

Abstract
Cerebellar granule neurons cultured in medium containing a physiological concentration of KCl (5 mm) undergo apoptosis. The cells can be rescued by the in vitroaddition of NMDA. The protective effect of NMDA is thought to reflect the in vivo innervation of developing cerebellar granule neurons by glutamatergic afferents. In the current work, we investigated the mechanism of the anti-apoptotic (protective) effect of NMDA. NMDA treatment reduced caspase-3-like activity in cerebellar granule neurons, and the time course and concentration dependence of the protective effect of NMDA mirrored the ability of NMDA to induce brain-derived neurotrophic factor (BDNF) expression. Furthermore, a Trk receptor antagonist, K252a, as well as a blocking antibody to BDNF, attenuated the protective effects of both NMDA and BDNF. These results suggest that NMDA-induced BDNF expression mediates the anti-apoptotic effect of NMDA. The protective effects of NMDA and BDNF were reduced by inhibitors of the phosphatidylinositol 3′-OH kinase (PI 3-kinase) signal transduction cascade (wortmannin and LY29004) but not by a MAP kinase kinase (MEK) inhibitor (PD98059) or a protein kinase A inhibitor (Rp-cAMPS). BDNF increased phosphorylation of Akt, a target of PI 3-kinase, and NMDA also induced Akt phosphorylation, but only after an exposure that was long enough to induce BDNF expression. Furthermore, ethanol, which interferes with NMDA receptor function, inhibited the NMDA-induced increase in BDNF levels but did not block the protective effect of BDNF. These findings further support the role of BDNF in the anti-apoptotic effect of NMDA in cerebellar granule neurons and suggest that the NMDA–BDNF interaction may play a key role inin vivo cerebellar granule neuron development, as well as in the deleterious effects of ethanol on the developing cerebellum.
Keywords: cerebellar granule neurons, apoptosis, NMDA, BDNF, PI 3-kinase, MAP kinase, ethanol
Cerebellar granule neurons obtained from neonatal rats and maintained in culture medium containing a physiological concentration of KCl (e.g., 5 mm) undergo apoptotic death (Balázs et al., 1988; D’Mello et al., 1993; Yan et al., 1994). This death can be prevented or reduced if the cells are grown in the presence of a depolarizing concentration of KCl (e.g., 25 mm) or if the glutamate receptor agonist NMDA is included in the culture medium (Balázs et al., 1988; Hack et al., 1993; Yan et al., 1994). The protective, anti-apoptotic effect of NMDAin vitro has been postulated to mimic the in vivoinnervation of the cerebellar granule neurons by glutamatergic mossy fiber afferents during development (Altman, 1982) [i.e., the innervated neurons are protected against apoptosis (Balázs et al., 1988)].
We have shown recently that ethanol can attenuate the protective effect of NMDA and thereby promote apoptosis of cultured cerebellar granule neurons (Bhave and Hoffman, 1997). Our results suggested that this action of ethanol was mediated by inhibition of NMDA receptor function (i.e., inhibition of the initial response to NMDA) measured as an increase in intracellular Ca2+ (Bhave and Hoffman, 1997). However, although the NMDA-induced Ca2+ influx and activation of a calcium/ calmodulin-dependent protein kinase have been implicated in the protective effect of NMDA (Balázs et al., 1992; Hack et al., 1993), little is known regarding the subsequent signal transduction pathways that mediate this action of NMDA.
Neurotrophins, including brain-derived neurotrophic factor (BDNF) as well as insulin-like growth factor-1 (IGF-1), have also been found to protect cultured cerebellar granule neurons against apoptosis (D’Mello et al., 1993; Lindholm et al., 1993; Harper et al., 1996;Nonomura et al., 1996; Courtney et al., 1997; Dudek et al., 1997;Miller et al., 1997; Suzuki and Koike, 1997; Ichikawa et al., 1998;Zhang et al., 1998), and the signal transduction cascades mediating the actions of these agents, including the phosphatidylinositol 3′-OH kinase (PI 3-kinase) and mitogen-activated protein kinase (MAPK) pathways, have been investigated (Nonomura et al., 1996; Dudek et al., 1997; Gunn-Moore et al., 1997; Miller et al., 1997). Activation of PI 3-kinase seems to be necessary for the protective effect of IGF-1 (Dudek et al., 1997; Miller et al., 1997), but the pathway(s) mediating the protective effect of BDNF is less clear (Courtney et al., 1997;Shimoke et al., 1997).
It is of particular interest that treatment of cerebellar granule neurons with NMDA has been reported to increase the level of mRNA for BDNF (Favaron et al., 1993). This finding suggests the possibility that BDNF could be involved in the protective effect of NMDA. In the present work, we compared the role of the various signal transduction cascades in the protective effects of NMDA and BDNF and evaluated the interactions between the anti-apoptotic effects of these agents. We also investigated further the mechanism of ethanol-induced inhibition of the protective effect of NMDA.
MATERIALS AND METHODS
Materials. NMDA, dizocilpine, and Rp-cAMPS were obtained from Research Biochemicals (Natick, MA). K252a was obtained from LC Laboratories (Woburn, MA). LY294002, PD98059, and wortmannin were obtained from Calbiochem (La Jolla, CA). Basal essential medium and fetal bovine serum were obtained from Life Technologies (Gaithersburg, MD). The ApopTag kit was obtained from Oncor (Gaithersburg, MD). The BDNF Emax immunoassay kit and anti-active (phosphorylated)-MAP kinase antibody were obtained from Promega (Madison, WI). The anti-BDNF blocking antibody was obtained from Research Diagnostics (Flanders, NJ), and the anti-IGF-1 blocking antibody was obtained from Upstate Biotechnology (Lake Placid, NY). The anti-Akt antibody was obtained from Stressgen Biotechnologies (Victoria, Canada), and the anti-phosphorylated Akt antibody was obtained from New England Biolabs (Beverly, MA). The ApoAlter CPP32 assay kit and DEVD–fmk were obtained from Clontech (Cambridge, UK). Enhanced chemiluminescence reagents were obtained from DuPont-NEN (Boston, MA). BDNF was a gift from Amgen (Thousand Oaks, CA). IGF-1 was a gift from Dr. Kim Heidenreich (Department of Pharmacology, University of Colorado Health Sciences Center, Denver, CO). All other products were obtained from Sigma (St. Louis, MO).
Cell culture. Primary cultures of cerebellar granule cells were prepared from 7-d-old Sprague Dawley rats as described previously (Iorio et al., 1992; Bhave and Hoffman, 1997), except that cells were maintained in medium containing 5 mm KCl unless otherwise noted. The percent of glial cells present in this preparation, as estimated visually, was 4.5 ± 0.4% (n = 3). For assessing apoptosis, cells were plated on glass coverslips (2 × 106 cells/well) or on eight-chambered microscope slides (Falcon culture slide; 0.5 × 106cells/well) coated with polyethyleneimine (100 μg/ml). Cerebellar granule cells (2 × 107 cells/100 mm dish) plated in tissue culture dishes coated with poly-l-lysine (10 μg/ml) were used for the extraction of total protein for analyzing BDNF levels. For assessing the levels of caspase-3 activity, phosphorylated Akt, total Akt, and active (phosphorylated) MAP kinase, cells (5 × 106 cells/well) were plated in poly-l-lysine-coated six-well dishes.
Measurement of apoptosis. In the experiments designed to assess the protective effect of NMDA and other agents against cerebellar granule neuron apoptosis, these agents were dissolved in conditioned medium containing 5 mm KCl, and 5–10 μl was added per milliliter of culture medium on day 4 in vitro to give final concentrations of 100 μm NMDA, 100 ng/ml BDNF, or 50 ng/ml IGF-1. Apoptosis was determined 12 or 24 hr later (day 5in vitro). Inhibitors of signal transduction pathways (PD98059, wortmannin, and LY294002 dissolved in DMSO and Rp-cAMPS dissolved in distilled water; 1–3 μl added per milliliter of culture medium), receptor antagonists (K252a dissolved in DMSO and dizocilpine dissolved in distilled water; 1–3 μl added per milliliter of culture medium), and ethanol were added 5 min before the protective agents, at concentrations noted in the Results and/or in the figure legends. Vehicle was added to control cultures as appropriate. Because of its reported lability (Kimura et al., 1994; Miller et al., 1997), wortmannin was replenished every 6 hr. Blocking antibodies to BDNF or IGF-1 were added to the cells 3 hr before the protective agents.
For the time course studies, NMDA (100 μm) was added to the culture medium on day 4 in vitro for different time periods. After these time periods, cells were washed with conditioned medium containing 5 mm KCl to remove NMDA, and cells were maintained in this conditioned medium until day 5 in vitro, when apoptosis was determined.
To assess apoptosis, we fixed the neurons and determined apoptotic cell death with the ApopTag kit, according to the manufacturer’s instructions (Bhave and Hoffman, 1997). This method provides forin situ fluorescent labeling of the 3′-OH ends of fragmented DNA. Total cell number is assessed by staining the fixed cells with propidium iodide. Fluorescence was detected with an epifluorescence microscope (Nikon; 100× objective). The total (propidium iodide-labeled) and apoptotic (fluorescein-labeled) cells were counted manually in three randomly chosen fields on each coverslip by an investigator who was unaware of the treatments.
Analysis of caspase-3-like activity. The activity of a caspase that cleaves the substrate DEVD–7-amino-4-trifluoromethyl coumarin (DEVD–AFC; “caspase-3-like activity”) in the cerebellar granule cells was determined using the ApoAlter CPP32 fluorescent assay kit, following the manufacturer’s instructions. In brief, cerebellar granule cells, maintained in medium containing 5 mm KCl, were treated with 100 μm NMDA on day 4 in vitro. On day 5 in vitro these neurons, as well as cells that had been maintained for 4 or 5 d in 5 mm KCl or for 5 d in 25 mm KCl in the absence of added NMDA, were extracted with the supplied cell lysis buffer, and caspase-3-like activity in the cell lysate was determined with DEVD–AFC. Proteolytic cleavage of this substrate releases free AFC that can be detected fluorimetrically (excitation, 400 nm; emission, 505 nm). The specificity of the enzyme activity measured was assessed using a selective inhibitor of caspase-3-like activity, DEVD–fmk (10 μm). The ability of DEVD–fmk to protect neurons against apoptosis was determined by treating the cells with 10 μm DEVD–fmk (dissolved in DMSO; 10 μl per milliliter of culture medium) on day 4 in vitro and measuring apoptosis, as described above, on day 5 in vitro.
Analysis of BDNF levels. The level of BDNF protein in the cerebellar granule cells after various treatments was determined using the BDNF Emax immunoassay kit in an antibody sandwich format as described by the manufacturer. Cerebellar granule cells were extracted in a lysis buffer (20 mm Tris, 137 mm NaCl, 1% NP-40, 10% glycerol, 1 mm PMSF, 10 μg/ml aprotinin, 1 μg/ml leupeptin, and 0.5 mm sodium vanadate), and determination of BDNF levels was performed after acid treatment according to the manufacturer’s instructions.
Western blot analysis. For analysis of the levels of phosphorylated Akt, total Akt, and active (phosphorylated) MAP kinase [extracellular-regulated kinase 1 and 2 (ERK1 and ERK2)], cerebellar granule cells on day 4 in vitro were treated with NMDA (100 μm), BDNF (100 ng/ml), or IGF-1 (50 ng/ml) for 5 min at 37°C. After this treatment, the neurons were washed twice with ice-cold PBS and harvested in a buffer containing 20 mm Tris, pH 7.4, 140 mm NaCl, 1% NP-40, 1 mm EDTA, 1 mm sodium vanadate, 20 mm NaF, 2 mm sodium pyrophosphate, 1 mm PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin. The amount of protein in each sample was estimated by the bicinchoninic acid method (Pierce, Rockford, IL), the membranes were solubilized, and 5 μg aliquots were subjected to SDS-PAGE on 10% polyacrylamide gels, according to the procedures described in Snell et al. (1996). After electrophoretic separation, the proteins were transferred to nitrocellulose membranes (0.22 μm; Schleicher & Schuell, Keene, NH). After blocking with 5% nonfat dry milk (for the anti-Akt antibody) or with 1% BSA (for the anti-phosphorylated Akt and anti-phosphorylated MAPK antibodies) in Tris-buffered saline containing 0.05% Tween-20, blots were probed with specific antibodies (anti-Akt, 1:5000; anti-phosphorylated Akt, 1:1000; and anti-phosphorylated MAPK, 1:20,000) for 1 hr and then incubated with horseradish peroxidase-conjugated goat IgG (1:20,000). Immunoreactive bands were visualized using a chemiluminescence method and were quantitated by image analysis using a Bio-Rad (Hercules, CA) GS-250 Molecular Imager and PhosphorAnalyst image analysis software. When more than one band was detected by the antibodies (phosphorylated ERK1 and ERK2; total Akt), the overall density of the two bands was quantitated to obtain a single value. The results are calculated as the volume (area × Phosphor counts) of the appropriate band(s) and are generally expressed as percent of control.
Statistical analysis. All values are presented as mean ± SEM. When data were expressed as ratios or percents, statistical significance was determined by the Kruskal–Wallis nonparametric ANOVA, followed by post hoc multiple comparisons; otherwise, ANOVA with post hoc comparisons was used. All analyses were performed using the SigmaStat 2.01 program (Jandel Scientific Software, San Rafael, CA); p < 0.05 was considered significant.
RESULTS
Characterization of the protective effect of NMDA
In our previous work, we found that treatment of cerebellar granule neurons with 100 μm NMDA for 24 hr, from day 4 to 5 in vitro, resulted in protection of ∼50% of the cells from apoptosis (Bhave and Hoffman, 1997). The amount of apoptosis observed at this time and the degree of protection afforded by NMDA were similar to that reported by others (Yan et al., 1994; Kharlamov et al., 1995). The effect of NMDA is receptor-mediated because it can be blocked by specific NMDA receptor antagonists (Yan et al., 1994). To characterize the anti-apoptotic effect of NMDA further, we evaluated the concentration–response relationship and the time course of the protective effect. As shown in Figure1A, NMDA, added to the cells for 24 hr, decreased apoptosis in a concentration-dependent manner, with 100 μm NMDA again producing ∼50% protection. The effect of NMDA was also dependent on the time that the cells were exposed to NMDA, with a maximum effect seen after 12 and 24 hr of exposure (Fig. 1B). It has been reported that caspase-3 or a caspase-3-like (DEVD-sensitive) enzyme mediates apoptosis in cultured cerebellar granule neurons (Armstrong et al., 1997; Ni et al., 1997; Marks et al., 1998). We found that caspase-3-like activity increased between day 4 and 5 in vitro, as apoptosis increased (Bhave and Hoffman, 1997), and was elevated in cells grown in medium containing 5 mm KCl compared with those grown in 25 mm KCl. Furthermore, 24 hr of exposure of the cells to 100 μm NMDA significantly reduced caspase-3-like activity (Fig. 2). The role of the caspase-3-like, DEVD-sensitive activity in cerebellar granule neuron apoptosis was also supported by the finding of a significant 41% reduction of apoptosis after treatment of the cells with the caspase inhibitor DEVD–fmk, similar to that in a previous report (D’Mello et al., 1998) (data not shown).
Fig. 1.

Concentration and time dependence of the anti-apoptotic effect of NMDA. A, Cerebellar granule neurons were maintained in medium containing 5 mm KCl for 4 d in vitro and were then treated with the indicated concentrations of NMDA for 24 hr, as described in Materials and Methods. Apoptosis was assessed with the ApopTag kit on day 5in vitro. Results are expressed as the percent decrease in apoptosis produced by NMDA. In the absence of NMDA, apoptosis was detected in 41% of the cells. Values represent the mean ± SEM of 11–33 observations in four separate experiments. Kruskal–Wallis ANOVA was performed on the raw data (percent apoptotic cell death) and revealed a significant effect of NMDA (H = 46.9; df = 3; p < 0.001). Post hoccomparisons showed significant effects with 10, 30, and 100 μm NMDA compared with that with no NMDA (control).B, NMDA (100 μm) was added to the culture medium of the cerebellar granule neurons on day 4 in vitro, and at the indicated times after addition, cells were washed to remove NMDA. The cells were maintained until day 5 in vitro in conditioned medium containing 5 mm KCl. Cells were then fixed for determination of apoptosis using the ApopTag kit. Results are expressed as the number (percent) of apoptotic (fluorescein-positive) cells per total cell number (propidium iodide-labeled cells). Values represent the mean ± SEM of 12–33 observations in four separate experiments. Kruskal–Wallis ANOVA revealed a significant effect of time of exposure to NMDA (H = 74.9; df = 4; p < 0.001); *p < 0.001 compared with all other groups (post hoc comparisons).
Fig. 2.

Effect of NMDA on caspase-3-like activity in cerebellar granule neurons. Cerebellar granule neurons were prepared as described in Materials and Methods and maintained in medium containing either 5 or 25 mm KCl. On day 4 in vitro, NMDA (100 μm) was added to cells grown in 5 mm KCl. Caspase-3-like activity was also measured in neurons maintained in 5 mm KCl and treated on day 4in vitro with the specific caspase-3-like inhibitor DEVD–fmk (10 μm). Caspase-3-like activity was measured fluorimetrically on the day in vitro(DIV) indicated, as described in Materials and Methods (i.e., cells were treated with NMDA or inhibitor for 24 hr). Values are expressed as the percent of caspase activity in cells grown in 5 mm KCl for 5 d in vitro in the absence of NMDA (control cells) and represent the mean ± SEM of three observations. Kruskal–Wallis ANOVA revealed a significant effect of treatment (H = 13.5; df = 4;p = 0.009); *p < 0.05 compared with all other groups (post hoccomparisons).
Characterization of signal transduction cascades involved in the protective effects of NMDA, BDNF, and IGF-1
To evaluate the importance of various signal transduction pathways in the protective effects of NMDA and the other trophic factors, we used specific inhibitors of steps in each pathway. Figure3A shows that pretreatment of the cells with Rp-cAMPS, a protein kinase A (PKA) inhibitor, at a concentration shown previously to inhibit PKA activity (Colwell and Levine, 1995) did not alter the protective effect of NMDA. Similarly, treatment of the cells with the MEK inhibitor PD98059 did not interfere with the protective effect of NMDA or with that of BDNF or IGF-1 (Fig. 3B). The concentration of PD98059 used (Miller et al., 1997) was sufficient to block the activation of MAP kinase (phosphorylation of ERK1 and ERK2) by BDNF (Fig. 3C).
Fig. 3.

Effect of inhibitors of PKA or MAP kinase on the anti-apoptotic actions of NMDA, BDNF, or IGF-1.A, Cerebellar granule neurons were prepared and cultured in medium containing 5 mm KCl, as described in Materials and Methods. On day 4 in vitro, cells were pretreated with vehicle or Rp-cAMPS (100 μm), a PKA inhibitor, 5 min before treatment with NMDA (100 μm). Twelve hours later, apoptosis was determined using the ApopTag kit, as described in Materials and Methods. Data are presented as the percent of cells showing apoptosis. Values represent the mean ± SEM of 9–12 observations in three separate experiments. Kruskal–Wallis ANOVA revealed a significant effect of treatment (H = 28.7; df = 3; p < 0.001); *p < 0.001 compared with the 5 mm KCl alone group (post hoc comparisons).B, Cerebellar granule neurons were prepared and cultured in medium containing 5 mm KCl, as described in Materials and Methods. On day 4 in vitro, cells were pretreated with vehicle or PD98059 (50 μm) 5 min before treatment with NMDA (100 μm), BDNF (100 ng/ml), or IGF-1 (50 ng/ml). Twelve hours later, apoptosis was determined using the ApopTag kit, as described in Materials and Methods. Data are presented as the percent of cells showing apoptosis. Values represent the mean ± SEM of three to six observations in two separate experiments. Kruskal–Wallis ANOVA revealed a significant effect of treatment (H = 33.5; df = 7;p < 0.001); *p < 0.001 compared with the 5 mm KCl alone group (post hoc comparisons). C, On day 4 in vitro, cells were treated with 100 μm NMDA, 100 ng/ml BDNF in the absence or presence of 50 μm PD98059, or 50 ng/ml IGF-1 for 5 min. Cells were extracted, and immunoblotting was performed as described in Materials and Methods.Inset, A representative blot of phosphorylated ERK1 and ERK2 is shown. Data are presented as the mean ± SEM percent of control values (level of phosphorylated ERK1 and ERK2 in vehicle-treated cells) obtained from six to eight observations in three separate experiments. Kruskal–Wallis ANOVA revealed a significant effect of treatment (H = 24.9; df = 4;p < 0.001); *p < 0.05 compared with control (post hoccomparisons).
In contrast to these results, treatment of cells with two different inhibitors of PI 3-kinase, wortmannin and LY294002, at concentrations shown previously to inhibit the activation of PI 3-kinase effectively (Dudek et al., 1997; Miller et al., 1997) did antagonize the protective effects of NMDA and BDNF, as shown in Figure4. These inhibitors also attenuated the protective effect of IGF-1, as expected (Dudek et al., 1997; Miller et al., 1997) (Fig. 4).
Fig. 4.

Effect of PI 3-kinase inhibitors on the anti-apoptotic action of NMDA, BDNF, or IGF-1. Cerebellar granule neurons were prepared and grown in medium containing 5 mmKCl, as described in Materials and Methods. On day 4 in vitro, cells were pretreated with vehicle or one of the PI 3-kinase inhibitors, wortmannin (100 nm; A) or LY294002 (10 μm; B), 5 min before treatment with NMDA (100 μm), BDNF (100 ng/ml), or IGF-1 (50 ng/ml). Wortmannin was replenished after 6 hr. Twelve hours after addition of the protective agents, apoptosis was assessed using the ApopTag kit, as described in Materials and Methods. The number of apoptotic cells is expressed as a percent of total cells. Values represent the mean of four to six observations in two separate experiments. A, Kruskal–Wallis ANOVA revealed a significant effect of treatment (H = 39.1; df = 7; p < 0.001). Post hoccomparisons showed that NMDA, BDNF, and IGF-1 significantly inhibited apoptotic cell death (*p < 0.001 compared with the 5 mm KCl group in the absence of wortmannin), wortmannin significantly decreased these effects (**p < 0.001 compared with the appropriate treatment in the absence of wortmannin), and wortmannin alone increased apoptosis (*p < 0.001 compared with the 5 mm KCl group in the absence of wortmannin). B, Kruskal–Wallis ANOVA revealed a significant effect of treatment (H = 34.9; df = 7; p < 0.001). Post hoccomparisons showed that NMDA, BDNF, and IGF-1 significantly inhibited apoptotic cell death (*p < 0.001 compared with the 5 mm KCl group in the absence of LY294002) and that LY294002 significantly reversed these effects (**p< 0.001 compared with the appropriate treatment in the absence of LY294002).
A downstream target of PI 3-kinase that has been suggested to be a mediator of cerebellar granule neuron survival is the kinase Akt (protein kinase B) (Dudek et al., 1997). When we compared the ability of NMDA, BDNF, and IGF-1 to phosphorylate (activate) Akt after a 5 min exposure, only BDNF and IGF-1 produced measurable phosphorylation of the kinase (Fig.5A). Four hours of exposure of the neurons to NMDA, which was necessary to observe a protective effect of NMDA (Fig. 1B), also resulted in increased Akt phosphorylation (Fig. 5B). The Akt phosphorylation induced by BDNF or by the 4 hr exposure to NMDA was prevented by LY294002 at the concentration that blocked the protective effects of NMDA, BDNF, and IGF-1 and by the Trk antagonist K252a at a concentration that blocked the protective effect of BDNF and NMDA (see below) (Fig.5).
Fig. 5.
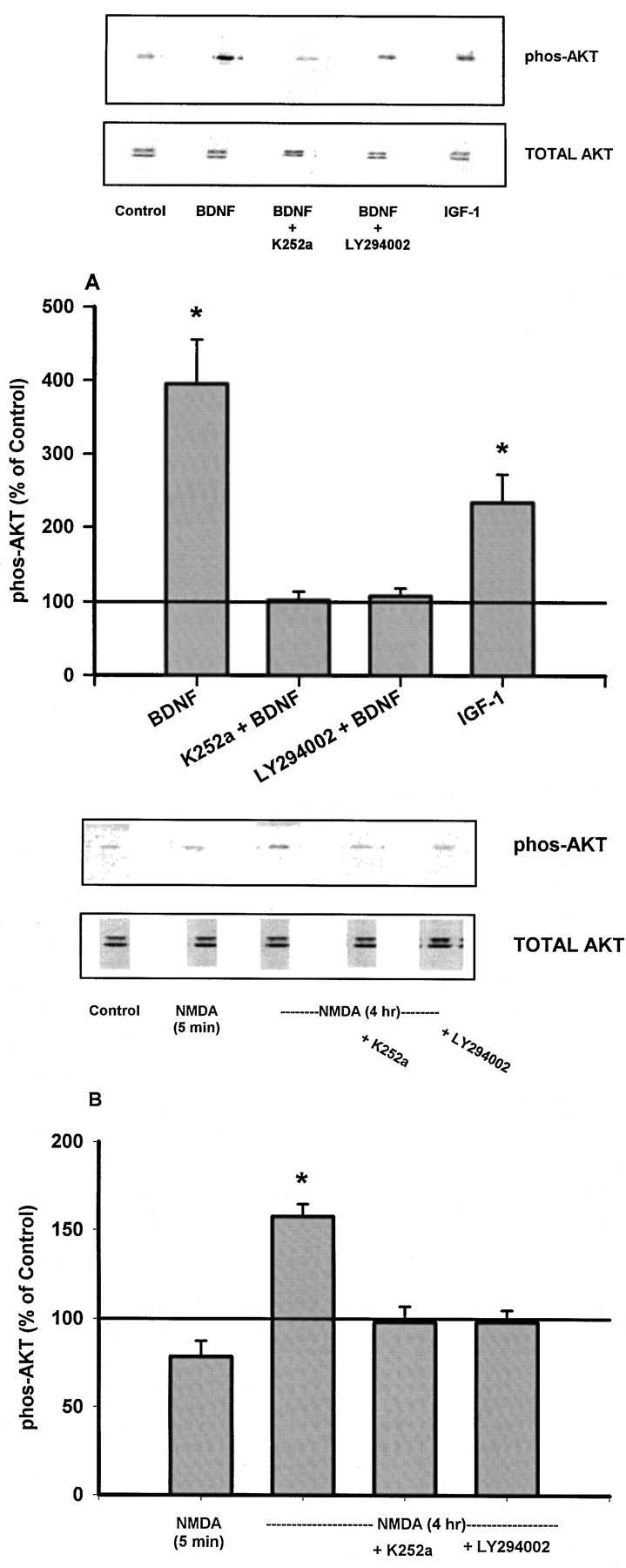
Effect of NMDA, BDNF, and IGF-1 on phosphorylation of Akt. Cerebellar granule neurons were prepared and grown in medium containing 5 mm KCl, as described in Materials and Methods. On day 4 in vitro, cells were treated with 100 ng/ml BDNF for 5 min in the presence or absence of LY294002 (10 μm) or K252a (300 nm), with 50 ng/ml IGF-1 for 5 min, or with 100 μm NMDA for 5 min or for 4 hr in the presence or absence of LY294002 (10 μm) or K252a (300 nm). Cells were extracted, and immunobloting was performed as described in Materials and Methods. A, Effects of BDNF and IGF-1 on Akt phosphorylation. Inset, A representative immunoblot of phosphorylated (phos-AKT) and total Akt. Quantitation of phosphorylated Akt was performed as described in Materials and Methods. Data are presented as the mean ± SEM percent of the control value (level of phosphorylated Akt in vehicle-treated cells) obtained from 6–10 observations in three separate experiments. Kruskal–Wallis ANOVA revealed a significant effect of treatment (H = 39.6; df = 4;p < 0.001); *p < 0.05 compared with control (post hoc comparisons).B, Effect of NMDA on Akt phosphorylation.Inset, A representative immunoblot of phosphorylated and total Akt. Data are presented as the mean ± SEM percent of the control value (level of phosphorylated Akt in vehicle-treated cells) obtained from 6–10 observations in three separate experiments. Kruskal–Wallis ANOVA revealed a significant effect of treatment (H = 19.8; df = 2; p < 0.001); *p < 0.05 compared with control (post hoc comparison).
Interaction of the protective effects of NMDA and BDNF
The above results, indicating a delayed effect of NMDA to phosphorylate Akt, suggested that the ability of NMDA to activate the PI 3-kinase pathway might require an intermediate step. The previous finding that NMDA increases the expression of BDNF mRNA in cerebellar granule neurons (Favaron et al., 1993) suggested that BDNF synthesis might be necessary to observe the response to NMDA. Figure6, A and B, shows that NMDA increased the level of BDNF protein in cerebellar granule neurons in a concentration- and time-dependent manner that was reminiscent of the protective effect of NMDA, as characterized in Figure 1, and was also compatible with the time course for NMDA-induced activation of Akt. The ability of NMDA to increase BDNF expression was receptor-mediated because it was blocked by the NMDA receptor antagonist dizocilpine (Fig. 6C).
Fig. 6.
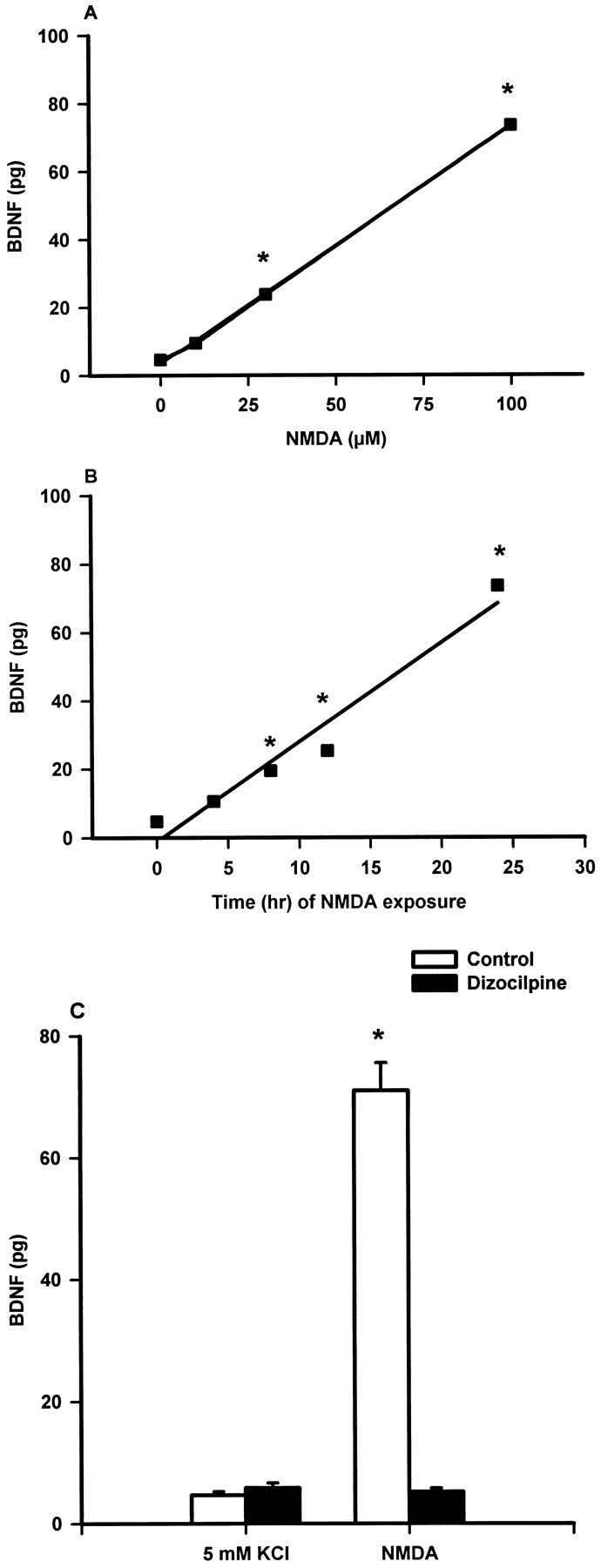
NMDA-induced BDNF expression in cerebellar granule cells. Cerebellar granule cells were prepared and maintained in medium containing 5 mm KCl, as described in Materials and Methods.A, On day 4 in vitro, cells were treated with buffer or the indicated concentration of NMDA. Twenty-four hours later, on day 5 in vitro, cells were extracted for analysis of BDNF levels as described in Materials and Methods. Values represent the mean ± SEM of 4–19 observations in three separate experiments. ANOVA revealed a significant effect of NMDA (F = 139.1; df = 3,38; p< 0.001); *p < 0.001 compared with the group with no NMDA (post hoc comparisons). B, NMDA (100 μm) was added to the culture medium of the cerebellar granule neurons on day 4 in vitro, and at the indicated times after addition, cells were washed to remove NMDA. Cells were maintained until day 5 in vitro in conditioned medium containing 5 mm KCl and were then extracted for the determination of BDNF levels as described in Materials and Methods. Values represent the mean ± SEM of 4–19 observations in three separate experiments. ANOVA revealed a significant effect of time of exposure to NMDA (F = 118.9; df = 4,49;p < 0.001); *p < 0.001 compared with the 0 time group (post hoccomparisons). C, On day 4 in vitro, vehicle or dizocilpine (1 μm) was added to the culture medium of the cerebellar granule neurons 5 min before addition of 100 μm NMDA. Cells were extracted 24 hr later for analysis of BDNF levels as described in Materials and Methods. Values represent the mean ± SEM of 3–19 observations in three separate experiments. ANOVA revealed a significant effect of treatment (F= 140.5; df = 3,36; p < 0.001); *p < 0.001 compared with all other groups (post hoc comparisons).
To investigate the possibility that the NMDA-induced increase in BDNF levels played a role in the protective effect of NMDA, we first determined whether a BDNF receptor antagonist could block the effect of NMDA. As shown in Figure 7, the nonselective Trk antagonist K252a effectively blocked the protective effect of both BDNF and NMDA but had no effect on the response to IGF-1. K252a did not affect the ability of NMDA to increase intracellular Ca2+ in the cerebellar granule neurons (i.e., did not interfere directly with NMDA receptor function) (data not shown). Blockade of the Trk receptor could also reduce the effects of both BDNF and NMDA if endogenous or exogenous BDNF increased the release of glutamate (i.e., if glutamate mediated the protective effect of BDNF). However, the NMDA receptor antagonist dizocilpine blocked only the effect of NMDA but not that of BDNF (data not shown).
Fig. 7.

Effect of K252a on the anti-apoptotic action of NMDA, BDNF, or IGF-1. Cerebellar granule neurons were prepared and grown in medium containing 5 mm KCl, as described in Materials and Methods. On day 4 in vitro, the Trk antagonist K252a (300 nm) was added to the culture medium 5 min before the addition of NMDA (100 μm), BDNF (100 ng/ml), or IGF-1 (50 ng/ml). The number of apoptotic cells was determined 24 hr later using the ApopTag kit, as described in Materials and Methods. Data are expressed as the percent of cells showing apoptosis. Values represent the mean ± SEM of 6–12 observations in two separate experiments. Kruskal–Wallis ANOVA revealed a main effect of treatment (H = 36.4; df = 7;p < 0.001); *p < 0.001 compared with the 5 mm KCl group in the absence of K252a, and **p < 0.001 compared with the appropriate treatment in the absence of K252a (post hoccomparisons).
We also evaluated the ability of a blocking antibody against BDNF to reduce the protective effects of NMDA and BDNF. As shown in Figure8A, pretreatment of cerebellar granule neurons with this antibody reduced the protective effects of both NMDA and BDNF. In contrast, treatment of the cells with a blocking antibody to IGF-1 reduced only the effect of IGF-1 and not that of NMDA (Fig. 8B). Both of these antibodies alone increased apoptosis, suggesting a role for endogenous IGF-1 and BDNF in cell survival, although only the effect of the anti-IGF-1 antibody was statistically significant.
Fig. 8.

Effect of anti-BDNF and anti-IGF-1 blocking antibodies on the protective effects of NMDA, BDNF, or IGF-1. Cerebellar granule neurons were prepared and grown in medium containing 5 mm KCl, as described in Materials and Methods.A, On day 4 in vitro, the anti-BDNF blocking antibody (1 μg/ml) was added 3 hr before the addition of NMDA (100 μm) or BDNF (100 ng/ml). The number of apoptotic cells was determined 24 hr after NMDA or BDNF addition using the ApopTag kit, as described in Materials and Methods. The number of apoptotic cells is reported as the percent of total cells. Values represent the mean ± SEM of 6–12 observations in three separate experiments. Kruskal–Wallis ANOVA revealed a significant effect of treatment (H = 57.8; df = 5;p < 0.001); *p < 0.001 compared with the 5 mm KCl group in the absence of antibody, and **p < 0.001 compared with the appropriate treatment in the absence of antibody (post hoc comparisons). B, The protective effect of NMDA (100 μm) and IGF-1 (50 ng/ml) was assessed in the absence and presence of an anti-IGF-1 blocking antibody (20 μg/ml), exactly as described above for the anti-BDNF antibody. Values represent the mean ± SEM of 6–12 observations in three separate experiments. Kruskal–Wallis ANOVA revealed a significant effect of treatment (H = 55.4; df = 5;p < 0.001); *p < 0.001 compared with the 5 mm KCl group in the absence of antibody, and **p < 0.001 compared with the IGF-1 group in the absence of antibody (post hoccomparisons).
Effect of ethanol treatment on the responses to NMDA and BDNF
We showed previously that ethanol, added to cerebellar granule cells in the presence of NMDA, attenuated the protective effect of NMDA in a concentration-dependent manner (Bhave and Hoffman, 1997). In the present study, as in the previous work, we found that ethanol alone increased apoptosis of cerebellar granule neurons (Fig.9). This effect is probably caused by inhibition of the protective effect of endogenous glutamate (Bhave and Hoffman, 1997). Ethanol also reduced the protective effect of IGF-1, as reported previously (Zhang et al., 1998) (Fig. 9). In contrast, ethanol did not attenuate the protective effect of BDNF (Fig. 9). However, treatment of the cells with ethanol did reduce NMDA-induced BDNF expression (Fig. 10).
Fig. 9.

Effect of ethanol on the anti-apoptotic action of BDNF and IGF-1. Cerebellar granule neurons were prepared and grown in medium containing 5 mm KCl, as described in Materials and Methods. On day 4 in vitro, cells were treated with 100 mm ethanol (6.2 μl of 95% ethanol/ml) 5 min before the addition of BDNF (100 ng/ml) or IGF-1 (50 ng/ml). Twenty-four hours later, apoptosis was assessed with the ApopTag kit, as described in Materials and Methods. The number of apoptotic cells is expressed as a percent of total cells. Values represent the mean ± SEM of eight observations in three separate experiments. Kruskal–Wallis ANOVA revealed a significant effect of treatment (H = 39.5; df = 5; p < 0.001); *p < 0.001 compared with the 5 mm KCl group in the absence of ethanol, and **p < 0.05 compared with the IGF-1 group in the absence of ethanol (post hoc comparisons).
Fig. 10.

Effect of ethanol on the NMDA-induced expression of BDNF. Cerebellar granule neurons were prepared and grown in medium containing 5 mm KCl, as described in Materials and Methods. On day 4 in vitro, cells were treated with 100 mm ethanol (as described in the legend to Fig. 9) 5 min before the addition of 100 μm NMDA. Twenty-four hours later, cells were extracted for determination of BDNF levels as described in Materials and Methods. Values represent the mean ± SEM of 4–19 observations in two separate experiments. ANOVA revealed a significant effect of treatment (F = 120.4; df = 3,41; p < 0.001); *p < 0.001 compared with the 5 mm KCl group, and **p < 0.001 compared with the NMDA group in the absence of ethanol (post hoc comparisons).
DISCUSSION
The current studies have characterized in detail the protective effect of NMDA against cerebellar granule neuron apoptosis, including inhibition of caspase-3-like activity and involvement of the PI 3-kinase signal transduction cascade. The results presented are compatible with the hypothesis that NMDA protects cerebellar granule neurons against apoptosis by increasing the expression of BDNF, which then acts as an autocrine agent to reduce apoptosis.
Treatment of cerebellar granule neurons with NMDA had been reported previously to increase mRNA levels for BDNF (Favaron et al., 1993) and has been shown very recently to increase BDNF protein levels (Marini et al., 1998). In all of those studies, the neurons were grown in a depolarizing concentration of KCl, under conditions in which glutamate and NMDA are toxic to the cells (e.g., Manev et al., 1989; Iorio et al., 1993). It was suggested that the NMDA-induced increase in BDNF under these conditions may mediate the protective effect provided by NMDA pretreatment against glutamate-induced toxicity (Marini et al., 1998). However, in this study, it was not determined whether glutamate toxicity was caused by necrosis, apoptosis, or both (e.g., Ankarcrona et al., 1995). We have now shown that NMDA treatment increases the levels of BDNF protein in cerebellar granule cells grown in the presence of a physiological KCl concentration, under conditions in which NMDA protects the cells from apoptosis. However, induction of BDNF expression by NMDA does not necessarily indicate that BDNF is responsible for the protective effect of NMDA. Although BDNF mRNA levels (and protein levels; S. V. Bhave and P. L. Hoffman, unpublished observations) are higher in cerebellar granule cells grown in the presence of a depolarizing concentration of KCl, which protects the neurons from apoptosis (Condorelli et al., 1998), an antibody to BDNF did not affect the survival of cerebellar granule neurons grown under depolarizing conditions (Miller et al., 1997; Shimoke et al., 1997). In addition, the survival-promoting effect of exogenous BDNF on cells grown in low KCl was less than the effect of growth in the presence of a high KCl concentration (Condorelli et al., 1998; Ichikawa et al., 1998). Ghosh et al. (1994), using cultured cerebral cortical neurons, found that both a depolarizing concentration of KCl and NMDA induced expression of BDNF mRNA but that BDNF only mediated the protective effect of depolarization. In spite of the BDNF induction, NMDA did not protect the cortical cells against apoptosis.
Our conclusion that BDNF mediates the protective effect of NMDA in cerebellar granule neurons is based on our findings of a parallel concentration dependence and time course for the protective effect of NMDA and for NMDA induction of BDNF expression, as well as on studies showing that the nonselective Trk antagonist K252a, as well as a specific blocking antibody to BDNF, attenuates the protective effects and effects on signal transduction cascades not only of BDNF but also of NMDA. The results of studies using specific inhibitors of various signal transduction cascades also support the proposed interaction (i.e., the PI 3-kinase pathway, but not the MAP kinase pathway, is involved in the protective effects of both BDNF and NMDA). On the other hand, although IGF-1 also protects cerebellar granule neurons against apoptosis, our findings do not support a role for IGF-1 in the protective effect of NMDA.
As mentioned above, several studies have shown that BDNF can protect cerebellar granule neurons from apoptosis. Conflicting results have been reported regarding the effect of wortmannin, a PI 3-kinase inhibitor, on neuroprotection by BDNF (Nonomura et al., 1996; Courtney et al., 1997; Shimoke et al., 1997). Our finding that both wortmannin and the structurally unrelated inhibitor of PI 3-kinase LY294002 reduced the protective effects of NMDA and BDNF provides confidence that these protective effects involve activation of PI 3-kinase. Further support for this hypothesis is provided by the data showing that treatment of the cells with NMDA (after a delay), BDNF, or IGF-1 results in the phosphorylation of Akt, one of the downstream targets of PI 3-kinase (Duronio et al., 1998). One other target of PI 3-kinase that may mediate anti-apoptotic effects is p70S6 kinase. However, we found that rapamycin did not alter the ability of BDNF to prevent apoptosis in the cerebellar granule neurons (data not shown), in agreement with previous work (Dudek et al., 1997; Gunn-Moore et al., 1997).
PI 3-kinase enzymes are involved in many different cell regulatory pathways, including mitogenesis and protection against apoptosis (Duronio et al., 1998). Isozymes of this enzyme can bind directly to the platelet-derived growth factor (PDGF) receptor (Yao and Cooper, 1995) and mediate the anti-apoptotic effect of PDGF, e.g., in pheochromocytoma (PC12) cells. However, although PI 3-kinase also seems to be necessary for the anti-apoptotic effect of NGF in PC12 cells, PI 3-kinase does not bind directly to the TrkA (NGF) receptor (Ohmichi et al., 1992). Recent work suggests that the Grb2-associated binder-1 protein serves as a docking protein that mediates the association of PI 3-kinase with TrkA (Holgado-Madruga et al., 1997). This interaction is similar to the situation with the IGF-1 receptor, which requires phosphorylation of intermediate docking proteins that can bind and activate PI 3-kinase isozymes [i.e., insulin receptor substrates 1 and 2 (LeRoith et al., 1995)]. Little is known regarding the BDNF-associated signal transduction pathways, but it seems likely that intermediate proteins [possibly insulin receptor substrates 1 and 2 (Yamada et al., 1997)] will also be involved in the association of TrkB and PI 3-kinase.
Activation of the ERK subgroup of MAP kinases is associated with cell survival and/or growth (Xia et al., 1995). NMDA appeared to activate ERK1 and ERK2 directly (i.e., after a 5 min treatment), consistent with a previous report (Xia et al., 1996). NGF activates the MAP kinase pathway via activation of the small GTP-binding protein Ras and the subsequent phosphorylation and activation of the kinases Raf, MEK, ERK1, and ERK2 (D’Arcangelo and Halegoua, 1993), and BDNF activation of ERK1 and ERK2 presumably involves a similar pathway. Although experiments with PD98059 indicate that ERK activation does not play a role in the protective effect of either BDNF or NMDA in cerebellar granule cells (also see Gunn-Moore et al., 1997), Ras can be an upstream activator of PI 3-kinase (Kodaki et al., 1994), and activation of Ras by BDNF (or NMDA) could thus play a role in the protective effects of these agents.
It has been reported that treatment of cerebellar granule neurons with pituitary adenylyl cyclase-activating peptide or increasing cAMP levels via other means can prevent apoptosis, either via activation of protein kinase A or MAP kinase (D’Mello et al., 1993; Cavallaro et al., 1996;Chang et al., 1996; Villalba et al., 1997; Vaudry et al., 1998). However, a protective effect of cAMP or PKA activation has not been universally reported (Balázs et al., 1992; Yan et al., 1995). We also found that inhibition of protein kinase A did not alter the protective effect of NMDA on cerebellar granule neurons.
We had shown previously that ethanol treatment can promote cerebellar granule neuron apoptosis, apparently by inhibiting the function of the NMDA receptor (Bhave and Hoffman, 1997). In the current work, we wanted to determine whether ethanol also acted downstream of the receptor to promote apoptosis. The mechanism by which NMDA receptor activation results in increased BDNF expression in cerebellar granule neurons is likely to involve NMDA-induced increases in intracellular Ca2+ concentration. It has been demonstrated that Ca2+ influx through NMDA receptors can increase mRNA levels for BDNF and release of BDNF protein from hippocampal and cortical neurons (Zafra et al., 1990, 1991; Ghosh et al., 1994). Our findings that ethanol inhibits NMDA-induced expression of BDNF but does not inhibit the protective effect of BDNF are therefore consistent with the hypotheses that (1) ethanol promotes apoptosis by acting at the NMDA receptor [i.e., inhibiting NMDA-induced increases in intracellular Ca2+ (Hoffman et al., 1989; Bhave and Hoffman, 1997)] and (2) ethanol is not acting downstream of the NMDA receptor, with regard to the pathways activated by BDNF. By inhibiting the response to NMDA, ethanol is, in essence, producing a state of growth factor (BDNF) withdrawal. We also found that ethanol reduces the protective effect of IGF-1, as reported recently by Zhang et al. (1998), who concluded that ethanol inhibited the catalytic activity of the IGF-1 receptor and did not act at a site downstream of the receptor, similar to our findings. These results reinforce the hypothesis that ethanol does not act nonspecifically on all systems but that instead there are “receptive elements” for ethanol in the brain, such as the NMDA receptor, that are particularly sensitive to pharmacologically relevant concentrations of ethanol (Tabakoff and Hoffman, 1987).
There is considerable evidence that NMDA and BDNF play key roles in cerebellar development in vivo (Komuro and Rakic, 1993;Schwartz et al., 1997), and our results suggest that it may be NMDA-induced BDNF expression that contributes, at least in part, to this development. Our results also indicate that the presence of ethanol in the CNS at a critical period of development would interfere with the effect of NMDA on BDNF expression, leading to inappropriate apoptosis of cerebellar granule neurons and granule cell loss that is associated with the fetal alcohol syndrome (Pierce et al., 1989;Miller, 1992).
Footnotes
This work was supported in part by the National Institute on Alcohol Abuse and Alcoholism (AA9005 and AA3527) and the Banbury Foundation. We thank Ms. Karin Nunley for expert technical assistance and Drs. Kim Heidenreich and Boris Tabakoff for invaluable discussion.
Correspondence should be addressed to Dr. Paula L. Hoffman, Department of Pharmacology, University of Colorado Health Sciences Center, 4200 East 9th Avenue, Box C236, Denver, CO 80262.
REFERENCES
- 1.Altman J. Morphological development of rat cerebellum and a source of its mechanism. In: Chan-Palay V, Palay S, editors. The cerebellum: new vistas. Springer; Berlin: 1982. pp. 8–49. [Google Scholar]
- 2.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong RC, Aja TJ, Hoang KD, Gaur S, Bai X, Alnemri ES, Litwack G, Karanewsky DS, Fritz LC, Tomaselli KJ. Activation of the CED3/ICE-related protease CPP32 in cerebellar granule neurons undergoing apoptosis but not necrosis. J Neurosci. 1997;17:553–562. doi: 10.1523/JNEUROSCI.17-02-00553.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balázs R, Jorgensen OS, Hack N. N-Methyl-d-aspartate promotes the survival of cerebellar granule cells in culture. Neuroscience. 1988;27:437–451. doi: 10.1016/0306-4522(88)90279-5. [DOI] [PubMed] [Google Scholar]
- 5.Balázs R, Hack N, Resink A, Aronica E, van der Valk JBF. Trophic effect of excitatory amino acids on differentiating granule cells: involvement of calcium and other second messengers. Mol Neuropharmacol. 1992;2:203–206. [Google Scholar]
- 6.Bhave SV, Hoffman PL. Ethanol promotes apoptosis in cerebellar granule cells by inhibiting the trophic effect of NMDA. J Neurochem. 1997;68:578–586. doi: 10.1046/j.1471-4159.1997.68020578.x. [DOI] [PubMed] [Google Scholar]
- 7.Cavallaro S, Copani A, D’Agata V, Musco S, Petralia S, Ventra C, Stivala F, Travali S, Canonico PL. Pituitary adenylate cyclase activating polypeptide prevents apoptosis in cultured cerebellar granule neurons. Mol Pharmacol. 1996;50:60–66. [PubMed] [Google Scholar]
- 8.Chang JY, Korolev VV, Wang J-Z. Cyclic AMP and pituitary adenylate cyclase-activating polypeptide (PACAP) prevent programmed cell death of cultured rat cerebellar granule cells. Neurosci Lett. 1996;206:181–184. doi: 10.1016/s0304-3940(96)12468-x. [DOI] [PubMed] [Google Scholar]
- 9.Colwell CS, Levine MS. Excitatory synaptic transmission in neostriatal neurons: regulation by cyclic AMP-dependent mechanisms. J Neurosci. 1995;15:1704–1713. doi: 10.1523/JNEUROSCI.15-03-01704.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Condorelli DF, Dell’Albani P, Timmusk T, Mudò G, Belluardo N. Differential regulation of BDNF and NT-3 mRNA levels in primary cultures of rat cerebellar neurons. Neurochem Int. 1998;32:87–91. doi: 10.1016/s0197-0186(97)00038-7. [DOI] [PubMed] [Google Scholar]
- 11.Courtney MJ, Akerman KEO, Coffey ET. Neurotrophins protect cultured cerebellar granule neurons against the early phase of cell death by a two-component mechanism. J Neurosci. 1997;17:4201–4211. doi: 10.1523/JNEUROSCI.17-11-04201.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Arcangelo G, Halegoua S. A branched signaling pathway for nerve growth factor is revealed by Src-, Ras-, and Raf-mediated gene inductions. Mol Cell Biol. 1993;13:3146–3155. doi: 10.1128/mcb.13.6.3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D’Mello SR, Aglieco F, Roberts MR, Borodezt K, Haycock JW. A DEVD-inhibited caspase other than CPP32 is involved in the commitment of cerebellar granule neurons to apoptosis induced by K+ deprivation. J Neurochem. 1998;70:1809–1818. doi: 10.1046/j.1471-4159.1998.70051809.x. [DOI] [PubMed] [Google Scholar]
- 15.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 16.Duronio V, Scheid MP, Ettinger S. Downstream signalling events regulated by phosphatidylinositol 3-kinase activity. Cell Signal. 1998;10:233–239. doi: 10.1016/s0898-6568(97)00129-0. [DOI] [PubMed] [Google Scholar]
- 17.Favaron M, Manev RM, Rimland JM, Candeo P, Beccaro M, Manev H. NMDA-stimulated expression of BDNF mRNA in cultured cerebellar granule neurones. NeuroReport. 1993;4:1171–1174. [PubMed] [Google Scholar]
- 18.Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- 19.Gunn-Moore FJ, Williams AG, Toms NJ, Tavaré JM. Activation of mitogen-activated protein kinase and p70S6 kinase is not correlated with cerebellar granule cell survival. Biochem J. 1997;324:365–369. doi: 10.1042/bj3240365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hack N, Hidaka H, Wakefield MJ, Balázs R. Promotion of granule cell survival by high K+ or excitatory amino acid treatment and Ca2+/calmodulin-dependent protein kinase activity. Neuroscience. 1993;57:9–20. doi: 10.1016/0306-4522(93)90108-r. [DOI] [PubMed] [Google Scholar]
- 21.Harper SJ, Macaulay AJ, Hill RG, Priestley T. The effects of insulin-like growth factor analogues on survival of cultured cerebral cortex and cerebellar granule neurons. Brain Res. 1996;709:303–310. doi: 10.1016/0006-8993(95)01355-5. [DOI] [PubMed] [Google Scholar]
- 22.Hoffman PL, Rabe CS, Moses F, Tabakoff B. N-Methyl-d-aspartate receptors and ethanol: inhibition of calcium flux and cyclic GMP production. J Neurochem. 1989;52:1937–1940. doi: 10.1111/j.1471-4159.1989.tb07280.x. [DOI] [PubMed] [Google Scholar]
- 23.Holgado-Madruga M, Moscatello DK, Emlet DR, Dieterich R, Wong AJ. Grb2-associated binder-1 mediates phosphatidylinositol 3-kinase activation and the promotion of cell survival by nerve growth factor. Proc Natl Acad Sci USA. 1997;94:12419–12424. doi: 10.1073/pnas.94.23.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ichikawa D, Tabuchi A, Taoka A, Tsuchiya T, Tsuda M. Attenuation of cell death mediated by membrane depolarization different from that by exogenous BDNF in cultured mouse cerebellar granule cells. Mol Brain Res. 1998;56:218–226. doi: 10.1016/s0169-328x(98)00062-x. [DOI] [PubMed] [Google Scholar]
- 25.Iorio KR, Reinlib L, Tabakoff B, Hoffman PL. Chronic exposure of cerebellar granule cells to ethanol results in increased NMDA receptor function. Mol Pharmacol. 1992;41:1142–1148. [PubMed] [Google Scholar]
- 26.Iorio KR, Tabakoff B, Hoffman PL. Glutamate-induced neurotoxicity is increased in cerebellar granule cells exposed chronically to ethanol. Eur J Pharmacol. 1993;248:209–212. doi: 10.1016/0926-6917(93)90045-r. [DOI] [PubMed] [Google Scholar]
- 27.Kharlamov E, Cagnoli CM, Atabay C, Ikonomovi S, Grayson DR, Manev H. Opposite effect of protein synthesis inhibitors on potassium deficiency-induced apoptotic cell death in immature and mature neuronal cultures. J Neurochem. 1995;65:1395–1398. doi: 10.1046/j.1471-4159.1995.65031395.x. [DOI] [PubMed] [Google Scholar]
- 28.Kimura K, Hattori S, Kabuyama Y, Shizawa Y, Takayanagi J, Nakamura S, Toki S, Matsuda Y, Onodera K, Fukui Y. Neurite outgrowth of PC12 cells is suppressed by wortmannin, a specific inhibitor of phosphatidylinositol 3-kinase. J Biol Chem. 1994;269:18961–18967. [PubMed] [Google Scholar]
- 29.Kodaki T, Woscholski R, Hallberg B, Rodriguez-Viciana P, Downward J, Parker PJ. The activation of phosphatidylinositol 3-kinase by Ras. Curr Biol. 1994;4:798–806. doi: 10.1016/s0960-9822(00)00177-9. [DOI] [PubMed] [Google Scholar]
- 30.Komuro H, Rakic P. Modulation of neuronal migration by NMDA receptors. Science. 1993;260:95–97. doi: 10.1126/science.8096653. [DOI] [PubMed] [Google Scholar]
- 31.LeRoith D, Werner H, Beitner-Johnson D, Roberts CT., Jr Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16:143–163. doi: 10.1210/edrv-16-2-143. [DOI] [PubMed] [Google Scholar]
- 32.Lindholm D, Dechant G, Heisenberg C-P, Thoenen H. Brain-derived neurotrophic factor is a survival factor for cultured rat cerebellar granule neurons and protects them against glutamate-induced neurotoxicity. Eur J Neurosci. 1993;5:1455–1464. doi: 10.1111/j.1460-9568.1993.tb00213.x. [DOI] [PubMed] [Google Scholar]
- 33.Manev H, Favaron M, Guidotti A, Costa E. Delayed increase of Ca2+ influx elicited by glutamate: role in neuronal death. Mol Pharmacol. 1989;36:106–112. [PubMed] [Google Scholar]
- 34.Marini AM, Rabin SJ, Lipsky RH, Mocchetti I. Activity-dependent release of brain-derived neurotrophic factor underlies the neuroprotective effect of N-methyl-d-aspartate. J Biol Chem. 1998;273:29394–29399. doi: 10.1074/jbc.273.45.29394. [DOI] [PubMed] [Google Scholar]
- 35.Marks N, Berg MJ, Guidotti A, Saito M. Activation of caspase-3 and apoptosis in cerebellar granule cells. J Neurosci Res. 1998;52:334–341. doi: 10.1002/(SICI)1097-4547(19980501)52:3<334::AID-JNR9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 36.Miller MW. Effects of prenatal exposure to ethanol on cell proliferation and neuronal migration. In: Miller MW, editor. Development of the central nervous system: effects of alcohol and opiates. Wiley; New York: 1992. pp. 47–69. [Google Scholar]
- 37.Miller TM, Tansey MG, Johnson EM, Jr, Creedon DJ. Inhibition of phosphatidylinositol 3-kinase activity blocks depolarization- and insulin-like growth factor I-mediated survival of cerebellar granule cells. J Biol Chem. 1997;272:9847–9853. doi: 10.1074/jbc.272.15.9847. [DOI] [PubMed] [Google Scholar]
- 38.Ni B, Wu X, Du Y, Su Y, Hamilton-Byrd E, Rockey PK, Rosteck P, Jr, Poirier GG, Paul SM. Cloning and expression of a rat brain interleukin-1beta-converting enzyme (ICE)-related protease (IRP) and its possible role in apoptosis of cultured cerebellar granule neurons. J Neurosci. 1997;17:1561–1569. doi: 10.1523/JNEUROSCI.17-05-01561.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nonomura T, Kubo T, Oka T, Shimoke K, Yamada M, Enokido Y, Hatanaka H. Signaling pathways and survival effects of BDNF and NT-3 on cultured cerebellar granule cells. Dev Brain Res. 1996;97:42–50. doi: 10.1016/s0165-3806(96)00130-7. [DOI] [PubMed] [Google Scholar]
- 40.Ohmichi M, Decker SJ, Saltiel AR. Activation of phosphatidylinositol-3 kinase by nerve growth factor involves indirect coupling of the trk proto-oncogene with src homology 2 domains. Neuron. 1992;9:769–777. doi: 10.1016/0896-6273(92)90039-g. [DOI] [PubMed] [Google Scholar]
- 41.Pierce DR, Goodlett CR, West JR. Differential neuronal loss following early postnatal alcohol exposure. Teratology. 1989;40:113–126. doi: 10.1002/tera.1420400205. [DOI] [PubMed] [Google Scholar]
- 42.Schwartz PM, Borghesani PR, Levy RL, Pomeroy SL, Segal RA. Abnormal cerebellar development and foliation in BDNF−/− mice reveals a role for neurotrophins in CNS patterning. Neuron. 1997;19:269–281. doi: 10.1016/s0896-6273(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 43.Shimoke K, Kubo T, Numakawa T, Abiru Y, Enokido Y, Takei N, Ikeuchi T, Hatanaka H. Involvement of phosphatidylinositol-3 kinase in prevention of low K(+)-induced apoptosis of cerebellar granule neurons. Brain Res Dev Brain Res. 1997;101:197–206. doi: 10.1016/s0165-3806(97)00065-5. [DOI] [PubMed] [Google Scholar]
- 44.Snell LD, Nunley KR, Lickteig RL, Browning MD, Tabakoff B, Hoffman PL. Regional and subunit specific changes in NMDA receptor mRNA and immunoreactivity in mouse brain following chronic ethanol ingestion. Mol Brain Res. 1996;40:71–78. doi: 10.1016/0169-328x(96)00038-1. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki K, Koike T. Brain-derived neurotrophic factor suppresses programmed death of cerebellar granule cells through a posttranslational mechanism. Mol Chem Neuropathol. 1997;30:101–124. doi: 10.1007/BF02815153. [DOI] [PubMed] [Google Scholar]
- 46.Tabakoff B, Hoffman PL. Biochemical pharmacology of alcohol. In: Meltzer HY, editor. Psychopharmacology—the third generation of progress. Raven; New York: 1987. pp. 1521–1526. [Google Scholar]
- 47.Vaudry D, Gonzalez BJ, Basille M, Anouar Y, Fournier A, Vaudry H. Pituitary adenylate cyclase-activating polypeptide stimulates both c-fos gene expression and cell survival in rat cerebellar granule neurons through activation of the protein kinase A pathway. Neuroscience. 1998;84:801–812. doi: 10.1016/s0306-4522(97)00545-9. [DOI] [PubMed] [Google Scholar]
- 48.Villalba M, Bockaert J, Journot L. Pituitary adenylate cyclase-activating polypeptide (PACAP-38) protects cerebellar granule neurons from apoptosis by activating the mitogen-activated protein kinase (MAP kinase) pathway. J Neurosci. 1997;17:83–90. doi: 10.1523/JNEUROSCI.17-01-00083.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 50.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamada M, Ohnishi H, Sano S, Nakatani A, Ikeuchi T, Hatanaka H. Insulin receptor substrate (IRS)-1 and IRS-2 are tyrosine-phosphorylated and associated with phosphatidylinositol 3-kinase in response to brain-derived neurotrophic factor in cultured cerebral cortical neurons. J Biol Chem. 1997;272:30334–30339. doi: 10.1074/jbc.272.48.30334. [DOI] [PubMed] [Google Scholar]
- 52.Yan G-M, Ni B, Weller M, Wood KA, Paul SM. Depolarization or glutamate receptor activation blocks apoptotic cell death of cultured cerebellar granule neurons. Brain Res. 1994;656:43–51. doi: 10.1016/0006-8993(94)91364-1. [DOI] [PubMed] [Google Scholar]
- 53.Yan G-M, Lin S-Z, Irwin RP, Paul SM. Activation of G proteins bidirectionally affects apoptosis of cultured cerebellar granule neurons. J Neurochem. 1995;65:2425–2431. doi: 10.1046/j.1471-4159.1995.65062425.x. [DOI] [PubMed] [Google Scholar]
- 54.Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- 55.Zafra F, Hengerer B, Leibrock J, Thoenen H, Lindholm D. Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J. 1990;9:3545–3550. doi: 10.1002/j.1460-2075.1990.tb07564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zafra F, Castrén E, Thoenen H, Lindholm D. Interplay between glutamate and gamma-aminobutyric acid transmitter systems in the physiological regulation of brain-derived neurotrophic factor and nerve growth factor synthesis in hippocampal neurons. Proc Natl Acad Sci USA. 1991;88:10037–10041. doi: 10.1073/pnas.88.22.10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang FX, Rubin R, Rooney TA. Ethanol induces apoptosis in cerebellar granule neurons by inhibiting insulin-like growth factor 1 signaling. J Neurochem. 1998;71:196–204. doi: 10.1046/j.1471-4159.1998.71010196.x. [DOI] [PubMed] [Google Scholar]