

Abstract
We have investigated the regulation of calcitonin gene-related peptide (CGRP) release from trigeminal neurons by the serotonergic antimigraine drug sumatriptan. Serum levels of the neuropeptide CGRP are elevated during migraine. Treatment with the drug sumatriptan returns CGRP levels to normal coincident with the alleviation of headache. However, despite this clinical efficacy, the cellular target and mechanism of sumatriptan action are not well understood beyond the pharmacology of its recognition of the 5-HT1 class of serotonin receptors. We have used cultured trigeminal neurons to demonstrate that sumatriptan can directly repress CGRP secretion from sensory neurons. The stimulated secretion in response to depolarization or inflammatory agents was inhibited, but not the basal secretion rate. Unexpectedly, sumatriptan did not lower cAMP levels, in contrast to the classical role ascribed to the 5-HT1 receptors. Instead, activation of 5-HT1 receptors caused a slow and remarkably prolonged increase in intracellular calcium. The inhibition of CGRP secretion is attenuated by the phosphatase inhibitor okadaic acid, suggesting that sumatriptan action is mediated by calcium-recruited phosphatases. These results suggest that 5-HT1 agonists may block a deleterious feedback loop in migraine at the trigeminal neurons and provide a general mechanism by which this class of drugs can attenuate stimulated neuropeptide release.
Keywords: CGRP, serotonin receptors, trigeminal neurons, calcium, phosphatase, migraine, neuropeptide
Calcitonin gene-related peptide (CGRP) is a 37 amino acid regulatory neuropeptide derived from alternative splicing of the calcitonin/CGRP gene (Rosenfeld et al., 1983). During migraine, a painful neurological disorder afflicting 16% of the general population (Stewart et al., 1994), activation of trigeminal neurons leads to increased secretion of CGRP (Moskowitz, 1993). Together with substance P and neurokinin A, CGRP helps mediate neurogenic inflammation, a condition characterized by vasodilation, plasma protein extravasation, and mast cell degranulation (Buzzi et al., 1995). CGRP is the most potent vasodilatory neuropeptide known (McCulloch et al., 1986) and recently has been shown to cause dural mast cell degranulation (Ottosson and Edvinsson, 1997). CGRP is also believed to convey nociceptive information from the vasculature to the CNS (Van Rossum et al., 1997). On the basis of these data, CGRP is believed to play a key role in the painful phase of migraine.
This belief has been strongly supported by the clinical efficacy of the selective 5-HT1 receptor drug sumatriptan (Ferrari, 1998). Sumatriptan has been shown to decrease the elevated CGRP levels in migraine patients, coincident with relief of headache pain (Goadsby and Edvinsson, 1993). Trigeminal nerves play an important role in the regulation of cerebral blood flow during normal and disease states and are the major source of sensory and CGRP innervation to the cerebral vasculature (McCulloch et al., 1986; O’Conner and Van Der Kooy, 1988). However, because all of the previous studies have used in vivo model systems and 5-HT1 receptors are expressed by both cerebral blood vessels and trigeminal nerves (Bouchelet et al., 1996), the site of sumatriptan’s action, let alone the cellular mechanism, has remained unclear.
In this study, we have demonstrated that sumatriptan and other 5-HT1 receptor agonists can directly repress the stimulated, but not basal, release of CGRP from cultured trigeminal neurons. Somewhat surprisingly, we found that sumatriptan did not mediate a decrease in intracellular cAMP levels, a function typically associated with activation of the 5-HT1 receptors (Boess and Martin, 1994). Rather, sumatriptan treatment resulted in a slow and remarkably prolonged increase in intracellular calcium in trigeminal neurons. We also demonstrated that a phosphatase inhibitor effectively blocked the inhibitory effect of sumatriptan on stimulated CGRP release. These data are suggestive that sumatriptan mediates an increase in phosphatase activity via a calcium-dependent pathway. On the basis of our results, we have elucidated a novel mechanism by which the antimigraine drug sumatriptan may block a deleterious feedback loop in migraine and restore CGRP to normal levels.
MATERIALS AND METHODS
Cell culture. Trigeminal ganglia primary cultures were established as described previously (Durham et al., 1997). Briefly, ganglia from <1-week-old Sprague Dawley rats were dissociated with Dispase II. The cells from three to four ganglia were plated on glass coverslips coated with mouse Engelbreth-Holm-Swarm laminin or plastic tissue culture dishes coated with poly-d-lysine and laminin. Cells were incubated in L15 medium, 10% fetal bovine serum (FBS), and 10 ng/ml mouse 2.5 S nerve growth factor at 37°C in 5% CO2. Penicillin and streptomycin were added to all media. Cultures of trigeminal ganglia used for CGRP secretion and calcium studies were subcultured for 24 hr in serum-free medium (Durham et al., 1997). HeLa cells stably expressing the 5-HT1B receptor (HeLa1B) were kindly provided by Dr. Mark Hamblin (Seattle Veterans Affairs Medical Center, Seattle, WA) (Hamblin et al., 1992) and were maintained in F-12 medium supplemented with 10% FBS. CGS 12066A monomaleate (CGS) and L-694,294 were purchased from RBI (Natick, MA). Sumatriptan succinate was obtained from the University of Iowa Pharmacy, methiothepin was from RBI, and okadaic acid was from Sigma (St. Louis, MO).
Immunohistochemistry. Trigeminal ganglion cells at various times in culture were fixed and stained as described previously for neurofilament protein using anti-rat NF-M monoclonal antibodies (Boehringer Mannheim Biochemicals, Indianapolis, IN) and FITC-conjugated secondary antibodies (Sigma). Expression of CGRP in trigeminal cultures was detected using CGRP-specific polyclonal antibodies (RBI) and Cy-3-conjugated secondary antibodies (Sigma).
Calcium measurements. Intracellular calcium levels in cultured trigeminal neurons were measured essentially as described previously (Durham et al., 1997). Briefly, dissociated trigeminal ganglia grown on laminin-coated 25 mm glass coverslips were maintained in phenol- and serum-free medium 24 hr before the start of the calcium imaging procedure. Cells were incubated in DMEM (high glucose) containing 0.2% BSA and 1 μm fura-2 AM for 25–30 min at 37°C in 5% CO2. After the cells were washed twice with DMEM/BSA, they were incubated in the same buffer for 30 min before analysis. Basal calcium levels were measured for a minimum of 180 sec before addition of 5-HT1 receptor agonists or other agents. Statistical analyses were performed using the Student’s ttest (two-tailed, unpaired samples).
CGRP and cAMP assays. For the CGRP secretion studies, cells were incubated in HBS (22.5 mm HEPES, 135 mmNaCl, 3.5 mm KCl, 1 mm MgCl, 2.5 mmCaCl, 3.3 mm glucose, and 0.1% BSA, pH 7.4) (Vasko et al., 1994), and the amount of CGRP released from trigeminal neurons into the culture media was determined using a specific CGRP radioimmunoassay (Peninsula Labs, Belmont, CA). As a control, the basal (unstimulated) rate of CGRP secreted into the media in 1 hr was determined, and these values were used to normalize for differences between dishes. Cells were pretreated with the indicated concentrations of 5-HT1receptor agonists, with or without the 5-HT1 antagonist methiothepin, or appropriate vehicle for 30 min before addition of either HBS (control), KCl, or inflammatory cocktail. The inflammatory cocktail (Strassman et al., 1996) contained 10 μm each of bradykinin, prostaglandin, and serotonin, and 50 μmhistamine in HBS at pH 5.5. This combination and concentration of agents was based on previous studies that elicited physiological responses (Steen et al., 1992; Strassman et al., 1996). Although it is difficult to know what the local concentrations of these agents would be during neurogenic inflammation, high hydrogen ion concentrations have been found in inflammation, pH 5.4, and the relatively high, perhaps unphysiological, concentrations of the chemical agents have been reported to be necessary for in vitro responses (Steen et al., 1992).
For the cAMP measurements, trigeminal cultures were incubated with either 2 μm forskolin (Sigma) in the presence or absence of sumatriptan (20 μm) or CGS (10 μm) for 30 min at 37°C. The HeLa1B cell line was incubated with 100 μm forskolin and 0.1 μm sumatriptan or CGS under the same conditions. These concentrations were chosen on the basis of previous studies with this cell line (Hamblin et al., 1992). Intracellular cAMP was determined using a cAMP-specific radioimmunoassay (Peninsula Labs). Each experimental condition was repeated in at least three independent experiments, and statistical analyses were performed using the Student’s t test (two-tailed, unpaired samples).
RESULTS
Regulated release of CGRP from cultured trigeminal ganglia neurons
To determine whether sumatriptan could directly repress CGRP secretion from trigeminal neurons, we took a reductionist approach by establishing primary cultures of rat trigeminal ganglia enriched for neuronal cells (estimated to be >80%). Expression of a neuron-specific protein, 160 kDa neurofilament subunit was detected in all cells exhibiting a neuronal morphology. A unique feature of our culture conditions is that almost all of the neuronal cells are CGRP positive (Fig. 1A), although it is estimated that only 23% of the neurons in the trigeminal ganglia express CGRP in vivo (O’Conner and Van Der Kooy, 1988). A likely reason for this bias is that only nerve growth factor, and not BDNF or NT-3, which are required for survival of other neurons, was included in the culture media (Buchman and Davies, 1993). CGRP secretion occurred at a steady-state rate of 148 pg/hr per dish of cells (approximately two ganglia). Treatment with potassium chloride (KCl) to mimic neuronal depolarization caused approximately a sevenfold increase in the rate of CGRP release (Fig.1B). Treatment with capsaicin, which selectively activates sensory C-fibers via a vanilloid receptor (Caterina et al., 1997), resulted in a similar sevenfold increase (Fig.1B). The rate of CGRP release was also markedly stimulated by a mixture of agents known to mediate physiological responses of neurogenic inflammation and sensitization of nociceptive afferents (Strassman et al., 1996) (Fig. 1B). Because the release of CGRP during migraine is thought to result in the production and/or release of agents that escalate and sustain the inflammatory response, our results indicate that these agents can also act to maintain CGRP secretion after the initial nerve activation.
Fig. 1.
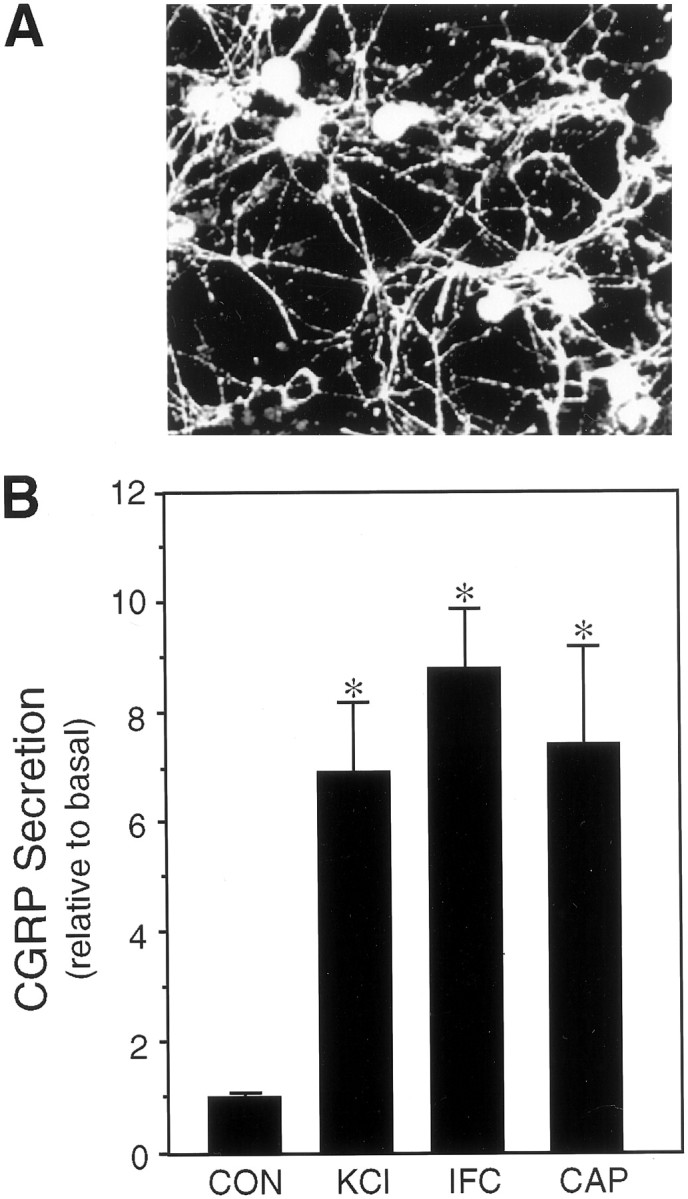
Expression and regulated release of CGRP from cultured trigeminal ganglia neurons. A, Fluorescent micrograph of CGRP-immunoreactive trigeminal neurons 7 d after plating on poly-d-lysine and laminin. B, The relative amount of CGRP secreted in 1 hr from untreated control cells (CON) or cells treated with 60 mmpotassium chloride (KCl), a cocktail of inflammatory agents (IFC), or 10 μmcapsaicin (CAP) is shown. The mean basal rate of CGRP release was 148 ± 5 pg/hr per dish (SE, n = 36). The secretion rate for each condition was normalized to the basal rate for each dish. The means and the SE from at least four independent experiments are shown. *p < 0.001 when compared with control levels.
Sumatriptan represses stimulated CGRP release
Having established that cultured trigeminal neurons exhibit regulated CGRP secretion, we then asked whether sumatriptan could directly inhibit this release. We found that sumatriptan inhibited potassium-stimulated CGRP secretion in a dose-dependent manner (Fig.2A). The secretion rate remained relatively stable throughout 4 hr of potassium stimulation, and a single dose of sumatriptan was able to maintain a steady inhibition throughout this period (Fig. 2A). In contrast, sumatriptan had no significant effect on the basal secretion of CGRP from unstimulated trigeminal neurons (Fig.2A). This finding is consistent with clinical reports that sumatriptan does not lower serum CGRP levels in normal, nonmigranuer individuals (Goadsby and Edvinsson, 1993). The specificity of sumatriptan action via the 5-HT1 receptors was confirmed by addition of a 5-HT1 receptor antagonist, methiothepin. Methiothepin completely blocked the action of sumatriptan on CGRP secretion from the cultured neurons (Fig.2B). Methiothepin treatment alone had little or no effect on secretion. We have shown previously that this antagonist is able to block the elevation of intracellular calcium by the 5-HT1 agonist CGS (Durham et al., 1997), and it can also block the increase in intracellular calcium after the sumatriptan treatment described below (data not shown). These results demonstrate that sumatriptan activation of trigeminal 5-HT1 receptors is sufficient to directly inhibit CGRP release.
Fig. 2.
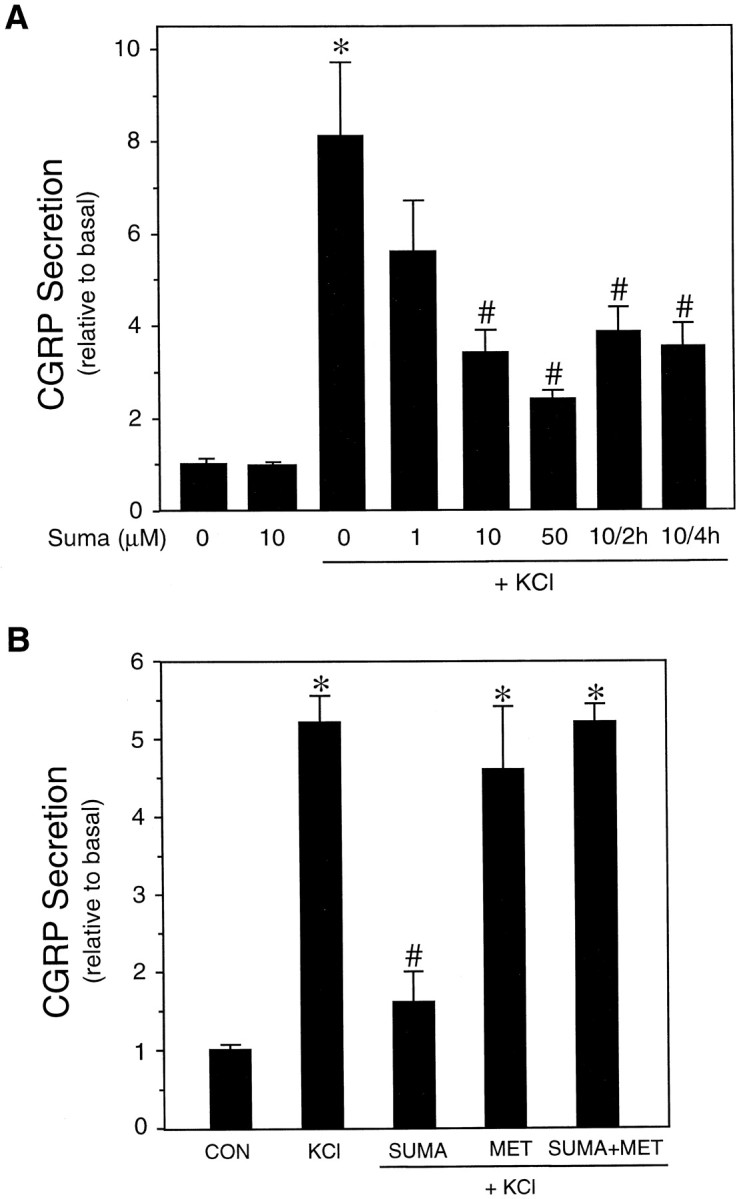
Effect of 5-HT1 receptor agonists on CGRP release. A, CGRP secretion as a function of sumatriptan concentration and treatment time. The effect of sumatriptan was determined on unstimulated and KCl-stimulated trigeminal neurons (cultured for 4–7 d). The amount secreted per hour was normalized to the basal rate determined before addition of buffer, 60 mmKCl, or sumatriptan (Suma) for 1 hr. Where indicated, 10 μm sumatriptan was added for 2 or 4 hr, and the amount per hour was normalized to basal. The mean basal CGRP secretion rate was 131 ± 4 pg/hr per dish. The means and the SE from at least three independent experiments are shown. *p < 0.001 when compared with control values.# p < 0.05 when compared with KCl-only values. B, The 5-HT1 receptor antagonist methiothepin blocks inhibitory effect of sumatriptan on KCl-stimulated CGRP release. The relative rates after addition of buffer (CON), KCl, KCl plus 10 μmsumatriptan (SUMA) and/or 20 μmmethiothepin (SUMA+MET) are shown. The mean basal rate was 122 ± 5 pg/hr per dish. The means and the SE from the two independent experiments are shown for each study. *p < 0.01 when compared with control values or sumatriptan values. # p < 0.05 when compared with KCl values.
We also investigated whether sumatriptan could inhibit the release of the vasoactive neuropeptide substance P, because it has been reported to colocalize with CGRP in sensory neurons (Edvinsson and Goadsby, 1994) and is involved in mediating neurogenic inflammation (Buzzi et al., 1995). In preliminary studies, we determined that sumatriptan could also inhibit the KCl-stimulated release of substance P (data not shown). Thus, the effectiveness of sumatriptan in reducing or abolishing the pain associated with migraine is likely caused by its ability to coordinately inhibit the release of vasoactive neuropeptides from trigeminal ganglion nerves.
Because we had found that a mixture of inflammatory agents caused a marked increase in the rate of CGRP secretion (Fig. 1), we wanted to determine whether sumatriptan could also block this type of stimulated CGRP release. The increase in CGRP release caused by the inflammatory cocktail was inhibited more than twofold by pretreatment with sumatriptan (Fig. 3). In addition, we showed that pretreatment with two other 5-HT1 receptor agonists, CGS and L-694,294, caused a similar inhibition of CGRP secretion (Fig. 3). These results demonstrate that multiple 5-HT1 agonists can repress CGRP secretion by at least two different stimuli, suggesting that these agents target a common downstream step.
Fig. 3.

Effect of 5-HT1 agonists on stimulated CGRP release. 5-HT1 repression of CGRP release after stimulation by inflammatory agents. The amount of CGRP secreted per hour was normalized to the basal rate determined for 1 hr before addition of 60 mm KCl or 5-HT1 agonists. The relative rates after addition of buffer (CON), inflammatory cocktail (IFC), and IFC plus 10 μm sumatriptan (Suma), 10 μmCGS, or 10 μm L-694,247 (L694) are shown. The mean basal rate was 137 ± 2 pg/hr per dish. The means and the SE from at least three independent experiments are shown. *p < 0.001 when compared with control values.# p < 0.05 when compared with IFC values.
Sumatriptan does not cause a decrease in intracellular cAMP levels
We then characterized the signaling pathway(s) used by sumatriptan in primary trigeminal ganglion cultures. Pharmacological studies have demonstrated that sumatriptan has high selectivity and potency at the 5-HT1B, 1D, and 1Freceptors, all of which are expressed by trigeminal neurons (Martin, 1997). The classical 5-HT1 signaling pathway based on studies using brain slices and non-neuronal cell lines overexpressing 5-HT1 receptors has been that these receptors inhibit adenylate cyclase and decrease cAMP levels via pertussis toxin-sensitive Gi/o proteins (Boess and Martin, 1994). However, in contrast to these reports, neither sumatriptan nor CGS inhibited forskolin-stimulated cAMP accumulation (Table1). In addition, treatment with the mixture of inflammatory agents that stimulated CGRP release did not elevate cAMP levels, nor did the cAMP levels change after cotreatment with sumatriptan (Table 1). As a positive control, we confirmed that we would be able to detect inhibition of cAMP production in cells known to couple 5-HT1B receptors to Gi/Go. Sumatriptan or CGS treatment essentially blocked forskolin-induced elevation of cAMP levels in HeLa cells stably expressing the 5-HT1B receptor (Table2). The degree of inhibition is similar to previously published results with this cell line (Hamblin et al., 1992). Thus, sumatriptan could lower cAMP levels in HeLa1B cells but did not cause a decrease in either the stimulated or unstimulated cAMP levels in trigeminal neurons.
Table 1.
cAMP levels in cultured trigeminal neurons
| Sample | cAMP (fmol/dish) | Fold increase |
|---|---|---|
| Control | 354 ± 25 | |
| FSK | 11283 ± 916* | 32 |
| FSK + sumatriptan | 10930 ± 1029 | 31 |
| FSK + CGS | 11047 ± 720 | 31 |
| IFC | 364 ± 10 | 1.0 |
| IFC + sumatriptan | 356 ± 18 | 1.0 |
cAMP levels were measured from untreated control cultures or cells treated for 30 min with forskolin (FSK) or with a cocktail of inflammatory agents (IFC). The cultures were cotreated with vehicle, sumatriptan, or CGS. The means and SE from five independent experiments with duplicate samples, and the fold increases in cAMP levels relative to control cells are shown.
*p < 0.001 when compared with control levels.
Table 2.
cAMP levels in 5-HT1B-expressing HeLa cells
| Sample | cAMP (fmol/dish) | Fold increase |
|---|---|---|
| Control | 234 ± 8.9 | |
| FSK | 4950 ± 244* | 21 |
| FSK + sumatriptan | 348 ± 26 | 1.5 |
| FSK + CGS | 318 ± 26 | 1.4 |
cAMP levels were measured from untreated control cultures or cells treated for 15 min with forskolin (FSK). The cultures were cotreated with vehicle, sumatriptan, or CGS. The means and SE from two independent experiments with duplicate samples, and the fold increases in cAMP levels relative to control cells are shown.
*p < 0.02 when compared with control levels.
Sumatriptan mediates an increase in intracellular calcium
We had shown previously that activation of 5-HT1receptors by sumatriptan and other 5-HT1 receptor agonists caused a sustained increase in calcium in the neuronal-like CA77 thyroid C-cell line (Durham et al., 1997). With this in mind, we then tested whether a similar pathway was activated by sumatriptan in cultured trigeminal neurons. We found that sumatriptan caused a slow, but markedly prolonged increase in intracellular calcium in the neurons. There was approximately a fivefold increase in intracellular calcium when compared with basal calcium levels (Fig.4, Table3). The increased calcium levels did not reach the maximal levels until ∼8 min after treatment but were maintained for at least 30 min (longest time sampled). The calcium reached a maximum concentration of ∼600 nm on average and as high as 1 μm in some cells. We estimate that ∼40% of the neuronal cells did not respond to sumatriptan treatment. The viability of these neuronal cells was confirmed after the sumatriptan treatment by the elevation of calcium levels in response to high concentrations of KCl. The reason for this heterogeneity is not known but may indicate that not all of the neurons are expressing sufficient levels of 5-HT1 receptors. In contrast to the delayed increase in calcium after sumatriptan treatment, addition of depolarizing levels of KCl caused a very rapid and transient increase in calcium (Fig. 4). These data demonstrate that activation of endogenous trigeminal neuron 5-HT1 receptors is coupled to a calcium-signaling pathway and not to a Gi/o-coupled decrease in cAMP.
Fig. 4.
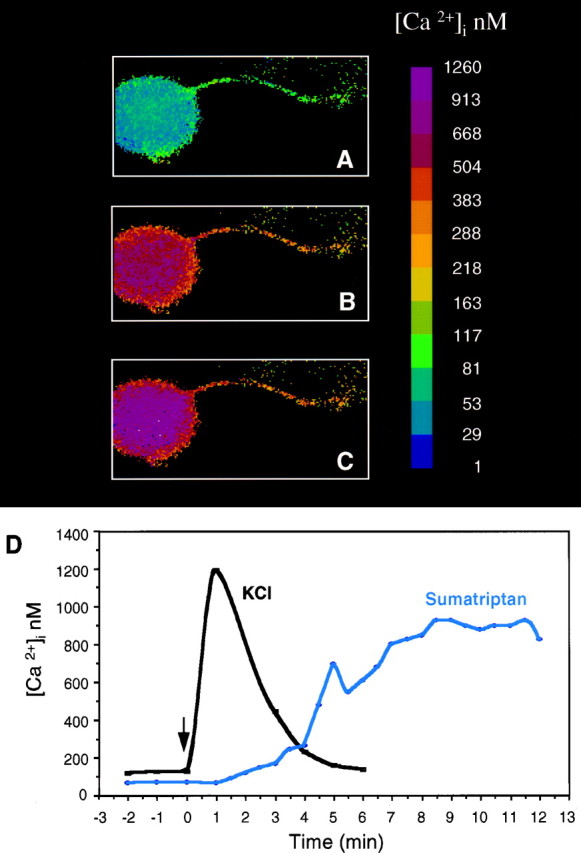
Sumatriptan increases the concentration of intracellular calcium in trigeminal neurons. A, Intracellular calcium concentrations, [Ca2+]i, from day 4 cultures were measured using fura-2 and a microscopic digital imaging system. The pseudo-color scale indicates the [Ca2+]i. Basal levels are in a single neuron with a neurite. B, The same cell 6 min after addition of 10 μm sumatriptan. C, The same cell after 12 min. D, A graphic representation of the change in [Ca2+]i as a function of time after sumatriptan treatment of a representative cell (same cell as above). For comparison, a trace of a different cell treated with only KCl (60 mm) is superimposed.
Table 3.
Effect of sumatriptan on calcium levels in cultured trigeminal neurons
| Sample | n | Basal (nm) | Peak (nm) | Fold increase | Peak time (sec) |
|---|---|---|---|---|---|
| Control | 11 | 119 ± 5 | 129 ± 73-a | 1.1 ± 0.83-a | a |
| Sumatriptan | 15 | 113 ± 7 | 621 ± 53* | 5.5 ± 0.6 | 488 ± 27 |
Intracellular calcium levels were measured from untreated control neurons or neurons treated with 10 μm sumatriptan. The means and SE are given.
*p < 0.001 when compared with control levels.
There was no significant change in calcium in control cells, so for comparison with the sumatriptan-treated cells, calcium levels at 488 sec were used to calculate peak and fold increase values for the control cells.
Okadaic acid blocks inhibitory effect of sumatriptan
How does a prolonged calcium elevation inhibit secretion? One possibility is that protein phosphorylation states are changed. It is generally accepted that changes in calcium can alter protein phosphorylation and that phosphorylation plays an important role in regulating neuropeptide release from sensory neurons (Greengard et al., 1993). We have used okadaic acid, a potent inhibitor of serine threonine protein phosphatases, especially PP1 and PP2A (Denhardt, 1996), to test the possibility that 5-HT1 agonists may be activating a phosphatase to attenuate stimulated secretion. Okadaic acid treatment blocked the inhibitory effect of sumatriptan on stimulated CGRP release (Fig. 5). Okadaic acid treatment alone increased CGRP release that was similar in magnitude to that caused by depolarization (Fig. 5), which is in agreement with previous studies by Vasko and colleagues (Hingtgen and Vasko, 1994) using cultured sensory neurons from dorsal root ganglia. Cotreatment with okadaic acid and KCl did not result in a greater increase in CGRP release than observed with each agent alone. These results indicate that sumatriptan acts by stimulating a serine threonine phosphatase. Although the identity of this phosphatase is not known, it is intriguing that okadaic acid has recently been reported to inhibit a MAP kinase phosphatase activity (Runden et al., 1998), and we have shown previously that 5-HT1 agonists cause a long-term increase in MAP kinase phosphatase activity in the CA77 cells (Durham and Russo, 1998).
Fig. 5.

Okadaic acid treatment blocks sumatriptan-mediated inhibition of potassium-stimulated CGRP release. The relative amount of CGRP secreted from trigeminal neurons stimulated with 60 mmKCl, 600 nm (unless indicated as 300 nm) okadaic acid (OA), or the combination of KCl and OA, with or without cotreatment with 10 μm sumatriptan (Suma) is shown. The mean basal rate of CGRP secretion was 99 ± 4 pg/hr per dish. The means and SE from at least three independent experiments are shown. *p < 0.001 when compared with control values. # p < 0.05 when compared with KCl values.+ p < 0.05 when compared with KCl plus sumatriptan values.
DISCUSSION
Our results support a model in which the trigeminal ganglion nerves are activated during migraine and release CGRP to cause vasodilation and mast cell degranulation leading to the release of inflammatory agents (Fig. 6). On the basis of our data using CA77 cells (Durham and Russo, 1998), these agents may stimulate MAP kinase pathways leading to an increase in CGRP synthesis and secretion that could potentially maintain elevated CGRP levels for the long duration (up to 72 hr) of a migraine. Activation of this pathway ultimately leads to sensitization of the trigeminal neurons and nociceptive transmission to the CNS contributing to the pain, nausea, and photophobia associated with migraine (Buzzi et al., 1995). It is likely that sumatriptan is able to block this pathway via activation of the 5-HT1 receptors leading to a prolonged elevation in calcium that mediates the recruitment of phosphatases. The concentration of sumatriptan required for inhibition in vitro is higher than the estimated plasma concentration in patients (∼0.2 μm) (Fowler et al., 1991). Possible explanations are that the effective receptor number may be low because of the culture conditions and/or lack of colocalization of receptors and secretory machinery at nerve terminals. Alternatively, higher concentrations may be required to counteract chronic stimulation of the cultures. In either case, the ability to block stimulated CGRP secretion in the absence of vascular contributions strongly supports the neurogenic model of migraine.
Fig. 6.

Model of 5-HT1 receptor-mediated inhibition of CGRP release from trigeminal neurons. A depolarizing stimulus causes the initial release of CGRP from trigeminal nerves, leading to neurogenic inflammation, which then further stimulates the release of CGRP. Activation of 5-HT1 receptors blocks this cycle by inhibiting CGRP release via an increase in phosphatase activity that is likely mediated by a sustained elevated level of intracellular calcium.
In the process of demonstrating this point, we have uncovered several unexpected findings. First, activation of endogenous trigeminal ganglion neuron 5-HT1 receptors did not decrease cAMP levels, which contradicts the commonly held belief (Boess and Martin, 1994). These observations are consistent with our findings that the 5-HT1 receptor agonist CGS also did not decrease cAMP levels in a model neuronal cell line and that CGS actions were pertussis toxin independent (Durham et al., 1997). The simplest explanation is that the cellular context is critical for assigning second messenger pathways to receptors. In support of this conclusion, others have reported that terminal 5-HT1 autoreceptors in hippocampal neurons may not be coupled to Gi/o proteins (Blier, 1991).
The second and perhaps most intriguing finding is that the inhibition of neuropeptide secretion by 5-HT1 receptor activation is paradoxically coupled to an unusually prolonged intracellular calcium signal. At face value, this observation is paradoxical because increased calcium is well known to be a signal to increase secretion (Matthews, 1996). Indeed, this dogma held true for the potassium treatment, which caused a more typical transient increase in calcium, with increased CGRP release from cultured trigeminal ganglia neurons. There is precedence in parathyroid endocrine cells for coupling of elevated intracellular calcium with inhibition of peptide secretion (Shoback et al., 1984). Our data demonstrate that activation of endogenous trigeminal neuron 5-HT1 receptors is coupled to a calcium-dependent signaling pathway that differs from depolarization-induced changes in calcium. This raises the possibility that the amplitude and duration of increased calcium can differentially regulate neuropeptide secretion from sensory neurons, analogous to recent evidence that the amplitude, duration, and localization of increased calcium can differentially activate transcription factors (Dolmetsch et al., 1997).
Our working model is that there is a balance between kinase and phosphatase activity that controls CGRP secretion (Fig. 6). Support for this type of mechanism is provided by our data showing that the protein phosphatase inhibitor okadaic acid blocks the inhibitory effect of sumatriptan on stimulated CGRP release. Under basal conditions the phosphorylation state is at an intermediate level. Depolarization, inflammatory agents, and okadaic acid change the balance, leading to increased secretion. In agreement with this model, okadaic acid treatment alone stimulated neuropeptide release from cultured trigeminal (this study) and dorsal root ganglia neurons (Vasko et al., 1994). Sumatriptan is able to blunt the increased secretion in response to depolarization and inflammatory agents, but not okadaic acid, suggesting that specific phosphatases are recruited by 5-HT1 receptor activation. The possibility of coordinated regulation by phosphatases is suggested by our previous studies showing that CGRP promoter activity was repressed in CA77 cells by a calcium-dependent increase in MAP kinase phosphatase-1 activity via 5-HT1 receptor activation (Durham and Russo, 1998). A remaining question is how sumatriptan selectively inhibits stimulated but not basal release of CGRP. To our knowledge, this is the first report of a drug that selectively targets only one of these events. One possible explanation would be if sumatriptan causes dephosphorylation of proteins responsible for the assembly, fusion, and/or recycling of vesicles in response to depolarization or inflammatory agents.
In conclusion, our results have demonstrated that activation of the 5-HT1 receptor class of antimigraine drugs is able to directly block CGRP release from trigeminal nerves. The inhibitory effect of sumatriptan occurs via a paradoxical elevation in calcium and activation of an okadaic acid-sensitive phosphatase. During migraine, CGRP helps mediate neurogenic inflammation that may result in the release of inflammatory agents. These agents could in turn feed back to sensitize the trigeminal ganglia neurons to sustain an elevated rate of CGRP release (Fig. 6). On the basis of our data, the effectiveness of sumatriptan is attributable in part to its ability to break this deleterious feedback loop at trigeminal ganglia nerve terminals by inhibiting CGRP secretion.
Footnotes
This work was supported by grants from National Institutes of Health (HD25969, NS37386, HL14388) and the American Heart Association (96013860) to A.R., with tissue culture support provided by the Diabetes and Endocrinology Center (DK25295) and an Iowa Cardiovascular Interdisciplinary Research Fellowship (HL07121) to P.D. We thank members of the lab and K. Campbell for comments on this manuscript and discussions and M. Hamblin for generously providing reagents.
Correspondence should be addressed to Dr. Andrew F. Russo, Department of Physiology and Biophysics, University of Iowa College of Medicine, Iowa City, IA 52242.
REFERENCES
- 1.Blier P. Terminal serotonin autoreceptor function in the rat hippocampus is not modified by pertussis and cholera toxins. Naunyn Schmiedebergs Arch Pharmacol. 1991;344:160–166. doi: 10.1007/BF00167213. [DOI] [PubMed] [Google Scholar]
- 2.Boess FG, Martin IL. Molecular biology of 5-HT receptors. Neuropharmacology. 1994;33:275–317. doi: 10.1016/0028-3908(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 3.Bouchelet I, Cohen Z, Case B, Seguela P, Hamel E. Differential expression of sumatriptan-sensitive 5-hydroxytryptamine receptors in human trigeminal ganglia and cerebral blood vessels. Mol Pharmacol. 1996;502:219–223. [PubMed] [Google Scholar]
- 4.Buchman VL, Davies AM. Different neurotrophins are expressed and act in a developmental sequence to promote the survival of embryonic sensory neurons. Development. 1993;118:989–1001. doi: 10.1242/dev.118.3.989. [DOI] [PubMed] [Google Scholar]
- 5.Buzzi MG, Bonamini M, Moskowitz MA. Neurogenic model of migraine. Cephalagia. 1995;15:277–280. doi: 10.1046/j.1468-2982.1995.1504277.x. [DOI] [PubMed] [Google Scholar]
- 6.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 7.Denhardt DT. Signal-transducing protein phosphorylation cascades mediated by Ras/Rho proteins in the mammalian cell: the potential for multiplex signaling. Biochem J. 1996;318:729–747. doi: 10.1042/bj3180729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dolmetsch RE, Lewis RS, Goddnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- 9.Durham PL, Russo AF. Serotonergic repression of mitogen-activated protein kinase control of the calcitonin gene-related peptide enhancer. Mol Endocrinol. 1998;12:1000–1008. doi: 10.1210/mend.12.7.0135. [DOI] [PubMed] [Google Scholar]
- 10.Durham PL, Sharma R, Russo AF. Repression of the calcitonin gene-related peptide promoter by 5-HT1 receptor activation. J Neurosci. 1997;17:9545–9553. doi: 10.1523/JNEUROSCI.17-24-09545.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edvinsson L, Goadsby PJ. Neuropeptides in migraine and cluster headache. Cephalagia. 1994;14:320–327. doi: 10.1046/j.1468-2982.1994.1405320.x. [DOI] [PubMed] [Google Scholar]
- 12.Ferrari MD. Migraine. Lancet. 1998;351:1043–1051. doi: 10.1016/S0140-6736(97)11370-8. [DOI] [PubMed] [Google Scholar]
- 13.Fowler PA, Lacey LF, Thomas M, Keene ON, Tanner RJ, Baber NS. The clinical pharmacology, pharmacokinetics and metabolism of sumatriptan. Eur Neurol. 1991;31:291–294. doi: 10.1159/000116756. [DOI] [PubMed] [Google Scholar]
- 14.Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol. 1993;33:48–56. doi: 10.1002/ana.410330109. [DOI] [PubMed] [Google Scholar]
- 15.Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- 16.Hamblin MW, Metcalf MA, McGuffin RW, Karpells S. Molecular cloning and functional characterization of a human 5-HT1B serotonin receptor: a homologue of the rat 5-HT1B receptor with 5-HT1D-like pharmacological specificity. Biochem Biophys Res Commun. 1992;184:752–759. doi: 10.1016/0006-291x(92)90654-4. [DOI] [PubMed] [Google Scholar]
- 17.Hingtgen CM, Vasko MR. The phosphatase inhibitor, okadaic acid, increases peptide release from rat sensory neurons in culture. Neurosci Lett. 1994;178:135–138. doi: 10.1016/0304-3940(94)90308-5. [DOI] [PubMed] [Google Scholar]
- 18.Martin GR. Serotonin receptor involvement in the pathogenesis and treatment of migraine. In: Goadsby PJ, Silberstein SD, editors. Headache. Butterworth-Heinemann; Boston: 1997. pp. 25–39. [Google Scholar]
- 19.Matthews G. Neurotransmitter release. In: Cowan WM, editor. Annual review of neuroscience. Annual Reviews, Inc; Palo Alto, CA: 1996. pp. 219–233. [DOI] [PubMed] [Google Scholar]
- 20.McCulloch J, Uddman R, Kingman TA, Edvinsson L. Calcitonin gene-related peptide: functional role in cerebrovascular regulation. Proc Natl Acad Sci USA. 1986;83:5731–5735. doi: 10.1073/pnas.83.15.5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moskowitz MA. Neurogenic inflammation in the pathophysiology and treatment of migraine. Neurology. 1993;43:S16–20. [PubMed] [Google Scholar]
- 22.O’Conner TP, Van Der Kooy D. Enrichment of vasoactive neuropeptide calcitonin gene-related peptide in the trigeminal sensory projections to the intracranial arteries. J Neurosci. 1988;8:2468–2476. doi: 10.1523/JNEUROSCI.08-07-02468.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ottosson A, Edvinsson L. Release of histamine from dural mast cells by substance P and calcitonin gene-related peptide. Cephalagia. 1997;17:166–174. doi: 10.1046/j.1468-2982.1997.1703166.x. [DOI] [PubMed] [Google Scholar]
- 24.Rosenfeld MG, Mermod J-J, Amara SG, Swanson LW, Sawchenko PE, Rivier J, Vale WW, Evans RM. Production of a novel neuropeptide encoded by the calcitonin gene via tissue-specific RNA processing. Nature. 1983;304:129–135. doi: 10.1038/304129a0. [DOI] [PubMed] [Google Scholar]
- 25.Runden E, Seglen PO, Haug F, Ottersen OP, Wieloch T, Shamloo M, Laake JH. Regional selective neuronal degeneration after protein phosphatase inhibition in hippocampal slice cultures: evidence for a MAP kinase-dependent mechanism. J Neurosci. 1998;18:7296–7305. doi: 10.1523/JNEUROSCI.18-18-07296.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shoback DM, Thatcher J, Leombruno R, Brown EM. Relationship between parathyroid hormone secretion and cytosolic calcium concentration in dispersed bovine parathyroid cells. Proc Natl Acad Sci USA. 1984;81:3113–3117. doi: 10.1073/pnas.81.10.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steen KH, Reeh PW, Anton F, Handwerker HO. Protons selectively induce lasting excitation and sensitization to mechanical stimulation of nociceptors in rat skin, in vitro. J Neurosci. 1992;12:86–95. doi: 10.1523/JNEUROSCI.12-01-00086.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stewart WF, Schechter A, Rasmussen BK. Migraine heterogeneity: disability, pain intensity, and attack frequency and duration. Neurology. 1994;44:S24–39. [PubMed] [Google Scholar]
- 29.Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996;384:560–564. doi: 10.1038/384560a0. [DOI] [PubMed] [Google Scholar]
- 30.Van Rossum D, Hanisch U-K, Quirion R. Neuroanatomical localization, pharmacological characterization and functions of CGRP, related peptides and their receptors. Neurosci Biobehav Rev. 1997;21:649–678. doi: 10.1016/s0149-7634(96)00023-1. [DOI] [PubMed] [Google Scholar]
- 31.Vasko MR, Campbell WB, Waite KJ. Prostaglandin E2 enhances bradykinin-stimulated release of neuropeptides from rat sensory neurons in culture. J Neurosci. 1994;14:4987–4997. doi: 10.1523/JNEUROSCI.14-08-04987.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]