

Abstract
Activation of the arachidonic acid cascade is an essential step for the development of fever during brain inflammation. We investigated the brain sites where this activation takes place by use of a rat model of brain inflammation. Intracerebroventricular administration of lipopolysaccharide but not of its vehicle evoked fever. The fever was markedly suppressed when the rats had been pretreated with a cyclooxygenase-2-specific inhibitor. In situhybridization and immunohistochemical studies revealed that cyclooxygenase-2 mRNA and its protein were induced by lipopolysaccharide in blood vessels near the cerebral ventricles and in those in the subarachnoidal space. Double immunohistochemical staining revealed that these cyclooxygenase-2-positive cells were mostly endothelial cells. The time course of fever and that of cyclooxygenase-2 induction in the endothelial cells were in parallel. Cyclooxygenase-2 mRNA in a certain type of telencephalic neurons was also upregulated by the intracerebroventricular administration, but this neuronal response occurred both in vehicle-injected rats and in lipopolysaccharide-injected ones to the same extent. Therefore, the neuronal response was not essential to the development of fever. These results suggest that brain endothelial cells play a crucial role in the development of fever during brain inflammation by activating their arachidonic acid cascade.
Keywords: brain inflammation, fever, meningitis, cyclooxygenase, prostaglandin, endothelial cells
Fever is a common symptom associated with various types of cerebral inflammation, including meningitis and encephalitis. Under such inflammatory conditions, one essential process leading the patients to fever is the activation of the arachidonic acid cascade in the brain (for review, see Milton, 1982), in which membrane phospholipid is enzymatically converted to several kinds of bioactive prostaglandins (PGs). Among the PGs, PGE2 is most likely to be the mediator of fever because it is highly pyrogenic when injected into the cerebral ventricle or into the preoptic area, the thermoregulatory center and a presumed locus of PGE2 action (Milton, 1982; Stitt, 1986). The PGE2 level in the CSF indeed increases during fever (Bernheim et al., 1980;Coceani et al., 1983). Little is known, however, where in the brain such activation of the arachidonic acid cascade takes place during the cerebral inflammation. Although there are several cell groups potentially to be the source of PGs, including neurons (Fujimoto et al., 1992; Van Dam et al., 1993), astrocytes (Katsuura et al., 1989;O’Banion et al., 1996), microglia (Matsuo et al., 1995; Minghetti and Levi, 1995; Fiebich et al., 1996; Bauer et al., 1997; Slepko et al., 1997), and endothelial cells (Moore et al., 1988; Van Dam et al., 1993;de Vries et al., 1995), it has not been clarified yet which of these cell groups plays the major role in the production of PGs during cerebral inflammation in vivo.
To answer the above question, we focused our attention on cyclooxygenase-2 in the brain. Cyclooxygenases (COXs), i.e., cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2), are rate-limiting enzymes in the biosynthesis of PGs and are the targets of the nonsteroidal anti-inflammatory drugs such as aspirin and indomethacin (Vane, 1971; Goppelt-Struebe, 1995; Herschman, 1996). COX-2 is distinct from COX-1 in that COX-2 is immediately induced by inflammatory stimuli and, therefore, is considered to be involved in the PG biosynthesis during inflammation (Goppelt-Struebe, 1995;Herschman, 1996). In the present study, we used an experimental model of fever associated with brain inflammation, in which rats received an intracerebroventricular injection of lipopolysaccharide (LPS). The pathophysiological events occurring in this experimental model are particularly relevant to those of meningitis (McAllister et al., 1975;Sanna et al., 1995). In this experimental model, we examined whether COX-2 is involved in this type of fever, and if so, where COX-2 is induced in the brain.
MATERIALS AND METHODS
Materials. Male Wistar rats of 7 weeks of age were purchased from Shizuoka Laboratory Animal Cooperative (Shizuoka, Japan). They were housed four or five to a cage in the room at 26 ± 2°C with a standard 12 hr light/dark cycle until they had surgery. Other materials used and their sources are as follows: autoradiographic14C microscales (RPA511 and RPA504); [35S]UTP and x-ray films (βmax) (Amersham, Arlington Heights, IL); emulsion for microautoradiography (NTB2) (Eastman Kodak, Rochester, NY); lipopolysaccharide of Escherichia coli O26:B6 (Sigma, St. Louis, MO); RNA transcription kit (Stratagene, La Jolla, CA); NS-398, a COX-2-specific inhibitor (a generous gift from Dr. S. Higuchi of Taisho Pharmaceutical Co. Ltd, Tokyo, Japan); rabbit polyclonal antibody against murine COX-2 (Cayman Chemical); rabbit polyclonal antibody against human von Willebrand (vW) factor (Dako, Carpinteria, CA); and mouse monoclonal antibody OX-42 against rat microglia/macrophages (Serotec, Oxford, UK).
Surgery. Under pentobarbital anesthesia, temperature transmitters (Mini-Mitter, Sunriver, OR) were implanted in the abdominal cavity of the rats. Their heads were then fixed in a stereotaxic apparatus (Narishige, Tokyo, Japan) according to a brain atlas (Paxinos and Watson, 1986). The skin of the skull was incised in the midline, and two holes (0.8 mm in diameter) for intracerebroventricular injection were drilled into the skull so that they were located above the right and left lateral ventricles (0.8 mm posterior and 1.2 mm lateral to the bregma). The incision of the skin was closed, and the rats were housed individually in cages placed on the receivers of the temperature telemetry system (Data Sciences, St. Paul, MN).
Experimental protocol. Intracerebroventricular injections were made after a recovery period of at least 1 week. Between 9:00 and 9:30 A.M. on the day of the experiment, the rats were lightly anesthetized with halothane (3% in the air) and received an intracerebroventricular injection of either LPS (50 ng/rat in 15 μl of saline) or saline alone. The injections were made through one of the holes in the skull by inserting a stainless steel needle (27 gauge) that was connected to a 1 ml disposable syringe via a polyethylene (PE) tubing (PE20, Cray Adams) of 50 cm length. The syringe, PE tubing, and the needle were filled with either saline or LPS solution. Once the needle had been inserted into the brain 4 mm from the skull, the syringe was removed from the PE tubing, and the free end of the PE tubing was lifted up so that it was located 50 cm above the rat’s head. If the tip of the needle was properly located in the lateral ventricle, the solution in the PE tubing was delivered into the ventricle by hydrostatic pressure. This was confirmed by a drop in the level of meniscus of the solution in the PE tubing. The volume of the solution injected was checked with the precalibrated scale put on the PE tubing. If the meniscus of the solution did not drop, the needle was slightly moved until the meniscus started to drop. After the injection, the rats were returned to their home cages. The whole process of this injection took ∼5 min for each rat. The other hole in the skull was left unused. Abdominal temperature (Tab) of each rat was monitored every 10 min during a 24 hr period before and after the injection.
To investigate the effect of COX-2 inhibitor on the febrile response, we injected rats intraperitoneally with either NS-398 (4 mg/kg, i.p.) or its vehicle [500 μl of 50% dimethyl sulfoxide (DMSO) in saline] 1 hr before the injection of LPS. NS-398 possesses a high selectivity for COX-2 as shown by several in vitro and in vivo studies (Futaki et al., 1993a,b, 1994; Masferrer et al., 1994; Gierse et al., 1995).
In situ hybridization and immunohistochemistry.Based on the time course of fever, two time points, i.e., 1.5 and 3.5 hr after the intracerebroventricular injections, were chosen for sacrificing the animals for in situ hybridization of COX-2 mRNA. For each time point, four or five rats were anesthetized with diethylether and perfused via the left ventricle with 50 ml of ice-cold saline followed by 200 ml of 4% paraformaldehyde solution, pH 6.5. The brains were put in a solution containing 20% sucrose and 4% paraformaldehyde overnight, frozen in dry-ice powder, and stored at −80°C until used. In situ hybridization of COX-2 was done with 35S-labeled rat COX-2 antisense riboprobe as described in our previous study (Cao et al., 1995, 1997). A sense strand35S-labeled COX-2 probe of identical length was used for negative-control experiments.
For immunohistochemical detection of COX-2 protein, four time points, i.e., 2, 4, 6, and 24 hr after the intracerebroventricular injection, were chosen for killing the animals. At each time point, three rats were anesthetized with diethylether and perfused via the left ventricle with 50 ml of ice-cold 20 mm PBS, pH 7.4. Immunostainings of COX-2, vW factor (an endothelial marker), and a microglia/macrophage marker were performed as reported previously (Cao et al., 1998; Matsumura et al., 1998a).
Analysis and statistics. Relative changes in the COX-2 mRNA level in the telencephalic regions were analyzed on autoradiographic films by use of an image analysis program (NIH Image, version 1.55), as described in a previous study (Cao et al., 1997). The number of COX-2 immunoreactive (COX-2-IR) cells in the blood vessels was counted in three consecutive brain sections prepared from each rat. Coronal sections used for this evaluation corresponded to the plane of bregma around 0 mm according to a stereotaxic atlas (Paxinos and Watson, 1986). Data were expressed as mean ± SE. The Mann–WhitneyU test was used to evaluate the significance of differences.
RESULTS
Fever evoked by intracerebroventricular injection of LPS
Figure 1 shows a 14 hr recording of the change in Tab after intracerebroventricular injection of LPS or saline, as well as recordings under the untreated condition of both groups 1 d before the injection. The average Tabs at the time of injection, which was made between 9:00 and 9:30 A.M., were 37.0 ± 0.17°C for the saline-injected group (n = 4) and 36.9 ± 0.18°C for the LPS-injected one (n = 5). These values were not significantly different from their average Tab (36.9 ± 0.12°C) measured at 9:00 A.M. 1 d before the injection. The Tab of the untreated group showed a typical circadian fluctuation, being low during the day and high during the night. When the rats received the intracerebroventricular injections, Tabs of both LPS-injected and saline-injected rats initially decreased because of the light halothane anesthesia and then recovered to the preanesthesia level until 1.5 hr after the injection. After this time point, the Tab of LPS-injected rats continued to rise steeply until 4 hr after the injection and stayed at this higher level (∼1.5°C above the preinjection level) during the next 6 hr, whereas the Tab of the saline-injected rats increased slowly and reached a level only slightly higher than that under the untreated condition.
Fig. 1.

Changes in abdominal temperature (Tab) after intracerebroventricular injection of LPS (closed circle) or saline (triangle). LPS (50 ng/rat) or saline (15 μl) was injected at time point 0, which was between 9:00 and 9:30 A.M. Each value represents the mean ± SE of four (saline) or five (LPS) rats. The change in Tab was significantly different between LPS- and saline-injected rats in the time period denoted by theasterisk (p < 0.05). Their Tab 1 d before the injection was also shown as the control (open circle).
Involvement of COX-2 in the fever generation
To examine whether COX-2 is involved in fever evoked by intracerebroventricular injection of LPS, we pretreated rats with either NS-398 (4 mg/kg), a COX-2-specific inhibitor, or its vehicle (500 μl of 50% DMSO in saline) intraperitoneally 1 hr before intracerebroventricular injection of LPS. The average Tabbefore the injection was 36.6 ± 0.19°C for the NS-398-pretreated group (n = 4) and 36.9 ± 0.26°C for the vehicle-pretreated group (n = 5). In the NS398-pretreated rats, the fever was significantly suppressed as compared with that in the vehicle-pretreated rats (Fig.2). This result implies that COX-2 is involved in fever evoked by intracerebroventricular injection of LPS.
Fig. 2.

Changes in Tab of NS-398- or vehicle-pretreated rats after the intracerebroventricular injection of LPS. NS-398 (4 mg/kg; n = 4) or its vehicle (500 μl of 50% DMSO; n = 5) was injected intraperitoneally at time point 0. LPS (50 ng/rat) was injected via the intracerebroventricular route at the time point of 60 min. Each value represents the mean ± SE of four or five rats. The change in Tab was significantly different between NS-398- and vehicle-pretreated rats in the time period denoted by theasterisk (p < 0.05).
Induction of COX-2 mRNA
Figure 3 shows the distribution of COX-2 mRNA in coronal brain sections of the rats under untreated (Fig.3A1,A2), saline-injected (Fig.3B1–C2), and LPS-injected (Fig.3D1–E2) conditions. The coronal planes of Figure3, A1–E1, contained the cerebral cortex, striatum, septum, the organum vasculosum laminae terminalis, the rostral part of the preoptic area, and optic chiasma, corresponding to the stereotaxic plane of bregma 0 mm (Paxinos and Watson, 1986). Coronal planes of Figure 3, A2–E2, contained the cerebral cortex, dorsal hippocampus, amygdala, thalamus, and mediobasal hypothalamus, corresponding to the stereotaxic plane of bregma 3.6 mm. Under the untreated condition (Fig.3A1,A2), COX-2 mRNA was constitutively expressed in the neocortex (cingulate-frontal cortices, layers II-III and V), piriform cortex, hippocampus, and lateral amygdala. Consistent with previous reports (Yamagata et al., 1993; Cao et al., 1995), these COX-2 mRNA signals were from neurons as revealed by light microscopic examination of COX-2 mRNA signals (Fig.4A). When saline was injected intracerebroventricularly, the level of COX-2 mRNA increased in the neocortex and piriform cortex on the injection side (right side in each brain section) until 1.5 hr after the injection (Fig.3B1,B2). Semiquantitative image analysis of the COX-2 mRNA level in these regions revealed a threefold to fivefold increase in COX-2 mRNA from the untreated condition (Fig.5). This increase was mainly caused by an enhanced COX-2 mRNA induction in the neurons as revealed by light microscopic examination (Fig. 4B). By 3.5 hr after the saline injection, the COX-2 mRNA level had become reduced, although it was still high compared with that on the opposite side of the cortex or with that in the untreated cortex (Figs.3C1,C2, 4C, 5). On the other hand, COX-2 mRNA levels in the other side of the cerebral cortices did not differ very much from those of untreated rats.
Fig. 3.
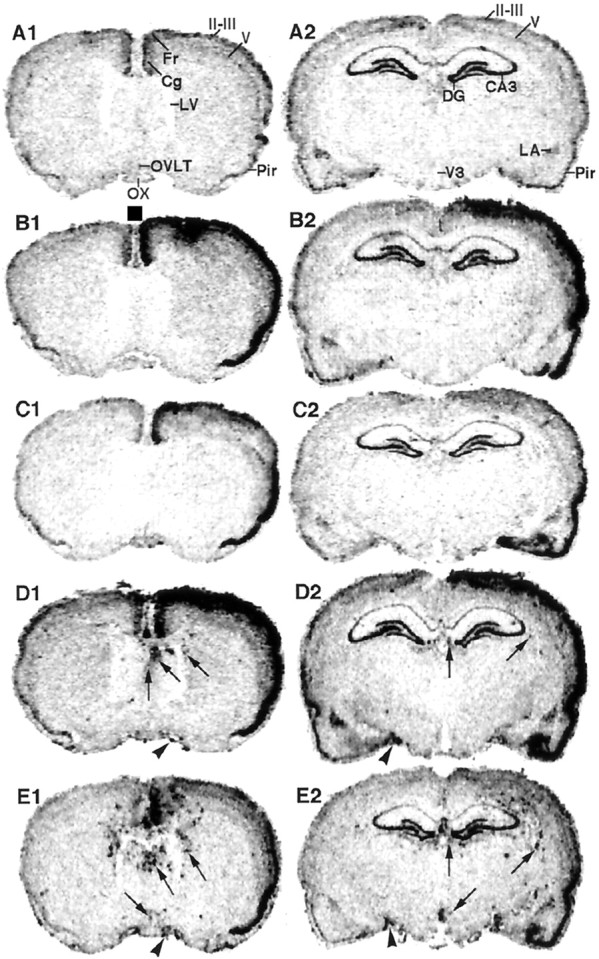
Macroautoradiographic images of COX-2 mRNA signals in coronal sections of the rat brain. InB1–E2, the sections were arranged so that the side in which intracerebroventricular injection was made was shown on the right. In the brain sections of untreated rats (A1, A2), the COX-2 mRNA signal was observed in the cingulate/frontal cortices, layer II-III and V of the neocortex, piriform cortex, lateral amygdala, and hippocampus. The mRNA signals were markedly enhanced in one side of the cerebral cortex where the injection of either saline (B1, C2) or LPS (D1, E2) had been made. Until 1.5 hr after LPS injection (D1, D2), but not saline injection, the spot-like COX-2 mRNA signals appeared in the brain parenchyma near the cerebral ventricles (arrows) and in the subarachnoidal space (arrowheads). Until 3.5 hr after LPS injection (E1, E2), the spot-like COX-2 mRNA signals markedly increased in the number and intensity. OX, Optic chiasma; Cg, cingulate cortex; Fr, frontal cortex;Pir, piriform cortex;II-III, the second and third layers of the neocortex; V, the fifth layer of the neocortex;LV, lateral ventricle; V3, third ventricle; DG, dentate gyrus; CA3, field CA3-in Ammon’s horn; LA, lateral amygdala.
Fig. 4.

Light microscopic views of localization of neuronal COX-2 mRNA signals in the untreated rats and saline- or LPS-injected ones. A–E were obtained from the layer II/III of the neocortex of injection side (except forA) in the plane of Figure2A1–E1. A, Constitutive COX-2 mRNA signals were observed in neuron-like cells (arrowheads). COX-2 mRNA signal was markedly enhanced in the neurons 1.5 hr after saline (B) or LPS (D) injection. The mRNA signals decreased until 3.5 hr after the injections (C, E). Scale bar (A–E), 20 μm.
Fig. 5.

Change in COX-2 mRNA level in the cortical areas. The optical densities of the autoradiographic films in each region were converted to the relative amount of radioactivity using a standard scale for 14C, the particles of which isotope possess almost the same energy as those of 35S, allowing us to quantitate the relative amount of 35S using the14C scale as the reference. The value in each region and time point was expressed as the ratio to that of untreated rats. The values for the neocortex (NC), including the cingulate/frontal cortex and the layers II-III of the neocortex, and piriform cortex (PC) were obtained in the plane of Figure 2A1–E1(n = 4). Significant differences from the untreated group (N) were denoted by *p < 0.05; significant differences between the injection side and the opposite side were denoted by #p < 0.05.
When LPS was injected via the intracerebroventricular route, the cortical COX-2 mRNA level was elevated in a similar manner to that observed after intracerebroventricular injection of saline in terms of its distribution, magnitude, and time course (Figs.3D1–E2, 4D,E, 5). Such elevation of cortical COX-2 mRNA is unlikely to be essential to fever because intracerebroventricular injection of saline did not result in fever. On the other hand, another type of COX-2 mRNA signal appeared only after intracerebroventricular injection of LPS. By 1.5 hr after the LPS injection, spot-like COX-2 mRNA signals appeared in the brain parenchyma near the cerebral ventricles (Fig.3D1,D2, arrows) and in the subarachnoidal space (Fig. 3D1,D2,arrowheads). This type of signal had further increased in number and intensity by 3.5 hr after the injection (Fig.3E1,E2).
Figure 6 shows light microscopic views of such spot-like COX-2 mRNA signals that were specifically induced by intracerebroventricular injection of LPS. These signals were, in most cases, associated with blood vessels near the lateral ventricle (Fig.6A–D) and the third ventricle (Fig.6E–G), and in the subarachinoidal space (Fig. 6H). The mRNA signals associated with blood vessels were more intense at 3.5 hr after the LPS injection (Fig.6D,F) than at 1.5 hr after it (Fig. 6B,E) and were very faint, if present at all, after the intracerebroventricular injection of saline (Fig. 6A,C). Sometimes, clusters of COX-2 mRNA signals were also found in sites where a vascular structure was not apparent, for example in the bottom right of Figure 6B, leaving it uncertain whether they were associated with blood vessels. The following immunohistochemical study provided more anatomical details of the COX-2-positive cells.
Fig. 6.

Light microscopic views of COX-2 mRNA signals in the blood vessels. A small amount of COX-2 mRNA (arrows) was induced in the cells of blood vessels near the lateral ventricles 1.5 (A) and 3.5 (C) hr after saline injection. Marked COX-2 mRNA signals were induced in blood vessels of the same area 1.5 (B) and 3.5 (D) hr after LSP injection. COX-2 mRNA (arrows) was also induced in blood vessels near the third ventricle 1.5 hr after LPS injection (E) and was further enhanced 3.5 hr after LPS injection (F,G). COX-2 mRNA was also induced in blood vessels of the subarachnoidal space (H).BV, Blood vessel; V3, third ventricle. Scale bars: A–D, G, 50 μm; E, F, 100 μm; H, 25 μm.
Induction of COX-2 protein
After intracerebroventricular injection of LPS, COX-2-IR cells were found in the subarachinoidal space (Fig.7A–C) and in the brain parenchyma near the cerebral ventricles (Fig.7G–I). The distribution of COX-2-positive blood vessels, but not that of individual cells, at 4 hr after LPS injection is summarized in Figure 8. In the cisterna chiasmatis, where the subarachinoidal space is enlarged lateral to the optic chiasma, COX-2-IR cells appeared by 2 hr after intracerebroventricular injection of LPS (Fig. 7A), increased in number and signal intensity until 4 hr (Fig.7B), slightly decreased until 6 hr (Fig. 7C), and almost disappeared by 24 hr after the injection (Fig. 7D). In contrast, intracerebroventricular injection of saline did not induce COX-2-IR cells in the same area until 4 hr (Fig. 7E) and 6 hr (Fig. 7F) after the injection. Essentially the same response occurred in other parts of the subarachnoidal space, although the number of blood vessels varied among them. COX-2-IR cells also appeared in the brain parenchyma near the third ventricle (Fig.7G) and the lateral ventricles (Fig.7H,I) with a time course similar to that in the subarachnoidal space. This time course was shown as the total number of COX-2-IR cells near the third ventricle and in the cisterna of the chiasma (Fig.9A) and as that near the lateral ventricles (Fig. 9B). COX-2-IR cells were most abundant at 4 hr after the LPS injection, at which time the fever was just before the plateau. Although 24 hr after LPS injection there was still a small number of COX-2-IR cells only near the lateral ventricle, the staining was faint, and there was no apparent fever at this time point. Injection of saline also induced weak COX-2-like immunoreactivity in a small number of cells only in the brain parenchyma near the lateral ventricle (Fig. 9B). The intensity of COX-2 staining in the blood vessels was generally higher in the subarachnoidal space than near the third ventricle.
Fig. 7.

COX-2-IR cells appeared in the cisterna chismatis at 2 (A), 4 (B), 6 (C), and 24 (D) hr after LPS injection. Little COX-2-IR cell was observed at 4 (E) and 6 (F) hr after saline injection. COX-2-IR cells also appeared in the blood vessels near the third ventricle (G) 4 hr after LPS injection and in those near the lateral ventricle (H,I) 4 and 6 hr after LPS injection, respectively.OX, Optic chiasma; V3, third ventricle. Scale bar, 50 μm.
Fig. 8.

Distribution of COX-2-IR blood vessels in LPS-injected rats. Each symbol represents one blood vessel associated with one to several COX-2-IR cells. Results from three rats, which were killed 4 hr after LPS injection, were plotted on the same plane. The three different symbolsrepresent the result from each rat. COX-2-positive blood vessel was mainly observed the near lateral ventricles, third ventricle, and the cisterna chismatis. The outline of the section represents the pia mater.
Fig. 9.

Time course of the number of COX-2-IR cells after LPS injection. The COX-2-IR cells were counted near the third ventricle, including the preoptic area and cisterna chismatis (A), and near lateral ventricles (B). The numbers represent the average number of cells in three consecutive brain sections.
Identification of the COX-2-IR cells
To determine the type of cells that express COX-2 after intracerebroventricular injection of LPS, we conducted double-immunostaining of COX-2 and cell-specific markers. Figure10, A–D, shows double-immunostaining of COX-2 (red) and vW factor (green) in blood vessels 4 hr after intracerebroventricular injection of LPS. In large subarachnoidal blood vessels, as shown in Figure 10A, a COX-2-IR structure (red) was surrounded by a vW factor-positive structure (green). This was also the case for small and large parenchymal blood vessels, as shown in Figure 10, B andC1, respectively. An additional staining with a nuclear-specific dye, TOTO-3, showed that the COX-2-IR structure overlapped the cellular nucleus (Fig.10C2,D2). Confocal microscopic imaging of thinner optical sections further demonstrated that the COX-2-IR structure was mainly located in the nuclear membrane but not inside the nucleus (Fig. 10E). On the other hand, vW factor is a protein specifically expressed in the endothelial cytosol (Wagner and Marder, 1983). Taken together, these results indicate that COX-2 was expressed in the nuclear membrane of the endothelial cells. Although TOTO-3 staining showed a number of cells located near the blood vessel, those positive for COX-2 were, in most cases, stained with anti-vW factor (Fig. 10C2). However, little evidence was obtained for expression of COX-2 in microglia/macrophage, as revealed by double-immunostaining with OX-42, a monoclonal antibody that recognizes rat complement receptor 3 (Fig. 10E).
Fig. 10.

Double immunostaining of COX-2 (red) and von Willebrand factor (green), an endothelial cell marker in a large subarachnoidal blood vessel (A), in a small parenchymal blood vessel (B), and in a large parenchymal blood vessel (C1, C2). An additional staining for nucleus was done with TOTO-3 (blue) in C2, and the magnified views were shown inD1 and D2. Note that COX-2-IR structure overlapped in a large part with nuclear staining resulting inpinkish color. Double immunostaining of COX-2 (red) and OX-42 (green) provided little evidence for the expression of COX-2 in microglial/macrophages (E). Scale bars: A, B, 10 μm; C1, C2, 25 μm; D1,D2, 7.5 μm; E, 15 μm.
DISCUSSION
To unveil the molecular mechanism of fever accompanying cerebral inflammation, the present study was aimed at elucidating the role of COX-2 and sites of its induction in rats that had received intracerebroventricular administration of LPS, resulting in pathophysiological events relevant to those seen in central inflammation, particularly to those of meningitis (McAllister et al., 1975; Sanna et al., 1995).
Involvement of COX-2 in fever evoked by intracerebroventricular injection of LPS
It is highly likely that the small amount of LPS (50 ng) injected via the intracerebroventricular route evoked fever through its action within the brain, because the same amount of LPS, when injected peripherally, did not evoke fever of similar magnitude (our unpublished observations). The fever induced by the intracerebroventricular injection of LPS was markedly suppressed by pretreatment with a COX-2-specific inhibitor, NS-398. These results suggest that NS-398 exerted its antipyretic action through an inhibition of COX-2 activity within the brain. This idea is also in line with previous reports that fever evoked by intracerebroventricular injection of LPS was suppressed by intracerebroventricular injection of indomethacin (Morimoto et al., 1987; Hashimoto et al., 1988), although indomethacin is a nonspecific inhibitor for both COX-1 and COX-2. We therefore further searched for the sites of COX-2 expression in the brain as the sites of activation of the arachidonic acid cascade.
Upregulation of COX-2 mRNA in neuronal cells
In situ hybridization study for COX-2 mRNA showed that, in both LPS- and saline-injected rats, COX-2 mRNA was elevated in the neurons of the cerebral cortex on the injection side. Staining of COX-2-like immunoreactivity was also enhanced on the same side of the cerebral cortex (data not shown). The extent of this response was similar in LPS- and saline-injected rats, whereas the fever was evident only in the LPS-injected ones. These results clearly indicate that COX-2 induction in the cortical neurons is not essential to the fever. This conclusion is in line with the following two previous findings of ours. In rats that had received intraperitoneal injection of LPS, the level of COX-2 mRNA in the telencephalic neurons was not correlated with the magnitude of fever (Cao et al., 1997). Furthermore, lowering the level of neuronal COX-2 mRNA by a general anesthesia did not prevent the development of fever (Cao et al., 1997).
At present, little is known about the physiological significance of COX-2 induction in the cortical neurons after the intracerebroventricular injection of either saline or LPS. As for its mechanism, we currently speculate that mechanical stimulus in the cortex evoked by insertion of the needle into the lateral ventricle triggered a response known as cortical spreading depression, in which a depolarizing wave spreads over one side of the cortex. Cortical spreading depression is reported to enhance the level of COX-2-like immunoreactivity in cortical neurons (Caggiano et al., 1996).
Induction of COX-2 in the endothelial cells
The sites where the intracerebroventricular injection of LPS, but not that of saline, specifically induced COX-2 were blood vessels in the periventricular parenchyma and those in the subarachnoidal space. As revealed by the double immunostaining of COX-2 and vW factor, the latter being a cytosolic marker protein of endothelial cells (Wagner and Marder, 1983), most of these COX-2-positive cells were endothelial cells. On the other hand, double immunostaining of COX-2 and microglial/macrophage marker provided little evidence for the expression of COX-2 in these phagocytic cells. Thus, under the present experimental conditions, the endothelial cells seem to be the major sites where activation of the arachidonic acid cascade took place in response to intracerebroventricular administration of LPS in a COX-2-dependent manner.
The above results were somewhat unexpected because a number of previousin vitro studies had demonstrated that glial cells expressed COX-2 in response to inflammatory stimuli. For example, astrocytes (Katsuura et al., 1989; O’Banion et al., 1996) and microglia (Minghetti and Levi, 1995; Bauer et al., 1997; Slepko et al., 1997) in culture upregulated COX-2 in response to LPS or to IL-1β and enhanced the release of PGE2 into the culture medium. Elmquist et al. (1997) also demonstrated that COX-2 was induced in perivascular microglia, but not in parenchymal ones, in response to intravenous injection of LPS. To our knowledge, however, COX-2 expression in the parenchymal glia has not been reported in in vivopreparations yet. These results imply that glial cells may express COX-2 under certain conditions but that, at least under the present experimental conditions, the endothelial cells are more sensitive to LPS in terms of COX-2 induction.
Significance of endothelial COX-2 in fever
The predominant expression of COX-2 in brain endothelial cells was also the case in another experimental model of fever, in which LPS was injected intraperitoneally (Cao et al., 1997; Matsumura et al., 1998a); this model was considered to represent pathophysiological events accompanying peritonitis. In both experimental models, i.e., intracerebroventricular and intraperitoneal administrations of LPS, the time courses of COX-2 expression in brain endothelial cells were in parallel with that of fever, and inhibition of COX-2 activity by its specific inhibitor suppressed the fever (Cao et al., 1997). These results strongly suggest that COX-2 in the brain endothelial cells plays an essential role in the development of fever both in peritonitis and in central inflammation.
How does the endothelial expression of COX-2 lead to the development of fever? It was reported that brain endothelial cells produce and secrete PGE2, the most pyrogenic PG, in response to LPS or inflammatory cytokines (de Vries et al., 1995). We also have an unpublished result that shows that the PGE2 level in the CSF dramatically increased when the brain endothelial cells expressed COX-2 in response to intraperitoneally administration of LPS and that this increase in PGE2 as well as fever was suppressed by the COX-2-specific inhibitor. These lines of evidence, although still fragmental, brought us to hypothesize that PGE2 is produced in the endothelial cells in a COX-2-dependent manner and is released into the brain parenchyma and/or into the CSF. The secreted PGE2 presumably then acts on the neurons in the thermoregulatory center to evoke fever.
Several lines of evidence have suggested that one of the brain loci at which PGE2 acts to evoke fever is the anteroventral part of the preoptic area. The evidence includes the following: (1) the density of PGE2-binding sites, presumably its receptors, were found to be particularly high in that region (Watanabe et al., 1988;Matsumura et al., 1990, 1998b); (2) that region was most sensitive to PGE2 in terms of fever response (Scammell et al., 1996); and (3) microinjection of COX inhibitors into that region suppressed fever that was evoked by systemic injection of LPS (Scammel et al., 1998). In line with this idea, we found a large number of COX-2-IR endothelial cells there. In particular, it should be emphasized that the endothelial cells in the subarachnoidal space just ventral to the anteroventral part of the preoptic area were higher in number and in intensity of COX-2 staining than those in the parenchymal region.Scammel et al. (1998) also demonstrated that microinjection of a COX inhibitor into the subarachnoidal space ventral to the anteroventral part of the preoptic area effectively suppressed fever. These results suggest that COX-2-positive endothelial cells in the subarachnoidal space play a significant role in the development of the fever during central inflammation.
It is well agreed that fever is a consequence of immune-brain communication. There are, however, several hypotheses as to how the immune signals are transmitted to the brain (Tilders et al., 1994;Watkins et al., 1995; Blatteis and Sehic, 1997). The present study as well as our previous ones (Cao et al., 1995, 1997; Matsumura et al., 1998a) strongly suggest that LPS signal is converted in the brain endothelial cells to a PG signal, which could directly affect the neuronal activity of the brain. Recently, another hypothesis for the mechanism of immune-brain communication was proposed by Watkins et al. (1995). Their hypothesis emphasizes the importance of vagal sensory nerves in the transmission of peripheral immune signals to the brain and, consequently, in the development of fever. Under the present experimental condition, however, the involvement of vagal sensory system is very unlikely because LPS was administrated within the brain, where no vagal sensory system exists. Further studies will be required to understand how LPS administered via the intracerebroventricular route induced COX-2 in the endothelial cells. In other words, does LPS act on the endothelial cells directly, or does it act via induction of cytokines, such as interleukin-1 and/or tumor necrosis factor? In fact, brain endothelial cells, but not microglial cells, possess interleukin-1 type 1 receptor and also express COX-2 mRNA in response to these cytokines (Cunningham et al., 1992; Cao et al., 1996,1998).
Fever is a common symptom associated with various types of inflammatory diseases, as has been known since the age of Hippocrates. The understanding of its mechanism at the molecular level was facilitated by two important findings made in the early 1970s. Milton and Wendlandt (1970) reported that PGE1 was pyrogenic when injected into the cat brain. In 1971, Vane (1971) reported that nonsteroidal anti-inflammatory drugs exert their antipyretic action through the inhibition of PG biosynthesis, i.e., inhibition of cyclooxygenase. After these findings, the mechanism and site of PGE2biosynthesis have been two of the central issues of fever research. The present study as well as our previous ones (Cao et al., 1995, 1996,1997, 1998; Matsumura et al., 1998a) have provided answers to these questions by demonstrating the seminal role of brain endothelial COX-2 in the development of fever. We believe this information to be crucial for an understanding of fever at the molecular level and to be helpful when considering the clinical management of fever.
Footnotes
This work was supported in part by Research for the Future Program JSPS-RFTF 98L00201 from the Japan Society for the Promotion of Science. The initial phase of this work was supported in part by Subfemtomole Biorecognition Project, ICORP, and Japan Science Technology Corporation. We thank Dr. Larry D. Frye for editorial help with this manuscript, and Dr. Kanato Yamagata for providing us with the rat COX-2 cDNA.
Correspondence should be addressed to Dr. Kiyoshi Matsumura, Department of Neuroscience, Osaka Bioscience Institute, 6-2-4 Furue-dai, Suita, Osaka 565, Japan.
REFERENCES
- 1.Bauer MKA, Lieb K, Schulze-Osthoff K, Berger M, Gebicke-Haerter PJ, Bauer J, Fiebich BL. Expression and regulation of cyclooxygenase-2 in rat microglia. Eur J Biochem. 1997;243:726–731. doi: 10.1111/j.1432-1033.1997.00726.x. [DOI] [PubMed] [Google Scholar]
- 2.Bernheim HA, Gilbert TM, Stitt JT. Prostaglandin E levels in third ventricular cerebrospinal fluid of rabbits during fever and changes in body temperature. J Physiol (Lond) 1980;301:69–78. doi: 10.1113/jphysiol.1980.sp013189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blatteis CM, Sehic E. Fever: how may circulating pyrogens signal the brain? News Physiol Sci. 1997;12:1–9. [Google Scholar]
- 4.Caggiano AO, Breder CD, Kraig RP. Long-term elevation of cyclooxygenase-2, but not lipoxygenase, in regions synaptically distant from spreading depression. J Comp Neurol. 1996;376:447–462. doi: 10.1002/(SICI)1096-9861(19961216)376:3<447::AID-CNE7>3.0.CO;2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao C, Matsumura K, Yamagata K, Watanabe Y. Induction by lipopolysaccharide of cyclooxygenase-2 mRNA in rat brain: its possible role in the febrile response. Brain Res. 1995;697:187–196. doi: 10.1016/0006-8993(95)00839-i. [DOI] [PubMed] [Google Scholar]
- 6.Cao C, Matsumura K, Yamagata K, Watanabe Y. Endothelial cells of the rat brain vasculature express cyclooxygenase-2 mRNA in response to systemic interleukin-1β: a possible site of prostaglandin synthesis responsible for fever. Brain Res. 1996;733:263–272. doi: 10.1016/0006-8993(96)00575-6. [DOI] [PubMed] [Google Scholar]
- 7.Cao C, Matsumura K, Yamagata K, Watanabe Y. Involvement of cyclooxygenase-2 in LPS-induced fever and regulation of its mRNA in the rat brain by LPS. Am J Physiol. 1997;272:R1712–R1725. doi: 10.1152/ajpregu.1997.272.6.R1712. [DOI] [PubMed] [Google Scholar]
- 8.Cao C, Matsumura K, Yamagata K, Watanabe Y. Cyclooxygenase-2 is induced in brain blood vessels during fever evoked by peripheral or central administration of tumor necrosis factor. Mol Brain Res. 1998;56:45–56. doi: 10.1016/s0169-328x(98)00025-4. [DOI] [PubMed] [Google Scholar]
- 9.Coceani F, Bishai I, Dinarello CA, Fitzpatrick FA. Prostaglandin E2 and thromboxane B2 in cerebrospinal fluid of the febrile and afebrile cat. Am J Physiol. 1983;244:R785–R793. doi: 10.1152/ajpregu.1983.244.6.R785. [DOI] [PubMed] [Google Scholar]
- 10.Cunningham JET, Wada E, Carter DB, Tracey DE, Battey JF, De Souza EB. In situ histochemical localization of type I interleukin-1 receptor messenger RNA in the central nervous system, pituitary, and adrenal gland of the mouse. J Neurosci. 1992;12:1101–1114. doi: 10.1523/JNEUROSCI.12-03-01101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Vries HE, Hoogendoorn KH, van Dijk J, Zijlstra FJ, van Dam A-M, Breimer DD, van Berkel TJC, de Boer AG, Kuiper J. Eicosanoid production by rat cerebral endothelial cells: stimulation by lipopolysaccharide, interleukin-1 and interleukin-6. J Neuroimmunol. 1995;59:1–8. doi: 10.1016/0165-5728(95)00009-q. [DOI] [PubMed] [Google Scholar]
- 12.Elmquist JK, Breder CD, Sherin JE, Scammell TE, Hickey WF, Dewitt D, Saper CB. Intravenous lipopolysaccharide induces cyclooxygenase 2-like immunoreactivity in rat brain perivascular microglia and meningeal macrophages. J Comp Neurol. 1997;381:119–129. doi: 10.1002/(sici)1096-9861(19970505)381:2<119::aid-cne1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 13.Fiebich BL, Biber K, Lieb K, van Calker D, Berger M, Bauer J, Gebicke-Haerter PJ. Cyclooxygenase-2 expression in rat microglia is induced by adenosine A2a-receptors. Glia. 1996;18:152–160. doi: 10.1002/(SICI)1098-1136(199610)18:2<152::AID-GLIA7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 14.Fujimoto N, Kanneko T, Eguchi N, Urade Y, Mizuno N, Hayaishi O. Biochemical and immunohistochemical demonstration of a tightly bound form of prostaglandin E2 in the rat brain. Neuroscience. 1992;49:591–606. doi: 10.1016/0306-4522(92)90229-u. [DOI] [PubMed] [Google Scholar]
- 15.Futaki N, Takahashi S, Yokoyama M, Arai I, Higuchi S, Otomo S. NS-398, a new anti-inflammatory agent, selectively inhibits prostaglandin G/H synthase/cyclooxygenase (COX-2) activity in vitro. Prostaglandins. 1994;47:55–59. doi: 10.1016/0090-6980(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 16.Futaki N, Arai I, Hamasaka Y, Takahashi S, Higuchi S, Otomo S. Selective inhibition of NS-398 on prostanoid production in inflamed tissue in rat carrageenan-air-pouch inflammation. J Pharm Pharmacol. 1993a;45:753–755. doi: 10.1111/j.2042-7158.1993.tb07103.x. [DOI] [PubMed] [Google Scholar]
- 17.Futaki N, Yoshikawa K, Hamasaka Y, Arai I, Higuchi S, Iizuka H, Otomo S. NS-389, a novel non-steroidal anti-inflammatory drug with potent analgesic and antipyretic effects, which causes minimal stomach lesions. Gen Pharmacol. 1993b;24:105–110. doi: 10.1016/0306-3623(93)90018-s. [DOI] [PubMed] [Google Scholar]
- 18.Gierse JK, Hauser SD, Creely DP, Koboldt C, Rangwala SH, Isakson PC, Seibert K. Expression and selective inhibition of the constitutive and inducible forms of human cyclo-oxygenase. Biochem J. 1995;305:479–484. doi: 10.1042/bj3050479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goppelt-Struebe M. Regulation of prostaglandin endoperoxide synthase (cyclooxygenase) isozyme expression. Prostaglandins Leukot Essent Fatty Acids. 1995;52:213–222. doi: 10.1016/0952-3278(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 20.Hashimoto M, Bando T, Iriki M, Hashimoto K. Effect of indomethacin on febrile response to recombinant human interleukin 1-a in rabbits. Am J Physiol. 1988;255:R527–R533. doi: 10.1152/ajpregu.1988.255.4.R527. [DOI] [PubMed] [Google Scholar]
- 21.Herschman HR. Prostaglandin synthase 2. Biochim Biophys Acta. 1996;1299:125–140. doi: 10.1016/0005-2760(95)00194-8. [DOI] [PubMed] [Google Scholar]
- 22.Katsuura G, Gottschall PE, Dahl RR, Arimura A. Interleukin-1 beta increases prostaglandin E2 in rat astrocyte cultures: modulatory effect of neuropeptides. Endocrinology. 1989;124:3125–3127. doi: 10.1210/endo-124-6-3125. [DOI] [PubMed] [Google Scholar]
- 23.Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci USA. 1994;91:3228–3232. doi: 10.1073/pnas.91.8.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsumura K, Watanabe Y, Onoe H, Watanabe Y, Hayaishi O. High density of prostaglandin E2 binding sites in the anterior wall of the 3rd ventricle: a possible site of its hyperthermic action. Brain Res. 1990;533:147–151. doi: 10.1016/0006-8993(90)91808-t. [DOI] [PubMed] [Google Scholar]
- 25.Matsumura K, Cao C, Ozaki M, Morii H, Nakadate K, Watanabe Y. Brain endothelial cells express cyclooxygenase-2 during lipopolysaccharide-induced fever: light and electron microscopic immunocytochemical studies. J Neurosci. 1998a;18:6279–6289. doi: 10.1523/JNEUROSCI.18-16-06279.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsumura K, Cao C, Watanabe Y, Watanabe Y. Prostaglandin system in the brain: sites of biosynthesis and sites of action under normal and hyperthermic conditions. In: Sharma HS, Westman J, editors. Brain function in hot environment, Prog Brain Res 115. Elsevier; Amsterdam: 1998b. pp. 275–295. [DOI] [PubMed] [Google Scholar]
- 27.Matsuo M, Hamasaki Y, Fujiyama F, Miyazaki S. Eicosanoids are produced by microglia, not by astrocytes, in rat glial cell cultures. Brain Res. 1995;685:201–204. doi: 10.1016/0006-8993(95)00490-h. [DOI] [PubMed] [Google Scholar]
- 28.McAllister CK, O’Donoghue JM, Beaty HN. Experimental pneumococcal meningitis. II. Characterization and quantitation of the inflammatory process. J Infect Dis. 1975;132:355–360. doi: 10.1093/infdis/132.4.355. [DOI] [PubMed] [Google Scholar]
- 29.Milton AS. Prostaglandins in fever and the mode of action of antipyretic drugs. In: Milton AS, editor. Pyretics and antipyretics. Springer; Berlin: 1982. pp. 259–267. [Google Scholar]
- 30.Milton AS, Wendlandt S. A possible role for prostaglandin E1 as a modulator for temperature regulation in the central nervous system of the cat. J Physiol (Lond) 1970;207:76–77P. [PubMed] [Google Scholar]
- 31.Minghetti L, Levi G. Induction of prostanoid biosynthesis by bacterial lipopolysaccharide and isoproterenol in rat microglial cultures. J Neurochem. 1995;65:2690–2698. doi: 10.1046/j.1471-4159.1995.65062690.x. [DOI] [PubMed] [Google Scholar]
- 32.Moore SA, Spector AA, Hart MN. Eicosanoid metabolism in cerebromicrovascular endothelium. Am J Physiol. 1988;254:C37–C44. doi: 10.1152/ajpcell.1988.254.1.C37. [DOI] [PubMed] [Google Scholar]
- 33.Morimoto A, Murakami N, Nakamori T, Watanabe T. Evidence for separate mechanisms of induction of biphasic fever inside and outside the blood-brain barrier in rabbits. J Physiol (Lond) 1987;383:629–637. doi: 10.1113/jphysiol.1987.sp016433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Banion MK, Miller JC, Chang JW, Kaplan MD, Coleman PD. Interleukin-1b induces prostaglandin G/H synthase-2 (cyclooxygenase-2) in primary murine astrocyte cultures. J Neurochem. 1996;66:2532–2540. doi: 10.1046/j.1471-4159.1996.66062532.x. [DOI] [PubMed] [Google Scholar]
- 35.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; London: 1986. [DOI] [PubMed] [Google Scholar]
- 36.Sanna PP, Weiss F, Samson ME, Bloom FE, Pich EM. Rapid induction of tumor necrosis factor a in the cerebrospinal fluid after intracerebroventricular injection of lipopolysaccharide revealed by a sensitive capture immuno-PCR assay. Proc Natl Acad Sci USA. 1995;92:272–275. doi: 10.1073/pnas.92.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scammell TE, Elmquist JK, Griffin JD, Saper CB. Ventromedial preoptic prostaglandin E2 activates fever-producing autonomic pathways. J Neurosci. 1996;16:6246–6254. doi: 10.1523/JNEUROSCI.16-19-06246.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scammel TE, Griffin JD, Elmquist JK, Saper CB. Microinjection of a cyclooxygenase inhibitor into the anteroventral preoptic region attenuates LPS fever. Am J Physiol. 1998;274:R783–R789. doi: 10.1152/ajpregu.1998.274.3.R783. [DOI] [PubMed] [Google Scholar]
- 39.Slepko N, Minghetti L, Polazzi E, Nicolini A, Levi G. Reorientation of prostanoid production accompanies “activation” of adult microglial cells in culture. J Neurosci Res. 1997;49:292–300. doi: 10.1002/(sici)1097-4547(19970801)49:3<292::aid-jnr4>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 40.Stitt JT. Prostaglandin E as the neural mediator of the febrile response. Yale J Biol Med. 1986;59:137–149. [PMC free article] [PubMed] [Google Scholar]
- 41.Tilders FJH, DeRijk RH, van Dam A-M, Vincent VAM, Schotanus K, Persoons JHA. Activation of the hypothalmus-pituitary-adrenal axis by bacterial endotoxins: routes and intermediate signals. Psychoneuroendocrinology. 1994;19:209–232. doi: 10.1016/0306-4530(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 42.Van Dam A-M, Brouns M, Man-A-Hing W, Berkenbosch F. Immunocytochemical detection of prostaglandin E2 in microvasculature and in neurons of rat brain after administration of bacterial endotoxin. Brain Res. 1993;613:331–336. doi: 10.1016/0006-8993(93)90922-a. [DOI] [PubMed] [Google Scholar]
- 43.Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- 44.Wagner DD, Marder VJ. Biosynthesis of von Willebrand protein by human endothelial cells. J Biol Chem. 1983;258:2065–2067. [PubMed] [Google Scholar]
- 45.Watanabe Y, Watanabe Y, Hayaishi O. Quantitative autoradiographic localization of prostaglandin E2 binding sites in monkey diencephalon. J Neurosci. 1988;8:2003–2010. doi: 10.1523/JNEUROSCI.08-06-02003.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watkins LR, Maier SF, Goehler LE. Cytokine-to-brain communication: a review & analysis of alternative mechanisms. Life Sci. 1995;57:1011–1026. doi: 10.1016/0024-3205(95)02047-m. [DOI] [PubMed] [Google Scholar]
- 47.Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]