

Abstract
The trkC locus encodes several receptors for neurotrophin-3, including the well studied full-length tyrosine kinase isoform, in addition to receptor isoforms lacking the kinase active domain. TrkC receptors are widely expressed throughout mouse development in many different organs. To investigate the function of truncated receptors in vivo and to identify cell types that are biologically responsive to this gene product, we have overexpressed a physiological truncated trkC isoform in the mouse. Mice overexpressing this receptor develop to term but die in the first postnatal days. High levels of transgene expression result in severe developmental defects in the peripheral nervous system and in the heart. The severity of neuronal losses observed in these animals suggests that truncated receptors may act by sequestering neurotrophin, thus, closely relating this mouse model to the neurotrophin-3-deficient one. Lower levels of exogenous truncated receptor in transgenic mice result in a more modest phenotype and, in some neuronal populations, do not cause neural deficits. Taken together, these data suggest that truncated trkC receptor isoforms may have modulatory functions in development.
Keywords: trkC, truncated receptor, development, neurotrophin, trk receptors, transgenic
Neurotrophins and their receptors are highly expressed throughout mouse ontogenesis in both neuronal and non-neural cell types (for review, see Tessarollo, 1998). These families of genes play crucial roles in the development and function of the nervous system (Korsching, 1993; Snider, 1994; Bothwell, 1995;Lewin and Barde, 1996). Neurotrophin activation of trk tyrosine kinase receptors results in the initiation of signal transduction cascades, including the ras pathway, and the induction of proliferation, survival, and differentiation of peripheral nervous system (PNS) and CNS neurons. Many functions of these tyrosine kinase receptors and their signaling mechanisms have been described (Segal and Greenberg, 1996). The trkB and trkC loci additionally encode, by alternative splicing, receptor isoforms that lack the catalytic kinase domain (Klein et al., 1990; Middlemas et al., 1991; Tsoulfas et al., 1993; Valenzuela et al., 1993; Garner and Large, 1994). The unique intracellular domains of truncated forms of both trkB and trkC are highly conserved in human, mouse, rat, and chicken (Garner and Large, 1994; Baxter et al., 1997). The high conservation of these intracellular domains has raised the notion that truncated isoforms may have signaling ability.
Indeed, recent data obtained in vitro have shown that truncated trkB receptors can induce a ligand-mediated increase in the release of cellular acidic metabolites (Baxter et al., 1997), and that activated truncated trkC receptors can promote differentiation of chicken neural crest cells (Hapner et al., 1998). Mice lacking all trkC isoforms, including the truncated ones, have a more severe phenotype, including reduced viability and a more pronounced neuronal deficit, compared with mice lacking only the full-length tyrosine kinase receptors, suggesting a positive role for truncated trkC receptorsin vivo (Klein et al., 1994; Minichiello et al., 1995;Tessarollo et al., 1997). However, no signaling molecules downstream of truncated receptors have been identified to date.
The presence of abnormalities in mice heterozygous for a specific neurotrophin null allele has demonstrated that precise levels of neurotrophins are critical for normal mouse development (Ernfors et al., 1994; Korte et al., 1995; Donovan et al., 1996; Erickson et al., 1996; Chen et al., 1997). Expression analysis of truncated trkB receptors and results obtained from coexpressing full-length and truncated trkB receptors have suggested that truncated receptors could act by sequestering excessive neurotrophins or as naturally occurring dominant negative elements of the full length receptor (Biffo et al., 1995; Eide et al., 1996; Ninkina et al., 1996). However, there are as yet no in vivo data available in support of such nonsignaling functions of truncated receptors.
To investigate functions of truncated trkC receptors in vivoand to identify cell types that respond to these receptors, we have overexpressed a truncated trkC isoform in mouse. Ectopic expression of this truncated receptor results in perinatal lethality. Severe deficiencies were observed in sites that normally express endogenous truncated trkC receptors, such as the heart and the PNS (Hiltunen et al., 1996; Menn et al., 1998). The effects on PNS development vary with the amount of transgenic mRNA expressed, suggesting that truncated trkC receptors may have pleiotropic modulatory functions in vivo.
MATERIALS AND METHODS
Generation of transgenic mice. A trkC truncated fusion gene containing the human B-actin promoter and the SV40 poly(A) signal was constructed by inserting a 2.2 kb rat trkC truncated cDNA (Tsoulfas et al., 1993) into the pHB APr-1 vector (Gunning et al., 1987). This gene was microinjected into fertilized (C57BL/6J × C3H/HeJ) F2 mouse eggs as previously described (Osborn et al., 1987).
RNase protection analysis. RNA was extracted using RNAzol (Cinna/Biotecx) following the manufacturer’s recommendations. RNase protection experiments were performed as previously described (Tessarollo et al., 1992) using a ribonuclease protection assay kit (Ambion). A genomic TrkC-specific probe that spans nucleotides 1238–1424 of the extracellular domain (trkC-all) of the rat sequence (Tsoulfas et al., 1993) was used to generate an antisense RNA probe used in RNase protection analysis to detect all trkC isoforms. The trkC truncated (TK−)-specific probe was obtained by subcloning a region of the rat trkC truncated receptor (nucleotides 1732–1909;Tsoulfas et al., 1993). The glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-specific probe (Ambion) was included in the same reaction as a mean of assessing relative levels of RNA present in each hybridization.
Histological techniques and neuronal cell counts. After microinjection, transgenic mice were checked for the presence of the transgene. Mutant and wild-type littermates were killed by decapitation, and the bodies and heads were immediately fixed in Bouin’s fixative overnight. The next day, after rinsing for a few hours in water, the heads and spinal cords were transferred to 70% ethanol and processed for paraffin embedding. For neuronal counts, the heads of one transgenic littermate and one-wild type littermate were embedded in the same block, sectioned sagitally at 5 μm, and Nissl-stained with 0.1% cresyl violet. To assure unbiased analysis, neuronal counts were performed in a blinded manner.
Immunohistochemical neurofilament staining. For immunohistochemistry, embryos fixed in 4% paraformaldehyde were bleached for 1 hr on ice in 3:1 methanol/hydrogen peroxide (30% solution in water), rehydrated, washed in PBS containing 0.1% Tween 20 (PBT), treated with proteinase K (20 μg/ml) for 5 min, and refixed. Embryos were then incubated overnight at 4°C in 1% serum and mouse monoclonal anti-neurofilament 160 antibody (1:1000; Sigma Chemical Co., St. Louis, MO), washed for 1 d in PBT, and incubated overnight at 4°C in PBT containing 1% serum and peroxidase-conjugated rabbit anti-mouse Ig (1:1000; Sigma). The color reaction was developed by incubation in 0.3 mg/ml diaminobenzidine, 0.03% H2O2, and 0.05% NiCl2 and stopped by washing in PBT.
RESULTS
Generation of truncated trkC transgenic mice
The trkC gene is expressed in many tissues and at almost all stages of development (Tessarollo et al., 1993; Tsoulfas et al., 1993;Lamballe et al., 1994). The specific pattern of expression of the different isoforms encoded by this locus is not known. However, truncated trkC receptors are expressed in both the PNS and CNS and, although at lower levels, in non-neural tissues, including the cardiovascular system (Tsoulfas et al., 1993; Hiltunen et al., 1996;Menn et al., 1998) (Figure1A). Thus, we have used the human β-actin promoter (Gunning et al., 1987) to drive the expression of a rat truncated trkC receptor that has a 36 amino acid domain that is 100% conserved between rat and chicken (Tsoulfas et al., 1993). Mouse and rat truncated trkC receptors are very conserved and are biologically indistinguishable; therefore, we used a rat truncated isoform to distinguish between the endogenous and the transgenic truncated receptor (P. Tsoulfas, personal communication) (Menn et al., 1998). RNase protection experiments showed that indeed transgenic truncated trkC (Tg trkC TK−) RNA could be detected at high levels in both neural and non-neural tissues (Fig.1A,B).
Fig. 1.

Expression of trkC receptors in truncated isoform transgenic animals. A, RNase protection analysis performed with a probe specific for the truncated trkC receptor (trkC TK−) reveals higher expression of endogenous trkC TK− in the brain compared with the leg in wild-type animals. Levels of expression of trkC TK− gene in transgenic animals are also shown.B, RNase protection analysis performed with an extracellular rat trkC antisense cDNA probe (all-trkC) that detects all isoforms of trkC RNA including the transgenic one (Tg). A polymorphism allows the distinction between the transgenic (bottom trkC band) from the wild-type mRNA (top band). C, High levels of expression of the transgene are associated with early postnatal lethality. RNA from legs of mice that were moribund at the indicated age (P0, P1, P3) was analyzed by RNase protection for the presence of the transgene (Tg TrkC TK−) by using the all-trkC probe. The endogenous trkC transcripts are also detected (Wt TrkC). Note that the time of death correlated with particular levels of expression of the transgene. In all experiments, a GAPDH antisense probe was included in the hybridization mixture to control for RNA loading.
Expression analysis of transgenic animals indicated that overexpression of this gene, even at high levels, did not impair embryo viability, because embryos could develop to term. However, transgenic mice fail to thrive when compared with their wild-type littermates, and several newborn pups died immediately after birth (Fig.2). Tissue analysis from mice that died immediately after birth revealed that they had the highest copy number of transgenic DNA (data not shown) and that they had the highest levels of the Tg trkC TK− transcript when compared with other transgenic animals that could live up to 9 d [postnatal day 9 (P9); Fig.1C]. Although we do not know the cause of death of these mice, their dusky appearance and immediate death suggested cardiac deficits as a potential cause. Histological evaluation of trkC TK− transgenic mouse hearts revealed abnormalities in the valvular architecture and cardiac septation. Some of the defects, including abnormally thickened aortic and semilunar valves and atrial septal defects, are shown in Figure 3. These findings strongly suggest that cardiac dysfunction similar to that observed in the neurotrophin-3 (NT-3)- or trkC-deficient animals could play a role in the perinatal lethality associated with overexpression of the trkC truncated receptor (Donovan et al., 1996; Tessarollo et al., 1997).
Fig. 2.
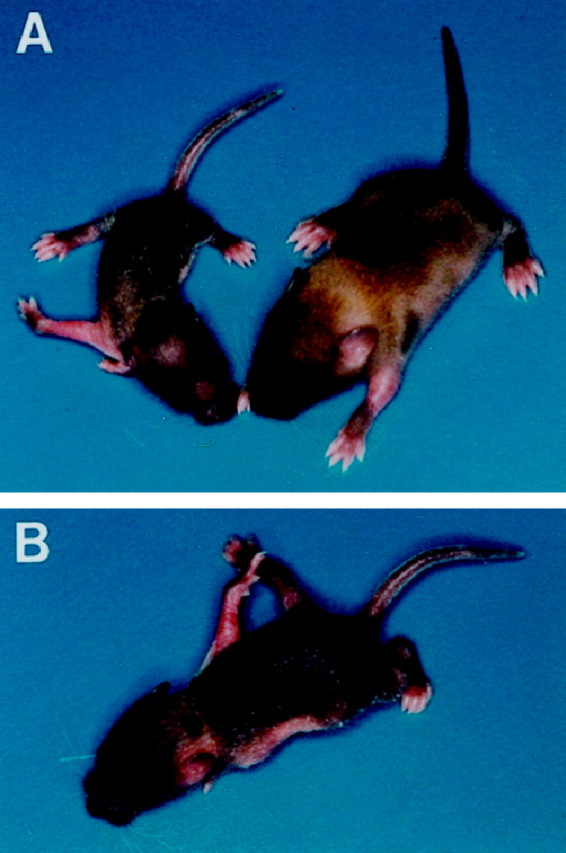
Mice overexpressing a truncated trkC receptor fail to thrive and display a behavioral phenotype characteristic of proprioception deficiency. A, A transgenic animal (left) is compared with a wild-type littermate (right). B, Transgenic mice display severe movement impairments.
Fig. 3.
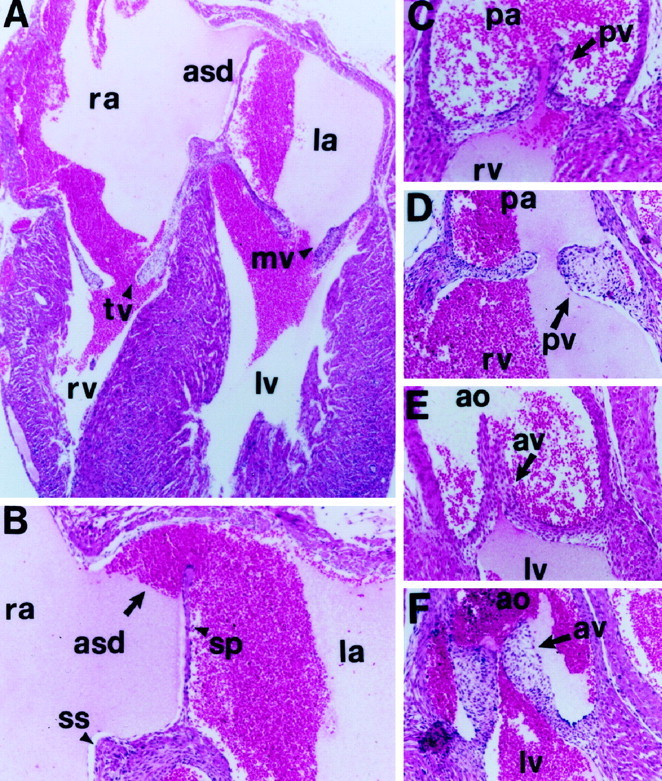
Cardiac defects in truncated trkC transgenic animals. A, Section of a newborn trkC TK− transgenic heart demonstrating an atrial septal defect (asd), markedly dilated atria with decreased trabeculations, and large septum primum. Magnification, 50×. B, Higher magnification ofA with asd, septum primum (sp), and septum secundum (ss) indicated by arrows. Magnification, 100×. C–F, Sections through the pulmonic (C, D) and aortic (E, F) valves of a normal (C, E) and a transgenic (D, F) mouse heart. Magnification, 100×. Note the thickening of the pulmonic (pv) and aortic (av) valve leaflets (arrows) of the mutant animal compared with the control littermate. rv, Right ventricle; lv, left ventricle; ra, right atrium; la, left atrium; pa, pulmonic artery; ao, aorta; tv, tricuspid valve; mv, mitral valve.
Peripheral nervous system deficiencies in mice expressing high levels of trkC truncated receptor
Newborn transgenic mice exhibited deficiencies in their movements and postures that resembled the proprioception defects observed in trkC and NT-3 mutant animals (Fig. 2). To address whether these animals had PNS defects, we evaluated selected sensory and sympathetic neuronal populations of mice with the most severe phenotype (moribund at P0) compared with wild-type littermates. Severe reductions were observed in the trigeminal, geniculate, vestibular, and cochlear ganglia, whereas the petrosal–nodose ganglia displayed only a modest reduction. Also the superior cervical ganglion showed substantial neuronal losses when compared with controls. Thus, neuronal deficits in the transgenic animals are present in both sensory and sympathetic neuronal populations similar to the NT-3 mutant mouse (Table1). The development of an NT-3-like phenotype suggests that truncated receptors act by sequestering soluble neurotrophins. If truncated trkC receptor would act exclusively as dominant negative inhibitors of the full-length kinase active isoforms, the phenotype should be more similar to trkC-deficient mice. However, in contrast to mice lacking either the kinase active or all trkC receptors, the trkC TK-overexpressing mice develop severe sympathetic neuronal losses like the NT-3 knock-out mice (Table 1) (Tessarollo et al., 1997).
Table 1.
Neuronal cell counts in sensory and sympathetic populations of wild-type and Tg trkC TK− mice at P0
| Wild type (3) | Tg trkC TK− (3) | Reduction (%) Tg trkC TK− | Reduction (%) NT-3−/−1-a | Reduction (%) trkC−/−1-a | |
|---|---|---|---|---|---|
| Sensory ganglia | |||||
| Trigeminal | 62,242 ± 2652 | 38,098 ± 1193 | 39* | 62 | 21 |
| Geniculate | 1916 ± 90 | 1370 ± 139 | 29** | 35 | 11 |
| Vestibular | 5844 ± 210 | 4272 ± 341 | 27** | 19 | 15 |
| Cochlear | 10,974 ± 198 | 3324 ± 672 | 70* | 87 | 70 |
| Petrosal–nodose | 11,364 ± 606 | 10,356 ± 1338 | 9 | 34 | 14 |
| L5 dorsal root | 10,140 ± 887 | 4776 ± 356 | 53* | 69 | 33 |
| Sympathetic ganglion | |||||
| Superior cervical | 27,838 ± 1302 | 18,460 ± 1116 | 34* | 44 | 0 |
Reduction in neuronal cell counts of transgenic mice relative to wild-type mice were compared with those of trkC−/− and NT-3−/− mutant mice (Tessarollo et al., 1997).
Tissue sections of newborn (P0) wild-type and mutant littermates were prepared as described in Materials and Methods. Neurons identified by a clear nucleus and nucleolus were counted in every sixth section, and the sum of counts was multiplied by 6. Values are not corrected and are expressed as mean number of neurons ± SEM. Differences were evaluated using a one-tailed Student’s test. In parenthesis, the numbers of animals analyzed are indicated.
From Tessarollo et al. (1997).
*p < 0.005.
**p < 0.02.
PNS deficiencies arise before neuronal target innervation
We then asked at what stage of development the PNS of transgenic mice expressing high levels of the truncated trkC gene exhibited cell losses. Whole-mount analysis using an antibody that detects neurofilament 160 protein suggested normal development of the PNS in embryonic day 11 (E11) transgenic embryos when compared with wild-type littermates (Fig.4A,B). However, by E13, reductions in the size of dorsal root ganglia (DRG) could be observed (Fig. 4C,D). These results suggest that the deficits observed in newborn animals develop between E11 and E13 before neuronal target innervation occurs. Interestingly, NT-3 mutant mice also display reductions in trigeminal and DRG neurons at this time of development (Tessarollo et al., 1994; Fariñas et al., 1996; Wilkinson et al., 1996). Therefore, the onset of neuronal deficits in the DRG of our transgenic mice is comparable with that observed in NT-3 null mutant mice.
Fig. 4.

Neuronal losses in mice overexpressing a truncated trkC receptor occur between embryonic days 11 and 13. The PNS of control (A) and transgenic (B) mice at day E11 of embryonic development was visualized by immunohistochemistry using an anti-neurofilament 160 antibody. No significant differences were detected between the PNS of the two embryos at this developmental stage. By E13, transgenic (D) dorsal root ganglia (drg, arrow) are reduced in size when compared with wild-type drg (C, arrow) in sections at the same thoracic level. Cranial ganglia are indicated: TG, trigeminus with ophtalmic (op), maxillary (mx), and mandibular (md) branches;g, geniculate; V, vestibulocochlear;p, petrosal; n, nodose; O, optic nerve; SC, spinal cord; lu, lung.
PNS in mice expressing low levels of the trkC truncated receptor
Truncated trkC receptor expressed at high levels may act by neutralizing neurotrophins, as suggested by the fact that these transgenic mice develop a phenotype similar to that caused by NT-3 deficiency. As a result, this may mask the potential effects of a signaling function by the overexpressed truncated receptor in responsive cells. Thus, we analyzed mice with lower levels of the transgene. Such animals displayed a less-severe behavioral phenotype and prolonged viability (up to 9 d). Histopathological analysis of a variety of organs from mice that died at P9 showed no obvious abnormalities (data not shown). Neuronal counts of four sensory ganglia from this group of animals revealed reductions in the geniculate and the cochlear ganglia, although less severe than in mice expressing higher levels of the transgene. However, neuronal losses were higher in the petrosal–nodose ganglion. Surprisingly, we even observed a mild increase in the vestibular neuronal population (Table2). To investigate the specificity of this effect, we analyzed additional transgenic litters. Mice that died at P0–P1 expressed high levels of the truncated trkC transgene as reported above. However, by P3 we were able to identify animals that displayed weight losses but only mild behavioral phenotypes. To avoid losing these mice, we killed them for analysis of their peripheral nervous system at P3 (Table 3). We again observed neuronal losses in almost all investigated ganglia compared with controls. However, they were less severe than in the transgenic pups analyzed at P9. Most importantly, we observed again an increase in neuronal counts of the vestibular ganglia. This increase is not statistically significant; however, the reproducibility of this result suggests that this trkC truncated effect on the vestibular ganglia may be biologically significant.
Table 2.
Neuronal cell counts in sensory populations of wild-type and transgenic Tg trkC TK− mice at postnatal day 9
| Sensory ganglia | Wild type (3) | Tg trkC TK− (3) | Reduction (%) |
|---|---|---|---|
| Geniculate | 1194 ± 72 | 816 ± 72 | 30* |
| Vestibular | 3246 ± 366 | 3444 ± 410 | +6 |
| Cochlear | 8424 ± 1,220 | 3696 ± 428 | 56* |
| Petrosal–nodose | 8184 ± 816 | 5262 ± 452 | 34* |
Tissue sections from P9 wild-type and mutant littermates were prepared as described in Materials and Methods. Neurons having a clear nucleus and nucleolus were counted in every sixth section, and the sum of counts multiply by 6. Values are not corrected and are expressed as mean number of neurons ± SEM. In parenthesis, the numbers of animals analyzed are indicated. Differences were evaluated using two-tailed Student’s test.
*p < 0.05.
Table 3.
Neuronal cell counts in sensory populations of wild-type and transgenic Tg trkC TK− mice at postnatal day 3
| Sensory ganglia | Wild type (4) | Tg trkC TK− (4) | Reduction (%) |
|---|---|---|---|
| Trigeminal | 40,584 ± 2626 | 31,290 ± 3388 | 23* |
| Geniculate | 1158 ± 156 | 996 ± 84 | 14 |
| Vestibular | 3852 ± 396 | 4218 ± 380 | +10 |
| Cochlear | 8976 ± 958 | 5112 ± 1044 | 43** |
| Petrosal–nodose | 7704 ± 806 | 6696 ± 946 | 13 |
| DRG C1 | 3594 ± 168 | 1668 ± 316 | 53* |
Tissue sections from P3 wild-type and mutant littermates were prepared as described in Materials and Methods. Neurons having a clear nucleus and nucleolus were counted in every sixth section, and the sum of counts multiply by 6. Values are not corrected and are expressed as mean number of neurons ± SEM. In parenthesis, the numbers of animals analyzed are indicated. Differences were evaluated using two-tailed Student’s test.
*P < 0.02.
**P < 0.05.
DISCUSSION
Ectopic expression of truncated trkC receptors interferes with normal mouse development
The function of truncated trk neurotrophin receptor isoforms (lacking a functional cytoplasmic kinase domain) is still unknown. Expression of truncated trkC receptors in cell lines, e.g., of the sympathoadrenal (PC12) and mesoderm (NIH3T3) lineages, failed to induce any apparent change in the cell cycle or cellular morphology (Tsoulfas, personal communication). This result suggested that either these cells are lacking the signal transduction machinery downstream of this receptor, or the mechanism of action cannot be revealed because of the limitations of these in vitro systems. One goal of this study was to identify cell types responsive to truncated receptor functions in vivo. The truncated trkC receptor isoform that we used to generate transgenic mice is widely expressed in normal mouse development (Tsoulfas et al., 1993; Valenzuela et al., 1993; Hiltunen et al., 1996; Menn et al., 1998). In particular, it has been found throughout the CNS and PNS, including the DRGs and cranial ganglia (Menn et al., 1998). Furthermore, its expression has also been detected in several organs outside the nervous system, including the cardiovascular system and mesodermal derivatives such as muscles (Tsoulfas et al., 1993; Hiltunen et al., 1996; Menn et al., 1998) (Fig.1A). Here we have shown that mice overexpressing this truncated trkC receptor at high levels die perinatally because of severe cardiac defects and PNS abnormalities. All the tissues for which we report defects normally do express endogenous truncated trkC receptors, suggesting specific functions for these receptors in the development of those organs (Hiltunen et al., 1996; Menn et al., 1998). Despite the ubiquitous expression of trkC TK− in the transgenic animals, histological analysis to date has failed to detect abnormalities in other organs (data not shown), suggesting that truncated receptors do not affect cell survival, proliferation, or differentiation per se.
NT-3-deficient mice and trkC TK− transgenic mice develop similar PNS and cardiac deficiencies
Expression analysis in rodents and chicken during development has shown the presence of truncated trkB and trkC receptors in non-neuronal tissues, and it has been suggested that these receptors act by limiting neurotrophic factors available to bind kinase active receptors (Klein et al., 1990; Tsoulfas et al., 1993; Valenzuela et al., 1993; Garner and Large, 1994; Biffo et al., 1995). In fact, truncated trkB receptors in the ventricular zone of the spinal cord and the ependimal layer of ventricles in the brain provide a barrier to the diffusion of intraventricular injections of brain-derived neurotrophic factor, consistent with the idea that this type of receptor could remove neurotrophins (Yan et al., 1994; Armanini et al., 1995; Biffo et al., 1995). However, no data are available supporting such function during development. Here, we show that ubiquitous high-level expression of a truncated trkC isoform in mouse causes cardiac defects that resemble those of mice lacking the NT-3 or trkC gene (Donovan et al., 1996;Tessarollo et al., 1997). However, NT-3-deficient mice develop more severe neuronal losses than trkC-deficient mice (Tessarollo et al., 1997; Table 1). Overexpression of the trkC TK− transgene in the mouse results in PNS neuronal losses that most resemble the NT-3 null phenotype (Table 1). Furthermore, the establishment of neuronal deficits occurs between E11.5 and E13.5, as reported for the NT-3 mutant mice (Fariñas et al., 1996; Wilkinson et al., 1996). These results support the hypothesis that truncated receptors function in neutralizing NT-3. However, we cannot rule out that the truncated receptor also inhibits the full-length tyrosine kinase receptor directly by acting as a dominant negative receptor (Eide et al., 1996;Ninkina et al., 1996). In fact, the observed co-expression of truncated and full-length trkC receptors in developing peripheral sensory neurons suggests such a role (Menn et al., 1998). However, if this putative dominant negative function was the only mechanism of action by truncated isoforms, the phenotype of the transgenic mice would never be more severe than the one seen in trkC-deficient mice, which is not the case. Additionally, truncated trkC receptors could function as dominant negative inhibitors of trkA or trkB receptors. However, heterodimerization of different trk receptors has not been demonstratedin vivo, and none of the defects observed in the trkC TK− transgenic animal is characteristic of trkA or trkB null mutants (Klein et al., 1993; Smeyne et al., 1994). For example, the proprioception deficits observed in the transgenic mice (Fig. 2) suggest that NT-3–trkC is the main ligand–receptor system affected in the trkC TK− overexpressing mice (Tessarollo, 1998). Furthermore, trkA and trkB full-length receptors are not expressed in the heart during development (Hiltunen et al., 1996). Thus, only inhibition of the full-length trkC receptors by truncated isoforms could account for the defects observed in the transgenic animals.
Modulatory effects of trkC TK− isoforms on PNS development
A noncatalytic receptor that controls NT-3 access to specific neuronal targets and that regulates the level of neurotrophins interacting with kinase active trk receptors can provide the organism with additional ways to increase functional diversity. Such a mechanism is even more relevant for a neurotrophin such as NT-3, which may have the ability to activate all trk family members and which is widely distributed during development (Bothwell, 1995; Lewin and Barde, 1996;Tessarollo, 1998). However, it seems unlikely that an organism would need different types of truncated receptors just to regulate neurotrophin levels during ontogenesis. The high degree of conservation of intracellular domains of truncated receptors among species suggests other functions, such as signaling, for these receptors (Klein et al., 1990; Middlemas et al., 1991; Valenzuela et al., 1993; Garner and Large, 1994).
Indeed recently, Hapner et al. (1998) have shown that ectopic overexpression of truncated trkC receptors in neural crest cellsin vitro, although lacking any mitogenic effect, induces neural differentiation with participation of p75 LANR. This differentiation response is elicited only by a small subset of cells and is induced exclusively by truncated trkC receptors, which contain the evolutionarily conserved 39 amino acid intracellular domain, suggesting that specific intracellular domains of truncated receptors are required for activation of the differentiation pathway (Hapner et al., 1998). Furthermore, it has been reported that truncated trkB receptors can induce a ligand-mediated increase in the rate of acidic metabolites released from the cells, a common physiological consequence of many signaling pathways. Again, a conserved, truncated, isoform-specific domain is required to induce this response (Baxter et al., 1997). These data, although not providing information on the type of pathway activated by truncated receptors, do represent very intriguing results and suggest that both trkB and trkC truncated receptors activate intracellular signaling pathways.
The variations observed in the PNS neuronal populations of trkC TK− transgenic mice, including a tendency to an increase in the number of neurons in the vestibular ganglia, are reciprocally related to the level of expression of the transgene, suggesting a modulatory role of truncated trkC on ganglia of the PNS (Tables 2, 3). The data are also consistent with the finding that mice lacking the truncated trkC isoforms, in addition to the full-length receptors, are missing 15% more DRG and 20% more cochlear ganglia neurons compared with mice lacking only the tyrosine kinase isoforms (Minichiello et al., 1995;Tessarollo et al., 1997). The observation that changes in neuron numbers are limited in both transgenic (only a 10% increase in neurons in the vestibular ganglia) and knock-out animal models [20% fewer cochlear and DRG neurons in mice lacking both kinase and truncated trkC receptors compared with the ones lacking only the kinase isoforms (Minichiello et al., 1995; Tessarollo et al., 1997)] indicates that only a subset of neurons is responsive to the presence of truncated receptors. Indeed, the expression of endogenous truncated trkC receptors is restricted to a subset of DRG neurons at some stages of embryonic development. The identity and functional role of these neurons remain to be determined (Menn et al., 1998).
Different mechanisms, such as the ability of truncated receptors to remove ligand, inhibit the kinase active isoforms, or promote survival and/or differentiation, may not be mutually exclusive. The cellular environment may determine the mechanisms by which truncated receptors will exert their functions. Mice with conditional targeted alleles for truncated trkC receptor mutations may serve as tools to address specific signaling functions in vivo. Furthermore, such mouse strains would enable the investigation of a role of truncated receptors in the maturation process of the nervous system, an analysis that is now precluded by the limited life span of mice in which NT-3 and trkC receptor levels have been altered during development.
Footnotes
This work was supported by the National Cancer Institute under contract with Advanced Bioscience Laboratories. We thank Esta Sterneck, Eileen Southon, and Barbara Hempstead for critical reading of this manuscript; Frances Lefcort and Pantelis Tsoulfas for sharing unpublished results and for useful discussions; Debbie Swing, Susan Reid, and Janet Blair-Flynn for excellent technical assistance; and Carmen Birchmeier and Alistair Garratt for the whole-mount immunohistochemistry protocol.
Correspondence should be addressed to Dr. Lino Tessarollo, ABL-Basic Research Program, National Cancer Institute-Frederick Cancer Research and Development Center, P.O. Box B, Frederick, MD 21702.
REFERENCES
- 1.Armanini MP, McMahon SB, Sutherland J, Shelton DL, Phillips HS. Truncated and catalytic isoforms of trkB are co-expressed in neurons of rat and mouse CNS. Eur J Neurosci. 1995;7:1403–1409. doi: 10.1111/j.1460-9568.1995.tb01132.x. [DOI] [PubMed] [Google Scholar]
- 2.Baxter GT, Radeke MJ, Kuo RC, Makrides V, Hinkle B, Hoang R, Medina-Selby A, Coit D, Valenzuela P, Feinstein SC. Signal transduction mediated by the truncated trkB receptor isoforms, trkB.T1 and trkB.T2. J Neurosci. 1997;17:2683–2690. doi: 10.1523/JNEUROSCI.17-08-02683.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biffo S, Offenhauser N, Carter BD, Barde YA. Selective binding and internalisation by truncated receptors restrict the availability of BDNF during development. Development. 1995;121:2461–2470. doi: 10.1242/dev.121.8.2461. [DOI] [PubMed] [Google Scholar]
- 4.Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Ann Rev Neurosci. 1995;18:223–253. doi: 10.1146/annurev.ne.18.030195.001255. [DOI] [PubMed] [Google Scholar]
- 5.Chen KS, Nishimura MC, Armanini MP, Crowley C, Spencer SD, Phillips HS. Disruption of a single allele of the nerve growth factor gene results in atrophy of basal forebrain cholinergic neurons and memory deficits. J Neurosci. 1997;17:7288–7296. doi: 10.1523/JNEUROSCI.17-19-07288.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donovan MJ, Hahn R, Tessarollo L, Hempstead BL. Identification of an essential nonneuronal function of neurotrophin-3 in mammalian cardiac development. Nat Genet. 1996;14:210–213. doi: 10.1038/ng1096-210. [DOI] [PubMed] [Google Scholar]
- 7.Eide FF, Vining ER, Eide BL, Zang K, Wang X-Y, Reichardt LF. Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J Neurosci. 1996;16:3123–3129. doi: 10.1523/JNEUROSCI.16-10-03123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Erickson JT, Conover JC, Borday V, Champagnat J, Barbacid M, Yancopoulos G, Katz DM. Mice lacking brain-derived neurotrophic factor exhibit visceral sensory neuron losses distinct from mice lacking NT4 and display a severe development deficit in control of breathing. J Neurosci. 1996;16:5361–5371. doi: 10.1523/JNEUROSCI.16-17-05361.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ernfors P, Lee K-F, Kucera J, Jaenisch R. Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell. 1994;77:503–512. doi: 10.1016/0092-8674(94)90213-5. [DOI] [PubMed] [Google Scholar]
- 10.Fariñas I, Yoshida CK, Backus C, Reichardt LF. Lack of neurotrophin-3 results in death of spinal sensory neurons and premature differentiation of their precursors. Neuron. 1996;17:1065–1078. doi: 10.1016/s0896-6273(00)80240-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garner AS, Large TH. Isoforms of the avian TrkC receptor: a novel kinase insertion dissociates transformation and process outgrowth from survival. Neuron. 1994;13:457–472. doi: 10.1016/0896-6273(94)90360-3. [DOI] [PubMed] [Google Scholar]
- 12.Gunning P, Leavitt J, Muscat G, Ng SY, Kedes L. A human beta-actin expression vector system directs high-level accumulation of antisense transcripts. Proc Natl Acad Sci USA. 1987;84:4831–4835. doi: 10.1073/pnas.84.14.4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hapner SJ, Boeshore K, Large TH, Lefcort L. Neural differentiation promoted by truncated trkC receptors in collaboration with p75NTR. Dev Biol. 1998;201:90–100. doi: 10.1006/dbio.1998.8970. [DOI] [PubMed] [Google Scholar]
- 14.Hiltunen JO, Arumae U, Moshnyakov M, Saarma M. Expression of mRNAs for neurotrophins and their receptors in developing rat heart. Circ Res. 1996;79:930–939. doi: 10.1161/01.res.79.5.930. [DOI] [PubMed] [Google Scholar]
- 15.Klein R, Conway D, Parada LF, Barbacid M. The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell. 1990;61:647–656. doi: 10.1016/0092-8674(90)90476-u. [DOI] [PubMed] [Google Scholar]
- 16.Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75:113–122. [PubMed] [Google Scholar]
- 17.Klein R, Silos-Santiago I, Smeyne RJ, Lira SA, Brambilla R, Bryant S, Zhang L, Snider WD, Barbacid M. Disruption of the neurotrophin-3 receptor gene trkC eliminates Ia muscle afferents and results in abnormal movements. Nature. 1994;368:249–251. doi: 10.1038/368249a0. [DOI] [PubMed] [Google Scholar]
- 18.Korsching S. The neurotrophic factor concept: a reexamination. J Neurosci. 1993;13:2739–2748. doi: 10.1523/JNEUROSCI.13-07-02739.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamballe F, Smeyne RJ, Barbacid M. Developmental expression of trkC, the neurotrophin-3 receptor, in the mammalian nervous system. J Neurosci. 1994;14:14–28. doi: 10.1523/JNEUROSCI.14-01-00014.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewin GR, Barde Y-A. Physiology of neurotrophins. Annu Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- 22.Menn B, Timsit S, Calothy G, Lamballe F. Differential expression of TrkC catalytic and noncatalytic isoforms suggests that they act independently or in association. J Comp Neurol. 1998;401:47–64. doi: 10.1002/(sici)1096-9861(19981109)401:1<47::aid-cne4>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 23.Middlemas DS, Lindberg RA, Hunter T. trkB, a neural receptor protein-tyrosine kinase: evidence for a full-length and two truncated receptors. Mol Cell Biol. 1991;11:143–153. doi: 10.1128/mcb.11.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minichiello L, Piehl F, Vasquez E, Schimmang T, Hökfelt T, Represa J, Klein R. Differential effects of combined trk receptor mutations on dorsal root ganglion and inner ear sensory neurons. Development. 1995;121:4067–4075. doi: 10.1242/dev.121.12.4067. [DOI] [PubMed] [Google Scholar]
- 25.Ninkina N, Adu J, Fischer A, Pinon L, Buchman V, Davies A. Expression and function of TrkB variants in developing sensory neurons. EMBO J. 1996;15:6385–6393. [PMC free article] [PubMed] [Google Scholar]
- 26.Osborn L, Rosenberg M, Keller S, Meisler M. Tissue-specific and insulin-dependent expression of a pancreatic amylase gene in transgenic mice. Mol Cell Biol. 1987;7:326–334. doi: 10.1128/mcb.7.1.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Segal R, Greenberg M. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 28.Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature. 1994;368:246–248. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- 29.Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 30.Tessarollo L. Pleiotropic functions of neurotrophins in development. Cytokine Growth Factor Rev. 1998;9:125–137. doi: 10.1016/s1359-6101(98)00003-3. [DOI] [PubMed] [Google Scholar]
- 31.Tessarollo L, Nagarajan L, Parada LF. c-ros: the vertebrate homolog of the sevenless tyrosine kinase receptor is tightly regulated during organogenesis in mouse embryonic development. Development. 1992;115:11–20. doi: 10.1242/dev.115.1.11. [DOI] [PubMed] [Google Scholar]
- 32.Tessarollo L, Tsoulfas P, Martin-Zanca D, Gilbert DJ, Jenkins NA, Copeland N, Parada LF. trkC, a receptor for neurotrophin-3, is widely expressed in the developing nervous system and in non-neuronal tissues. Development. 1993;118:463–475. doi: 10.1242/dev.118.2.463. [DOI] [PubMed] [Google Scholar]
- 33.Tessarollo L, Vogel KS, Palko ME, Reid SW, Parada LF. Targeted mutation in the neurotrophin-3 gene results in loss of muscle sensory neurons. Proc Natl Acad Sci USA. 1994;91:11844–11848. doi: 10.1073/pnas.91.25.11844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tessarollo L, Tsoulfas P, Donovan MJ, Palko ME, Blair-Flynn J, Hempstead BL, Parada LF. Targeted deletion of all isoforms of the trkC gene suggests the use of alternate receptors by its ligand neurotrophin-3 in neuronal development and implicates trkC in normal cardiogenesis. Proc Natl Acad Sci USA. 1997;94:14776–14781. doi: 10.1073/pnas.94.26.14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsoulfas P, Soppet D, Escandon E, Tessarollo L, Mendoza-Ramirez J-L, Rosenthal A, Nikolics K, Parada LF. The rat trkC locus encodes multiple neurogenic receptors that exhibit differential response to neurotrophin-3 in PC12 cells. Neuron. 1993;10:975–990. doi: 10.1016/0896-6273(93)90212-a. [DOI] [PubMed] [Google Scholar]
- 36.Valenzuela DM, Maisonpierre PC, Glass DJ, Rojas E, Nuñez L, Kong Y, Gies DR, Stitt TN, Ip NY, Yancopoulos GD. Alternative forms of rat TrkC with different functional capabilities. Neuron. 1993;10:963–974. doi: 10.1016/0896-6273(93)90211-9. [DOI] [PubMed] [Google Scholar]
- 37.Wilkinson GA, Fariñas I, Backus C, Yoshida CK, Reichardt LF. Neurotrophin-3 is a survival factor in vivo for early mouse trigeminal neurons. J Neurosci. 1996;16:7661–7669. doi: 10.1523/JNEUROSCI.16-23-07661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan Q, Matheson C, Sun J, Radeke MJ, Feinstein SC, Miller JA. Distribution of intracerebral ventricularly administered neurotrophins in rat brain and its correlation with trk receptor expression. Exp Neurol. 1994;127:23–36. doi: 10.1006/exnr.1994.1076. [DOI] [PubMed] [Google Scholar]