

Abstract
Presenilin-1 (PS1) and presenilin-2 (PS2), the major genes of familial Alzheimer’s disease, are homologous tosel-12, a Caenorhabditis elegans gene involved in cell fate decision during development. Recently, wild-type and mutant presenilins have been associated also with apoptotic cell death. By using stable transfection of antisense cDNAs, we studied the functions of PS1 and PS2 during neuronal differentiation in the NTera2 human teratocarcinoma (NT2) cell line. Expression of antisense PS1 resulted in a failure of the clones to differentiate into neurons after retinoic acid induction, whereas cells transfected with antisense PS2 differentiated normally. Concomitantly, antisense PS1 clones were associated with increased apoptosis both under basal conditions and during the early period of neuronal differentiation after retinoic acid treatment. Overexpression of bcl-2 in antisense PS1 clones reduced cell death and resulted in a recovery of neuronal differentiation. These studies suggest that PS1 plays a role in differentiation and cell death and that PS1 and PS2 have differing physiological roles in this experimental paradigm.
Keywords: presenilins, apoptosis, neuronal differentiation, Bcl-2, antisense, NT2 cell line
Alzheimer’s disease (AD), the most common age-related neurodegenerative disorder, is associated with several genetic risk factors. Two genes, presenilin-1 (PS1) and presenilin-2 (PS2), have recently been shown to be the major causes of early onset familial Alzheimer’s disease (FAD) (Levy-Lahad et al., 1995; Rogaev et al., 1995; Sherrington et al., 1995). The proteins encoded by PS1 and PS2 share 67% identity to each other and are predicted to be integral membrane proteins, with six to eight transmembrane domains (Doan et al., 1996; De Strooper et al., 1997;Lehman et al., 1997). Presenilin homologs to both PS1 and PS2 have been identified in Drosophila, Xenopus, and C. elegans, the latter to a gene called sel-12, which has been shown to facilitate notch/lin-12 signaling during development (Levitan and Greenwald 1995; Boulianne et al., 1997;Hong and Koo, 1997; Tsujimura et al., 1997). Consistent with this observation, the in vivo developmental expression of presenilins and notch are closely matched to each other (Berezovska et al., 1997). Moreover, functional homology between the presenilins and sel-12 has been established by the ability of human PS1 and PS2 to substitute for sel-12 function inC. elegans (Levitan et al., 1996; Baumeister et al., 1997). Therefore, these studies suggest that the presenilin gene represents a widely conserved gene family and that the putative interaction with thenotch/lin-12 genes is preserved.
In addition to the potential involvement of presenilins in thenotch pathway, recent studies have suggested that presenilins also play a role in cell death. The evidence is better established for PS2 than PS1 (Vito et al., 1996a). Specifically, overexpression of PS2 in pheochromocytoma cell line (PC12) cells led to increased cell death in response to a variety of apoptotic stimuli (Deng et al., 1996; Vito et al., 1996b; Wolozin et al., 1996). Expression of a PS2 mutation associated with FAD led to increased levels of apoptosis at both basal and stimulated conditions (Wolozin et al., 1996). The role of PS1 in cell death is less clear. Overexpression of mutant PS1 (L286V mutation) but not wild-type PS1 resulted in increased susceptibility to cell death induced by trophic factor withdrawal and Aβ-mediated neurotoxicity (Guo et al., 1997). Therefore, both mutant PS1 and PS2 appear to acquire proapoptotic properties. Disruption of calcium homeostasis and pathways involving heterotrimeric G-proteins may mediate the increased susceptibility to cell death (Guo et al., 1996; Wolozin et al., 1996).
To further understand the normal biological functions of presenilins and whether they contribute to the pathophysiology of AD, we examined the consequences of inhibiting presenilin expression in the NT2 neuronal precursor cell line. The rationale for this approach is the putative relationship between presenilins and the notch gene family, the latter having been shown to play a role in cell fate decisions during neuronal development (Artavanis-Tsakonas et al., 1995). Furthermore, presenilins appear to be expressed predominantly in neurons in a developmentally regulated pattern (Lee et al., 1996;Takahashi et al., 1996; Berezovska et al., 1997; Lah et al., 1997). Thus, inhibiting presenilin expression during differentiation may reveal novel biological properties of these proteins. Our results showed that in NT2 human teratocarcinoma cells PS1 expression is related to neuronal development and that inhibiting expression of this protein is correlated with both increased cell death and an inability of the cells to differentiate into neurons. Surprisingly, in this experimental paradigm, PS2 appeared to function independently and did not share similar roles in neuronal differentiation.
MATERIALS AND METHODS
Cell culture. NT2 cells were cultured in 50% Opti-MEM–50% DMEM containing 10% fetal bovine serum (FBS). The procedure for neuronal differentiation was as described previously (Pleasure et al., 1992; Pleasure and Lee, 1993). Briefly, NT2 cells plated in T-75 flasks were grown to 50% confluency, and the medium was changed to DMEM with 10% FBS containing 10 μm retinoic acid (RA) for 4 weeks. After 4 weeks, the cultures were replated twice, the second time onto matrigel-coated dishes, and then treated with mitotic inhibitors (10 μm fluorodeoxyuridine and 10 μm uridine) (Pleasure et al., 1992). For Northern blot analysis, cell cultures were lysed with Trizol reagent (Life Technologies, Grand Island, NY), and total RNA (20 μg) was separated by 2.2 m formaldehyde–agarose gel and transferred to nylon membrane. Hybridization was performed in Quikhyb solution (Stratagene, La Jolla, CA) at 65°C and washed at 60°C with 0.1% SDS–0.2× SSC.
Transfections. For antisense experiments, PS1 cDNA (from 140 bp upstream of the first AUG site to 200 bp downstream of the AUG) was amplified by PCR and subcloned into pcDNA3 (Invitrogen, San Diego, CA) vector in the antisense orientation by using BamHI andEcoRI linkers. Anti-PS2 cDNA encompasses a region from −100 bp to +230 bp from the first AUG site and subcloned into pcDNA3 vector, as above. For rescuing experiments, bcl-2 cDNA was subcloned into pBabe-puro vector containing puromycin resistance marker (Morgenstern and Land, 1990). For stable transfections, undifferentiated NT2 cells were transfected with Lipofectamine reagent (Life Technologies) according to the manufacturer’s instructions. Stable transfectants were selected by either G418 (200μ gm/ml) or puromycin (200 ng/ml).
Immunoblotting. Cells were lysed with 1% NP-40 containing 10 μg/ml leupeptin and 240 μg/ml amino ethyl benzenesulfonyl fluoride for 30 min on ice, and then 0.1% SDS and 1% deoxycholate were added into the lysates. Before SDS-PAGE separation, the concentration of proteins were measured by Micro BCA Protein Assay (Pierce, Rockford, IL). Cell lysates (30 μg of total proteins) were denatured in Laemmeli sample buffer at 70°C for 20 min, separated on 14% SDS-PAGE, and transferred to nitrocellulose membranes. For quantitation of PS1 and PS2 levels in antisense experiments, serial twofold dilutions of control samples were fractionated in parallel to estimate the amount of protein reduction in experimental samples. PS1 polyclonal antibody J27 was raised against residues 27–45 of PS1 (Xia et al., 1997), and monoclonal antibody PSN2 was raised against residues 31–56 of PS1 (Okochi et al., 1997). Both antibodies recognize the N-terminal fragment of PS1. A monoclonal antibody PS2Loop was raised against residues 320–352 within the putative hydrophilic “loop” domain of PS2 (Y. Nomata and H. Mori, unpublished observations). PS2loop recognizes the PS2 C-terminal fragment. Additional monoclonal antibodies include β-tubulin (Developmental Studies Hybridoma Bank, Iowa City, IA), Bcl-2 (Santa Cruz Biotechnology, Santa Cruz, CA), and a 68 kDa neurofilament triplet (NF-L) (Sigma). All immunoblots were developed with enhanced chemiluminescence (Pierce) and quantitated where indicated by densitometry.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay. For quantitative cell death assay, fluorescein-12-dUTP labeling system (Promega, Madison, WI) with propidium iodide (10 ng/ml) counterstaining were used, the latter for scoring the total number of cells within a microscopic field. Cells were counted from three random microscopic fields from three different coverslips. The experiments were performed three times, and the results were expressed as an average of the percentage of terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL)-positive cells (±SEM). A t test was used to determine statistical significance of the experimental values.
RESULTS
Expression of presenilins during neuronal differentiation
Human teratocarcinoma NT2 cells were used to determine whether the presenilins play a role in neuronal differentiation. NT2 is a teratocarcinoma cell line, which under basal conditions resembles neuroepithelial stem cells, thus expressing low levels of several neuronal markers, such as microtubule-associated proteins, NFs, and N-cadherins (Pleasure and Lee, 1993). After RA treatment, a portion of the NT2 cells differentiate into postmitotic neurons. During the differentiation phase, these cells show characteristic morphological changes. Two distinct cell populations are discerned after 2 weeks of treatment: islands of epitheloid cells surrounded by clumps of multilayered neuronal progenitor cells (Fig.1A–C). By 4 weeks of RA treatment, the neurons within the dividing multilayered cells show long neuritic processes (Fig. 1C). Further evidence of neuronal differentiation is seen by the presence of differentiated CNS neuronal markers, such as high levels of the light subunit of NF-L mRNA and protein (Figs. 2,3). As an initial effort to define the possible role of presenilins in neuronal differentiation, we examined the presenilin expression through the period of RA-induced differentiation (Fig. 2). PS1 mRNA expression increased dramatically during RA treatment, starting after 3 d and peaking at 1–2 weeks, with a slight decrement during the remainder of the differentiation period (Fig. 2A). Interestingly, the increase in PS1 expression preceded the increase in NF-L expression. PS2 expression also increased during this period, but in comparison to PS1, the increment was slightly delayed. Finally, the increase in PS1 and PS2 mRNA expression was correlated with that of presenilin protein expression during the differentiation (Fig. 2B). This finding of increased presenilin expression during neuronal differentiation in vitro is consistent with the developmental regulation of these transcripts in humans seen in vivo by in situ hybridization (Lee et al., 1996;Berezovska et al., 1997; Capell et al., 1997; Hartmann et al., 1997).
Fig. 1.
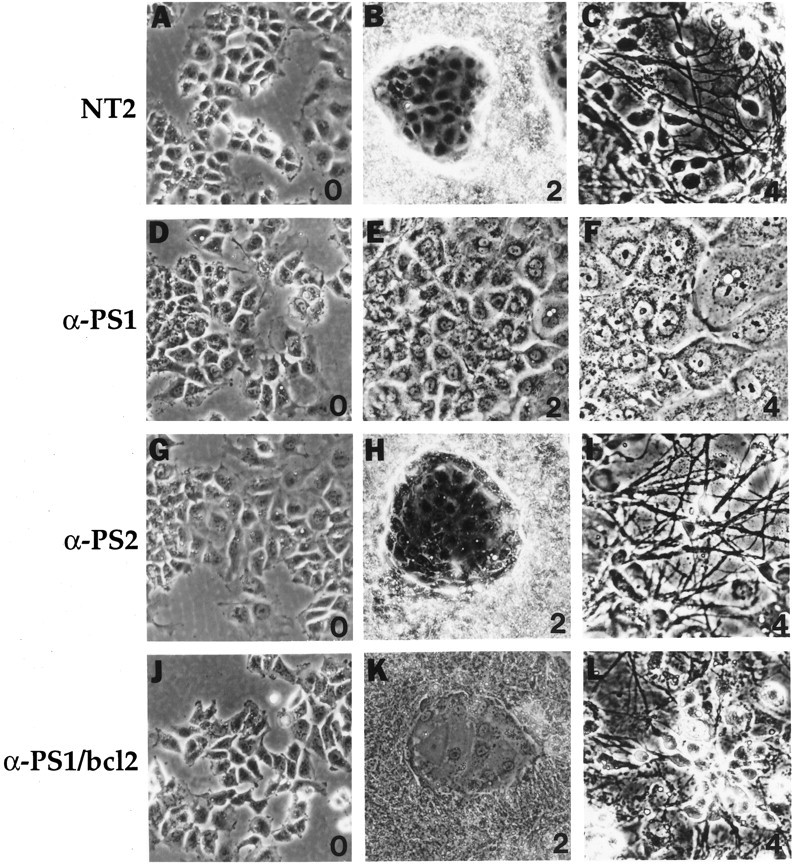
Morphological changes of NT2 cells and transfected NT2 cells during neuronal differentiation. A–C, NT2 cells in basal conditions (0) and after2 and 4 weeks of RA treatment. Note that the 2 week RA culture shows a characteristic bimodal pattern with large groups of clumped multilayered cells surrounding small islands of epitheloid cells. The latter cells are flat and apparently nondividing and contrast sharply to the rapidly dividing neuronal progenitor cells. The 4 week RA culture shows a dense neuritic network extending from neurons that overlay the epitheloid cell layer. D–F, Representative anti-PS1 cell line in basal culture conditions (0) and after 2 and4 weeks of RA induction. In comparison to normal NT2 cells, the anti-PS1 cells consist exclusively of flat epitheloid cells, even after 4 weeks of RA treatment, without any evidence of neuronal differentiation. The neuritic processes are conspicuously absent. Note that these cells are strikingly similar to the epitheloid cells in normal NT2 cells. G–I, Representative anti-PS2 cell line in basal culture conditions (0) and after2 and 4 weeks of RA induction. The anti-PS2 cells resemble control NT2 cells at basal and RA-treated conditions. These cultures differentiate normally into neurons after 4 weeks. J–L, Representative anti-PS1 cell line transfected with bcl-2 cDNA (anti-PS1/bcl-2 cells) at basal culture conditions (0) and after 2 and4 weeks of RA treatment. In comparison with anti-PS1 cells, the anti-PS1/bcl-2 cells resemble control NT2 cells showing characteristic morphology and differentiate into neurons. Magnification, 240×.
Fig. 2.
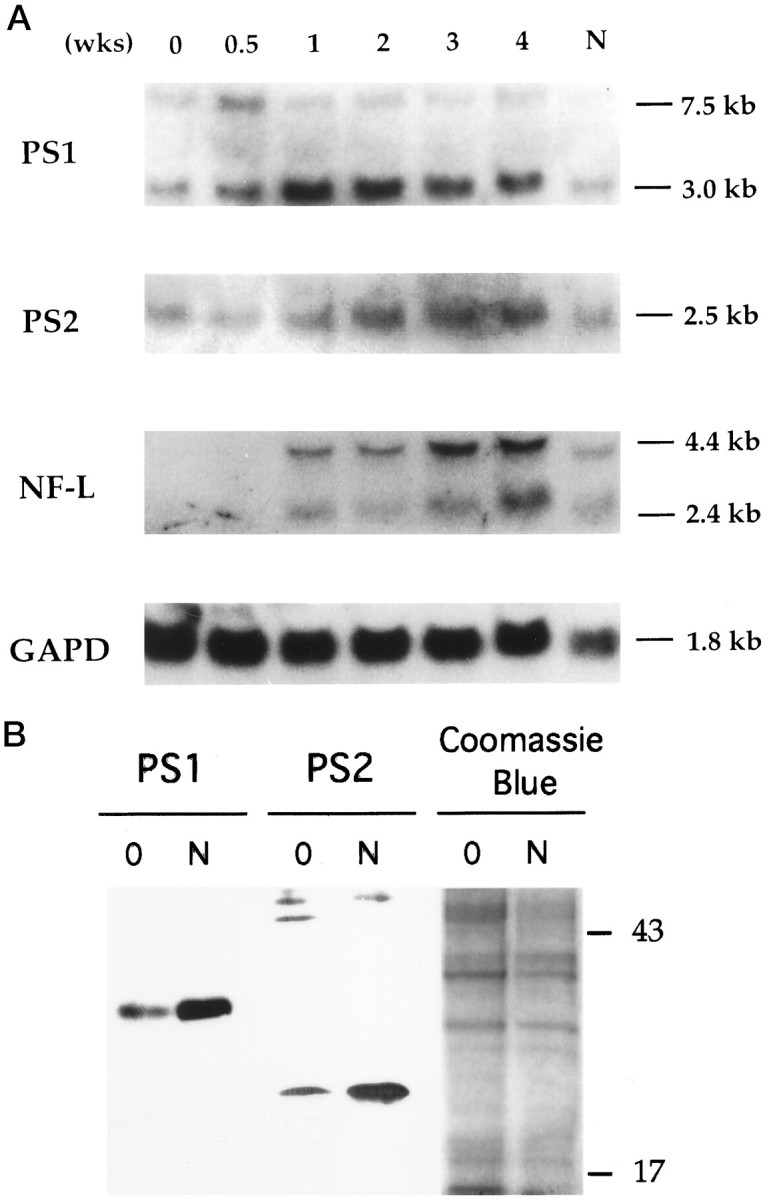
Presenilin gene expression during neuronal differentiation of NT2 cells. A, Total RNAs were isolated from NT2 cell cultures staged with RA treatment (top, 0–4 weeks and N after isolation of >95% pure neurons). After transfer to nylon membrane, the Northern blot analysis was sequentially performed with cDNA probes of PS1, PS2, NF-L, and glyceraldehyde-3-phosphate dehydrogenase (GAPD) as control for RNA loading. PS1 probe recognized a major transcript of 3.0 kb and a minor transcript of 7.0 kb (Sherrington et al., 1995). NF-L probe detected two transcripts of 2.4 and 4.4 kb (Pleasure and Lee, 1993). B, Immunoblotting analysis of PS1 and PS2 expression in NT2 cells. Equal amounts of total protein from lysates of uninduced NT2 cells (0) or isolated neurons after RA induction (N) were separated by SDS-PAGE and blotted with PS1 and PS2 monoclonal antibodies. The PS1 N-terminal proteolytic fragment (∼28 kDa) is significantly higher in neuronally differentiated NT2 (N) cultures than in the uninduced NT2 cells. Expression of the PS2 C-terminal proteolytic fragment (∼20 kDa) is similarly increased in the neuronally differentiated cultures. The respective PS1 and PS2 full-length proteins were not detected in any NT2 cell lysates (see Fig. 4). The ∼43–50 kDa bands in the PS2 immunoblot are nonspecific bands. Right panel, Coomassie blue staining of a duplicate gel run in parallel is shown for control of sample loading.
Fig. 3.

Immunoblot analysis of NF-L expression in NT2 cells and transfected NT2 cells during RA-induced neuronal differentiation. Cell lines were induced with 10 μm RA for 4 weeks. NT2 and anti-PS2 cell lines were further replated and treated with mitotic inhibitors (see Materials and Methods) to isolate pure neurons (N). Uninduced cells (0), neurons (N), or cultures treated with RA for 4 weeks without replating (4) were lysed and analyzed with NF-L monoclonal antibody. The differentiated NT2, anti-PS2, and anti-PS1/bcl-2 cells all express high levels of NF-L (∼68 kDa), which are absent in RA-treated anti-PS1 cells.Bottom panels, Coomassie blue-stained SDS-PAGE gels of duplicate samples run in parallel for control of sample loading.
Functional involvement of presenilins in neuronal differentiation
We next established NT2 cell lines stably transfected with antisense-oriented PS1 cDNA or control vector cDNA. To inhibit PS1 but not PS2 expression, the antisense cDNA construct consisted of an ∼300 bp fragment encompassing a portion of the 5′ untranslated and coding regions, with no homology to PS2. After antibiotic selection, a total of 35 independent cell lines were isolated and analyzed. All of the isolated cell lines showed normal morphology under basal culture conditions. Six of these cell lines failed to differentiate into neurons after RA treatment (Fig. 1D–F). From 2 to 8 weeks, only large flat epithelial-appearing cells were present in the cultures. The characteristic multilayered cells and associated neuritic processes in which neurons are derived were conspicuously absent from cultures treated for 2 or more weeks with RA (Fig.1E,F). Furthermore, in contrast to control NT2 cells, NF-L protein was undetectable by Western blotting in cultures after 4 weeks of RA treatment (Fig. 3). In addition, abundant, detached, and dead cells were evident in these cultures within several days of RA treatment. The remaining clones, as well as the vector control transfected cells (30 clones), all differentiated normally into neurons after RA induction. Immunoblotting showed that the levels of PS1 N-terminal fragment in the antisense transfected cells, defective in differentiation, were maximally reduced to 30% of the normal untransfected NT2 cells (Fig.4A). In contrast, in the antisense PS1 transfected cell lines with normal neuronal phenotype after RA treatment, PS1 protein levels were not diminished compared with controls (Fig. 4A). Further analyses were therefore focused on the cell lines defective in the neuronal differentiation (referred to as anti-PS1 cell lines).
Fig. 4.
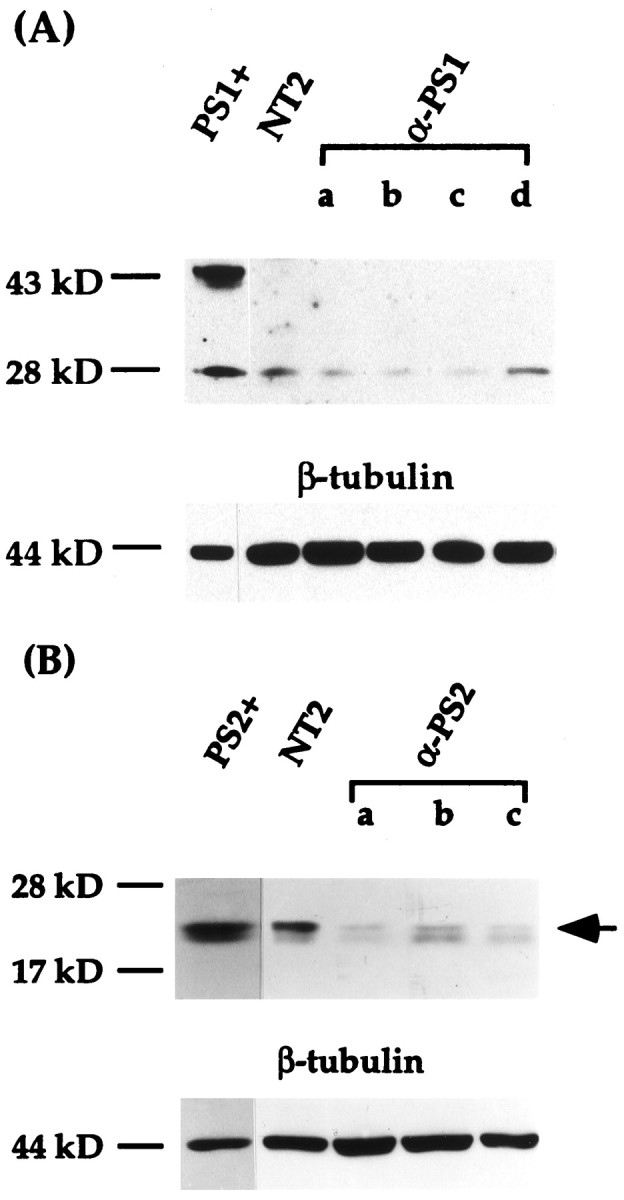
Immunoblot analysis of PS1 and PS2 proteins in transfected NT2 cell lines. NT2 cells were transfected with either antisense-oriented PS1 or PS2 cDNA. Equal amounts of total proteins (30 μg) were separated by SDS-PAGE, transferred to nitrocellulose membranes, and incubated with either J27 polyclonal antibody to PS1 or PS2 monoclonal antibody. A, The amount of N-terminal PS1 proteolytic fragment is reduced in the differentiation-defective anti-PS1 cell lines (a–c) compared with that of control NT2 cell line but not from a PS1 antisense transfected cell line with normal differentiation (d). Note that in addition to the ∼28 kDa N-terminal proteolytic fragment, full-length PS1 protein (∼43 kDa) is present in a Chinese hamster ovary (CHO) cell line overexpressing PS1 (PS1+) but not in any of the NT2 cell lines. Bottom panel, The filter was stripped and incubated with β-tubulin antibody for normalization of the amount of protein loaded in each lane. B, PS2 expression of anti-PS2 cell lines. The ∼20 kDa PS2 C-terminal fragment (arrow) was detected in a CHO cell line stably transfected with PS2 (PS2+), control NT2 cells, and representative anti-PS2 cell lines. PS2 is reduced to different levels in the anti-PS2 cells (a–c), but all the cell lines differentiated into neurons by RA. In this autoradiogram, there is a faint band just below the PS2 C-terminal fragment that is present throughout the samples to varying levels (see Fig.2B). Because this band is neither increased in overexpressing cells nor decreased in antisense transfected cells, it appears to be a cross-reactive species. Bottom panel, The filter was incubated with β-tubulin antibody after antibody stripping.
To determine whether inhibition of PS2 expression leads to similar results regarding neuronal differentiation in NT2 cells, the above experiments were repeated using an antisense-oriented PS2 cDNA construct. A total of 65 independent stable transfectants were isolated and induced with RA. However, in marked contrast to the anti-PS1 cells, all the antisense PS2 transfectants differentiated normally without overt growth retardation or cell death. Neurite formation and NF-L protein expression were normal in these cultures (Figs.1G–I, 3). Compared with untransfected controls, immunoblotting showed that the level of PS2 protein was reduced in a subset of these transfectants to a level (25–30%) comparable to that seen in anti-PS1 cells (Fig. 4B). Therefore, reduction of PS2 expression did not result in a mutant phenotype with regard to neuronal differentiation and was distinctly different to that seen after inhibition of PS1 expression. This result is consistent with PS2 studies of fas ligand-mediated cell death in which antisense PS2 or a dominant negative C-terminal PS2 construct protected cells from apoptosis (Wolozin et al., 1996).
Enhanced cell death by blocking PS1 expression
As mentioned above, the anti-PS1 cell lines consistently showed more cellular debris, especially during the initial period of RA treatment. This led us to postulate that cell death may be enhanced after RA induction in these cell lines. Cell death was therefore assayed by the TUNEL staining method before and after RA treatment (Fig. 5). As anticipated, the anti-PS1 cell lines showed a fourfold increase in the percentage of cell death compared with normal NT2 cells analyzed after 3 d of RA treatment (p < 0.005). Interestingly, from this analysis, it was evident that even at basal culture conditions, the anti-PS1 cell lines showed an approximately threefold increase in cell death (p < 0.005) compared with the untransfected NT2 cells. The cell death is consistent with apoptosis by the presence of characteristic DNA laddering (data not shown). Therefore, these findings indicate that inhibiting PS1 expression in NT2 cells results in greater cell death, particularly after RA-induced differentiation. Interestingly, control NT2 cells also showed an approximately threefold increase in cell death (p < 0.005) after RA treatment, suggesting that this cell death process usually accompanies neuronal differentiation in NT2 cells. Therefore, it is possible that the inability of anti-PS1 cells to differentiate into neurons is attributable to enhanced apoptotic cell death, especially after RA treatment, rather than the failure of the cells to differentiate into neurons per se.
Fig. 5.

Cell death in anti-PS1 cell lines after RA induction. Normal NT2 cells and representative anti-PS1 and anti-PS1/bcl-2 cell lines were cultured on poly-d-lysine-coated glass coverslips with (+) and without (−) 10 μm RA for 3 d, and apoptotic nuclei were detected by TUNEL assay. The percentage (±SEM) of cells undergoing apoptosis was measured by scoring the number of TUNEL-positive cells divided by the total number of cells (detected by propidium iodide staining) from three random microscopic fields performed in triplicate. The data are an average from three independent experiments.
Functional replacement of PS1 by bcl-2
If increased apoptosis underlies the loss of neuronal differentiation after RA administration, then the activity of genes that modulate neural death may alter the mutant phenotype of anti-PS1 cells. Overexpression of the proto-oncogene bcl-2 has been shown to inhibit apoptosis in a variety of cell types, including cell death in neural cells induced by a number of insults (Garcia et al., 1992; Allsopp et al., 1993). Immunoblotting showed that the levels of Bcl-2 are normally increased during neuronal differentiation of NT2 cells resulting from RA induction (Fig.6). In both control untransfected NT2 cells and anti-PS1 cell lines, the levels of Bcl-2 were not detectable by immunoblotting at basal conditions. After RA induction, the Bcl-2 level increased in both untransfected and anti-PS1 cells, although substantially less in the anti-PS1 cells. This result suggests that either the enhanced vulnerability of anti-PS1 cells to cell death may be related to lower expression of Bcl-2 or that the increased apoptosis during RA-induced differentiation led to lower expression of Bcl-2. We therefore stably transfected anti-PS1 cell lines with bcl-2cDNA to examine the effect of bcl-2 overexpression in the NT2 system. As expected, these anti-PS1/bcl-2 doubly transfected cells showed increased Bcl-2 under basal conditions. Interestingly, these cells also showed an increase in Bcl-2 after RA treatment to a level exceeding that in the control NT2 cells (Fig. 6). Surprisingly, however, these bcl-2 transfected cell lines were able to differentiate into neurons by RA treatment (Figs.1J–L, 3). Finally, analysis of apoptosis in anti-PS1/bcl-2 cells show that cell death was reduced by ∼40% and 20% compared with the parental anti-PS1 cells after RA treatment and during basal conditions (p < 0.005), respectively (Fig. 5). Despite this reduction, cell death is still higher than control NT2 cells under both basal and RA treatment conditions. Nevertheless, this result demonstrated that inhibition of cell death by overexpression of bcl-2 led to both a rescue of neuronal differentiation and decreased cell death in the anti-PS1 cell lines.
Fig. 6.

Bcl-2 expression at uninduced and induced conditions. Untransfected NT2, anti-PS1, and anti-PS1/bcl-2 cells were analyzed before (−) and after (+) 2 weeks of RA treatment. Equal amounts of total protein were fractionated by SDS-PAGE, transferred to nitrocellulose membrane, and incubated with a monoclonal antibody to Bcl-2.
DISCUSSION
In this study, we used the approach of stable transfection of antisense constructs to examine the biological roles of PS1 and PS2 in cultured cells. Using the experimental paradigm of NT2 cells, we showed that PS1 participates in neuronal differentiation and cell death. During neuronal differentiation induced by RA in NT2 cells, expression of PS1 increased in a pattern that recapitulates what has been observedin vivo (Lee et al., 1996; Berezovska et al., 1997). Moreover, inhibition of PS1 expression by stable transfection of an antisense construct resulted in a failure of the cells to differentiate into neurons after RA induction. Instead, only large flat epithelial-appearing cells were evident after RA treatment. Unexpectedly, increased apoptotic cell death over untransfected control cells was observed at basal conditions, but this increased markedly during RA treatment. Because of this finding, the cells were further transfected with bcl-2, an anti-apoptotic oncogene. Concomitant with a decrease in cell death, overexpression ofbcl-2 reversed the inability of mutant anti-PS1 clones to differentiate into neurons. Therefore, our results showed that loss of PS1 but not PS2 expression is associated with both a loss of neuronal differentiation and increased cell death in this experimental paradigm.
Our study was not designed to resolve or delineate the apparent dual roles of PS1 in differentiation and apoptosis. On the one hand, it is possible that increased apoptosis in anti-PS1 cells, in and of itself, is sufficient to result in a failure of neuronal differentiation. That is to say, there are simply insufficient numbers of neuronal progenitor cells remaining in the cultures to differentiate into neurons after the fourfold increase in cell death after RA treatment. The rescue by Bcl-2 overexpression would favor this interpretation. On the other hand, this supposition would not adequately explain the abrupt increase in PS1 expression associated with neuronal differentiation induced by RA. Moreover, despite increased apoptosis in the anti-PS1 cells, large epitheloid cells persisted in these cultures for up to 8 weeks without apparent cell division. These cells resemble one of the two cell types normally seen in control NT2 cells after RA treatment. In addition, these epitheloid cells have been shown to represent postmitotic mesenchymal cells induced by overexpression of RA receptors (Moasser et al., 1995). Consequently, the results also suggest that PS1 plays a role in choosing cell fate, specifically with regard to neuronal differentiation. This interpretation is consistent with the putative interaction of notch/lin-12 and with the recent findings described in transgenic mice deficient in PS1 expression (Wong et al., 1997; Shen et al., 1997). In addition to gross deformities of the axial skeleton, neurogenesis appears to be impaired in the targeted PS1 null mutants (Shen et al., 1997). It has also been documented in other studies that proteolytic processing of PS1 is developmentally regulated, findings that have been interpreted to suggest that PS1 plays a direct role in neuronal differentiation (Capell et al., 1997; Hartmann et al., 1997). Together, it might be argued that activation of the apoptotic pathway in the anti-PS1 cells may, in part, be secondary to the inability of the cells to differentiate into neurons after RA treatment.
To date, overexpression of wild-type PS1 has not been reported to be associated with apoptosis. Only overexpression of mutant PS1 has resulted in increased cell death in response to Aβ-induced toxicity and growth factor withdrawal but not at unstimulated basal conditions (Guo et al., 1997). Rather than increasing presenilin expression, the strategy of inhibiting expression by antisense approach was used in our studies to determine the biological functions of PS1. It was by reducing the levels of PS1 protein at a time when PS1 expression normally increased during differentiation that our findings became evident. In contrast, the reported studies overexpressed PS1 in cells in which endogenous levels of the protein are likely to be low. Therefore, testing PS1 function at the point when expression is normally increased may have been critical in our observations.
In contrast to PS1, inhibition of PS2 expression in NT2 cells did not result in a similar failure of NT2 cells to differentiate into neurons. It is possible that this observation was attributable to a quantitative rather than qualitative phenomenon. That is to say, the levels of PS2 protein suppression from transfection of the antisense construct were inadequate to achieve the biological effect. However, a number of observations argue that this is not likely to be the case. First, the levels of PS2 in the antisense experiments were as low as those achieved in anti-PS1 cells. Second, twice the number of antisense PS2 clones compared with anti-PS1 clones were analyzed without finding a similar mutant phenotype. Third, the result is entirely consistent with the series of experiments showing a proapoptotic effect of full-length PS2 in both T cells and PC12 cells (Guo et al., 1996; Vito et al., 1996a,b; Wolozin et al., 1996). In these studies, overexpression of wild-type and mutant PS2 resulted in increased apoptosis induced by a variety of stimuli, but expression of an antisense PS2 construct conferred protection against apoptosis from the same stimuli (Wolozin et al., 1996). Finally, in striking contrast to PS1, it was recently reported that there were no developmental abnormalities or any obvious phenotype in transgenic animals deficient in PS2 (Boeve et al., 1998). Therefore, we hypothesize that PS2 does not play a significant role in neuronal differentiation and that inhibiting PS2 expression, as has been pointed out, results in properties of antiapoptosis and protection from otherwise toxic stimuli.
The results of this study highlighted the contrasting effects of the loss of PS1 and PS2 in NT2 cells during neuronal differentiation in vitro. First, impairment of only PS1 and not PS2 expression with an antisense construct affected normal differentiation of NT2 cells induced by RA treatment. Second, our results reinforce the concept that PS1 and PS2 may have different effects on apoptosis. In other words, only PS2 and not PS1 may function as a proapoptotic gene. Related to this hypothesis is that all our attempts to compensate for the loss of PS1 function in anti-PS1 cells with PS2 overexpression have been unsuccessful and that PS2 expression was not increased in anti-PS1 cell lines (data not shown). However, in view of the ability of both PS1 and PS2 to substitute forsel-12 function in C. elegans (Levitan et al., 1996; Baumeister et al., 1997), this postulate may be too simplistic at this time.
Together, our results suggest that presenilins may have multiple physiological roles involving neuronal differentiation and cell death. Furthermore, the homologous gene products PS1 and PS2 at times appear to function similarly but may, at times, function independently. Interestingly, the property of multiple independent functions has been described in members of the bcl-2 gene family (Reed, 1997). Whether the biological functions of PS1 and PS2 require coordinate activity, such as heterodimerization as has been suggested by others (Vito et al., 1996b), remains an interesting mechanism to resolve. Finally, it will be important to determine whether these physiological properties of presenilins or perturbations of these properties, such as in cell death or in affecting Aβ production (Scheuner et al., 1996), contribute to the pathogenesis of Alzheimer’s disease.
Footnotes
This work was supported by National Institutes of Health Grants NS01812 and AG12282 and The Paul Beeson Physician Faculty Scholar in Aging Research from American Federation for Aging Research. We thank D. Kang for reviewing this manuscript.
Correspondence should be addressed to Edward H. Koo, Department of Neurosciences 0691, University of California, San Diego, La Jolla, CA 92093-0691.
Dr. Hong’s present address: Department of Molecular Genetics and Biochemistry, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261.
REFERENCES
- 1.Allsopp TE, Wyatt S, Paterson HF, Davies AM. The proto-oncogene Bcl-2 can selectively rescue neurotrophic factor-dependent neurons from apoptosis. Cell. 1993;73:295–307. doi: 10.1016/0092-8674(93)90230-n. [DOI] [PubMed] [Google Scholar]
- 2.Artavanis-Tsakonas S, Matsuno K, Fortini ME. Notch signaling. Science. 1995;268:225–232. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- 3.Baumeister R, Leimer U, Zweckbronner I, Jakubek C, Grunberg J, Haass C. Human presenilin-1, but not familial Alzheimer’s disease (FAD) mutants, facilitate Caenorhabditis elegans Notch signaling independently of proteolytic processing. Genes Funct. 1997;1:149–159. doi: 10.1046/j.1365-4624.1997.00012.x. [DOI] [PubMed] [Google Scholar]
- 4.Berezovska O, Xia MQ, Page K, Wasco W, Tanzi RE, Hyman BT. Developmental regulation of presenilin mRNA expression parallels notch expression. J Neuropathol Exp Neurol. 1997;56:40–44. doi: 10.1097/00005072-199701000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Boeve C, Craessaerts K, Vanderstichele H, Serneels L, Checler F, von Figura K, Van Leuven F, Annaert W, Saftig P, De Strooper B. Processing of APP in presenilin 1- and presenilin 2-deficient neurons. Neurobiol Aging. 1998;19:S283. [Google Scholar]
- 6.Boulianne GL, Livne-Bar I, Humphreys JM, Liang Y, Lin C, Rogaev E, St. George-Hyslop P. Cloning and characterization of the Drosophila presenilin homologue. NeuroReport. 1997;8:1025–1029. doi: 10.1097/00001756-199703030-00041. [DOI] [PubMed] [Google Scholar]
- 7.Capell A, Saffrich R, Olivo JC, Meyn L, Walter J, Grunberg J, Mathews P, Nixon R, Dotti C, Haass C. Cellular expression and proteolytic processing of presenilin proteins is developmentally regulated during neuronal differentiation. J Neurochem. 1997;69:2432–2440. doi: 10.1046/j.1471-4159.1997.69062432.x. [DOI] [PubMed] [Google Scholar]
- 8.De Strooper B, Beullens M, Contreras B, Levesque L, Craessaerts K, Cordell B, Moechars D, Bollen M, Fraser P, St. George-Hyslop P, Van Leuven F. Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer’s disease-associated presenilins. J Biol Chem. 1997;272:3590–3598. doi: 10.1074/jbc.272.6.3590. [DOI] [PubMed] [Google Scholar]
- 9.Deng G, Pike CJ, Cotman CW. Alzheimer-associated presenilin-2 confers increased sensitivity to apoptosis in PC12 cells. FEBS Lett. 1996;397:50–54. doi: 10.1016/s0014-5793(96)01142-8. [DOI] [PubMed] [Google Scholar]
- 10.Doan A, Thinakaran G, Borchelt DR, Slunt HH, Ratovitsky T, Podlisny M, Selkoe DJ, Seeger M, Gandy SE, Price DL, Sisodia SS. Protein topology of presenilin 1. Neuron. 1996;17:1023–1030. doi: 10.1016/s0896-6273(00)80232-9. [DOI] [PubMed] [Google Scholar]
- 11.Garcia I, Martinou I, Tsujimoto Y, Martinou JC. Prevention of programmed cell death of sympathetic neurons by the Bcl-2 proto-oncogene. Science. 1992;258:302–304. doi: 10.1126/science.1411528. [DOI] [PubMed] [Google Scholar]
- 12.Guo Q, Furukawa K, Sopher BL, Pham DG, Robinson N, Martin GM, Mattson MP. Alzheimer’s PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid β-peptide. NeuroReport. 1996;8:379–383. doi: 10.1097/00001756-199612200-00074. [DOI] [PubMed] [Google Scholar]
- 13.Guo Q, Sopher BL, Furukawa K, Pham DG, Robinson N, Martin GM, Mattson MP. Alzheimer’s presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid β-peptide. J Neurosci. 1997;17:4212–4222. doi: 10.1523/JNEUROSCI.17-11-04212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartmann H, Bursciglio J, Baumann KH, Staufenbiel M, Yankner BA. Developmental regulation of presenilin-1 processing in the brain suggests a role in neuronal differentiation. J Biol Chem. 1997;272:14505–14508. doi: 10.1074/jbc.272.23.14505. [DOI] [PubMed] [Google Scholar]
- 15.Hong C-S, Koo EH. Isolation and characterization of Drosophila presenilin homolog. NeuroReport. 1997;8:665–668. doi: 10.1097/00001756-199702100-00017. [DOI] [PubMed] [Google Scholar]
- 16.Lah JJ, Heilman CJ, Nash NR, Rees HD, Yi H, Counts SE, Levey AI. Light and electron microscopic localization of presenilin-1 in primate brain. J Neurosci. 1997;17:1971–1980. doi: 10.1523/JNEUROSCI.17-06-01971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee MK, Slunt HH, Martin LJ, Thinakaran G, Kim G, Gandy SE, Seeger M, Koo E, Price DL, Sisodia SS. Expression of presenilin 1 and 2 (PS1 and PS2) in human and murine tissues. J Neurosci. 1996;16:7513–7525. doi: 10.1523/JNEUROSCI.16-23-07513.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehmann S, Chiesa R, Harris D. Evidence for a six-transmembrane domain structure of presenilin 1. J Biol Chem. 1997;272:12047–12051. doi: 10.1074/jbc.272.18.12047. [DOI] [PubMed] [Google Scholar]
- 19.Levitan D, Greenwald I. Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature. 1995;377:351–354. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- 20.Levitan D, Doyle TG, Brousseau D, Lee MK, Thinakaran G, Slunt HH, Sisodia SS, Greenwald I. Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc Natl Acad Sci USA. 1996;93:14940–14944. doi: 10.1073/pnas.93.25.14940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu C, Jondro PD, Schmidt WD, Wang K, Crowley AC, Fu Y-H, Guenette SY, Galas D, Nemens E, Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 22.Moasser MM, Reuter VE, Dmitrovsky E. Overexpression of the retinoic acid receptor-gamma directly induces terminal differentiation of human embryonal carcinoma cells. Oncogene. 1995;10:1537–1543. [PubMed] [Google Scholar]
- 23.Morgenstern JP, Land H. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;8:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okochi M, Ishii K, Usami M, Sahara N, Kametani F, Tanaka K, Fraser PE, Ikeda M, Saunders AM, Hendriks L, Shoji S, Nee LE, Martin JJ, Van Broeckhoven C, St, George-Hyslop PH, Roses AD, Mori H. Proteolytic processing of presenilin-1 (PS-1) is not associated with Alzheimer’s disease with or without PS-1 mutations. FEBS Lett. 1997;418:162–166. doi: 10.1016/s0014-5793(97)01378-1. [DOI] [PubMed] [Google Scholar]
- 25.Pleasure SJ, Lee VM-Y. NTera 2 cells: a human cell line which displays characteristics expected of a human committed neuronal progenitor cell. J Neurosci Res. 1993;35:585–602. doi: 10.1002/jnr.490350603. [DOI] [PubMed] [Google Scholar]
- 26.Pleasure SJ, Page C, Lee VM-Y. Pure, postmitotic, polarized human neurons derived from NTera 2 cells provide a system for expressing exogenous proteins in terminally differentiated neurons. J Neurosci. 1992;12:1802–1815. doi: 10.1523/JNEUROSCI.12-05-01802.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387:773–776. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- 28.Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Lin HC, Holman K, Tsuda T, Mar L, Sorbi S, Nacmias B, Placentini S, Amaducci L, Chumakov I, Cohen D, Lannfelt L, Fraser PE, Rommens JM, St. George-Hyslop PH. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 29.Scheuner D, Eckamn C, Jensen M, Song X, Citron M, Suzuki N, Bird TC, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Vitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 30.Shen J, Bronson R, Chen D, Xia W, Selkoe D, Tonegawa S. Skeletal and CNS defects in presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 31.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin J-F, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HAR, Haines JL, Pericak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St. George-Hyslop PH. Cloning of a gene bearing missense mutations in early onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi H, Murayama M, Takashima A, Mercken M, Nakazato Y, Noguchi K, Imahori K. Molecular cloning and expression of the rat homologue of presenilin-1. Neurosci Lett. 1996;206:113–116. doi: 10.1016/s0304-3940(96)12449-6. [DOI] [PubMed] [Google Scholar]
- 33.Tsujimura A, Yasojima K, Hashimoto-Gotoh T. Cloning of Xenopus presenilin-α and -β cDNAs and their differential expression in oogenesis and embryogenesis. Biochem Biophys Res Commun. 1997;231:392–396. doi: 10.1006/bbrc.1996.6043. [DOI] [PubMed] [Google Scholar]
- 34.Vito P, Lacana E, D’Adamio L. Interfering with apoptosis: Ca2+-binding protein ALG-2 and Alzheimer’s disease gene ALG-3. Science. 1996a;271:521–525. doi: 10.1126/science.271.5248.521. [DOI] [PubMed] [Google Scholar]
- 35.Vito P, Wolozin B, Ganjei JK, Iwasaki K, Lacana E, D’Adamio L. Requirement of the familial Alzheimer’s disease gene PS2 for apoptosis. J Biol Chem. 1996b;271:31025–31028. doi: 10.1074/jbc.271.49.31025. [DOI] [PubMed] [Google Scholar]
- 36.Wolozin B, Iwasaki K, Vito P, Ganjei JK, Lacana E, Sunderland T, Zhao B, Kusiak JW, Wasco W, D’Adamio L. Participation of presenilin 2 in apoptosis: enhanced basal activity conferred by an Alzheimer mutation. Science. 1996;274:1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- 37.Wong P, Zheng H, Chen H, Becher M, Sirinathsinghji D, Trumbauer M, Chen H, Price DL, Van der Ploeg L, Sisodia SS. Presenilin 1 is required for Notch 1 and Dll1 expression in the paraxial mesoderm. Nature. 1997;38:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- 38.Xia W, Zhang J, Kholodenko D, Citron M, Polisny MB, Teplow DB, Haass C, Seubert P, Koo EH, Selkoe DJ. Enhanced production and oligomerization of the 42-residue amyloid β-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J Biol Chem. 1997;272:7977–7982. doi: 10.1074/jbc.272.12.7977. [DOI] [PubMed] [Google Scholar]