

Abstract
The neurotransmitter GABA has been proposed to play a role during nervous system development. We show that theCaenorhabditis elegans gene unc-25encodes glutamic acid decarboxylase (GAD), the GABA biosynthetic enzyme. unc-25 is expressed specifically in GABAergic neurons. Null mutations in unc-25 eliminate the UNC-25 protein or alter amino acids conserved in all known GADs, result in a complete lack of GABA, and cause defects in all GABA-mediated behaviors. In unc-25 mutants the GABAergic neurons have normal axonal trajectories and synaptic connectivity, and the size and shape of synaptic vesicles are normal. The number of synaptic vesicles at GABAergic neuromuscular junctions is slightly increased. Cholinergic ventral nerve cord neurons, which innervate the same muscles as GABAergic ventral cord neurons, have normal morphology, connectivity, and synaptic vesicles. We conclude that GAD activity and GABA are not necessary for the development or maintenance of neuromuscular junctions in C. elegans.
Keywords: GABA, γ-amino butyric acid, GAD, glutamate decarboxylase, neuromuscular junctions, C. elegans
Classical neurotransmitters are synthesized, loaded into synaptic vesicles, and released into the synaptic cleft when a neuron is depolarized. This role of neurotransmitters in the functioning of mature synapses is well characterized. Recently, a role for neurotransmitters as trophic factors during nervous system development has been proposed (for reviews, see Mattson, 1988; Lipton and Kater, 1989; Zheng et al., 1996a). Both glutamate and acetylcholine are shown to elicit positive turning responses of growth cones from cultured hippocampal and spinal neurons (Mattson et al., 1988; Zheng et al., 1994, 1996b). The inhibitory neurotransmitter GABA might also play a role in nervous system development. GABA and its receptors are expressed in the cerebral cortex and retina during the period of neuronal proliferation and differentiation (Chun and Shatz, 1989; Mitchell and Redburn, 1996). GABA appears to inhibit cortical neuron cell divisions (LoTurco et al., 1995) and can influence spinal neuroblast movement in an in vitro assay (Behar et al., 1995). Altering GABA action in vivo during early stages of retinal development using agonists or antagonists results in abnormal axonal morphology of cone photoreceptors (Messersmith and Redburn, 1993). These experiments suggest that GABA can have effects on nervous system development. However, an in vivo role for GABA in nervous system development has not been demonstrated definitively, in part because experiments that depend on pharmacological manipulations could be misleading. One direct test would be to analyze the nervous systems of mutant animals that lack GABA.
GABA is synthesized in a single step from glutamate by the enzyme glutamic acid decarboxylase (GAD) (for review, see Martin and Rimvall, 1993). GAD binds the cofactor pyridoxal phosphate (PLP) via the tetrapeptide NPHK. Vertebrates have two GAD genes, GAD65 and GAD67, which are highly conserved in their C-terminal portions and diverged in their amino-terminal 100 amino acids (Erlander and Tobin, 1991). GAD65 differs from GAD67 in at least two other ways: GAD65 is predominantly associated with synaptic vesicles, and the GAD65-PLP adduct converts rapidly to apoGAD (Erlander et al., 1991). Recently, GAD65 and GAD67 knock-out mouse mutants were obtained (Asada et al., 1996, 1997; Condie et al., 1997). Homozygous mice lacking either GAD65 or GAD67 have anatomically normal brains. Mutant GAD65 mice have normal levels of brain GABA and behave normally, except that they display slightly increased susceptibility to seizures. Mutant GAD67 mice have low levels of brain GABA and develop cleft palates.
In the nematode Caenorhabditis elegans 26 neurons express GABA (McIntire et al., 1993b). These neurons fall into several classes: the four RME neurons form neuromuscular junctions with head muscles to control foraging; the AVL and DVB neurons synapse onto enteric muscles and regulate defecation; the RIS interneuron has no known function; and the six DD neurons (which innervate the dorsal body muscles) and the 13 VD neurons (which innervate the ventral body muscles) cause muscle relaxation during locomotion (White et al., 1986). In addition to the GABAergic inhibitory input from the DD and VD neurons, the body muscles receive excitatory cholinergic input. Body muscle contraction depends on the balance of antagonistic inputs from cholinergic and GABAergic synapses.
Killing the DD and VD GABAergic motor neurons causes a locomotory behavior known as “shrinking,” in which the animal simultaneously hypercontracts both ventral and dorsal body muscles (Hodgkin, 1983;McIntire et al., 1993b). Shrinker mutants define several genes required for the development and function of these neurons (McIntire et al., 1993a). It was proposed that one of these genes, unc-25, encodes the biosynthetic enzyme for GABA for three reasons. First,unc-25 mutations abolish all GABA functions as defined by laser killing of GABA-expressing neurons (McIntire et al., 1993a). Second, the 26 GABAergic neurons lack GABA immunoreactivity inunc-25 mutant animals (McIntire et al., 1993a). Third, addition of exogenous GABA restores GABA immunoreactivity to AVL and DVB and rescues the defecation defect, suggesting that a lack of GABA is the only defect in these neurons in unc-25 mutant animals (McIntire et al., 1993a).
We show in this paper that unc-25 encodes GAD and is likely to be the only GAD gene in C. elegans and that the lack of GABA in unc-25 mutant animals does not affect axonal morphology or the ratio of excitatory to inhibitory neuromuscular junctions.
MATERIALS AND METHODS
Genetic methods. Worms were maintained at 20°C as described by Brenner (1974), unless noted otherwise. Allunc-25 mutations reported here were induced by ethyl methanesulfonate: e156, e265, and e591were isolated by Brenner (1974) in screens for locomotory-deficient mutants; n2323, n2324, n2328,n2569, and n2638 were isolated by J. Kaplan and E. Jorgensen; sa94 was isolated by J. Thomas in screens for defection-defective mutants; and n2379,n2380, n2381, n2384, n2383, and n2385 were isolated by Y. Jin in screens for shrinker mutants.
Isolation and subcloning of genomic DNAs. cm9e10 was generated by C. Martin and M. Chalfie and was obtained from theC. elegans Genome Sequencing Center at the Medical Research Council, Cambridge, UK. We first examined DNAs from cosmids covered by Y37D8 for hybridization with the cm9e10 insert and failed to identify any positive clones. We then used the cDNA insert in cm9e10 to probe a C. elegans genomic λ phage library, kindly provided by Browning and Strome (1996). Two positive clones, YJD2 and YJC6, were isolated from 75,000 phage plaques (20 × genome equivalents). Subsequent purification of phage DNAs and subcloning into plasmids were performed following standard procedures (Sambrook et al., 1989).
Characterizations of GAD cDNAs. We determined the complete sequences of both strands of the cDNA insert in cm9e10 by generating nested ExoIII deletion DNA fragments and using the ABI PRISM cycle sequencing kit and an ABI373A sequencer according to the manufacturer’s instructions. This cDNA clone contains an insert of 1400 bp with a poly(A+) tail at the 3′ end. On a Northern blot, this cDNA detected a 1.8 kb mRNA transcript (data not shown). To isolate full-length cDNAs for GAD, we used the cm9e10 DNA insert to probe two C. elegans cDNA libraries made from mixed-stage poly(A+) RNA: a λ ZAP II cDNA library constructed by Barstead and Waterston (1989) and a λ-gt11 cDNA library constructed by Okkema and Fire (1994). The longest GAD cDNA we isolated, pSC180, contained a 1.8 kb insert in which an in-frame ATG is present seven nucleotides from the beginning of the cDNA. The size of the cDNA insert corresponded to the size of the transcript observed on Northern blots. None of the cDNAs possessed a trans-spliced leader, consistent with our inability to amplify the 5′ end of the GAD mRNA using SL1 and SL2 splice leader sequences as primers for RT-PCR (data not shown). To confirm that the longest cDNA represents the full-length GAD mRNA, we used the 5′ RACE system of anchored RT-PCR (Life Technologies, Gaithersburg, MD). The analysis of PCR products from RACE revealed that the transcripts began about 10 nucleotides 5′ to the beginning in-frame ATG, consistent with the size of the GAD cDNA in pSC180. In addition, we showed that when DNA constructs in which a unc-25promoter drove the expression of the GAD cDNA from pSC180 were injected into the germline of unc-25 mutant animals, the transgene rescued the locomotory defects completely (data not shown), suggesting that the cDNA encodes a fully functional GAD gene.
Germline transformation. Germline transformation was performed using standard procedures (Mello et al., 1991). pRF4, which contains the dominant mutation rol-6(su1006) (Kramer et al., 1990), was used as a coinjection marker when either N2 orunc-25 animals were used as the host for transformation. plin-15EK, which contains the entire gene for lin-15 (Clark et al., 1994), was used as a coinjection marker whenlin-15(n765) was used as the host.
Sequence analysis of unc-25 alleles. The genomic structure of the wild-type unc-25 gene was determined by using primers corresponding to exonic sequences to amplify genomic DNA and cDNA by PCR. The sequences of the PCR products were then determined and compared, revealing that unc-25 is composed of eight exons. We then amplified genomic DNAs including all exonic and exon/intron boundaries sequences from unc-25 mutant animals and determined their sequences using the fmol DNA cycle sequencing system (Promega, Madison, WI). Specific primer sequences are available on request.
Reporter gene constructs. In general, all reporter constructs were prepared by simple ligation between desiredunc-25 DNA fragments and lacZ or green fluorescent protein (GFP) reporter vectors (Fire et al., 1990; Chalfie et al., 1994). To tag GAD with GFP at the amino terminus, we first amplified the GFP using a primer that changes the stop codon of GFP to an XhoI site along with a primer corresponding to the sequence upstream of the multiple cloning sites in Tu#62 (Chalfie et al., 1994). The resulting PCR products were then digested withSalI and XhoI, thus allowing the fragment to be cloned into the XhoI site in the first exon ofunc-25 and generating the plasmid pSC317. GFP was thereby inserted in-frame at the amino terminus of GAD after residue 12.
Electron microscopy. Adult worms were cut with a scalpel in 8% glutaraldehyde and 0.7% osmium tetroxide in 0.1 mcacodylate, pH 7.4, on ice. After 2 hr worms were moved to 2% osmium tetroxide in 0.1 m cacodylate and left at 4° overnight. Processing and sectioning were performed as described by McIntire et al. (1992). Worms were sectioned until the region between the pharynx and the reflex of the gonad had been reached. Thereafter, roughly 1000 serial sections of 60 nm thickness were cut, mounted on slot grids, and photographed. The connectivity of the C. elegans nervous system is largely invariant, and cells can be identified by comparing reconstructions to the published wild-type reconstructions. Moreover, synapses are en passant; thus, complex dendritic arbors and axonal termini are absent. In this study, motor neurons were first identified by the order of the cell bodies along the ventral cord, the orientation of axons in regard to the cell body, the positions of their axons in the ventral nerve cord, their connectivities, and the morphologies of their synapses. In Figure 6, only the VA, VB, VD, and DD processes are shown. The axons of these motor neurons cluster around the neuromuscular junctions but can be readily distinguished even in single sections. Specifically, motor neuron axons are ordered in typical ventral to dorsal positions at the edge of the ventral nerve cord, with the DD neuron dorsal-most, VD below DD, VA next, and VB ventral-most. Second, the connectivities of these neurons differ: the DD neurons receive inputs from the VA and VB neurons, the VDs only form neuromuscular junctions in the ventral nerve cord, and the VAs and VBs form dyadic synapses to the DD neurons and the muscles. Third, the morphologies of the synapses differ: VD neurons have large varicosities and small active zones centered and oriented directly on the muscle; and VA and VB neurons have small varicosities with large active zones that are oriented dorsally. Position along the ventral cord was confirmed by noting the positions and identities of the other ventral cord motor neurons. Data for synaptic morphology were collected by examining two N2 and three unc-25(e156) animals. Serial reconstruction was made from one N2 and one unc-25(e156)animal.
Fig. 6.

Comparison of the ventral nerve cord reconstructions of a wild-type and a unc-25(e156)animal. The ventral nerve cord was reconstructed from serial electron micrographs between the DD2 cell body and the DD2 commissure. Anterior is up. Cell bodies that are anterior to the segment are indicated above the axon; cell bodies that are posterior to the segment are indicated below the axon. Synapses are indicated by a dot on the process. Synaptic input is shown as an arrow pointing toward the dot; synaptic output is indicated as an arrow pointing away from the dot. Dyadic synapses are indicated with anasterisk. Gap junctions are indicated as vertical bars. The commissure is indicated as a broken horizontal process. A question mark indicates that the cell identity is undefined.
RESULTS
unc-25 is a C. elegans GAD gene
unc-25 was previously mapped genetically on the right arm of chromosome III (Brenner, 1974). A partial cDNA clone, cm9e10, encodes a protein with sequence similarity to GAD and hybridizes to the YAC clone Y37D8, which is in the region of unc-25 on the physical map (Fig. 1A) (Waterston et al., 1992). We used cm9e10 as a probe to screen aC. elegans genomic library constructed in λ phage and isolated two positive clones (see Materials and Methods). Injection of DNA from either phage clone into the germline ofunc-25(e156) mutant worms produced stable transgenic lines in which the Unc-25 mutant phenotype was restored to wild type, indicating that the genomic DNA in the phage clones contained theunc-25 gene. We localized the rescuing activity to a 12 kb genomic DNA fragment (Fig. 1B). This 12 kb DNA contains a predicted gene corresponding to the cm9e10 cDNA. In addition, all known unc-25 alleles contained mutations in this gene (see below and Table 1). We conclude that unc-25 encodes a C. elegansGAD-like protein.
Fig. 1.

The unc-25 locus. A, Genetic and physical maps of the right arm of chromosome III.unc-25 is ∼7.5 map units right ofdpy-18, and 2 map units right of pie-1. cm9e10 is a partial GAD cDNA clone and hybridizes to the YAC clone Y37D8. Y37D8 overlaps the cosmid clones shown below it, none of which hybridized to cm9e10. The vertical lines indicate gaps in the cosmid overlaps. Based on physical map information from ACeDB (Eeckman and Durbin, 1995), cm9e10 could lie within either of the two gaps marked with #. For simplicity, only one of the two possible locations of cm9e10 and YJD2 is shown. B, Genomic structure of the unc-25 locus. Shown above is the 12 kb minimal rescuing fragment of unc-25.Boxes represent exons. The black portionof the boxes marks protein regions conserved between C. elegans GAD and human GADs. * marks intron–exon boundaries conserved between unc-25 and human GAD genes. +, Rescue, wild-type locomotory movement and normal defecation; −, no rescue, transgenic worms displayed the shrinker phenotype and were constipated; ±, partial rescue, nearly wild-type locomotory movement and worms were weakly constipated. Independently established transgenic lines (10–20) were scored with each construct.
Table 1.
unc-25 alleles and molecular lesions
| Allele | Nucleotide change | Amino acid change | Mutant phenotype |
|---|---|---|---|
| n2638 | C136A | L43 M | Strong |
| n2323 | G659A | G217D | Strong |
| n2324 | G881A | W291amber | Strong |
| n2380 | G884A | G292D | Strong |
| n2381 | G889A | G294R | Strong |
| n2569 | C970T | L321F | Weak, ts |
| sa94 | G975A | M322I | Weak, ts |
| n2379 | G977A | G323E | Weak, ts |
| n2384 | G1100A | G364E | Strong |
| e156 | G1157A | W383amber | Strong |
| e265 | G1168A | G387E | Strong |
| n2385 | G1169A | G387R | Strong |
| e591 | G1177A | G390R | Strong |
| n2383 | G1397A | G463D | Strong |
| n2328 | C1465T | Q486amber | Strong |
C. elegans GAD is equally similar to GAD65and GAD67
We constructed a full-length cDNA for unc-25 and determined its sequence (see Materials and Methods). Theunc-25 cDNA predicts a protein of 508 amino acids. The predicted UNC-25 protein shares 44% amino acid identity with human GAD65 and 46% with human GAD67 (Fig.2). The C-terminal 440 amino acids are highly conserved, and in this region the identity between the C. elegans and human GAD proteins is close to 65%. The landmark structural feature of GAD, a tetrapeptide Asn-Pro-His-Lys (NPHK) involved in binding pyridoxal phosphate, is conserved in C. elegans GAD. The overall structure of C. elegans GAD is closer to that of the Drosophila GAD than to that of the vertebrate GADs in that the C. elegans GAD lacks a long N-terminal extension.
Fig. 2.

Sequence comparisons of GADs. Sequences of human GADs (hGAD65, hGAD67) andDrosophila GAD (dGAD) are from Erlander et al. (1990). The NPHK tetrapeptide is in bold face. Positions in which unc-25 mutations were found are indicated with the corresponding amino acid change above.X represents sites of nonsense mutations. Table 1 lists additional information about unc-25 mutations.
The similarity of unc-25 to human GADs is revealed further at the level of genomic structure. Most exon boundaries are conserved between the human GAD65 and GAD67 genes (Bu and Tobin, 1994). The unc-25 gene is composed of eight exons (Fig. 1B). Remarkably, four exon–intron boundaries occur at the same positions as those found in human GAD65and GAD67 (Bu and Tobin, 1994) (Fig. 1B), suggesting that GAD gene structure is conserved in evolutionarily distant phyla.
unc-25 mutations affect conserved residues in GAD
Fifteen unc-25 mutations were isolated from various genetic screens (see Materials and Methods). We determined the molecular lesions in these alleles (Fig. 2, Table 1). Twelve of theunc-25 mutant alleles cause severe defects in locomotion and defecation. The phenotype caused by these strong alleles is indistinguishable from the phenotype caused by a strong allele intrans to a deficiency (McIntire et al., 1993a). Three of these strong unc-25 alleles are nonsense mutations, whereas the other nine are missense mutations changing amino acid residues conserved among known GADs (Fig. 2). The nonsense mutationn2324 changes Trp291 to an amber stop codon and is likely to result in a premature protein that lacks the C-terminal half of the protein, including the NPHK tetrapeptide, the cofactor binding site. This mutation is thus likely to cause complete loss ofunc-25 function.
Three alleles, sa94, n2379, and n2569, caused temperature-sensitive locomotory defects: these mutants displayed nearly wild-type locomotion at 15°C but a shrinker phenotype at 25°C (Reiner and Thomas, 1995; our unpublished observations). These three mutants, however, were defective in defecation at all temperatures, suggesting that different classes of GABAergic neurons may require the function of GAD to different extents. Specifically, the type D neurons may be less sensitive to the level of GAD than are the AVL and DVB neurons. These three temperature-sensitive mutations are missense mutations clustered adjacent to the NPHK tetrapeptide (Fig. 2, Table 1). These mutations may interfere with the regulation of GAD activity but not abolish GAD function.
unc-25 is expressed exclusively in GABAergic neurons
To determine the expression pattern of unc-25, we made a series of reporter gene constructs using the GFP (Chalfie et al., 1994) (Fig. 3). We found that all of the GABAergic neurons and only these cells express unc-25 (Fig.4A), and theunc-25 expression was visible as soon as these neurons were generated. This expression pattern indicates that the GABA immunoreactive cells accumulate GABA via de novo synthesis rather than via uptake of GABA released by neighboring cells. Furthermore, our reporter gene analysis suggested that UNC-25 expression in different classes of GABAergic neurons is regulated at both the transcriptional and post-transcriptional levels. Specifically, reporter constructs containing either the entire unc-25genomic sequences (pSC317) or genomic sequences up to exon 6 (pSC100) were expressed in all 26 GABAergic neurons. Reporter constructs containing shorter genomic sequences that included the putative 5′ regulatory region and various lengths of genomic sequences up to exon 5 (pSC380, pSC379, pSC98, and pSC315) were not expressed in the RIS, AVL, and DVB neurons. However, the expression of unc-25 reporter gene constructs in these three neurons did not depend on specific intronic or exonic sequences. We used an unc-25 genomic fragment that contained only the 5′ regulatory region and the first 13 amino acid residues in exon 1 to drive a GFP reporter gene in which multiple synthetic introns were inserted into the GFP coding sequence (pSC381) (A. Fire, personal communication). This construct expressed GFP in all 26 GABAergic neurons (data not shown). Although these experiments are subject to the general caveat that overexpression of a reporter gene may not accurately represent endogenous gene expression, this analysis suggests that the 5′ region ofunc-25 contains the information required for expression in all GABAergic neurons and that the expression of unc-25 in AVL, DVB, and RIS may additionally require RNA processing. Such post-transcriptional regulation might be achieved through regulated nuclear RNA export and/or RNA stability (Johnson, 1994; Rethmeier et al., 1997).
Fig. 3.

unc-25 reporter gene constructs. Exons of unc-25 are indicated as in Figure 1. GFP reporter genes are shown as hatched boxes.Lines represent nonexonic sequences. +, GFP expression observed; −, no GFP expression.
Fig. 4.

The expression pattern ofunc-25. A, An adult worm of genotypelin-15(n765); juEx[pSC100+lin-15(EK)]. GFP is expressed in all and in only GABAergic neurons. RMEs, DVB, AVL, and RIS are marked by arrows; arrowheads point to several DD and VD neurons. Not all DD and VD neurons are seen as a consequence of the mosaic expression of the transgene.B, An adult worm of genotype unc-25(e156); lin-15(n765); juEx[pSC317+ plin-15(EK)]. UNC-25:: GFP is found in cell bodies (arrowheads), axons, and commissures (arrow). C, The ventral cord of an adult worm of genotype unc-25(e156); lin-15(n765); juEx[pSC317+ plin-15(EK)]. UNC-25:: GFP is associated with synaptic varicosities seen as punctate fluorescent clusters (arrows). Arrowheads mark neuronal cell bodies. D, The ventral cord of an adult worm of genotypeunc-104(e1265); lin-15(n765); juEx[pSC317+ plin-15(EK)]. No punctate synaptic fluorescent clusters are visible. The arrowhead marks a neuronal cell body. Scale bar: A, 50 μm; B–D, 170 μm.
The two isoforms of vertebrate GAD differ in their subcellular locations (Erlander et al., 1991). To determine where UNC-25 is localized within a cell, we inserted GFP in-frame into the amino terminus after amino acid Val12 (pSC317). The transgene containing this construct rescued the Unc-25 phenotype, indicating that the GFP insertion did not disrupt the function of UNC-25 and therefore that this transgene was expressed at sites at which UNC-25 function is needed. GFP was observed throughout cell bodies and axonal branches and was enriched in synaptic regions (Fig. 4B,C). To evaluate whether the synaptic localization of UNC-25:: GFP is caused by an association with synaptic vesicles, we examined the expression of this transgene in unc-104 mutant animals, which accumulate synaptic vesicles in cell bodies because of defects in a kinesin-like molecule (Hall and Hedgecock, 1991; Otsuka et al., 1991). We found that in unc-104 animals, the synaptic punctate expression of GFP diminished and GFP became uniformly distributed in the axonal branches and highly concentrated in the cell bodies (Fig. 4D), suggesting that some fraction of UNC-25:: GFP was associated with vesicles. Because all synaptic vesicles are retained in cell bodies in unc-104mutants (Hall and Hedgecock, 1991), this analysis suggests that UNC-25 is present in both nonvesicular- and vesicular-bound forms, although it is possible that the nonvesicular localization is caused by overexpression from the transgenic array.
Axonal outgrowth and synapse formation are normal inunc-25 mutants
To investigate whether GABA plays a role in axon guidance, we examined the morphology of the GABAergic neurons in unc-25mutant animals using the GFP reporter transgene with multiple synthetic introns (pSC381). GFP expressed from this transgene was found throughout GABAergic neuron cell bodies and axons. We found that all 26 GABAergic neurons displayed an axonal trajectory pattern indistinguishable from that of the wild type (n > 100 animals) (data not shown). We conclude that lack of GABA does not affect the axonal development of these GABAergic neurons. Moreover, inunc-25 mutant animals, the number and positions of the motor neurons in the ventral nerve cord, as examined using Nomarski optics, were the same as in wild-type animals (data not shown), indicating that lack of GABA has no effect on the divisions of the precursor cells that generate the GABAergic DD and VD neurons.
To determine whether GABA plays a role in neuronal connectivity, we first compared in unc-25(e156) and wild-type animals the expression of a transgene, juIs1, in which GFP was fused to the C. elegans SNB-1 protein, a homolog of the synaptic vesicle protein synaptobrevin (Nonet et al., 1998), and driven by theunc-25 promoter (Jorgensen et al., 1995). Punctate fluorescent clusters of GFP were seen along the dorsal and ventral nerve cords, corresponding in position to the synaptic varicosities of the DD and VD neurons, respectively (Jorgensen et al., 1995). We detected no abnormality in unc-25(e156) animals in either the shape or the density of the fluorescent clusters (Fig.5A,B), suggesting that the synaptic termini of these neurons were largely normal, although the intensity of the fluorescent clusters was slightly stronger inunc-25 mutants than that in wild-type animals. These experiments indicated that the distribution of GABAergic synapses was roughly normal in a unc-25 mutant. However, these experiments did not demonstrate that synaptic connectivity is normal in a unc-25 mutant. Specifically, they did not examine whether these synapses were directed to their normal muscle targets, nor did they determine whether the density of cholinergic synapses to the muscle was normal.
Fig. 5.
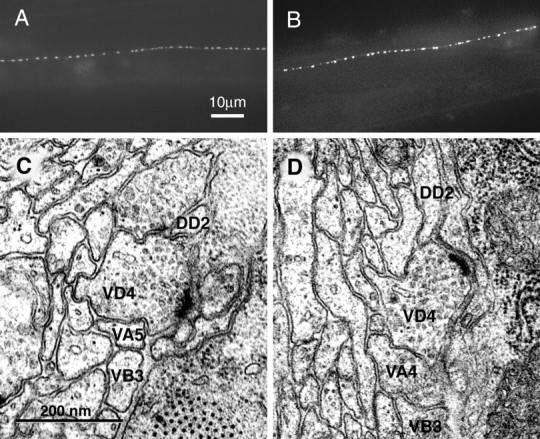
Neuromuscular junction morphology inunc-25(e156) and wild-type animals. A, Expression of juIs1, a GFP marker that labels the presynaptic termini of DD and VD neurons, in the dorsal cord of a wild-type young adult worm. In the juIs1 construct synaptobrevin was fused to the green fluorescent protein and expressed under the control of the unc-25 promoter (Jorgensen et al., 1995). The presynaptic zones of the GABAergic motor neurons are thus manifested as punctate fluorescent clusters in living worms (see Results).B, Expression of juIs1 in the dorsal cord of a unc-25(e156) young adult worm. The density and shape of synaptic varicosities is similar to that in the wild type, although the fluorescence is slightly more intense inunc-25 animals than in wild-type animals, possibly reflecting the slight increase of synaptic vesicles (see Results). C, An electron micrograph of the ventral nerve cord in a wild-type young adult animal. A neuromuscular junction (arrow) between the VD4 GABAergic motor neuron and muscle arms from the body muscles. D, An electron micrograph of the ventral nerve cord in a unc-25(e156)young adult animal. A neuromuscular junction (arrow) between the VD4 GABAergic motor neuron and muscle arms from the body muscles. Note that the VD active zone is neither hypertrophic nor diminished in comparison with that in the wild type.
To examine the synaptic connectivity of a unc-25 mutant, we fixed a unc-25(e156) animal and a wild-type animal and prepared electron micrographs from serial sections of each. The morphologies of the GABAergic and cholinergic neuromuscular junctions were normal in the unc-25 animal. Specifically, the cholinergic VA and VB and the GABAergic VD neuromuscular junctions were neither enlarged nor diminished in the unc-25 mutant compared with the wild type (Fig. 5C,D), and the diameters of synaptic vesicles were also unchanged (Table2).
Table 2.
Synaptic vesicle densities and diameters inunc-25 and wild-type animals
| SVs per midsynaptic profilea,b | Number of synapses | SV diameter (nm)b | Number of SVs | |
|---|---|---|---|---|
| Wild-type ACh synapses | 27.4 ± 2.3 | 32 | 30.1 ± 0.4 | 109 |
| Wild-type GABA synapses | 27.4 ± 3.4 | 14 | 29.6 ± 0.3 | 196 |
| unc-25(e156) ACh synapses | 26.9 ± 2.6 | 20 | 31.6 ± 0.5 | 170 |
| unc-25(e156) GABA synapses | 37.0 ± 3.3 | 14 | 31.6 ± 0.6 | 134 |
SVs, Synaptic vesicles.
Number of synaptic vesicles in a cross section at the center of the active zone.
Mean ± SEM.
In the absence of inhibitory input, the muscles in unc-25mutants should receive an excess of excitatory cholinergic input. Are cholinergic inputs pruned to restore the muscles to a normal level of excitation? To examine this question, we reconstructed a segment of the ventral nerve cord between the DD2 commissure and the DD2 cell body (Fig. 6) and counted the neuromuscular junctions from the cholinergic motor neurons VA4 and VB3 and the GABAergic motor neuron VD4. We found that the total number of synapses was approximately the same for the two strains in the reconstructed segment: VD4 formed 24 neuromuscular junctions in this interval in both the wild-type and the unc-25 animal. There were similar numbers of synapses from the cholinergic neurons in the wild-type (52) and unc-25 (48) individuals. Based on these numbers, the ratio of the VA and VB neuromuscular junctions to VD neuromuscular junctions was 2.2 in the wild-type and 2.0 in theunc-25(e156) animal. We conclude that there was no compensation in synaptic density of the GABAergic neurons or the cholinergic neurons in response to the lack of GABA in theunc-25 mutant.
Another possibility is that rather than remodeling neuromuscular junctions, the nervous system may compensate for reduced neurotransmission by changing the strength of existing synapses. Although the rate of release of synaptic vesicles cannot be measured inC. elegans at this time, the number of synaptic vesicles can be examined directly. We found that the mean number of synaptic vesicles at the midpoints of the GABAergic neuromuscular junctions was slightly increased in the unc-25 animals (37) compared with that in the wild-type animals (27) (Table 2) (p= 0.045). Although the increase is small, these data may indicate that a lack of transmission at these synapses causes a compensatory increase in the number of vesicles available for release.
DISCUSSION
The C. elegans gene unc-25 encodes a neuronal-specific GAD, the biosynthetic enzyme for the neurotransmitter GABA. Null mutations in unc-25 abolish GABA expression and cause animals to display behaviors indistinguishable from those in which the GABAergic neurons are killed by a laser. unc-25missense mutations affect amino acid residues that are conserved among members of the GAD family. Using both light and electron microscopy, we found that in unc-25 mutant animals GABAergic neurons exhibit normal axonal morphology and synaptic connectivity, and the size and shape of both synaptic vesicles and neuromuscular junctions are normal, indicating that GABA is not necessary for the development of these neurons and the maintenance of neuromuscular junctionsin vivo.
Regulation of GAD
Although GABA synthesis in the brain has been studied extensively (for review, see Martin and Rimvall, 1993), to date there has been no structure/function analysis of GAD or other decarboxylases besides the interaction between the NPHK tetrapeptide and the cofactor pyridoxal phosphate. For example, little is known about the residues needed for GAD catalytic activity. We have identified 12 unc-25missense mutations that indicate the functional importance of specific amino acids for GAD activity. Nine of these alleles are strong mutations and behave genetically as null mutations (McIntire et al., 1993a). All cause amino acid substitutions at positions conserved among known GADs. These residues might define sites important either for catalysis or for protein structure or stability. Three weakunc-25 alleles caused a constitutive loss of enteric muscle contractions but only a temperature-sensitive locomotory defect. These three alleles alter amino acids adjacent to the pyridoxal 5′-phosphate binding site, indicating that the amino acids around the cofactor binding site may contribute either to the binding of the cofactor or to the correct conformation of the catalytic site around the bound glutamate. These three unc-25 temperature-sensitive alleles may generate either temperature-sensitive proteins or proteins with lowered activity. In the latter case, more neurotransmitter would be required at higher temperatures, and these reduction of function mutations would simply be revealing this temperature-sensitive process.
unc-25 is transcribed and translated exclusively in the 26 GABAergic neurons. However, the regulation of unc-25expression among GABAergic cells may differ. Using reporter gene constructs, we found that unc-25 expression in the RIS, AVL, and DVB GABAergic neurons required the presence of introns in addition to the 5′ regulatory regions. This intron requirement does not seem to be sequence- or gene-specific, because adding synthetic introns into the GFP reporter gene driven by the unc-25 promoter caused GFP to be expressed in these neurons. Introns are known to facilitate RNA processing by regulating RNA export, splicing, and polyadenylation (Gallie and Young, 1994; Jarrous and Kaempfer, 1994; Damert et al., 1996; Rethmeier et al., 1997), and they also play roles in translation (Chapman and Walter, 1997). GABA is used in many different types of neurons in the nervous systems of many animals. The expression of vertebrate GADs in different types of neurons appears to be differentially regulated at both the mRNA and protein levels (Esclapez et al., 1994; Hendrickson et al., 1994; Houser and Esclapez, 1994). Although we do not know how post-transcriptional regulation is achieved in the AVL, DVB, and RIS neurons, the multi-level regulation ofunc-25 we observed in C. elegans may reflect a general mechanism used by different types of neurons in complex nervous systems.
Evolution of GAD
In contrast to vertebrates, C. elegans may have only a single GAD gene. This conclusion is based on three observations. First, mutations in unc-25 eliminate all GABA immunoreactivity and the known functions of all GABAergic neurons (McIntire et al., 1993a,b). Second, unc-25 is expressed in all 26 GABAergic neurons. Third, the sequence of 82% of the C. elegansgenome has been determined, and the sequences of many mRNAs have been partially determined as expressed sequence tags, yet no other gene is as similar to the vertebrate GADs as is unc-25. Most GAD activity in Drosophila can be attributed to a single gene, GAD1 (Jackson et al., 1990; Kulkarni et al., 1994), although other minor GADs may contribute to GABA synthesis in some tissues (Phillips et al., 1993) (M. Phillips, personal communication). Neither theC. elegans GAD gene nor the Drosophila GAD1 gene more closely resembles either the mammalian GAD65 or GAD67. Thus, the duplication and divergence of these mammalian genes probably occurred after the divergence of vertebrates from arthropods and nematodes.
The C. elegans GAD protein is strongly conserved with the vertebrate and Drosophila GAD proteins in its C portion. Even the locations of some exon/intron boundaries are maintained between species as distant as C. elegans and human. GAD65 and GAD67 each have a region of ∼100 amino acids at the amino terminus that is not conserved between the two forms. This amino-terminal region is absent in both the C. elegans and Drosophila GADs. In GAD65 this region contains two cysteines that can be palmitoylated and a region that is critical for membrane anchoring (Shi et al., 1994; Solimena et al., 1994). Moreover, the first 13 amino acids of GAD65include four serines that become phosphorylated when GAD65is associated with synaptic vesicles (Namchuk et al., 1997). Although the amino termini of the C. elegans andDrosophila GADs and of human GAD67 also contain multiple serines that could potentially be phosphorylated, GAD67 and UNC-25 are found in both synaptic regions and cytoplasm, suggesting that these serines may not be involved in an interaction with synaptic vesicles. Together, both the protein sequence comparisons and the subcellular expression patterns suggest that UNC-25 may resemble an ancestral member of the GAD family.
Function of GAD in nervous system development
Because there appears to be a single GAD gene in C. elegans, null mutations in unc-25 are likely to define all GABA-dependent functions. unc-25 animals exhibit hypercontraction of the body muscles, hyperflexions of the head during foraging, and a severe reduction in contractions of the enteric muscles. Our data indicate that these defects are a consequence of a lack of neurotransmitter function in the mature nervous system rather than of connectivity defects caused by absence of GABA during development. We reach this conclusion for several reasons. First, we detected no abnormalities in the axonal trajectories of the GABAergic neurons in unc-25 mutants. Second, the density of synaptic varicosities is normal in GABAergic neurons as analyzed by light microscopy. Third, we observed no abnormalities in neuromuscular connectivity or in the differentiation of neuromuscular junctions using electron microscopy. Fourth, we observed previously that the AVL and DVB neurons are capable of importing GABA and that the acute restoration of GABA to these cells by bath application can rescue the function of these neurons, suggesting that fully functional synapses are formed in unc-25 mutants and that neurotransmission fails only because GABA is absent (McIntire et al., 1993a). Fifth, inunc-49 mutant animals, which are defective in a GABA receptor (B. Bamber and E. J., unpublished observations) and hence likely to be defective in GABA function, the axonal morphology of GABAergic neurons (McIntire et al., 1993a) and the presynaptic termini of the DD and VD GABA neuromuscular junctions as revealed by a synapse-specific GFP marker are normal (Y. J., unpublished results). These data indicate that the behavioral defects ofunc-25 animals are caused by a lack of GABA function in an otherwise normal nervous system.
Although the connectivity of the nervous system is unchanged inunc-25 mutants, we noted that there is a slight increase in the number of synaptic vesicles at the GABAergic neuromuscular junctions in unc-25(e156) animals. This increase in synaptic vesicle number may be the result of a feedback mechanism. Specifically, the muscle cells may detect that GABA transmission is inadequate and hence may signal the motor neuron to make more synaptic vesicles and perhaps to increase the probability of release of these vesicles. However, because these vesicles lack GABA, increased synaptic release will not lead to increased transmission at these mutant synapses.
Our observations appear to contrast with several reports that GABA can affect the development and differentiation of the mammalian CNS. Exposing explants of rat embryonic cortex to GABA causes a decrease of the number of cortical cells synthesizing DNA and presumably undergoing cell divisions (LoTurco et al., 1995). However, we did not observe an increase or decrease in the number of ventral cord neurons inunc-25 mutants. Manipulations of GABA transmission in vivo by the addition of agonists or antagonists can alter axonal pathfinding during retinal development in the rabbit (Messersmith and Redburn, 1993). By contrast, we see no changes in the axonal trajectories of motor neurons in unc-25 mutants. Why might our results differ? One possibility is that our experiments were conducted in vivo in a mutant lacking GABA, whereas the other experiments were conducted either by adding exogenous GABA to cell cultures in vitro or by interfering with GABA transmission pharmacologically.
Our observations also appear to contrast with studies of the regulation of the size of mature vertebrate neuromuscular junctions. Pharmacological perturbations of vertebrate neuromuscular junctions cause the nervous system to be remodeled to compensate for these changes in neurotransmission. Specifically, reducing the effective level of neurotransmitter to the chick hindlimb muscles by blocking acetylcholine receptors with α-bungarotoxin causes the motor neuron to sprout and form additional synapses (Dahm and Landmesser, 1991). Increasing neurotransmitter activity by adding the acetylcholine agonist carbachol causes a compensatory reduction in the density of synapses made by the chick lumbosacral motor neurons (Lance-Jones and Landmesser, 1981). These experiments indicate that the density of synapses may change to maintain a constant level of input into the muscle. Given these data, we might have expected that in aunc-25 mutant a compensation for the lack of GABA neurotransmission would be hypertrophic arborization of the GABA neurons or an increase in the number of GABAergic neuromuscular junctions along the ventral cord. However, we found neither.
Differences between our studies and those of vertebrate neuromuscular junctions include the organism, the neurotransmitter examined, and whether pharmacological intervention was used. For example, unlike in vertebrates, in C. elegans muscles send processes to neurons, and neuromuscular junctions are formed en passant; chemotropic interactions between nerves and muscles could be different as a consequence. Second, although acetylcholine has been reported to have a role in the development of the vertebrate neuromuscular junction, no such function has been assigned to GABA for synaptic development in the CNS; perhaps GABA does not have such a role in either C. elegans or vertebrates. Finally, the vertebrate studies that indicated a role for neurotransmitters in the development of neuromuscular junctions were based on pharmacological manipulations, and it is conceivable that targets other than those intended were perturbed. A genetic study of a vertebrate, like our study of C. elegans, suggested that neurotransmitter function is not necessary for the formation of normal neuromuscular junction: in a zebrafish mutant lacking a muscle acetylcholine receptor, motor neurons have morphologically normal patterns of innervation and normal neuromuscular junctions (Westerfield et al., 1990).
Based on our findings, we conclude that neither synaptic development nor synaptic maintenance depends on GABA neurotransmission at neuromuscular junctions in C. elegans.
Footnotes
This work was supported by United States Public Health Service research Grants GM24663 (H.R.H), NS35546 (Y.J.), and NS34307 (E.J.). Y.J. was supported by the Jane Coffin Childs Foundation and the American Cancer Society (Massachusetts division). E.J. was supported by the Damon Runyon–Walter Winchell Cancer Research Fund and by the Howard Hughes Medical Institute. E.H. was supported by the Howard Hughes Medical Institute. H.R.H. is an Investigator of the Howard Hughes Medical Institute. We thank J. Kaplan and J. Thomas forunc-25 alleles, B. James for determining DNA sequences, A. Fire for pPD vectors, and M. Chalfie for Tu vectors. We thank A. Chisholm for comments concerning this manuscript.
Correspondence should be addressed to Dr. Yishi Jin, Department of Biology, University of California, Santa Cruz, CA 95064.
GenBank accession number for unc-25 cDNA isAF109378.
REFERENCES
- 1.Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji FY, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochem Biophys Res Commun. 1996;229:891–895. doi: 10.1006/bbrc.1996.1898. [DOI] [PubMed] [Google Scholar]
- 2.Asada H, Kawamura Y, Maruyama K, Kume H, Ding RG, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci USA. 1997;94:6496–6499. doi: 10.1073/pnas.94.12.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barstead RJ, Waterston RH. The basal component of the nematode dense-body is vinculin. J Biol Chem. 1989;264:10177–10185. [PubMed] [Google Scholar]
- 4.Behar TN, Schaffner AE, Tran HT, Barker JL. GABA-induced motility of spinal neuroblasts develops along a ventrodorsal gradient and can be mimicked by agonists of GABAA and GABAB receptors. J Neurosci Res. 1995;42:97–108. doi: 10.1002/jnr.490420111. [DOI] [PubMed] [Google Scholar]
- 5.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Browning H, Strome S. A sperm-supplied factor required for embryogenesis in C. elegans. Development. 1996;122:391–404. doi: 10.1242/dev.122.1.391. [DOI] [PubMed] [Google Scholar]
- 7.Bu D, Tobin A. The exon-intron organization of the genes (GAD1 and GAD2) encoding two human glutamate decarboxylases (GAD67 and GAD65) suggests that they derive from a common ancestral GAD. Genomics. 1994;21:222–228. doi: 10.1006/geno.1994.1246. [DOI] [PubMed] [Google Scholar]
- 8.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 9.Chapman RE, Walter P. Translational attenuation mediated by an mRNA intron. Curr Biol. 1997;7:850–859. doi: 10.1016/s0960-9822(06)00373-3. [DOI] [PubMed] [Google Scholar]
- 10.Chun JJ, Shatz CJ. The earliest-generated neurons of the cat cerebral cortex: characterization by MAP2 and neurotransmitter immunohistochemistry during fetal life. J Neurosci. 1989;9:1648–1667. doi: 10.1523/JNEUROSCI.09-05-01648.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark SG, Lu X, Horvitz HR. The Caenorhabditis elegans locus lin-15, a negative regulator of a tyrosine kinase signaling pathway, encodes two different proteins. Genetics. 1994;137:987–997. doi: 10.1093/genetics/137.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Condie BG, Bain G, Gottlieb DI, Capecchi ER. Cleft palate in mice with a targeted mutation in the gamma aminobutyric acid producing enzyme glutamic acid decarboxylase 67. Proc Natl Acad Sci USA. 1997;94:11451–11455. doi: 10.1073/pnas.94.21.11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dahm LM, Landmesser LT. The regulation of synaptogenesis during normal development and following activity blockade. J Neurosci. 1991;11:238–255. doi: 10.1523/JNEUROSCI.11-01-00238.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Damert A, Leibiger B, Leibiger IB. Dual function of the intron of the rat insulin I gene in regulation of gene expression. Diabetologia. 1996;39:1165–1172. doi: 10.1007/BF02658502. [DOI] [PubMed] [Google Scholar]
- 15.Eeckman FH, Durbin R. ACeDB and Macace. In: Epstein HF, Shakes DC, editors. Methods in cell biology. Caenorhabditis elegans: modern biological analysis of an organism. Academic; San Diego: 1995. pp. 583–605. [PubMed] [Google Scholar]
- 16.Erlander M, Tillakaratne NJ, Feldblum S, Patel N, Tobin AJ. Two genes encode distinct glutamate decarboxylases. Neuron. 1991;7:91–100. doi: 10.1016/0896-6273(91)90077-d. [DOI] [PubMed] [Google Scholar]
- 17.Erlander MG, Tobin AJ. The structural and functional heterogeneity of glutamic acid decarboxylase: a review. Neurochem Res. 1991;16:215–226. doi: 10.1007/BF00966084. [DOI] [PubMed] [Google Scholar]
- 18.Esclapez M, Tillakaratne NJ, Kaufman DL, Tobin AJ, Houser CR. Comparative localization of two forms of glutamic acid decarboxylase and their mRNAs in rat brain supports the concept of functional differences between the forms. J Neurosci. 1994;14:1834–1855. doi: 10.1523/JNEUROSCI.14-03-01834.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fire A, Harrison SW, Dixon D. A modular set of lacZ fusion vectors for studying gene expression in Caenorhabditis elegans. Gene. 1990;93:189–198. doi: 10.1016/0378-1119(90)90224-f. [DOI] [PubMed] [Google Scholar]
- 20.Gallie DR, Young TE. The regulation of gene expression in transformed maize aleurone and endosperm protoplasts: analysis of promoter activity, intron enhancement, and mRNA untranslated regions on expression. Plant Physiol. 1994;106:929–939. doi: 10.1104/pp.106.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall DH, Hedgecock EM. Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell. 1991;65:837–847. doi: 10.1016/0092-8674(91)90391-b. [DOI] [PubMed] [Google Scholar]
- 22.Hendrickson AE, Tillakaratne NJ, Mehra RD, Esclapez M, Erickson A, Vician L, Tobin AJ. Differential localization of two glutamic acid decarboxylases (GAD65 and GAD67) in adult monkey visual cortex. J Comp Neurol. 1994;343:566–581. doi: 10.1002/cne.903430407. [DOI] [PubMed] [Google Scholar]
- 23.Hodgkin J. Male phenotypes and mating efficiency in Caenorhabditis elegans. Genetics. 1983;103:43–64. doi: 10.1093/genetics/103.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houser CR, Esclapez M. Localization of mRNAs encoding two forms of glutamic acid decarboxylase in the rat hippocampal formation. Hippocampus. 1994;4:530–545. doi: 10.1002/hipo.450040503. [DOI] [PubMed] [Google Scholar]
- 25.Jackson FR, Newby LM, Kulkarni SJ. Drosophila GABAergic systems: sequence and expression of glutamic acid decarboxylase. J Neurochem. 1990;54:1068–1078. doi: 10.1111/j.1471-4159.1990.tb02359.x. [DOI] [PubMed] [Google Scholar]
- 26.Jarrous N, Kaempfer R. Induction of human interleukin-1 gene expression by retinoic acid and its regulation at processing of precursor transcripts. J Biol Chem. 1994;269:23141–23149. [PubMed] [Google Scholar]
- 27.Johnson LF. Posttranscriptional regulation of thymidylate synthase gene expression. J Cell Biochem. 1994;54:387–392. doi: 10.1002/jcb.240540405. [DOI] [PubMed] [Google Scholar]
- 28.Jorgensen EM, Hartwieg E, Schuske K, Nonet M, Jin Y, Horvitz HR. Defective recycling of synaptic vesicles in synaptotagmin mutants of C. elegans. Nature. 1995;378:196–199. doi: 10.1038/378196a0. [DOI] [PubMed] [Google Scholar]
- 29.Kramer JM, French RP, Park E-C, Johnson JJ. The Caenorhabditis elegans rol-6 gene, which interacts with the sqt-1 collagen gene to determine organismal morphology, encodes a collagen. Mol Cell Biol. 1990;10:2081–2089. doi: 10.1128/mcb.10.5.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulkarni SJ, Newby LM, Jackson FR. Drosophila GABAergic systems. II. Mutational analysis of chromosomal segment 64AB: a region containing the glutamic acid decarboxylase gene. Mol Gen Genet. 1994;243:555–564. doi: 10.1007/BF00284204. [DOI] [PubMed] [Google Scholar]
- 31.Lance-Jones C, Landmesser LT. Pathway selection by embryonic chick motoneurons in an experimentally altered environment. Proc R Soc Lond B Biol Sci. 1981;214:19–52. doi: 10.1098/rspb.1981.0080. [DOI] [PubMed] [Google Scholar]
- 32.Lipton SA, Kater SB. Neurotransmitter regulation of neuronal outgrowth, plasticity and survival. Trends Neurosci. 1989;12:265–270. doi: 10.1016/0166-2236(89)90026-x. [DOI] [PubMed] [Google Scholar]
- 33.LoTurco JJ, Owens DF, Heath MJS, Davis MB, Kriegstein AR. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron. 1995;15:1287–1289. doi: 10.1016/0896-6273(95)90008-x. [DOI] [PubMed] [Google Scholar]
- 34.Martin DL, Rimvall K. Regulation of γ-aminobutyric acid synthesis in the brain. J Neurochem. 1993;60:395–407. doi: 10.1111/j.1471-4159.1993.tb03165.x. [DOI] [PubMed] [Google Scholar]
- 35.Mattson MP. Neurotransmitters in the regulation of neuronal cytoarchitecture. Brain Res. 1988;472:179–212. doi: 10.1016/0165-0173(88)90020-3. [DOI] [PubMed] [Google Scholar]
- 36.Mattson MP, Dou P, Kater SB. Outgrowth-regulating actions of glutamate in isolated hippocampal pyramidal neurons. J Neurosci. 1988;8:2087–2100. doi: 10.1523/JNEUROSCI.08-06-02087.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McIntire SL, Garriga G, White J, Jacobson D, Horvitz HR. Genes necessary for directed axonal elongation or fasciculation in Caenorhabditis elegans. Neuron. 1992;8:307–322. doi: 10.1016/0896-6273(92)90297-q. [DOI] [PubMed] [Google Scholar]
- 38.McIntire SL, Jorgensen E, Horvitz HR. Genes required for GABA function in Caenorhabditis elegans. Nature. 1993a;364:334–337. doi: 10.1038/364334a0. [DOI] [PubMed] [Google Scholar]
- 39.McIntire SL, Jorgensen E, Kaplan J, Horvitz HR. The GABAergic nervous system of Caenorhabditis elegans. Nature. 1993b;364:337–341. doi: 10.1038/364337a0. [DOI] [PubMed] [Google Scholar]
- 40.Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Messersmith EK, Redburn DA. The role of GABA during development of the outer retina in the rabbit. Neurochem Res. 1993;18:463–470. doi: 10.1007/BF00967250. [DOI] [PubMed] [Google Scholar]
- 42.Mitchell CK, Redburn DA. GABA and GABA-A receptors are maximally expressed in association with cone synaptogenesis in neonatal rabbit retina. Dev Brain Res. 1996;95:63–71. doi: 10.1016/0165-3806(96)00064-8. [DOI] [PubMed] [Google Scholar]
- 43.Namchuk M, Lindsay L, Turck CW, Kanaani J, Baekkeskov S. Phosphorylation of serine residues 3, 6, 10, and 13 distinguishes membrane anchored from soluble glutamic acid decarboxylase 65 and is restricted to glutamic acid decarboxylase 65alpha. J Biol Chem. 1997;272:1548–1557. doi: 10.1074/jbc.272.3.1548. [DOI] [PubMed] [Google Scholar]
- 44.Nonet LM, Saifee O, Zhao H, Rand JB, Wei L. Synaptic transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J Neurosci. 1998;18:70–80. doi: 10.1523/JNEUROSCI.18-01-00070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Okkema PG, Fire A. The Caenorhabditis elegans NK-2 class homeoprotein CEH-22 is involved in combinatorial activation of gene expression in pharyngeal muscle. Development. 1994;120:2175–2186. doi: 10.1242/dev.120.8.2175. [DOI] [PubMed] [Google Scholar]
- 46.Otsuka AJ, Jeyaprakash A, Garcia-Anoveros J, Tang LZ, Fisk G, Hartshorne T, Franco R, Born T. The C. elegans unc-104 gene encodes a putative kinesin heavy chain-like protein. Neuron. 1991;6:113–122. doi: 10.1016/0896-6273(91)90126-k. [DOI] [PubMed] [Google Scholar]
- 47.Phillips AM, Salkoff LB, Kelly LE. A neural gene from Drosophila melanogaster with homology to vertebrate and invertebrate glutamate decarboxylases. J Neurochem. 1993;61:1291–1301. doi: 10.1111/j.1471-4159.1993.tb13621.x. [DOI] [PubMed] [Google Scholar]
- 48.Reiner DJ, Thomas JH. Reversal of a muscle response to GABA during c. elegans male development. J. Neurosci. 1995;15:6094–6102. doi: 10.1523/JNEUROSCI.15-09-06094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rethmeier N, Seurinck J, Van Montagu M, Cornelissen M. Intron-mediated enhancement of transgene expression in maize is a nuclear gene-dependent process. Plant J. 1997;12:895–899. doi: 10.1046/j.1365-313x.1997.12040895.x. [DOI] [PubMed] [Google Scholar]
- 50.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 51.Shi Y, Veit B, Baekkeskov S. Amino acid residues 24–31 but not palmitoylation of cysteines 30 and 45 are required for membrane anchoring of glutamic acid decarboxylase, GAD65. J Cell Biol. 1994;124:927–934. doi: 10.1083/jcb.124.6.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Solimena M, Dirkx R, Jr, Radzynski M, Mundigl O, De Camilli P. A signal located within amino acids 1–27 of GAD65 is required for its targeting to the Golgi complex region. J Cell Biol. 1994;126:331–341. doi: 10.1083/jcb.126.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waterston R, Martin C, Craxton M, Hunyh C, Coulson A, Hillier L, Durbin R, Green P, Shownkeen R, Halloran N. A survey of expressed genes in Caenorhabditis elegans. Nat Genet. 1992;1:114–123. doi: 10.1038/ng0592-114. [DOI] [PubMed] [Google Scholar]
- 54.Westerfield M, Liu DW, Kimmel CB, Walker C. Pathfinding and synapse formation in a zebrafish mutant lacking functional acetylcholine receptors. Neuron. 1990;4:867–874. doi: 10.1016/0896-6273(90)90139-7. [DOI] [PubMed] [Google Scholar]
- 55.White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- 56.Zheng JQ, Felder M, Connor JA, Poo MM. Turning of nerve growth cones induced by neurotransmitters. Nature. 1994;368:140–144. doi: 10.1038/368140a0. [DOI] [PubMed] [Google Scholar]
- 57.Zheng JQ, Poo MM, Connor JA. Calcium and chemotropic turning of nerve growth cones. Perspect Dev Neurobiol. 1996a;4:205–213. [PubMed] [Google Scholar]
- 58.Zheng JQ, Wan JJ, Poo MM. Essential role of filopodia in chemotropic turning of nerve growth cone induced by a glutamate gradient. J Neurosci. 1996b;16:1140–1149. doi: 10.1523/JNEUROSCI.16-03-01140.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]