 1 hr (Virdee and Tolkovsky, 1995). The MAPK pathway, however, is reported not to be a necessary component in NGF-mediated survival (Creedon et al., 1996; Virdee and Tolkovsky, 1996). To test whether ERK activity may play a role in delaying araC-induced apoptosis, we used PD98059, an inhibitor of the MAPK pathway. This compound inhibits activation of the kinase (MEK 1) immediately upstream of the ERKs, thereby preventing ERK activation (Dudley et al., 1995). Figure 7 shows the results of scoring for apoptosis after PD98059 treatment and parallel results obtained from immunoblots. When PD98059 was added to neurons treated with araC in the presence of NGF (Fig. 7A), the extent of apoptosis caused by araC was significantly enhanced (at 14 hr: NGF and araC, 32.7 ± 5; NGF, araC, and PD98059, 63.2 ± 11.2; mean ± SD;n = 5; p < 0.002), strongly suggesting a specific role for ERK1/2 activity in protecting against araC-induced apoptosis. Inhibition of ERK1/2 had no significant effect on either the ability of NGF to promote neuronal survival or the rate of death in the absence of NGF as shown previously (Creedon et al., 1996; Virdee and Tolkovsky, 1996). Figure 7B shows that PD98059 inhibited ERK1/2 phosphorylation in the presence of NGF to a level comparable with that seen in the absence of NGF (Fig. 2B), but Akt and c-Jun N-terminal phosphorylation were not significantly affected, whether or not araC was present. This further supports a role for ERK1/2 in antagonizing the specific mechanism by which araC causes apoptosis, because PD98059 had no effects on the other pathways investigated. In keeping with this observation, we found that PD98059 did not cause apoptosis when the cells were maintained with CNTF, nor did it further increase apoptosis in the presence of araC (apoptosis at 14 hr: CNTF, 8.9 ± 1.4%; CNTF and PD98059, 8.4 ± 1.3%; CNTF and araC, 69.6 ± 4.5%; CNTF, araC, and PD98059, 72.1 ± 2.5%; mean ± SD; n = 3). PD98059 also did not affect p53 levels in the absence of araC and did not impair the induction of p53 as a result of araC treatment (Fig. 7B), thus suggesting that ERK1/2 do not act by regulating the araC-induced elevation of p53 protein levels.
Because toxicity of araC in mitotic cells is dependent on the efficacy of its uptake and metabolism into triphosphonucleotides (Grant, 1998), it was possible that ERK activity decreased the uptake and/or metabolism of araC, so reducing its toxicity. We found, however, that PD98059 inhibited rather than increased the uptake of [3H]araC into the neurons by ∼50%, without affecting its relative metabolism into the mono-, di-, and triphosphate forms (measurements taken after 14 hr of [3H]araC uptake; data not shown). ERK1/2 may therefore be involved in controlling the uptake of arabinose nucleosides in some manner, but this is unlikely to be the cause of the increased toxicity of araC in the presence of PD98059.
DISCUSSION
We have compared changes in the levels of activity of signaling pathways during apoptosis induced by NGF-deprivation or araC treatment in the presence and absence of NGF to explore the mechanism of araC toxicity in sympathetic neurons. We found that (1) araC treatment did not interfere with any of the NGF-maintained signaling pathways examined, (2) araC treatment did not mimic signaling induced by NGF-withdrawal, (3) there was a sustained elevation of and requirement for p53 during apoptosis caused by araC but not NGF withdrawal, and (4) ERK activity protected against apoptosis caused by araC.
We have already reported some evidence that araC does not interfere with NGF-mediated signaling in which pretreatment of NGF-deprived neurons with araC for 3 hr did not inhibit the subsequent NGF-mediated induction of c-Fos protein (Tomkins et al., 1994). In this study, we have investigated the effect of araC on the phosphorylation and hence activation of the signaling kinases ERK1, ERK2, and Akt at a time when apoptosis was already occurring. Activation of the ERKs is mediated by Ras (for review, see Segal and Greenberg, 1996), which is critical for SCG sympathetic neuron survival (Nobes et al., 1996; Markus et al., 1997), but ERK activity is not required in NGF-mediated survival (Creedon et al., 1996; Virdee and Tolkovsky, 1996). Akt is also activated by NGF, in part via the PI 3-kinase pathway (Park et al., 1996), and is reported to mediate at least part of the NGF-induced survival of sympathetic neurons (Crowder and Freeman, 1998; Virdee and Tolkovsky, unpublished observations; but see Philpott et al., 1997). We report that araC does not act by inhibiting the pathways leading to either ERK or Akt activation, because apoptosis occurs without the reduction of the phosphorylation states of these proteins. Therefore, neither active Akt nor ERKs are sufficient to maintain survival in the presence of araC. It is possible that araC directly inhibits the signaling downstream of Akt or another Ras-dependent survival pathway, but the acceleration of apoptosis caused by araC in the absence of NGF suggests another mechanism of action.
After withdrawal of NGF from sympathetic neurons, various events have been found to take place, such as the induction of c-Jun gene expression (Estus et al., 1994), a transient increase in reactive oxygen species (ROS) (Greenlund et al., 1995), and an early and sustained increase in JNK activity, causing N-terminal phosphorylation of c-Jun (Virdee et al., 1997; Eilers et al., 1998). It has been reported that the transcription factor c-Jun is a necessary and possibly sufficient mediator of apoptosis induced by NGF-withdrawal (Estus et al., 1994; Ham et al., 1995). It has also been suggested that JNK activity and particularly phosphorylation of c-Jun at serine 63 (which enhances transcriptional activation by c-Jun) are requirements for induction of neuronal apoptosis after withdrawal of a survival stimulus (Eilers et al., 1998; Maroney et al., 1998; Watson et al., 1998). We have found that araC does not cause the activation of JNK or the phosphorylation of c-Jun at serine 63 either before or during apoptosis. Our data therefore suggest that (1) araC does not inhibit the ability of NGF to suppress JNK activity, and (2) enhancement of the transcriptional activation of c-Jun is not required for apoptosis induced by araC.
Although we analyzed the effects of araC on intracellular signaling when up to 40% of the population had undergone apoptosis, it is likely that our observations extend to most of the neurons, because (1) NGF-deprived SCG neurons commit apoptosis stochastically, as predicted for a homogenous population of cells (Edwards and Tolkovsky, 1994), so signaling events in early and late dying neurons would be expected to be similar, and (2) downregulation of ERK and Akt signals (Virdee and Tolkovsky, 1995; Virdee and Tolkovsky, unpublished observations) and upregulation in JNK activity–cJun phosphorylation (Virdee et al., 1997) precedes apoptosis by several hours. If, therefore, araC caused late death by mimicking NGF-withdrawal, such advance signaling changes should have already occurred in the yet-to-die population, but our data indicate that these changes did not occur.
The suggestion that araC causes apoptosis in postmitotic cerebellar granule neurons in a p53-dependent manner (Enokido et al., 1996a) prompted us to examine the requirement for p53 in araC-induced apoptosis in SCG neurons. We found that the response of sympathetic neurons to NGF withdrawal was independent of their p53 status, as reported previously (Davies and Rosenthal, 1994; Vogel and Parada, 1998), but that neurons from p53−/− mice were resistant to araC toxicity. One copy of the p53 gene was, however, sufficient to cause significant susceptibility to the toxin. We provided further evidence for a requirement for p53 in apoptosis caused by araC by demonstrating that the level of p53 protein in rat SCG neurons was elevated in response to araC after 4 hr of treatment and that p53 protein level remained elevated up to 14 hr (the longest time point tested), when significant apoptosis had already occurred. Furthermore, elevation of p53 protein levels by araC was independent of the presence of NGF, whereas p53 was not elevated by NGF withdrawal as described previously (Sadoul et al., 1996). Because overexpression of p53 using an adenovirus vector causes neuronal apoptosis (Slack et al., 1996; Jordan et al., 1997), we suggest that it is the elevation of endogenous p53 protein levels by araC that causes apoptosis in SCG neurons.
How araC causes this increase in p53 protein in postmitotic neurons remains unclear. Levels of the p53 protein may be increased by various stimuli, such as DNA damage, nucleotide pool imbalances, and hypoxia (for review, see Levine, 1997), but hypoxia is unlikely under the culture conditions used. It is also unlikely that araC mediates nucleotide pool imbalance, because high concentrations of deoxyribonucleosides, fluorodeoxyuridine, or hydroxyurea (Martin et al., 1990; Tomkins et al., 1994), which might be expected to affect nucleotide pool ratios, do not have the same toxic effect. AraC may therefore cause DNA damage, but the mechanism remains to be identified.
Although NGF and CNTF caused similar levels of survival of sympathetic neurons, the rate of apoptosis caused by araC in the presence of CNTF was not significantly different from that caused by araC added without any growth factors. In comparison, NGF significantly reduced this rate of apoptosis. This finding implied that one or more components of NGF signaling that are not shared with CNTF signaling have an inhibitory effect on the mechanism of araC toxicity. One difference in signaling pathways induced by these two neurotrophic factors is the duration of ERK activation; whereas NGF causes sustained activation of ERK activity, CNTF causes only a transient activation of <1 hr (Virdee and Tolkovsky, 1995). To test the involvement of the MAPK pathway in resistance to araC-induced apoptosis, we used the selective MAPK pathway inhibitor PD98059. We found that the rate of apoptosis caused by araC in the presence of NGF was substantially increased by PD98059, thereby suggesting a critical role for the MAPK pathway in NGF-mediated resistance to araC. This conclusion is reinforced by the lack of protection against araC toxicity observed when CNTF was the survival factor.
The mechanism of ERK-mediated protection against araC-induced apoptosis remains unclear. We found that PD98059 did not cause any detectable change in the level of p53 protein at 12 hr, suggesting that ERK primarily acts to inhibit araC-induced apoptosis downstream of p53 induction and also therefore downstream of araC metabolism. Because some of the targets of ERK are transcription factors (Segal and Greenberg, 1996), it is possible that downregulation of ERK activity may inhibit transcriptional activation and expression of unstable proteins involved in survival or protection against araC.
Evidence for an anti-apoptotic role for ERK has been reported in PC12 cells after growth factor withdrawal (Xia et al., 1995) and in Fas-inducible apoptosis in Jurkat cells (Holmstrom et al., 1998). ERK has also been reported to function as a suppressor of ROS in which the addition of PD98059 to SCG neurons caused a detectable but transient increase in ROS (Dugan et al., 1997). As it has been suggested that apoptosis caused by p53 is mediated by ROS (Johnson et al., 1996;Polyak et al., 1997) and araC induces ROS in lymphocytes (Hedley and McCulloch, 1996), the ability of ERK to suppress ROS would be a possible mechanism for inhibiting p53-mediated araC-induced apoptosis. In support, Skaper et al. (1998) showed that the glutamate-induced ROS-mediated apoptosis of young cerebellar granule cells could be inhibited by BDNF. This protection was reduced, however, by treatment with PD98059. The finding that different signaling pathways may be used to protect against different insults supports results obtained in fibroblasts (Ullrich et al., 1998) in which NGF suppression of apoptosis induced by different stimuli was mediated by different TrkA constructs.
Our results, summarized schematically in Figure9, provide a clear separation of the signaling cascades involved in NGF withdrawal-mediated apoptosis and those involved in araC-mediated apoptosis in sympathetic neurons. The data presented here also provide evidence for a role for the MAPK pathway in protection against apoptosis in primary neurons. Thus, although ERK activity is not required for NGF-mediated survival under certain conditions in vitro, it may be important for survival under some physiological or pathological conditions in vivo. More generally, our findings highlight the importance of sustaining multiple signaling pathways for conferring protection against a variety of insults.
Fig. 9.

Scheme summarizing proposed interactions between NGF signaling and apoptotic pathways. NGF induces at least three signals: the sustained activation of MAPK/ERK and Akt, and suppression of JNK and c-Jun phosphorylation. In the absence of NGF, JNK is activated, whereas ERK1/2 and Akt are inactivated. In the presence of NGF, araC treatment causes elevation of p53 and apoptosis without affecting NGF signaling. ERK1/2, however, suppresses signaling downstream of p53, thereby delaying apoptosis. Inhibition of the ERK pathway with PD98059 primarily eliminates this suppression, suggesting that the lack of ERK activity caused by NGF withdrawal may therefore account for the acceleration in the rate of apoptosis induced by araC.
Footnotes
This work was supported by a Wellcome Trust toxicology studentship to C.N.G.A. and a program grant to A.M.T. We thank Andreas Nelsbach (New England Biolabs, Bedford, MA) for the generous provision of anti-phospho-kinase antibodies used during the early part of this work, Alan Clarke (University of Edinburgh, Edinburgh, Scotland) for the kind gift of p53 null and heterozygote founder mice, Richard Farndale (University of Cambridge, Cambridge, UK) for help with statistical analysis, and members of the A.M.T. laboratory for critical discussions.
Correspondence should be addressed to A. M. Tolkovsky, Department of Biochemistry, Downing Site, Tennis Court Road, Cambridge, CB2 1QW, United Kingdom.
REFERENCES
- 1.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 2.Andjelkovic M, Jakubowicz T, Cron P, Ming XF, Han JW, Hemmings BA. Activation and phosphorylation of a pleckstrin homology domain-containing protein-kinase (RAC-PK/PKB) promoted by serum and protein phospatase inhibitors. Proc Natl Acad Sci USA. 1996;93:5699–5704. doi: 10.1073/pnas.93.12.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker WJ, Royer GLJ, Weiss RB. Cytarabine and neurologic toxicity. J Clin Oncol. 1991;9:679–693. doi: 10.1200/JCO.1991.9.4.679. [DOI] [PubMed] [Google Scholar]
- 4.Buckmaster A, Nobes CD, Edwards SN, Tolkovsky AM. NGF is required for induction of c-fos immunoreactivity by serum, depolarisation, cyclic AMP or trauma in cultured rat sympathetic neurons. Eur J Neurosci. 1991;3:698–707. doi: 10.1111/j.1460-9568.1991.tb00855.x. [DOI] [PubMed] [Google Scholar]
- 5.Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 6.Creedon DJ, Johnson EM, Jr, Lawrence JC. Mitogen-activated protein kinase-independent pathways mediate the effects of nerve growth factor and cAMP on neuronal survival. J Biol Chem. 1996;271:20713–20718. doi: 10.1074/jbc.271.34.20713. [DOI] [PubMed] [Google Scholar]
- 7.Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor dependent sympathetic neurons. J Neurosci. 1998;18:2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies AM, Rosenthal A. Neurons from mouse embryos with a null mutation in the tumour-suppressor gene p53 undergo normal cell death in the absence of neurotrophins. Neurosci Lett. 1994;182:112–114. doi: 10.1016/0304-3940(94)90219-4. [DOI] [PubMed] [Google Scholar]
- 9.Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dessi F, Pollard H, Moreau J, Ben-Ari Y, Charriaut-Marlangue C. Cytosine arabinoside induces apoptosis in cerebellar neurons in culture. J Neurochem. 1995;64:1980–1987. doi: 10.1046/j.1471-4159.1995.64051980.x. [DOI] [PubMed] [Google Scholar]
- 11.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 12.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascase. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dugan LL, Creedon DJ, Johnson EM, Holtzman DM., Jr Rapid suppression of free radical formation by nerve growth factor involves the mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA. 1997;94:4086–4091. doi: 10.1073/pnas.94.8.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edwards SN, Tolkovsky AM. Characterisation of apoptosis in cultured rat sympathetic neurons after nerve growth factor (NGF) withdrawal. J Cell Biol. 1994;124:537–546. doi: 10.1083/jcb.124.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eilers A, Whitfield J, Babij C, Rubin LL, Ham J. Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J Neurosci. 1998;18:1713–1724. doi: 10.1523/JNEUROSCI.18-05-01713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Enokido Y, Araki T, Shinichi A, Hatanaka H. p53 involves cytosine arabinoside-induced apoptosis in cultured cerebellar granule neurons. Neurosci Lett. 1996a;203:1–4. doi: 10.1016/0304-3940(95)12247-8. [DOI] [PubMed] [Google Scholar]
- 17.Enokido Y, Araki T, Tanaka K, Aizawa S, Hatanaka H. Involvement of p53 in DNA strand break-induced apoptosis in postmitotic CNS neurons. Eur J Neurosci. 1996b;8:1812–1821. doi: 10.1111/j.1460-9568.1996.tb01325.x. [DOI] [PubMed] [Google Scholar]
- 18.Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM., Jr Altered gene-expression in neurons during programmed cell-death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gotoh Y, Nishida E, Yamashita T, Hoshi M, Kawakami M, Sakai H. Microtubule-associated-protein (MAP) kinase activated by nerve growth factor and epidermal growth factor in PC12 cells. Identity with the mitogen activated MAP kinase of fibroblastic cells. Eur J Biochem. 1990;193:661–669. doi: 10.1111/j.1432-1033.1990.tb19384.x. [DOI] [PubMed] [Google Scholar]
- 20.Grant S. Ara-C: cellular and molecular pharmacology. Adv Cancer Res. 1998;72:197–233. doi: 10.1016/s0065-230x(08)60703-4. [DOI] [PubMed] [Google Scholar]
- 21.Greenlund LJS, Deckwerth TL, Johnson EM., Jr Superoxide-dismutase delays neuronal apoptosis—a role for reactive oxygen species in programmed neuronal death. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 22.Ham J, Babij C, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 23.Hawrot E, Patterson P. Long-term culture of dissociated sympathetic neurons. Methods Enzymol. 1979;58:574–584. doi: 10.1016/s0076-6879(79)58174-9. [DOI] [PubMed] [Google Scholar]
- 24.Hedley DW, McCulloch EA. Generation of reactive oxygen intermediates after treatment of blasts of acute myeloblastic leukemia with cytosine arabinoside: role of bcl-2. Leukemia. 1996;10:1143–1149. [PubMed] [Google Scholar]
- 25.Holmstrom TH, Chow SC, Elo I, Coffey ET, Orrenius S, Sistonen L, Eriksson JE. Suppression of Fas/APO-1-mediated apoptosis by mitogen activated kinase signaling. J Immunol. 1998;160:2626–2636. [PubMed] [Google Scholar]
- 26.Johnson TM, Yu Z-X, Ferrans VJ, Lowenstein RA, Finkel T. Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proc Natl Acad Sci USA. 1996;93:11848–11852. doi: 10.1073/pnas.93.21.11848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jordan J, Galindo MF, Prehn JHM, Weichselbaum RR, Beckett M, Ghadge GD, Roos RP, Leiden JM, Miller RJ. p53 expression induces apoptosis in hippocampal pyramidal neuron cultures. J Neurosci. 1997;17:1397–1405. doi: 10.1523/JNEUROSCI.17-04-01397.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 29.Markus A, von Holst A, Rohrer H, Heumann R. NGF-mediated survival depends on p21ras in chick sympathetic neurons from the superior cervical but not from lumbosacral ganglia. Dev Biol. 1997;191:306–310. doi: 10.1006/dbio.1997.8771. [DOI] [PubMed] [Google Scholar]
- 30.Maroney AC, Glicksman MA, Basma A, Walton KM, Knight EJ, Murphy CA, Bartlett BA, Finn JP, Angeles T, Matsuda Y, Neff NT, Dionne CA. Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J Neurosci. 1998;18:104–111. doi: 10.1523/JNEUROSCI.18-01-00104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin DP, Wallace TL, Johnson EM., Jr Cytosine arabinoside kills postmitotic neurons in a fashion resembling trophic factor deprivation: evidence that a deoxycytidine-dependent process may be required for nerve growth factor signal transduction. J Neurosci. 1990;10:184–193. doi: 10.1523/JNEUROSCI.10-01-00184.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mobley WC, Schenker A, Shooter EM. Characterization and isolation of proteolytically modified nerve growth factor. Biochemistry. 1976;15:5543–5551. doi: 10.1021/bi00670a019. [DOI] [PubMed] [Google Scholar]
- 33.Nobes CD, Tolkovsky AM. Neutralizing anti-p21(Ras) fabs suppress rat sympathetic neuron survival induced by NGF, LIF, CNTF and cAMP. Eur J Neurosci. 1995;7:344–350. doi: 10.1111/j.1460-9568.1995.tb01069.x. [DOI] [PubMed] [Google Scholar]
- 34.Nobes CD, Reppas JB, Markus A, Tolkovsky AM. Active p21ras is sufficient for rescue of NGF-dependent rat sympathetic neurons. Neuroscience. 1996;70:1067–1079. doi: 10.1016/0306-4522(95)00420-3. [DOI] [PubMed] [Google Scholar]
- 35.Park DS, Morris EJ, Stefanis L, Troy CM, Shelanski ML, Geller HM, Greene LA. Multiple pathways of neuronal death induced by DNA-damaging agents, NGF deprivation, and oxidative stress. J Neurosci. 1998;18:830–840. doi: 10.1523/JNEUROSCI.18-03-00830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park EK, Yang SI, Kang SS. Activation of Akt by nerve growth factor via phosphatidylinositol 3-kinase in PC12 pheochromocytoma cells. Mol Cells. 1996;6:494–498. [Google Scholar]
- 37.Philpott KL, McCarthy MJ, Klippel A, Rubin LL. Activated phoshpatidylinositol 3-kinase and Akt kinase promote survival of superior cervical neurons. J Cell Biol. 1997;139:809–815. doi: 10.1083/jcb.139.3.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Polyak K, Xia Y, Zweier JL, Kinzler KW. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 39.Ray LB, Sturgill TW. Insulin-stimulated microtubule-associated protein kinase is phosphorylated on tyrosine and threonine in vivo. Proc Natl Acad Sci USA. 1988;85:3753–3757. doi: 10.1073/pnas.85.11.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sadoul R, Quiquerez AL, Martinou I, Fernandez PA, Martinou JC. p53 protein in sympathetic neurons—cytoplasmic localization and no apparent function in apoptosis. J Neurosci Res. 1996;43:594–601. doi: 10.1002/(SICI)1097-4547(19960301)43:5<594::AID-JNR9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 41.Sanz-Rodriguez C, Boix J, Comella JX. Cytosine arabinoside is neurotoxic to chick embryo spinal motoneurons in culture. Neurosci Lett. 1997;223:141–144. doi: 10.1016/s0304-3940(97)13412-7. [DOI] [PubMed] [Google Scholar]
- 42.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 43.Skaper SD, Floreani M, Negro A, Facci L, Giusti P. Neurotrophins rescue cerebellar granule neurons from oxidative stress-mediated apoptotic death: selective involvement of phosphatidylinositol 3-kinase and the mitogen-activated protein kinase pathway. J Neurochem. 1998;70:1859–1868. doi: 10.1046/j.1471-4159.1998.70051859.x. [DOI] [PubMed] [Google Scholar]
- 44.Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Lochmuller H, Aloyz R, Haghighi A, Lach B, Seth P, Cooper E, Miller FD. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J Cell Biol. 1996;135:1085–1096. doi: 10.1083/jcb.135.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomkins CE, Edwards SN, Tolkovsky AM. Apoptosis is induced In postmitotic rat sympathetic neurons by arabinosides and topoisomerase-II inhibitors in the presence of NGF. J Cell Sci. 1994;107:1499–1507. doi: 10.1242/jcs.107.6.1499. [DOI] [PubMed] [Google Scholar]
- 46.Ullrich E, Duwel A, Kauffmann-Zeh A, Gilbert C, Lyon D, Rudkin B, Evan G, Martin-Zanca D. Specific TrkA survival signals interfere with different apoptotic pathways. Oncogene. 1998;16:825–832. doi: 10.1038/sj.onc.1201842. [DOI] [PubMed] [Google Scholar]
- 47.Virdee K, Tolkovsky AM. Activation of p44 and p42 MAP kinases is not essential for the survival of rat sympathetic neurons. Eur J Neurosci. 1995;7:2159–2169. doi: 10.1111/j.1460-9568.1995.tb00637.x. [DOI] [PubMed] [Google Scholar]
- 48.Virdee K, Tolkovsky AM. Inhibition of p42 and p44 mitogen activated protein kinase activity by PD98059 does not suppress nerve growth factor-induced survival of sympathetic neurones. J Neurochem. 1996;67:1801–1805. doi: 10.1046/j.1471-4159.1996.67051801.x. [DOI] [PubMed] [Google Scholar]
- 49.Virdee K, Bannister AJ, Hunt SP, Tolkovsky AM. Comparison between the timing and onset of JNK activation, c-Jun phosphorylation, and onset of death commitment in sympathetic neurones. J Neurochem. 1997;69:550–561. doi: 10.1046/j.1471-4159.1997.69020550.x. [DOI] [PubMed] [Google Scholar]
- 50.Vogel KS, Parada LF. Sympathetic neuron survival and proliferation are prolonged by loss of p53 and neurofibromin. Mol Cell Neurosci. 1998;11:19–28. doi: 10.1006/mcne.1998.0670. [DOI] [PubMed] [Google Scholar]
- 51.Wallace TL, Johnson EM., Jr Cytosine arabinoside kills postmitotic neurons: evidence that deoxycytidine may have a role in neuronal survival that is independent of DNA synthesis. J Neurosci. 1989;9:115–124. doi: 10.1523/JNEUROSCI.09-01-00115.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Watson A, Eilers A, Lallemand D, Kyriakis J, Rubin LL, Ham J. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J Neurosci. 1998;18:751–762. doi: 10.1523/JNEUROSCI.18-02-00751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]