

Abstract
Transgenic mice expressing exon 1 of the human Huntington’s disease (HD) gene carrying a 141–157 CAG repeat (line R6/2) develop a progressive neurological phenotype with motor symptoms resembling those seen in HD. We have characterized the motor deficits in R6/2 mice using a battery of behavioral tests selected to measure motor aspects of swimming, fore- and hindlimb coordination, balance, and sensorimotor gating [swimming tank, rotarod, raised beam, fore- and hindpaw footprinting, and acoustic startle/prepulse inhibition (PPI)]. Behavioral testing was performed on female hemizygotic R6/2 transgenic mice (n = 9) and female wild-type littermates (n = 22) between 5 and 14 weeks of age. Transgenic mice did not show an overt behavioral phenotype until around 8 weeks of age. However, as early as 5–6 weeks of age they had significant difficulty swimming, traversing the narrowest square (5 mm) raised beam, and maintaining balance on the rotarod at rotation speeds of 33–44 rpm. Furthermore, they showed significant impairment in prepulse inhibition (an impairment also seen in patients with HD). Between 8 and 15 weeks, R6/2 transgenic mice showed a progressive deterioration in performance on all of the motor tests. Thus R6/2 mice show measurable deficits in motor behavior that begin subtly and increase progressively until death. Our data support the use of R6/2 mice as a model of HD and indicate that they may be useful for evaluating therapeutic strategies for HD, particularly those aimed at reducing the severity of motor symptoms or slowing the course of the disease.
Keywords: transgenic mice, Huntington’s disease, CAG repeat, motor behavior, prepulse inhibition, sensorimotor gating, polyglutamine repeat diseases
Huntington’s disease (HD) is a progressive, genetic neurodegenerative disorder characterized by movement abnormalities, cognitive impairments, and an emotional disturbance (Harper, 1996). The predominant movement abnormality is chorea, although other motor abnormalities including dyskinesia, dystonia, rigidity, and tremor are also seen, the latter particularly in juvenile-onset HD.
An expanded unstable CAG trinucleotide repeat within the coding region of the HD gene has been identified as the genetic mutation responsible for the disease (The Huntington’s Disease Collaborative Group, 1993). It now appears that HD belongs to a family of neurodegenerative disorders associated with an expansion of CAG repeats in the causative gene. To date, seven additional CAG “triplet repeat” diseases have been identified, including spinal and bulbar muscular atrophy, spinocerebellar ataxia types 1, 2, 3, 6, and 7, and dentatorubral-pallidoluysian atrophy (for references, see Paulson and Fischbeck, 1997). The causative genes in each of these disorders are different and unrelated in function, although the expansion of the CAG repeats and the progressive neurological nature of these disorders suggests that they may share a common mechanism of pathogenesis (MacDonald and Gusella, 1996; Burright et al., 1997).
Over the past 20 years, many attempts have been made to generate animal models of HD. Of particular importance have been the excitotoxic and 3-nitropropionic acid (3-NP) models that replicate many of the histological features of HD in both rodents and primates (Beal et al., 1986, 1993; Brouillet et al., 1995). 3-NP models in particular reportedly model some of the motor symptoms of HD, including abnormalities in gait and impairments in sensorimotor gating (Borlongan et al., 1995; Brouillet et al., 1995; Kodsi and Swerdlow, 1997). However, because both models are neurochemically induced, the associated changes in behavior are not truly progressive. Thus they are limited in their usefulness for testing treatments aimed at preventing or slowing the motor dysfunction in HD. An animal model that exhibits a progressive neurological phenotype is essential before such studies can be usefully conducted.
A transgenic mouse model for HD (R6/2) that appears to have a progressive phenotype has been developed recently (Mangiarini et al., 1996). R6/2 mice, whose transgene contains the first exon of the humanHD gene carrying 141–157 CAG repeats, display a number of the key neuropathological features of HD (Davies et al., 1997; Cha et al., 1998). Critically, preliminary investigations showed that R6/2 transgenic mice develop several of the motoric characteristics of HD (Mangiarini et al., 1996), including a progressive hypoactivity similar to that seen in patients with HD (Carter et al., 1998; Dunnett et al., 1998). However, so far the behavioral phenotype of this mouse has not been analyzed in sufficient detail to determine its usefulness as a progressive model of HD. Therefore, in this study we have characterized the progressive motor deficits displayed by R6/2 transgenic mice. These data will provide a baseline against which to assess (1) the relevance of this mouse model to HD and (2) its potential usefulness for testing therapies aimed at treating this devastating disease.
MATERIALS AND METHODS
Animals (R6/2 transgenic mice). The R6/2 line of transgenic mice was generated as described previously (Mangiarini et al., 1996). A colony of R6/2 transgenic mice was established in the Department of Pharmacology, University of Cambridge, and the line was maintained by backcrossing to CBA × C57BL/6 F1 animals. Genotyping was confirmed by PCR based on a modification of the method of Mangiarini et al. (1996). The present study used 9 female hemizygotic transgenic mice and 22 female wild-type littermate control mice from six litters of the thirteenth generation, all born within 5 d of each other. Females were used because transgenic males were used for breeding and were not available for behavioral testing. The age of onset of overt symptoms in the R6/2 line is ∼2 months, with the severity of symptoms progressing rapidly over the following few weeks, as reported previously (Mangiarini et al., 1996). Here, behavioral testing began at 5–6 weeks of age, when transgenic mice were overtly phenotypically indistinguishable from their normal littermates. Tests were conducted regularly (see below) over a 10 week period to follow the progression of motor symptoms. One transgenic mouse died during the final week of the study; data from this mouse were included in the analysis.
Mice were housed together in numerical birth order in groups of mixed genotype, and data were recorded for analysis by mouse number. Until the appearance of the hindlimb grooming behavior, transgenic mice could not be distinguished from normal mice in their home cage. Therefore, until the grooming behavior appeared (between 8–9 weeks of age), the experimenters were blind to the genotype of the mice. Although data collected after the onset of an overt phenotype was not collected blind, it should be noted that the home cage observations for overt phenotype were performed and recorded separately from the behavioral tests. Because the abnormal grooming in its early stage is difficult to distinguish from normal grooming behaviors, this meant that until the abnormal grooming movements occurred regularly, the experimenters did not necessarily know the genotype of a particular mouse. Once the grooming behavior and other phenotypic changes became pronounced (usually between 10 and 14 weeks), it was impossible to conduct the experiments blind.
Body weight. Body weight was measured twice per week.
Motor tests. Mice were trained on a battery of motor and cognitive tests. Five motor tests of swimming, fore- and hindlimb coordination, balance, and sensorimotor gating are reported here. After the establishment of stable baselines, animals were retested at weekly intervals, with the exception of the acoustic startle test, in which animals were retested every 2 weeks. The mean age of mice midweek of each testing session was used for analysis and presentation purposes.
Swimming tank. To monitor swimming movements, mice were trained to swim from one end of a water-filled glass tank to a visible escape platform at the opposite end (Perry et al., 1995). The glass tank was 100 cm long and 6 cm wide and was filled to a depth of 20 cm with water at a temperature of 23°C. The visible escape platform was made from black perspex (6 cm square and 20.5 cm high), with the top surface 0.5 cm above the water level. A vertical black line on the side of the glass marked a horizontal distance 60 cm from the platform; this served as the start line for recording swimming performance. During the training period each mouse was placed in the water at one end of the tank, and within a couple of trials learned to swim straight to the visible escape platform at the opposite end. After training, mice were given two trials per day for 3 consecutive days, by which time they reached stable baseline performance levels. Mice were then given two consecutive trials on a weekly basis. Mice were videotaped from both sides, and the number of forelimb kicks, the number of hindlimb kicks, and the latency to swim the 60 cm distance were recorded. Analysis was based on the mean scores of the two trials for each measure.
Beam walking. Motor coordination and balance of mice were assessed by measuring the ability of the mice to traverse a graded series of narrow beams to reach an enclosed safety platform (Perry et al., 1995). The beams consisted of long strips of wood (1 m) with a 28-, 12-, or 5-mm-square cross-section or a 28, 17, or 11 mm round diameter. The beams were placed horizontally, 50 cm above the bench surface, with one end mounted on a narrow support and the other end attached to an enclosed box (20 cm square) into which the mouse could escape. Two anglepoise lights (60 W) were positioned above and to one side of the start of the beam. During training, mice were placed at the start of the 12-mm-square beam and trained over 3 d (4 trials per day) to traverse the beam to the enclosed box. Once the mice were trained (traversed the 12-mm-square beam in < 20 sec) they received two consecutive trials on each of the square beams and each of the round beams, in each case progressing from the widest to the narrowest beam. Mice were allowed up to 60 sec to traverse each beam. The latency to traverse each beam and the number of times the hind feet slipped off each beam were recorded for each trial. Analysis of each measure was based on the mean scores of the two trials for each beam.
Rotarod. The rotarod apparatus (Accelerating Model, Ugo Basile, Biological Research Apparatus, Varese, Italy) was used to measure fore- and hindlimb motor coordination and balance. During the training period, each mouse was placed on the rotarod at a constant speed (24 rpm) for a maximum of 60 sec, and the latency to fall off the rotarod within this time period was recorded. Mice received four trials per day for 3 consecutive days, by which time a steady baseline level of performance was attained. Mice then received two trials at 10 increasing speed levels, ranging from 5 to 44 rpm. The mean latency to fall off the rotarod (for the two trials at each speed level) was recorded and used in subsequent analysis.
Footprint test. The footprint test was used to compare the gait of R6/2 transgenic mice with that of wild-type control mice. To obtain footprints, the hind- and forefeet of the mice were coated with purple and orange nontoxic paints, respectively. The animals were then allowed to walk along a 50-cm-long, 10-cm-wide runway (with 10-cm-high walls) into an enclosed box. All mice had three training runs and were then given one run per week. A fresh sheet of white paper was placed on the floor of the runway for each run. The footprint patterns were analyzed for four step parameters (all measured in centimeters). (1) Stride length was measured as the average distance of forward movement between each stride. (2) Hind-base width and (3) front-base width were measured as the average distance between left and right hind footprints and left and right front footprints, respectively. These values were determined by measuring the perpendicular distance of a given step to a line connecting its opposite preceding and proceeding steps. (4) Distance from left or right front footprint/hind footprint overlap was used to measure uniformity of step alternation. When the center of the hind footprint fell on top of the center of the preceding front footprint, a value of zero was recorded. When the footprints did not overlap, the distance between the center of the footprints was recorded. For each step parameter, three values were measured from each run, excluding footprints made at the beginning and end of the run where the animal was initiating and finishing movement, respectively. The mean value of each set of three values was used in subsequent analysis.
Acoustic startle/prepulse inhibition (PPI). Acoustic startle and PPI responses were measured in a startle chamber (San Diego Instruments) adapted for mice. The chosen paradigm was adapted fromSwerdlow et al. (1995) and assessed startle amplitude and PPI using acoustic stimuli of 120 and 105 dB, a single prepulse interval (100 msec), and four different prepulse intensities [2, 4, 8, and 16 dB above background noise (white noise, 65 dB)]. A mouse was placed in the startle chamber and initially received a 5 min acclimatization period with background noise alone. The mouse was then presented with 72 startle trials, each trial consisting of one of three conditions: (1) a 30 msec 120 dB noise burst presented alone (S); (2) a 30 msec 120 dB noise burst preceded 100 msec by prepulses (30 msec noise bursts) that were 2, 4, 8, or 16 dB above background noise (PP2, PP4, PP8, or PP16, respectively); or (3) no stimulus (NS; background noise alone), which was used to measure baseline movement in the chamber. These six trial types (S, PP2, PP4, PP8, PP16, NS) were each repeated six times in a pseudorandom order (36 trials), such that each trial type was presented once within a block of six trials. This block of 36 trials was then repeated using an acoustic stimuli of 105 dB. Analysis was based on the mean of the six trials for each trial type.
Statistical analysis. Behavioral data were subjected to two-, three- or four-way ANOVA (SAS-RA software package) with one between-subject factor (genotype; transgenic or wild type) and with repeated measures on one or more factors depending on the test used (e.g., age, different beams, different rotarod speeds, etc.). Sidak’s test was used for multiple independent post hoc pair-wise comparisons between transgenic and wild-type mice at each relevant age and test level (Rohlf and Sokal, 1995). A critical value for significance of p < 0.05 was used throughout the study.
RESULTS
Genotyping
In all cases, mice genotyped as transgenic were those that exhibited a progressive deficit in the motor tests (see below).
Spontaneous appearance of motor deficits in R6/2 transgenic mice
At 5 weeks of age the home cage behavior of R6/2 transgenic mice was indistinguishable from that of normal littermate control mice. Overt signs of neurological abnormality were first evident by 8 weeks (58 ± 0.5 d) of age, similar to that reported previously (Mangiarini et al., 1996). The initial abnormal neurological signs included stereotypical hindlimb-grooming movements, dyskinesia of the hindlimbs when mice were suspended by the tail, and an irregular gait. The frequency and severity of these abnormalities worsened slowly over the following few weeks until by 12–13 weeks of age the animals showed clear signs of irregular gait, resting tremor, stereotypical grooming, abrupt and irregularly timed shuddering movements, occasional epileptic seizures, and body weight loss (see below).
R6/2 transgenic and control mice gained weight at a similar rate until 10 weeks of age (Fig. 1). The body weight of R6/2 transgenic mice stabilized at 10 weeks and then showed a gradual decline, whereas their wild-type littermates continued to grow throughout the study (genotype × age,F22,638 = 47.80, p < 0.0001). A significant decrease in total body weight in R6/2 transgenic mice was evident by 13 weeks of age.
Fig. 1.

Body weights of wild-type and R6/2 transgenic mice. Symbols indicate the means ± SEM body weight (gram) of wild-type (n = 22) and R6/2 transgenic (n = 9) mice. The body weight of R6/2 transgenic mice gradually declines from 12 weeks of age. Asterisksindicate significant differences between wild-type control R6/2 transgenic mice (**p < 0.01).
Measurement of progression of motor deficits in R6/2 transgenic mice
To assess in more detail the neurological consequences of the expression of the R6/2 transgene, R6/2 transgenic and wild-type littermate control mice were subjected to five tests of motor behavior.
Swimming tank
R6/2 transgenic mice displayed abnormalities in their swimming movements from the onset of testing, and these became more marked over subsequent weeks. When placed in the water, R6/2 transgenic mice often floated temporarily, adopted a twisted posture, and kicked in a splayed and uncoordinated manner with both hind- and forelimbs. In contrast, control mice swam with vigor immediately after being placed in the water, using mainly their hindlimbs to propel along. Although control mice maintained a constant swimming latency throughout the study, R6/2 transgenic mice swam significantly more slowly at 5–6 weeks of age (p < 0.0001) and swam even more slowly with an increase in age (genotype × age: F8,232 = 13.48, p < 0.0001) (Fig.2A). At 5–6 weeks of age, R6/2 transgenic mice also exhibited a significantly greater number of hind- and forelimb kicks to swim the 60 cm distance, compared with controls [hindlimb: p < 0.01 (Fig.2B); forelimb: p < 0.05 (Fig.2C)].
Fig. 2.
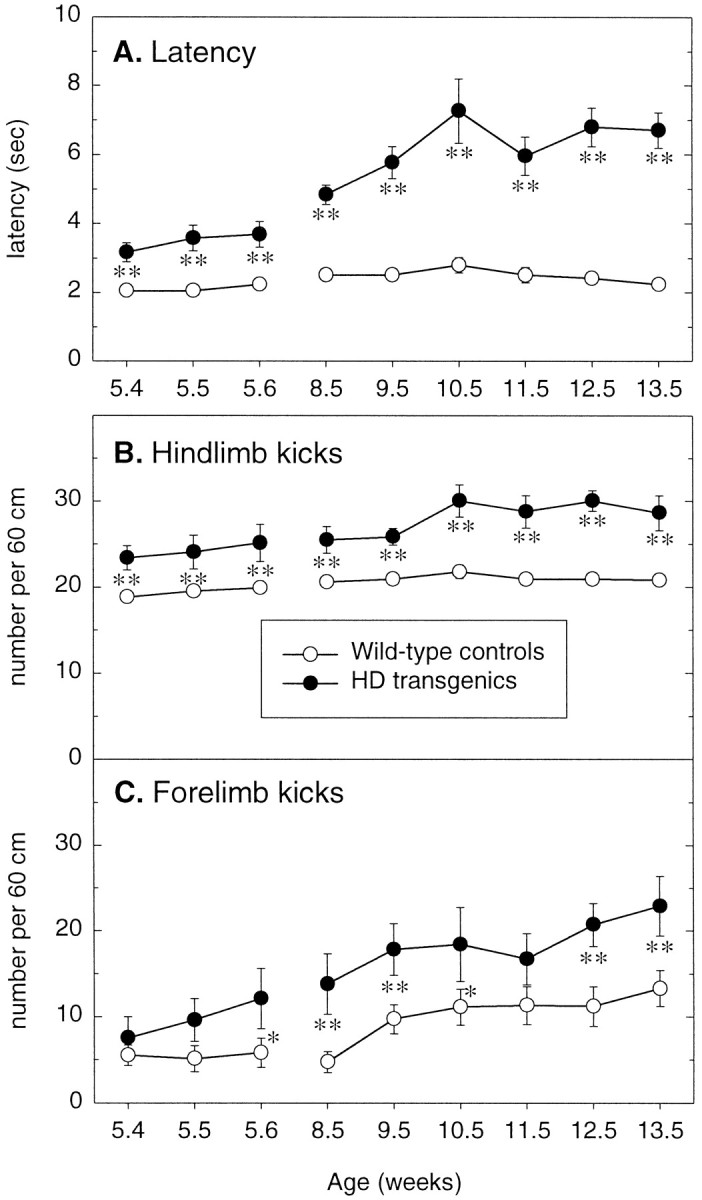
Swimming performance of wild-type and R6/2 transgenic mice in the swimming tank. Wild-type (n= 22) and R6/2 transgenic (n = 9) mice were tested in the swimming tank for two trials per day for 3 consecutive days at 5–6 weeks of age, retested at 8–9 weeks of age, and then weekly thereafter. Separate measurements were made of the latency (A), the number of hindlimb kicks (B), and the number of forelimb kicks (C) to swim 60 cm to a visible escape platform.Vertical bars indicate means ± SEM of each measure across all trials at each age. R6/2 transgenic mice display swimming impairments at the earliest age tested, 5–6 weeks, which progressively worsen over subsequent weeks. Asterisks indicate significant differences between wild-type control and R6/2 transgenic mice (*p < 0.05, **p < 0.01).
The frequency of hindlimb kicking increased significantly with the age of the R6/2 transgenic mice, in contrast with the control group, which sustained a consistent number of hindlimb kicks over the 10 week period (genotype × age: F18,232 = 2.24,p < 0.05). Although the number of forelimb kicks of R6/2 transgenic mice was invariably significantly greater than that of their wild-type littermates across sessions, no significant genotype × age interaction was observed (F8,232 = 2.02, p = 0.078), because both groups increased the frequency of forelimb kicking across sessions. The three-way interaction (genotype × limb × age:F8,464 = 1.34, p = 0.25) failed to reach significance, suggesting that the ratio of forelimb to hindlimb kicks was comparable for R6/2 transgenic and control mice (Fig. 2B,C). Swimming impairments, evident at 5–6 weeks of age, occurred at a time when the R6/2 transgenic animals were otherwise indistinguishable from their normal littermate controls.
Beam walking
Beam walking was used to compare the fine motor coordination and balance capabilities of R6/2 transgenic and control animals. Different levels of task difficulty were achieved by varying the shape and cross-section of the beams (Fig. 3). At the earliest age tested, 5–6 weeks, the control and R6/2 transgenic mice walked along all the beam types with ease, although the R6/2 transgenic mice were significantly slower in traversing the narrowest square beam (p < 0.0001) (Fig. 3C). However, by 8–9 weeks of age (when home cage phenotypic abnormalities first become apparent), R6/2 transgenic mice showed significant difficulty in traversing all but the widest round beam, as measured by their increased latency to traverse each beam, compared with control mice. By 13–14 weeks of age, R6/2 transgenic mice took up to 12 times as long as controls to traverse all the beams (p< 0.0001).
Fig. 3.
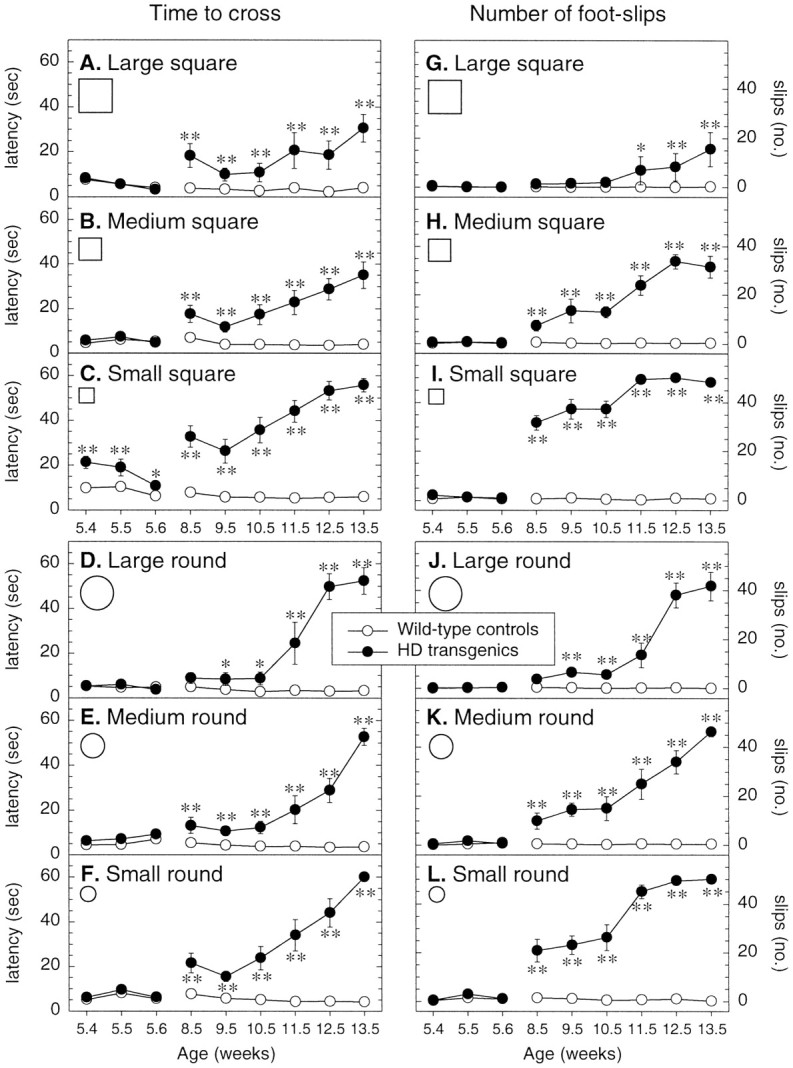
Balance and motor coordination on the raised beams. Wild-type (n = 22) and R6/2 transgenic (n = 9) mice were trained to walk across six progressively more difficult beams of square and round cross-section to reach an enclosed safety platform. The latency to cross (A–F) and the number of footslips made (G–L) were recorded on each trial. All animals were given two trials on each of the graded square and round beams, in each case progressing from the widest to the narrowest, for 3 consecutive days at 5–6 weeks of age, retested at 8–9 weeks of age, and then weekly thereafter. Individual symbols represent means ± SEM of the two groups on each test and age. R6/2 transgenic mice exhibit a progressive decline in beam-walking ability with age and beam difficulty. Asterisks indicate significant differences between wild-type control and R6/2 transgenic mice (*p < 0.05, **p < 0.01).
At 8–9 weeks of age, R6/2 transgenic mice also made significantly more footslips than control mice on all but the widest square and round beams, the frequency of which increased with age and as the beams became narrower. One obvious difference between R6/2 transgenic and wild-type control mice was the manner in which the R6/2 transgenic mice traversed the beams. At the onset of testing, R6/2 transgenic and wild-type control mice adopted a stable upright posture, but by 10–11 weeks of age most R6/2 transgenic mice displayed ventral recumbence: their thorax and abdomen were flattened against the upper surface of each beam, their hind- and forelimbs were wrapped around the lateral surface of each beam, and their forelimbs were used to drag themselves along the beam while their hindlimbs became redundant. By 13–14 weeks of age, some R6/2 transgenic mice failed to maintain balance on the beams and fell off. When this occurred on three consecutive trials the animal was no longer tested. In subsequent trials, for purposes of analysis, such animals were given a maximum latency of 60 sec and a footslip score of 50, which equates to the highest number of footslips before outright failure.
The significance of these observations was confirmed by ANOVA, which indicated a significant effect of genotype on both the latency and the number of footslips made while traversing the different beams (F1,29 = 528.00 and 1441.06, respectively; bothp < 0.0001) and a significant interaction between genotype × age × beam (shape and size) for both measures (latency: F16,1392 = 4.75; footslips:F16,1392 = 8.69; both p < 0.0001).
Rotarod
Motor coordination and balance of mice were measured using a rotarod. All mice used in this study showed learning of the rotarod test and reached a stable level of performance within 3 d, as measured by an increase in the mean duration time for mice to maintain balance on the rotarod (data not shown). Throughout the study control mice maintained balance on the rotarod for the maximum latency of 60 sec, on all rotation speeds, with the exception of 33 and 44 rpm, when they would occasionally fall off (Fig.4G,H).
Fig. 4.

Balance and motor coordination on the rotarod. Wild-type (n = 22) and R6/2 transgenic (n = 9) mice were subjected to eight different speeds on an accelerating rotarod (5–44 rpm), receiving two trials per speed. The means ± SEM of the duration of balance or latency to fall (maximum trial length = 60 sec) for the two trials at each speed level was recorded. Animals 5–6 weeks old were tested to establish baseline performance, and animals were retested at 8–9 weeks of age and then weekly thereafter. R6/2 transgenic mice 5–6 weeks old exhibit difficulty maintaining balance on the rotarod at the highest rotation speeds (G, H) and display a progressive decline in performance on the rotarod with increasing rotation speed and age. Asterisks indicate significant differences between wild-type control and R6/2 transgenic mice (*p < 0.05, **p < 0.01).
R6/2 transgenic mice 5–6 weeks old had difficulty maintaining balance on the rotarod at the highest rotation speeds of 33 and 44 rpm (p < 0.0001) (Fig. 4G,H). Thus these changes in rotarod rotation speeds identify significant deficits in motor coordination and balance of R6/2 transgenic mice even before any overt phenotype is detectable. There was a progressive decline in performance of R6/2 transgenic mice on the rotarod at all 10 speeds over ensuing weeks, and by 13–14 weeks of age R6/2 transgenic mice failed to maintain balance on the rotarod at any speed for >10 sec (Fig. 4A–H).
The significance of the difference between groups was confirmed by ANOVA, which revealed a highly significant main effect of genotype (F1,29 = 1868.70, p < 0.0001), genotype × speed (F10,319 = 18.21,p < 0.0001), and genotype × age (F6,174 = 350.54, p < 0.0001), and a significant genotype × speed × age interaction (F60,1914 = 3.57, p < 0.0001), reflecting a significant difference between the performance of R6/2 transgenic and control mice with an increase in rotarod rotation speed and age.
Footprint test
Gait abnormalities were assessed by analyzing the footprint pattern of mice while they walked along a narrow corridor. Footprint patterns of wild-type and R6/2 transgenic mice at 13–14 weeks of age are illustrated in Figure 5. At all ages, control mice walked in a straight line, with a regular even alternating gait, placing the hindpaw precisely at the position where the ipsilateral forepaw had been in the previous step (Fig. 5A). By contrast, with increasing age, R6/2 transgenic mice progressively weaved from side to side while walking along the runway, adopting unevenly spaced shorter strides, staggering movements, and a gait that lacked a normal, uniform, alternating left–right step pattern (Fig.5B).
Fig. 5.
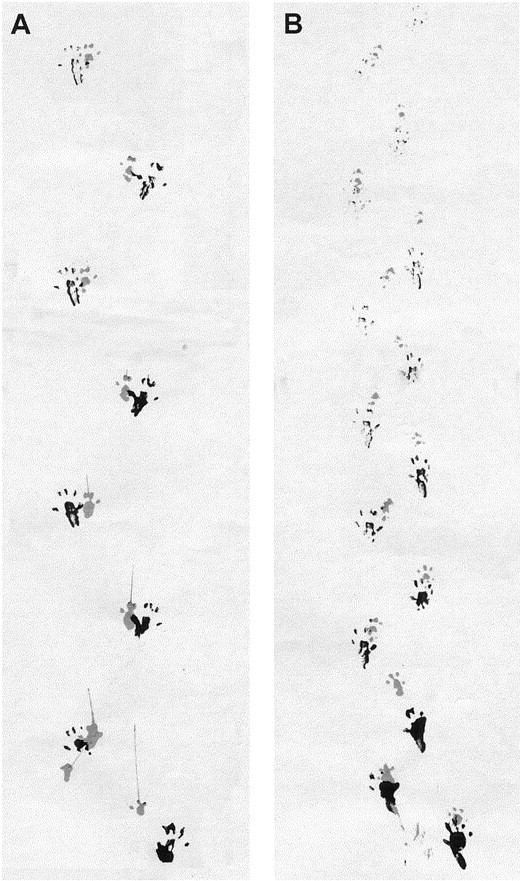
Representative walking footprint patterns of 13- to 14-week-old wild-type (A) and R6/2 transgenic (B) mice. Qualitatively, the generated patterns clearly differ showing that R6/2 transgenic mice display irregularly spaced shorter strides and an uneven left–right step pattern as compared with the evenly spaced and accurately positioned footprints of the wild-type control mice.
The resulting footprint patterns were assessed quantitatively by four measurements: stride length, hindbase width, frontbase width, and front footprint/hind footprint overlap. Although R6/2 transgenic and control mice exhibited comparable stride lengths at 5–6 weeks of age, these gradually deviated with age (genotype × age:F6,174 = 34.01, p < 0.0001) (Fig. 6A). The stride length of control mice increased significantly with age. By contrast, by 8–9 weeks, R6/2 transgenic mice displayed a significantly shorter stride length compared with control mice (p < 0.0001), and this effect became more pronounced as the mice got older (Fig. 6A).
Fig. 6.
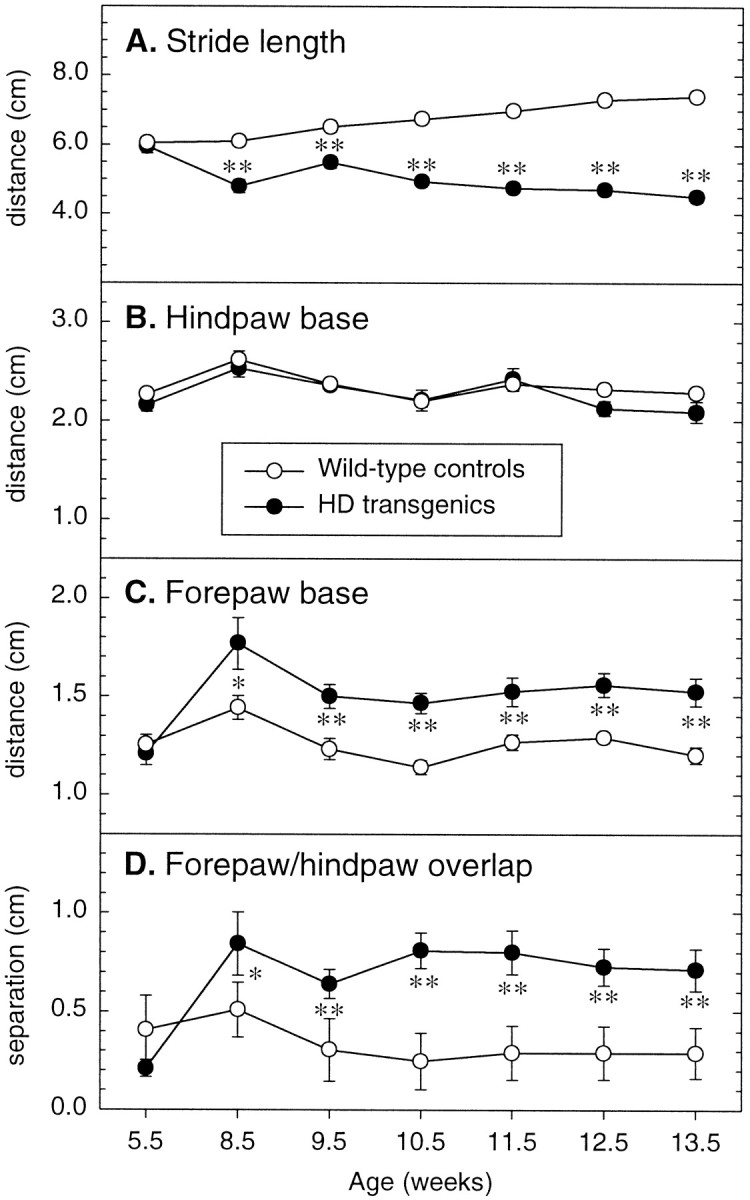
Quantitative analysis of the walking footprint patterns produced by wild-type and R6/2 transgenic mice, based on measurements of stride length (A), hindbase width (B), frontbase width (C), and distance between front and hind footprint placement or overlap (D). At 5–6 weeks of age the footprint patterns of R6/2 transgenic mice are indistinguishable from those produced by wild-type control mice, but by 8–9 weeks of age, R6/2 transgenic mice exhibit a significantly shorter stride length, a broader frontbase width, and a greater distance between front footprint/hind footprint overlap, as compared with wild-type control mice.Symbols indicate means ± SEM by mice of each group at each age on each measure. Asterisks indicate significant differences in the performance of wild-type control and R6/2 transgenic mice (*p < 0.05, **p < 0.01).
The hindbase width of R6/2 transgenic mice did not differ from that of their wild-type littermates throughout the study (genotype:F1,29 = 0.92, p = 0.345; genotype × age: F6,174 = 0.97,p = 0.442) (Fig. 6B). In contrast, the frontbase width of R6/2 transgenic mice was significantly broader and more splayed than that shown by controls at 8–9 weeks of age, and this difference was sustained over the five succeeding weeks (genotype: F1,29 = 23.60, p< 0.0001; genotype × age: F6,174 = 2.88,p = 0.013) (Fig. 6C).
The distance from left or right front footprint/hind footprint overlap provides an indication of uniformity of step alternation. As shown in Figure 6D, R6/2 transgenic mice displayed a similar uniformity in step alternation at 5–6 weeks of age compared with controls, but by 8–9 weeks of age the regular left–right step pattern began showing disruption, and they exhibited a progressively greater distance between front and hind footprint placement (i.e., a reduced overlap) as compared with controls. The progressive difference between R6/2 transgenic and control mice with age was highly significant (genotype × age: F6,174 = 8.06,p < 0.0001) (Fig. 6D).
Prepulse inhibition of the acoustic startle response
Acoustic startle is a motor reflex response to an intense loud noise stimulus. As shown in Figure7A,F, the acoustic startle response to 120 and 105 dB, respectively, did not differ between control and R6/2 transgenic mice over the first 10–11 weeks of age, but did decline in the R6/2 transgenic mice at the final test at 12.5 weeks of age (genotype × age: F3,87 = 4.88, p < 0.01 and F3,87=4.85, p < 0.007, respectively). It is noteworthy that the change in magnitude of acoustic startle responses during the course of the study appeared to parallel the change in body weight of R6/2 transgenic and control mice (Fig. 1).
Fig. 7.

Prepulse inhibition of the acoustic startle response in wild-type (n = 22) and R6/2 transgenic (n = 9) mice. Symbols indicate mean ± SEM of the response of the two groups at different ages to white noise stimuli of 120 dB (A–E) and 105 dB (F–J) with either the baseline startle response recorded to the primary acoustic startle stimulus alone (A, F, respectively) or with 100 msec prepulse warning stimuli of rising intensities (2, 4, 8, and 16 in B–Eand G–J, respectively). R6/2 transgenic mice exhibit impaired PPI by 8–9 weeks of age, and this impairment progressively declines with age. Asterisks indicate significant differences in performance between the two genotypes (*p < 0.05, **p < 0.01).
The acoustic startle response is reduced when a prepulse stimulus is presented before the main startle stimulus. The level of PPI, expressed as a percentage reduction of the baseline startle response, obtained with each of the four different prepulses in R6/2 transgenic and littermate control mice at different ages of testing is shown in Figure7B–E, G–J. At 5 weeks of age, R6/2 transgenic mice and wild-type littermate control mice displayed comparable PPI at all prepulse intensities. However, compared with control mice, a significant reduction of PPI was apparent in R6/2 transgenic mice by 8–9 weeks of age at prepulse intensity levels of 2 and 4 dB. By 12–13 weeks of age PPI was significantly disrupted in R6/2 transgenic mice at all four prepulse intensity levels.
Repeated-measures ANOVA of PPI to 120 and 105 dB startle stimuli revealed significant effects of genotype (F1,29= 23.23 and 35.96, respectively; both p < 0.001) and genotype × age (F3,87 = 8.02 and 16.18, respectively; both p < 0.001), confirming that the transgenic mice exhibited a significant progressively greater PPI deficit as they aged. However, neither the genotype × prepulse intensity (F3,87 = 0.30, p = 0.828 and F3,87 = 0.22, p = 0.925, respectively) nor the genotype × prepulse intensity × age (120 dB: F9,348 = 0.82, p= 0.597; 105 dB: F9,348 = 0.60,p = 0.794) approached significance, confirming the observation that the age-dependent deficit in the transgenic mice was apparent and of similar magnitude at all prepulse intensities, notwithstanding the fact that the higher intensity prepulses induced a greater inhibition than lower intensity prepulses in the transgenic and control mice alike.
DISCUSSION
Motor symptoms play a prominent role in the early stages of HD (Harper, 1996), and R6/2 transgenic mice have been reported to display a number of motor abnormalities reminiscent of those occurring in HD, including dyskinetic movements, resting tremor, seizures, and locomotor abnormalities (Mangiarini et al., 1996; Carter et al., 1998; Dunnett et al., 1998). In this study we tested motor function in R6/2 mice and showed that on all motor tests performed they display a marked and progressive deterioration in performance, compared with wild-type littermate control mice.
Behavioral testing commenced at 5–6 weeks of age. This age was chosen as the starting point because it was several weeks before overt symptoms first appear in R6/2 mice. However, even at 5–6 weeks it was apparent that R6/2 transgenic mice exhibited difficulties in a number of tasks (namely swimming, traversing the narrowest square raised beam, and maintaining balance on the rotarod at the fastest rotating speeds). For example, when R6/2 transgenic mice were placed in the swim tank, their abnormal posture and inappropriate kicking movements contrasted sharply with the coordinated, synchronized paddling movements shown by wild-type mice. Thus from 5–6 weeks the R6/2 transgenic mice swam more slowly and more inefficiently than wild-type control mice, although there were no deficits seen in the other tests (gait, sensorimotor gating, or motor performance on the lesser challenging wider beams and slower rotarod speeds). Furthermore, in their home cage at this age the transgenic mice were indistinguishable from their wild-type littermates by simple observation. There is therefore no clear “age of onset” of neurological phenotype in R6/2 transgenic mice; the age at which a phenotype is apparent depends on the sensitivity of the test and the difficulty of the particular task used to characterize it. Even if a mouse appears “normal” on inspection, a more sensitive test may reveal significant deficits in fine motor control and motor coordination.
By 8 weeks of age, signs of neurological abnormality in R6/2 mice were occasionally observed in the home cage, in particular stereotypical hindlimb grooming movements. The stereotypical grooming increased in frequency between 8 and 12 weeks, and at the same time, other deficits in motor activity became apparent. In particular, balance and coordination become increasingly impaired, and dyskinesia and resting tremor ensued. The frequency and severity of these abnormalities worsened gradually, until by 11 weeks of age the mice groomed frequently, there were clear signs of a constant resting tremor, and occasional epileptic seizures were evident. Concurrent with the onset of severe motor deficits, and consistent with the findings of Davies et al. (1997), R6/2 mice showed a progressive loss of body weight, compared with littermate controls. In our study this was not until 12 weeks of age, nearly 2 months after motor deficits were first observed. Therefore the motor deficits reported here cannot be attributed to loss of body weight. This suggests that the onset of motor symptoms is a consequence of the transgene rather than changes in body weight. It is interesting that although they developed the overt neurological phenotype at a similar time, mice in our colony in Cambridge began to lose weight nearly 4 weeks later than reported previously (Davies et al., 1997). Because their genetic background is the same as the original colony, we attribute the differences between the two colonies to changes in husbandry of our colony resulting in improved access to food and water.
Between 8 and 14 weeks of age, the performance of R6/2 transgenic mice in the swimming tank test worsened progressively. Performance on the beam and rotarod also deteriorated over this time, with the mice taking significantly longer to traverse all six beam types, exhibiting a greater number of footslips, and failing to maintain balance on the rotarod at all rotation speeds. Deficiencies in gait also became apparent from 8 weeks of age. By 12 weeks these deficits were pronounced; instead of walking in a straight line with evenly spaced and accurately positioned footprints, R6/2 transgenic mice often weaved from side to side, adopting staggering movements, irregularly spaced shorter strides, and a gait that lacked a normal, uniform, alternating left–right step pattern. Notably, these abnormalities were similar to gait disturbances described in patients with HD (Koller and Trimble, 1985; Harper, 1996) and also in 3-NP treated rats (Guyot et al., 1997). This latter observation is particularly interesting, because we have observed that the manner in which the R6/2 mice and 3-NP-treated rats traversed the beams is also very similar (R. J. Carter and A. J. Morton, unpublished observations). Because 3-NP treatment is a widely accepted model of the striatal pathology in HD, and the behavioral changes shown by 3-NP-treated rats have been attributed to selective striatal damage (Guyot et al., 1997), the striking similarities in the motor deficits of 3-NP-treated rats and R6/2 transgenic mice suggest that despite the absence of striatal neuronal loss, the motor deficits seen in R6/2 transgenic mice arise from striatal dysfunction.
R6/2 transgenic mice exhibited normal acoustic startle responses until relatively late in the study when 12- to 13-week-old R6/2 transgenic mice revealed a decline on both startle measures. In contrast, the degree to which a prepulse inhibits the acoustic startle reflex was significantly reduced in R6/2 transgenic mice at 8 weeks of age (at the most sensitive, i.e., lowest, prepulse intensities). The impairment in PPI in R6/2 mice increased with age, consistent with the progressive impairment in PPI seen in patients with HD (Swerdlow et al., 1995). PPI of the acoustic startle response has also been reported in rats after quinolinic acid lesions of the striatum or systemic 3-NP treatment (Kodsi and Swerdlow, 1995, 1997). Because the response of R6/2 transgenic mice to the basic acoustic startle only differed from that of control mice at a relatively late age (and the analysis of PPI is corrected for baseline level of startle), the loss of PPI in R6/2 transgenic mice is unlikely to be caused simply by a generalized muscular weakness but suggests that in R6/2 mice (as in humans) abnormalities in PPI are derived from sensorimotor deficits in the CNS.
There is at present no unequivocal neuropathological substrate for the progressive deterioration in the motor performance of R6/2 transgenic mice. Previous work has shown that although by 12 weeks of age the brains of R6/2 transgenic mice weigh ∼20% less than those of wild-type control mice, there is no selective striatal cell loss (Mangiarini et al., 1996; Davies et al., 1997). Recent studies, however, show that R6/2 transgenic mice have neuropathological changes; in particular they develop neuronal intranuclear inclusions (NIIs) (Davies et al., 1997) and display a decreased expression of dopamine (D1, D2) and mGluR1 receptor mRNA in the striatum (Cha et al., 1998), from as early as 4–5 weeks of age. Similar NIIs have been found in postmortem brains of patients with HD (Di Figlia et al., 1997), and postmortem changes in dopamine receptors in HD have been well documented (Harper, 1996). Thus, it seems possible that the motor abnormalities we observed in R6/2 transgenic mice are attributable to dysfunction of the striatum, despite the lack of neuronal cell death in the striatum.
It is not yet known why neuronal loss is not seen in brains of R6/2 mice. The most likely explanation is that the early and sudden death of these mice occurs before neuronal loss is evident. However, although at first sight the lack of striatal neurodegeneration is a weakness of the model, striatal dysfunction without cell death is also seen in HD. The onset of motor symptoms in HD patients does not correlate well with neuropathological grading, although this issue is complicated by the enormous variation in the presentation of motor symptoms in HD. Although few systematic studies relating clinical symptoms and neuropathology in early stage HD have been performed, Meyers et al. (1988) described five patients who died with clinical features of the disease, including chorea and other involuntary movements, yet had no discernible neuropathological abnormalities (Grade 0 pathology). Notably, one of these patients had shown choreiform motor abnormalities for 4 years before his death, yet still showed no neuronal loss. These findings are similar to our findings in the mice and support the suggestion that R6/2 transgenic mice may not only provide a relevant genetic model of HD, but may also demonstrate a progressive neurological phenotype that is directly comparable to that seen in early HD.
Our study describes detailed motor and sensorimotor changes in R6/2 transgenic mice that progressively worsen with age and with test difficulty. The quantifiable progression of these motor deficits makes this R6/2 transgenic mouse model particularly suitable for assessing the effectiveness of potential therapeutic agents and repair strategies for treating the motor symptoms of HD. Moreover, this model of HD offers a unique system in which the testing of experimental treatments for HD can be performed even before an overt phenotype has developed.
Note
Since this work was submitted, we have become aware that diabetes is present in R6/2 transgenic mice. We have tested the mice in our colony and confirm that we have diabetic mice; indeed, diabetes is common in R6/2 mice >14 weeks of age. However, we have found neither elevated blood glucose nor urinary glucose to be present under a normal feeding regime in mice <9 weeks of age. Furthermore, of the mice used in this study, at 13 weeks only two of eight transgenic mice tested had elevated blood glucose levels (14 mmol/l, 27.8 mmol/l) and glucose in the urine (14 mmol/l, 27.8 mmol/l). The remaining six mice had normal plasma glucose levels (4–8 mmol/l) and no glucose in their urine. Therefore, during the critical testing period (5–10 weeks of age) in our study, we believe that any deleterious effects that might be associated with diabetes are unlikely to have contributed to the behavioral phenotype we observed in R6/2 mice.
Footnotes
This work was supported by grants from the Hereditary Disease Foundation and the Wellcome Trust (UK). R.J.C. is supported by the Medical Research Council (UK); L.A.L. is supported by Parke-Davis Neuroscience Research (UK). We are grateful to Chris Riches for valuable technical assistance.
R.J.C. and L.A.L. contributed equally to this work.
Correspondence should be addressed to Dr. Jenny Morton, Department of Pharmacology, University of Cambridge, Tennis Court Road, Cambridge, CB2 1QJ, UK.
REFERENCES
- 1.Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature. 1986;321:168–171. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- 2.Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterisation of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borlongan CV, Koutouzis TK, Freeman TB, Cahill DW, Sanberg PR. Behavioral pathology induced by repeated systemic injections of 3-nitropropionic acid mimics the motoric symptoms of Huntington’s disease. Brain Res. 1995;697:254–257. doi: 10.1016/0006-8993(95)00901-2. [DOI] [PubMed] [Google Scholar]
- 4.Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal MF. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci USA. 1995;92:7105–7109. doi: 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burright EN, Orr HT, Clark HB. Mouse models of human CAG repeat disorders. Brain Pathol. 1997;7:965–977. doi: 10.1111/j.1750-3639.1997.tb00896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carter RJ, Hickey MA, Mangiarini L, Mahal A, Bates GP, Morton AJ. Abnormal motor response to (+) methamphetamine in mice transgenic for the Huntington’s disease mutation. Eur J Pharmacol. 1998;10:371. [Google Scholar]
- 7.Cha JJ, Kosinski CH, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington’s disease gene. Proc Natl Acad Sci USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies SW, Turmaine M, Cozens B, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 9.Di Figlia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of Huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;227:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 10.Dunnett SB, Carter RJ, Watts C, Torres EM, Mahal A, Mangiarini L, Bates GP, Morton AJ. Striatal transplantation in a transgenic mouse model of Huntington’s disease. Exp Neurol. 1998;154:31–40. doi: 10.1006/exnr.1998.6926. [DOI] [PubMed] [Google Scholar]
- 11.Guyot M, Hantraye P, Dolan R, Palfi S, Mazier M, Brouillet E. Quantifiable bradykinesia, gait abnormalities and Huntington’s disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience. 1997;79:45–56. doi: 10.1016/s0306-4522(96)00602-1. [DOI] [PubMed] [Google Scholar]
- 12.Harper PS. Huntington’s Disease. W. B. Saunders; London: 1996. [Google Scholar]
- 13.Huntington’s Disease Collaborative Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 14.Kodsi MH, Swerdlow NR. Prepulse inhibition in the rat is regulated by ventral and caudodorsal striatopallidal circuitry. Behav Neurosci. 1995;109:912–928. doi: 10.1037//0735-7044.109.5.912. [DOI] [PubMed] [Google Scholar]
- 15.Kodsi MH, Swerdlow NR. Mitochondrial toxin 3-nitropropionic acid produces startle reflex abnormalities and striatal damage in rats that model some features of Huntington’s disease. Neurosci Lett. 1997;231:103–107. doi: 10.1016/s0304-3940(97)00482-5. [DOI] [PubMed] [Google Scholar]
- 16.Koller WC, Trimble J. The gait abnormality of Huntington’s disease. Neurology. 1985;35:1450–1454. doi: 10.1212/wnl.35.10.1450. [DOI] [PubMed] [Google Scholar]
- 17.MacDonald ME, Gusella JF. Huntington’s disease: translating a CAG repeat into a pathogenic mechanism. Curr Opin Neurobiol. 1996;6:638–643. doi: 10.1016/s0959-4388(96)80097-3. [DOI] [PubMed] [Google Scholar]
- 18.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 19.Paulson HL, Fischbeck KH. Trinucleotide repeats in neurogenetic disorders. Annu Rev Neurosci. 1996;19:79–107. doi: 10.1146/annurev.ne.19.030196.000455. [DOI] [PubMed] [Google Scholar]
- 20.Perry TA, Torres EM, Czech C, Beyreuther K, Richards S, Dunnett SB. Cognitive and motor function in transgenic mice carrying excess copies of the 695 and 751 amino acid isoforms of the amyloid precursor protein gene. Alzheimer’s Res. 1995;1:5–14. [Google Scholar]
- 21.Rohlf FJ, Sokal RR. Statistical Tables, Ed 3. Freeman; New York: 1995. [Google Scholar]
- 22.Sanberg PR, Calderon SF, Giordano M, Tew JM, Norman AB. The quinolinic acid model of Huntington’s disease: locomotor abnormalities. Exp Neurol. 1989;105:45–53. doi: 10.1016/0014-4886(89)90170-2. [DOI] [PubMed] [Google Scholar]
- 23.Swerdlow NR, Paulson J, Braff DL, Butters N, Geyer MA, Swenson MR. Impaired prepulse inhibition of acoustic and tactile startle response in patients with Huntington’s disease. J Neurol Neurosurg Psychiatry. 1995;58:192–200. doi: 10.1136/jnnp.58.2.192. [DOI] [PMC free article] [PubMed] [Google Scholar]