

Abstract
In mouse thalamocortical slices in vitro, the potassium channel blocker 4-AP and GABAA receptor antagonist bicuculline together induced spontaneous prolonged depolarizations in layer VI neurons from postnatal day 2 (P2), in ventroposterior nucleus neurons (VP) from P7, and in reticular nucleus neurons (RTN) from P8. Dual whole-cell recordings revealed that prolonged bursts were synchronized in layer VI, VP, and RTN. Bursts were present in cortex isolated from thalamus, but not in thalamus isolated from cortex, indicating that bursts originated in cortex and propagated to thalamus. Prolonged bursts were synchronized in layer VI when vertical cuts extended from pia mater through layers IV or V, but were no longer synchronized when cuts extended through layer VI and white matter.
In voltage-clamp recordings before P10, burst conductance of all three neuronal populations was dominated by the NMDA receptor-mediated conductance, and therefore synapses were “silent”. In cortex and RTN, after P10, bursts were associated with strong AMPA/kainate receptor-mediated conductances, and synapses had become “functional”; silent synapses persisted in a large proportion of VP cells after P10.
Before P9, the NMDA receptor antagonist APV or the non-NMDA receptor antagonist CNQX blocked the prolonged bursts. After P9, CNQX continued to block the prolonged bursts, but APV merely shortened their duration.
Thus, NMDA receptor-based silent synapses are essential for paroxysmal corticothalamic activity during early postnatal development, and connections between layer VI neurons are sufficient for horizontal cortical synchronization.
Keywords: ventroposterior nucleus, reticular nucleus, somatosensory cortex, NMDA receptor, cortical oscillations, paroxysmal activity
Synchronous oscillatory activity of large populations of forebrain neurons is a concomitant of changes in conscious state during arousal and sleep (Steriade et al., 1993). Synchronization of large ensembles of cortical and thalamic cells during state-dependent changes in rhythmic activities of the forebrain is dependent on the corticothalamic projection (Contreras et al., 1996). The development of low- and medium-frequency, state-dependent oscillations in the thalamocortical network is delayed until maturation of the intrinsic membrane properties of thalamic cells (Warren and Jones, 1997). Until these properties mature, certain forms of seizure activity, notably the absence seizures of childhood, are uncommon. Nevertheless, in both the immature and adult nervous system, small perturbations of inhibitory or excitatory systems lead to highly synchronized, paroxysmal activity that can propagate across large distances.
Recently, the existence of silent synapses has been revealed in the developing optic tectum of Xenopus and in the hippocampus and layer IV of the cerebral cortex of mammals (Crair and Malenka, 1995; Durand et al., 1996; Wu et al., 1996; Isaac et al., 1997). These synapses are called silent because they induce excitatory postsynaptic conductances that are mediated solely by the NMDA receptor, and hence do not conduct current at the resting membrane potential of the cell. Silent synapses can be converted into functional synapses, i.e., synapses that induce both NMDA and AMPA/kainate-mediated EPSCs, by pairing presynaptic activation with postsynaptic depolarization. The selective transformation of silent synapses into functional synapses has been hypothesized to underlie the Hebbian, activity-dependent shaping of retinotectal (Wu et al., 1996) and thalamocortical (Isaac et al., 1997) circuitry and may determine the selective stabilization or retraction of synapses during development.
Although the existence of silent thalamocortical synapses during early development has been a topic of considerable discussion (Isaac et al., 1997; Malenka and Nicoll, 1997), there has been little consideration of the potential existence of silent corticocortical and corticothalamic synapses or of how they may support synchronized oscillatory or paroxysmal activity. It is well known that the neonatal brain is especially susceptible to induction of epileptiform activity (Blom et al., 1978; Ellenberg et al., 1984), but there has been little study of the mechanisms of seizure induction and propagation in immature cortical circuits.
Most in vitro studies of epileptiform activity in the cortex have been performed on isolated cortical slices that eliminate the corticothalamocortical loop. Although intrinsic cortical circuitry is rich in recurrent excitatory connections and can support the existence of epileptiform activity on its own, paroxysmal activity in vivo undoubtedly involves the integration of activity at both cortical and thalamic levels (Neckelmann et al., 1998, Steriade et al., 1998; Steriade and Contreras, 1998; Timofeev et al., 1998). In this study, using an in vitro slice preparation that maintains thalamocortical and corticothalamic connectivity (Agmon and Connors, 1991), we demonstrate that the corticothalamocortical circuit dominated by silent synapses can support the existence of synchronized paroxysmal activity and that connections between layer VI cells are sufficient for the propagation of paroxysmal activity.
MATERIALS AND METHODS
Postnatal day 0 (P0)–P17 ICR mice (Harlan Sprague Dawley, Indianapolis, IN) mice were anesthetized by hypothermia (P0–P4) or with ether (P5–P17) and decapitated. The brain was quickly removed and put in chilled artificial CSF (ACSF) containing in mm: NaCl 126, KCl 3, NaH2PO4 1.25, MgSO4 1.3, CaCl2 2.5, NaHCO3 26, and dextrose 20, pH 7.4 when bubbled with 95% O2 and 5% CO2, osmolarity 300–315 mOsm. Slices (400-μm-thick) containing the somatosensory cortex, reticular nucleus (RTN), and ventroposterior nucleus (VP) were cut at an angle that preserves corticothalamic and thalamocortical connectivity (Agmon and Connors, 1991); in three experiments, the thalamus and cortex were disconnected by cuts through the internal capsule and striatum. In one experiment, a vertical cut extending from the pia mater to the superficial white matter was made in the cortex. In two other experiments, vertical cuts extending from the pia to the layer IV/layer V border, or to the layer V/layer VI border were made in slices in which the cortex was disconnected from the thalamus. Cuts were made with a thin steel blade while slices were immersed in chilled ACSF. Slices were transferred to a submersion-type chamber, superfused with ACSF aerated with 95% O2 and 5% CO2, and allowed to recover for at least 1 hr before recording. Fourteen cells were recorded at 35°C, and the remaining 377 cells were recorded at room temperature (22–25°C). Because prolonged bursts were present at similar frequencies and were indistinguishable at both temperatures, results from the two sets of experiments have been pooled. All voltage-clamp data were recorded at room temperature.
Whole-cell recording pipettes were pulled from borosilicate glass on a Narishige PP-83 two-stage puller and had resistances of 2–5 MΩ. Internal solutions (in mm) included: either (1) potassium gluconate 120, HEPES 10, EGTA 1, MgCl2 2, CaCl20.1, NaCl 20, Na2ATP 2, NaGTP 0.5, pH adjusted to 7.2–7.4 with KOH; or (2) CsOH 120, d-gluconic acid 120, HEPES 10, EGTA 1.1, MgCl2 2, CaCl2 0.1, NaCl 20, Na2ATP 2, NaGTP 0.5, QX-314 3, pH adjusted to 7.2–7.4 with CsOH. Osmolarity was adjusted to 290–300 mOsm. Biocytin 0.5% was present in the internal solution during most recordings. Current-clamp recordings were performed with an Axoclamp-2A amplifier (Axon Instruments, Foster City, CA) and occasionally with an Axopatch 200B (Axon Instruments). During current-clamp recordings, series resistance was typically 5–25 MΩ and never >50 MΩ, and was compensated in bridge mode. Voltage-clamp recordings were always performed with the Axopatch 200B. During voltage-clamp recordings, series resistance was 5–25 MΩ, was monitored regularly, and was compensated by 70–80%. Extracellular field recordings were performed with whole-cell recording pipettes filled with ACSF. Data were digitized via a CED 1401plus interface (Cambridge Electronic Design, Cambridge, UK) at 150 Hz for the first nine current-clamp recordings and at 5000 Hz for the remaining recordings.
Drugs used included (in μm): 4-aminopyridine (4-AP) 70, bicuculline methchloride (BMC) 10,d-2-amino-5-phosphonovaleric acid (APV) 50, and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) 20. BMC and CNQX were purchased from Research Biochemicals (Natick, MA). APV was purchased from Research Biochemicals and from Sigma (St. Louis, MO). 4-AP was purchased from Tocris Cookson (Ballwin, MO). All drugs were applied in the bath.
RESULTS
This study is based on data collected during whole-cell recordings from 181 neurons in the VP nucleus of the thalamus, from 162 in layer VI of the somatosensory cortex, and from 48 neurons in the RTN at P2–P17. These data include dual whole-cell recordings from 20 VP neuron–cortical neuron pairs, 12 cortical neuron–cortical neuron pairs, 15 VP neuron–VP neuron pairs, 1 cortical neuron–RTN neuron pair, 1 RTN neuron–RTN neuron pair, and 1 RTN neuron–VP neuron pair. All cells recorded with a potassium gluconate-based solution in the pipette had membrane potentials more negative than −60 mV and fired overshooting action potentials in response to depolarizing current pulses or over-riding spontaneous EPSPs. Field potentials were recorded in layer VI simultaneously with whole-cell recordings from 5 VP neurons. Membrane potentials were corrected for a 10 mV junction potential.
Induction of synchronized prolonged bursts in cortical, VP, and RTN neurons by 4-AP and bicuculline
Current-clamp recordings were obtained with pipettes filled with a potassium gluconate-based internal solution from 101 VP, 81 layer VI, and 7 RTN neurons in thalamocortical slices from P2–P16 mice. From P2 in layer VI, P7 in VP, and P8 in RTN, joint application of 4-AP and bicuculline elicited prolonged depolarizations crested with trains of action potentials (Fig.1A–F). In VP and RTN, these prolonged bursts were present only in a subset of cells (50–100%) at all postnatal stages. Between P2 and P9, prolonged bursts were present in 20 of 56 (36%) layer VI cortical neurons and in all but one layer VI cell after P9. The duration of prolonged bursts (0.5–85.9 sec) and the intervals separating the prolonged bursts (0.8–454.0 sec) could remain relatively constant during the recording, but usually varied considerably. Bursts longer than 20 sec in duration were only recorded very rarely. Linear regression analysis demonstrated that the mean interval between the bursts decreased during postnatal development (data not shown). Furthermore, the duration of a bursts was often related to the interval of time separating it from the previous burst; bursts after a prolonged silence were usually but not invariably longer in duration than those after a short silent period.
Fig. 1.

Paired whole-cell recordings from a layer VI–layer VI neuron pair (A), a layer VI–VP neuron pair (B), a layer VI–RTN neuron pair (C), a VP–VP neuron pair (D), an RTN–RTN neuron pair (E), and a VP–RTN neuron pair (F) reveal that prolonged bursts are synchronized in layer VI, VP, and RTN. Schematic drawings illustrate the location of the paired recordings in each experiment.
In addition to eliciting prolonged bursts, 4-AP and BMC elicited short bursts in VP and RTN neurons (Figs. 1E,2A). Short bursts were typically <0.5 sec in duration and had characteristics that distinguished them from the prolonged bursts that were 5–1000 times longer in duration. In voltage clamp, the short bursts appeared as giant EPSCs characterized by a fast rise and smooth decay and reversing near 0 mV (data not shown). Because short bursts were absent from cortical neurons and did not occur synchronously within VP and RTN, they were not studied further.
Fig. 2.

A, Paired whole-cell recordings from two VP neurons in an isolated thalamic slice. Note the absence of prolonged bursts. Also note that short bursts are still present in thalamus isolated from cortex but do not appear synchronously.B, Paired whole-cell recordings from two layer VI neurons in cortex isolated from thalamus. Note that prolonged bursts are still present in cortex isolated from thalamus and are synchronized. C, Paired whole-cell recordings from two layer VI cortical neurons on either side of a vertical cut extending from the pia mater through layer IV; note that the prolonged bursts are present in both recordings and are still synchronized.D, Paired whole-cell recordings of two layer VI neurons on either side of a vertical cut extending from the pia mater through layer V; note that the prolonged bursts are present in both recordings and are still synchronized. E, Paired whole-cell recordings of two layer VI neurons on either sides of a cut extending from the pia mater through layer VI and white matter; note that the prolonged bursts are present in both recordings but are no longer synchronized.
Burst synchronization
To determine whether prolonged bursts in cortical cells were synchronized within the cortex, whole-cell recordings of pairs of layer VI cortical neurons were performed (n = 6 pairs). In all recordings in which prolonged bursts were present (n = 5), bursts appeared synchronously in the two cortical neurons (Fig. 1A), even when the cortical neurons were separated by 3–4 mm. The lag time between the beginning of a prolonged burst in one cortical neuron and the beginning of the synchronous bursts in the second cortical neuron was between 0 and 40 msec (n = 3 pairs) when the cortical neurons were separated by 100–500 μm, but was 100–300 msec when the cortical neurons were separated by 3–4 mm. The order in which the prolonged bursts were detected in the paired cells varied even within the same pair of cells, suggesting that a prolonged burst traveled as a propagating wave of activity across the cortex and that the location from which the wave started was random.
To determine whether prolonged bursts in VP cells were synchronized with prolonged bursts in the cortex, whole-cell recordings were performed in VP simultaneously with field potential recordings in deep layers of the cortex (n = 5) (data not shown). In all cases, prolonged bursts in VP neurons were synchronized with field potentials in the cortex. In addition, dual whole-cell recordings were obtained from pairs of cells in VP and layer VI of the cortex (n = 20 pairs). None of the cell pairs were monosynaptically coupled, as determined by the fact that action potentials elicited by the injection of depolarizing current pulses into either the thalamic or cortical neuron could not evoke EPSPs in the other recorded neuron. In all cases in which prolonged bursts were present in VP neurons, they were synchronized with prolonged bursts in the cortex (n = 11 pairs) (Fig.1B). In one case, prolonged bursts in the cortex were synchronized with barrages of subthreshold EPSPs in the VP neuron. The onset delay between the beginning of the prolonged burst in a cortical neuron and the beginning of the synchronous prolonged burst in the paired thalamic neuron varied between 5 and 100 msec. Bursts could appear first in the cortex or first in VP and, as in the cortex, the order in which the burst appeared varied even within the same pair of cells. However, in 85% (202 of 239) of the total number of burst pairs, the prolonged burst was first detected in the cortical cell.
To determine whether prolonged bursts in RTN were synchronized with prolonged bursts in the cortex, dual whole-cell recordings were performed in layer VI of the cortex and RTN (n = 1). The pair of cells was not monosynaptically coupled. Prolonged bursts in the layer VI cortical cell occurred synchronously with prolonged bursts in the RTN neuron (Fig. 1C).
To determine whether prolonged bursts were synchronized within VP, dual whole-cell recordings were performed in VP (n = 13 pairs). In all cases in which prolonged bursts were present in both VP neurons, they were always synchronized (n = 10 pairs) (Fig. 1D). In one case, prolonged bursts present in one VP neuron were synchronized with barrages of 2–4 mV EPSPs in the other VP neuron.
Dual whole-cell recordings from one RTN–VP pair and one RTN–RTN pair also revealed that prolonged bursts appeared synchronously (Fig.1E,F).
We attempted to define the site of origin of the prolonged bursts by completely cutting the cortex away from the thalamus. Whole-cell recordings of layer VI cortical neurons in the isolated cortex revealed that prolonged bursts were present in all neurons examined (n = 4). Paired recordings from layer VI neurons located in adjacent cortical columns revealed that the prolonged bursts remained synchronized in the isolated cortical preparation (n = 1 pair) (Fig. 2B). In contrast, prolonged bursts were never detected in whole-cell recordings of VP neurons in the isolated thalamus (n = 4), although short bursts were recorded in all cells (Fig. 2A). Paired whole-cell recordings in VP revealed that short bursts were not synchronized (n = 2 pairs).
To determine whether intracortical connections were critical for synchronizing prolonged bursts within the cortex, cuts extending from the pia mater to the superficial white matter were made in the cortex, and paired whole-cell recordings were performed in layer VI of the cortex on both sides of the cut. These recording sites were separated by 500–1000 μm, a range that matches the distances separating the recording sites in control experiments. In all three pairs of cells examined, prolonged bursts were present in all recordings, but bursts in the cell pairs were no longer synchronized, confirming that horizontal cortical connections synchronized the prolonged bursts in the cortex (Fig. 2E). To determine which cortical layers were critical for the horizontal spread of the prolonged bursts, partial vertical cuts were made in cortex isolated from the thalamus, and paired whole-cell recordings were performed in layer VI on either side of the cut. Cuts extending from the pial surface to the layer IV/layer V border did not disrupt the synchrony of the prolonged bursts (n = 4 pairs) (Fig.2C). Similarly, cuts extending from the pial surface to the layer V/layer VI border did not disrupt the synchrony of the prolonged bursts (n = 2 pairs) (Fig. 2D), suggesting that axons traversing layer VI and the white matter are sufficient for the synchronization of the prolonged bursts.
Voltage-clamp analysis of prolonged bursts
Prolonged synchronized corticothalamic bursts were further studied in voltage clamp in 80 VP neurons, 81 layer VI neurons, and 38 RTN neurons. Recordings were performed with pipettes filled with a cesium gluconate-based internal solution supplemented with QX-314 to reduce voltage-dependent potassium currents and block voltage-dependent Na+ currents and GABAB-mediated currents. Prolonged bursts were typically recorded at seven different holding potentials between −90 and +30 mV. At all postnatal ages, in all three cell populations, prolonged burst currents were recorded as compound inward currents at hyperpolarized membrane potentials and reversed to outward currents near 0 mV (layer VI neurons, −0.44 ± 4.38 mV; RTN neurons, +0.06 ± 4.53 mV; VP neurons, +1.07 ± 4.73 mV) (Fig. 3). There were no developmental changes in the reversal potentials in any of the three cell populations. There was a dramatic developmental shift in the voltage dependence of the peak conductance and charge transfer of the burst currents (Fig. 3). Between P2 and P10, at holding membrane potentials close to the resting membrane potential of the cell (around −70 mV), burst currents were relatively small in amplitude. Hyperpolarization of the membrane typically resulted in a further reduction of the peak current amplitude and charge transfer, whereas depolarization of the membrane potential led to a marked increase in the peak amplitude and charge transfer. Plotting the peak burst current amplitude versus the membrane potential made evident the strong voltage dependence of the burst peak conductance in all three cell populations and also revealed a region of negative conductance between −70 and −30 mV, typical of NMDA receptor-mediated conductances.
Fig. 3.

Voltage-clamp traces and current–voltage plots of synaptic currents underlying the prolonged bursts in layer VI (A), RTN (B), and VP (C) neurons at P7–P8 and P14. Note that at P7–P8 burst conductances show strong voltage dependence at the peak of the current, but that at P14, they show little or no voltage dependence.
After P12, at membrane potentials near the resting membrane potential of the cell, burst currents recorded from layer VI and RTN cells were very large in peak amplitude (up to several nanoamps) and displayed large charge transfer. Hyperpolarization of the membrane typically resulted in an increase, whereas depolarization resulted in a decrease of the peak amplitude and charge transfer of the burst currents. Current versus voltage plots typically revealed a linear or near-linear relationship between holding membrane potential and peak current amplitude typical of non-NMDA receptor-mediated conductances. After P12, burst currents recorded from a large proportion of VP neurons (25 of 31) still displayed strong voltage dependence, whereas the burst currents recorded from the remaining 6 VP neurons displayed linear or near linear current–voltage relationships.
To further quantify the change in voltage dependence in burst current peak amplitude during development, the ratio of the peak amplitude recorded at −90 mV to the peak amplitude recorded at +30 mV was calculated and plotted versus the age of the animals (Fig.4). Linear correlation analysis revealed a significant developmental shift in the voltage dependence of the peak burst current amplitude in layer VI (n = 56;R2 = 0.342; p < 0.0001) and RTN neurons (n = 28;R2 = 0.443; p = 0.0001). Changes were not proven to be linear. In VP, where a large number of cells did not show a decrease in the voltage dependence of the peak burst conductance (Fig. 4C), there was no statistically significant shift in the voltage dependence of the response (n = 41; R2 = 0.023;p > 0.1), although a few VP cells after P14 did show decreases in voltage dependence of burst currents. The strong voltage dependence of the peak conductance of the burst currents before P12 suggests that NMDA receptor-mediated currents are the dominant contributors to the total burst current during the early postnatal period. The lack of voltage dependence of the peak conductance of the burst currents in layer VI and RTN cells after P12 suggests that non-NMDA receptors, which typically show little voltage dependence at peak conductance, make a large contribution to the total burst current during more mature postnatal stages.
Fig. 4.
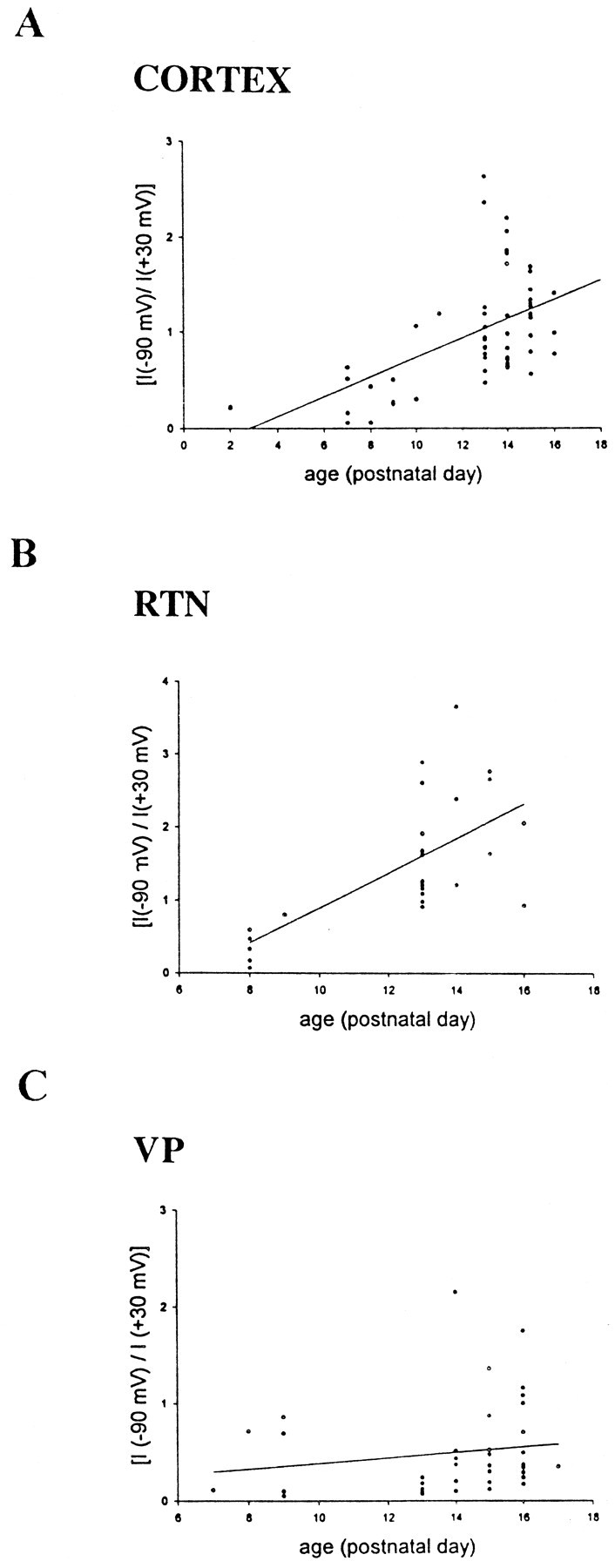
Scatter plot and linear regression analysis of the ratio of the peak burst current recorded at −90 mV to the peak burst current recorded at +30 mV versus developmental stage in layer VI (A), RTN (B), and VP (C) neurons. Note the increase in the ratio of the current at later developmental stages in layer VI and RTN neurons confirming that current–voltage relationships show voltage dependence at early stages but little or no voltage dependence at later developmental stages. In VP neurons, a large proportion of the neurons still show strong voltage dependence at later developmental stages.
Pharmacological analysis of prolonged corticothalamic bursts
The NMDA receptor antagonist APV and the non-NMDA glutamate receptor antagonist CNQX were applied together in the bath (n = 4 layer VI neurons, 1 VP neuron, and 1 RTN neuron). Application of APV and CNQX together always reversibly blocked the prolonged bursts, indicating that ionotropic glutamate receptors are essential for the genesis of the prolonged bursts (data not shown).
To determine which ionotropic glutamate receptor is essential for the genesis of the prolonged bursts at early and late developmental time points, CNQX and APV were applied separately to layer VI neurons, RTN neurons, and VP neurons at all postnatal ages examined. Application of CNQX alone reversibly blocked the prolonged bursts at all postnatal ages examined (P6–P15) (n = 2 VP neurons, 3 layer VI neurons), indicating that at both early and late postnatal stages non-NMDA ionotropic glutamate receptors are involved in the maintenance of the prolonged bursts (Fig. 5). The NMDA receptor antagonist APV, however, had developmentally specific effects on the synchronized corticothalamic bursts. APV reversibly blocked the prolonged bursts only when it was applied before postnatal day 8 (n = 2 of 2 cells) in layer VI cells and before postnatal day 10 in VP cells (n = 3 of 3 cells) (Fig.6A,C). APV reversibly blocked the prolonged bursts in two of three RTN cells before P10 and reduced the peak conductance in the third cell to <10% of the control value (data not shown). After P8 in cortical cells, and after P10 in VP and RTN cells, APV never blocked the prolonged bursts, only reducing the peak conductance of burst currents when recorded in voltage clamp (Fig. 6B,D).
Fig. 5.

Prolonged bursts recorded from P6 (A) and P14 (B) layer VI neurons before, during, and after application of CNQX. Note that CNQX blocks the prolonged bursts at both early and late postnatal ages.
Fig. 6.
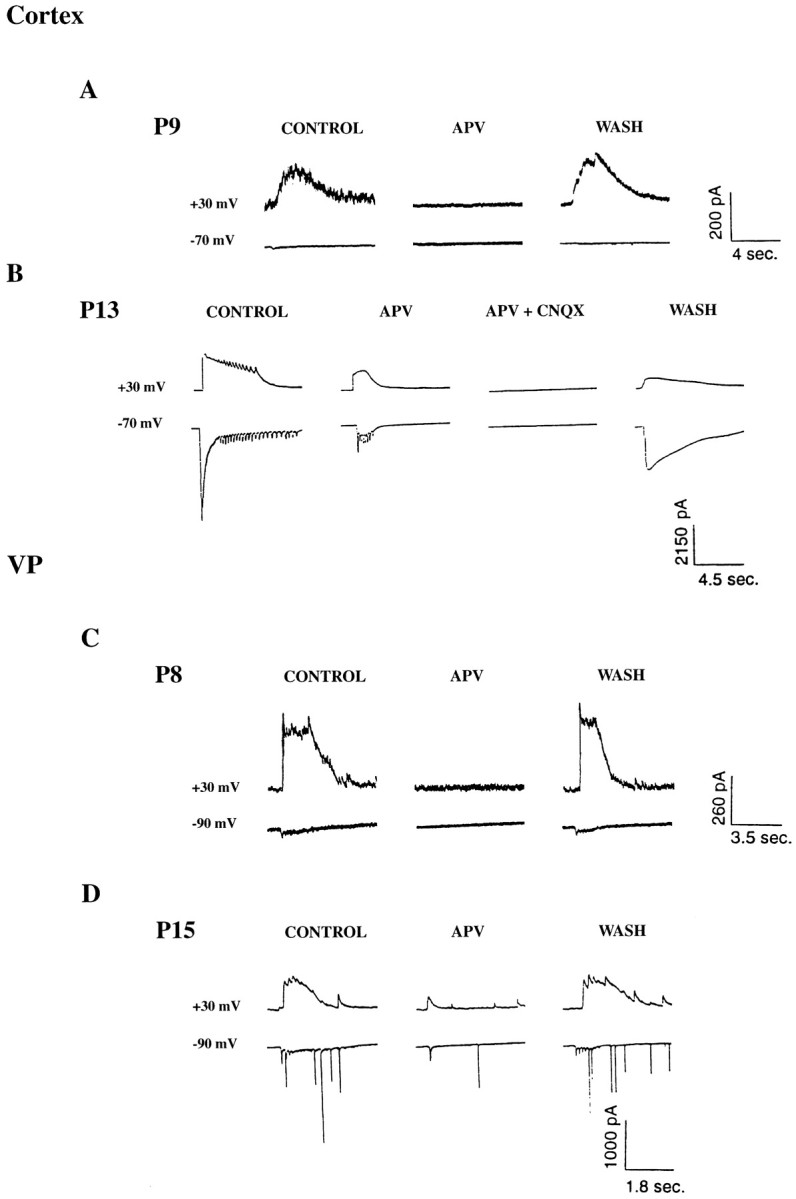
A, Whole-cell voltage-clamp recordings showing prolonged burst currents recorded from P9 layer VI neuron before, during, and after application of APV. Note that APV reversibly blocks the prolonged burst currents at this age.B, Whole-cell voltage-clamp recordings of prolonged burst currents recorded from a P13 layer VI neuron before, during, and after application of APV, and APV and CNQX. Note that APV no longer completely blocks the bursts but reduces the duration of the currents. Further addition of CNQX reversibly blocks the bursts.C, Whole-cell voltage-clamp recording of prolonged burst currents recorded from a P8 VP neuron before, during, and after application of APV. Note that APV reversibly blocks the burst currents at this age. D, Whole-cell voltage-clamp recording of prolonged burst currents recorded from a P15 VP neuron. Note that APV no longer blocks the burst currents and only reduces their duration.
To quantify further the developmentally specific effects of APV on the prolonged bursts, the proportion of the peak burst conductance remaining after APV application was plotted against the age of the animal. Linear correlation analysis revealed a significant decrease with increasing age in the proportion of the peak conductance blocked by APV in layer VI (n = 13;R2 = 0.536; p < 0.005), RTN (n = 7; R2 = 0.881;p < 0.005), and VP (n = 5;R2 = 0.865, p < 0.05), demonstrating that NMDA receptor-mediated currents are much more prominent in early postnatal development and are essential for the generation of the prolonged bursts during the early postnatal period (Fig. 7). During later postnatal ages, the APV-insensitive current typically displayed a linear peak current versus voltage relationship, again suggesting that the strong nonlinearities in the current–voltage relationships observed in slices from animals before P8–P10, resulted from the relatively large contribution of NMDA receptor-mediated conductances to the total burst conductance.
Fig. 7.
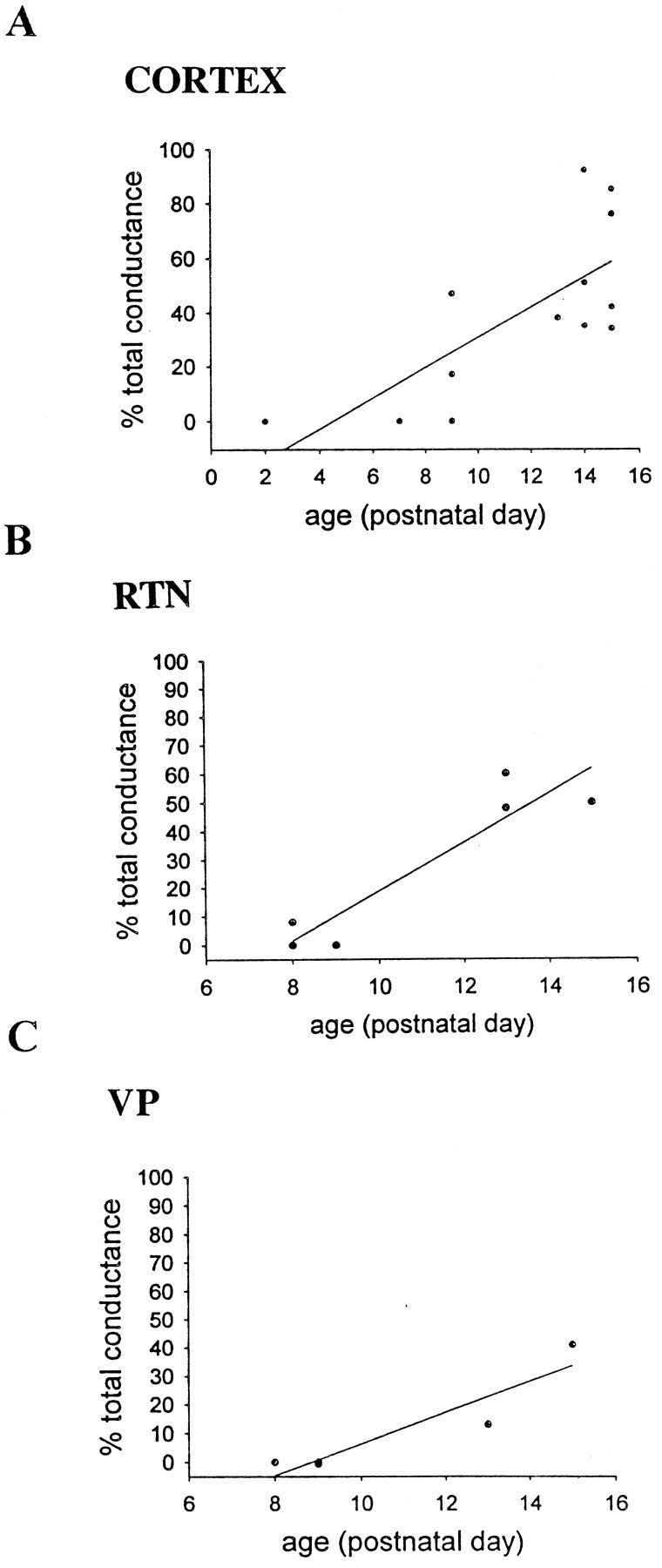
Scatter plots and linear regression analysis of percentage of the total peak conductance of the burst currents remaining after application of APV versus age in layer VI (A), RTN (B), and VP (C) neurons. Note that the percentage of the total peak conductance remaining after application of APV increases with increasing age of the animal in all three cell populations.
DISCUSSION
Induction of prolonged corticothalamic bursts by 4-AP and bicuculline
Application of 4-AP and bicuculline elicited prolonged bursts in layer VI, RTN, and VP neurons in P2–P16 thalamocortical slices. Paired whole-cell recordings demonstrated that the prolonged bursts appeared synchronously in the cortex, RTN, and VP. Synchronized prolonged bursts were still observed in cortex isolated from thalamus but were never observed in thalamus isolated from cortex, demonstrating that the prolonged bursts were generated in the cortex and propagated to RTN and VP via the corticothalamic projection. Because RTN neurons also receive strong excitatory input from collaterals of thalamocortical fibers, prolonged bursts could also have propagated from VP to RTN.
Paired whole-cell recordings in cortex demonstrated that even in the same pair of cells, there was a great deal of variability in the sequence of onset of the bursts. This suggests that prolonged bursts originated in different cortical loci with each burst and propagated through the two recording sites in a variable manner. Similar variability in the origin of spontaneous synchronized activity has been observed in the neocortex and hippocampus using voltage dye-imaging techniques (Colom and Saggau, 1994; Sutor et al., 1994). In a minority of cases, the onset of the prolonged burst was first observed in VP and then in the cortex. In these cases, synchronized activity probably propagated from an initial site in the cortex to the recording site in the thalamus before propagating to the recording site in the cortex.
Paired whole-cell recordings separated by vertical cuts of various depths through the cortex demonstrated that prolonged bursts remained synchronized in the recordings if the cut extended from the pia mater to the deep aspect of layer V but that prolonged bursts were no longer synchronized when cuts extended into layer VI and the white matter. This suggests that horizontal connections probably arising from layer VI cells are sufficient for the propagation of the prolonged bursts across the cortical slice. A subset of layer VI neurons possesses extensive horizontal collaterals restricted to infragranular layers (Ojima et al., 1992); excitation of these cells may play a cardinal role in the propagation of synchronized activity in the cortex.
The induction of synchronized activity by 4-AP and/or GABAAreceptor antagonists has been studied extensively in the hippocampus and neocortex (Gutnick et al., 1982; Traub and Wong, 1983; Connors, 1984; Hablitz, 1984; Voskuyl and Albus, 1985; Rutecki et al., 1989;Ives and Jeffreys, 1990; Hwa and Avoli, 1991; Lee and Hablitz, 1991a,b;Muller and Misgeld, 1991; Avoli et al., 1993, 1996a,b; Traub et al., 1993; Burke and Hablitz, 1994; Colom and Saggau, 1994; Scharfman, 1994a,b; Sutor et al., 1994; Bianchi and Wong, 1995; Hoffman and Prince, 1995; Bijak and Misgeld, 1996; Psarropoulou and Avoli, 1996;Barbarosie and Avoli, 1997; Benardo, 1997; Forti et al., 1997; Golomb and Amitai, 1997; Siniscalchi et al., 1997; Lopantsev and Avoli, 1998), but there has been little or no study of the propagation of paroxysmal activity from the cortex to the thalamus. At the low doses used in this study (70 μm), 4-AP is thought to selectively block the D-type potassium current (Wu and Barish, 1992). Suppression of the D current in axons or axon terminals prolongs the action potential and the period of calcium entry at the terminal and consequently increases transmitter release (Barish et al., 1996; Wheeler et al., 1996). Increased strength of excitatory transmission in a neocortical circuit rich in recurrent excitatory connections, accompanied by increases in the frequency of ectopic action potentials (Traub et al., 1995), leads to generation of spontaneous synchronous propagating discharges. In the present study, we blocked GABAA receptors to best study the development of excitatory circuitry in the corticothalamocortical loop, further promoting the generation and propagation of synchronized discharges. A recent study demonstrated that the GABAAreceptor antagonist used in this study, bicuculline methchloride, also directly blocks the afterhyperpolarization terminating the low-threshold spike burst in thalamic cells (Debarbieux et al., 1998). This effect could contribute to the generation of paroxysmal activity in this study.
In slices cut in an ideal plane, where both thalamocortical and corticothalamic connections are preserved, there are likely to be re-entrant loops of activity whereby a prolonged burst initiated in the cortex propagates to the thalamus through excitation of corticothalamic cells in layer V and VI and then backpropagates to cortex via the thalamocortical projection. Although the creation of corticothalamocortical “strong-loops” has been hypothesized to lead to epileptiform activity (Crick and Koch, 1998), thalamocortical activation of an already epileptic cortex may in fact act to disrupt the synchrony of cortical cells.
Voltage-clamp and pharmacological analysis of corticothalamic prolonged bursts
Voltage-clamp analysis of prolonged bursts in the layer VI, VP, and RTN cells demonstrated considerable developmental changes in the excitatory postsynaptic conductances underlying the prolonged bursts. The peak burst conductance was highly voltage-dependent during the early postnatal stages in all three types of neurons, but became nonvoltage-dependent or nearly so in most cortical and RTN neurons and in some VP neurons; a large proportion of VP cells still showed voltage-dependent burst conductances at later ages. These changes can be attributed to changes in the relative contributions of non-NMDA and NMDA receptors to the total burst conductance, because non-NMDA receptor-mediated conductances usually show little voltage dependence, whereas NMDA receptors show strong voltage dependence (Nowak et al., 1984; Mayer and Westbrook, 1987; Ascher and Nowak, 1988). This suggests that during the first 8–9 d of postnatal development, a very large majority of synapses activated during the prolonged bursts in layer VI, RTN, and VP are silent synapses, i.e., synapses in which only NMDA receptor-mediated currents can be evoked. Later in development, prolonged bursts activate functional synapses, i.e., synapses at which both AMPA and NMDA receptors are activated.
Pharmacological analysis further confirmed that the voltage-dependent conductance recorded during early postnatal times was an NMDA receptor-mediated conductance. APV reversibly blocked prolonged bursts only when it was applied before postnatal day 8 in layer VI cells and before postnatal day 10 in VP. In RTN cells, before P10, APV reversibly blocked prolonged bursts or reduced the peak conductance to <10% of the control value. Later in development, APV only reduced the amplitude and duration of burst currents and eliminated any voltage dependence in the current–voltage relation. The non-NMDA receptor antagonist CNQX, however, blocked the prolonged bursts at all ages examined. This indicates that activation of NMDA receptors at silent synapses was necessary but not sufficient for the existence of prolonged bursts at early postnatal ages. During the early developmental period, the relatively small contribution of non-NMDA receptors is critical for the genesis of the bursts, probably by causing an initial depolarization into the voltage range in which NMDA receptors can be activated. During later postnatal ages, activation of NMDA receptors was neither necessary nor sufficient for bursts, but activation of non-NMDA receptors was both necessary and sufficient. The larger proportion of synapses with active non-NMDA receptors after P9 is enough to cause recurrent excitation and propagation of the bursts.
Silent synapses have been demonstrated in developing Xenopustectal neurons, in hippocampal CA1 neurons, and in cortical layer IV neurons (Crair and Malenka, 1995; Durand et al., 1996; Wu et al., 1996;Isaac et al., 1997). In all these neurons, silent synapses can be converted to functional synapses by pairing presynaptic activity with postsynaptic depolarization, suggesting that calcium entry through NMDA receptors can enter the postsynaptic cell and activate AMPA receptors through activation of signal transduction intermediates. EPSCs at developing corticothalamic synapses are also dominated by NMDA receptor-mediated currents (Golshani et al., 1998). It will be interesting to determine whether sustained synchronous bursts can convert silent synapses between layer VI cortical neurons, VP neurons, and RTN neurons into functional synapses.
In the adult hippocampus, prolonged synchronized bursts that originate in CA3 and propagate to CA1 and are recorded in the presence of 4-AP, GABAA, and ionotropic glutamate receptor blockers, are mediated by activation of metabotropic glutamate receptors (mGluRs) (Bianchi and Wong, 1995). mGluR activation alone was incapable of sustaining the prolonged corticothalamic bursts recorded in our preparation (our unpublished observations), suggesting a higher density of recurrent excitatory connections containing mGluRs in CA3 compared with the cortex.
Silent synapses and neuronal synchronization: implications for juvenile epilepsies
We observed paroxysmal activity in the cortex as early as postnatal day 2, at a stage when only layers V and VI have formed, and synaptogenesis is only beginning. Spontaneous synaptic currents have been recorded in neocortical slices in very early postnatal stages (Blanton and Kriegstein 1991; Kim et al., 1995), but it has generally been assumed that synaptic connectivity at these stages is incomplete or insufficiently dense to maintain synchronized network or paroxysmal activity. By suppressing inhibition and increasing transmitter release, we have shown that very early postnatal cortical circuitry can indeed support the existence of synchronized activity, mainly through activation of silent synapses. Because synchronized presynaptic and postsynaptic activity can transform silent synapses into functional synapses, synchronous paroxysmal activity in time may transform a large number of silent synapses into functional synapses, thereby strengthening recurrent excitatory loops and promoting epileptogenesis. Synchronized paroxysmal activity may also interfere with activity-dependent formation of cortical, thalamocortical, and subcortical circuits and adversely affect the normal maturation of sensory, motor, and cognitive centers.
Footnotes
This study was supported by Grants NS21377 and NS30109 from the National Institutes of Health, United States Public Health Service. P.G. is an MD/PhD student and was supported by American Heart Association under Grant 96005020. We thank Dr. Richard Warren for a critical reading of this manuscript, Drs. Diane O’Dowd, Ivan Soltesz, Alberto Muñoz, and Greg Hollrigel for practical and theoretical advice during the experiments, and Vu Nguyen, Hyle Park, Phong Nguyen, and Hao Truong for technical assistance.
Correspondence should be addressed to Dr. Edward Jones, Center for Neuroscience, 1544 Newton Court, Davis, CA 95616.
REFERENCES
- 1.Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-j. [DOI] [PubMed] [Google Scholar]
- 2.Ascher P, Nowak L. The role of divalent cations in the N-methyl-d-aspartate responses of mouse central neurones in culture. J Physiol (Lond) 1988;399:247–266. doi: 10.1113/jphysiol.1988.sp017078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avoli M, Psarropoulou C, Tancredi V, Fueta Y. On the synchronous activity induced by 4-aminopyridine in the CA3 subfield of juvenile rat hippocampus. J Neurophysiol. 1993;70:1018–1029. doi: 10.1152/jn.1993.70.3.1018. [DOI] [PubMed] [Google Scholar]
- 4.Avoli M, Barbarosie M, Lucke A, Nagao T, Lopantsev V, Kohling R. Synchronous GABA-mediated potentials and epileptiform discharges in the rat limbic system in vitro. J Neurosci. 1996a;16:3912–3924. doi: 10.1523/JNEUROSCI.16-12-03912.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avoli M, Louvel J, Kurcewicz I, Pumain R, Barbarosie M. Extracellular free potassium and calcium during synchronous activity induced by 4-aminopyridine in the juvenile rat hippocampus. J Physiol (Lond) 1996b;493:707–717. doi: 10.1113/jphysiol.1996.sp021416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barbarosie M, Avoli M. CA3-driven hippocampal-entorhinal loop controls rather than sustains in vitro limbic seizures. J Neurosci. 1997;17:9308–14. doi: 10.1523/JNEUROSCI.17-23-09308.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barish ME, Ichikawa M, Tominaga T, Matsumoto G, Iijima T. Enhanced fast synaptic transmission and a delayed depolarization induced by transient potassium current blockade in rat hippocampal slices as studied by optical recording. J Neurosci. 1996;16:5672–5687. doi: 10.1523/JNEUROSCI.16-18-05672.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benardo LS. Recruitment of GABAergic inhibition and synchronization of inhibitory interneurons in rat neocortex. J Neurophysiol. 1997;77:3134–3144. doi: 10.1152/jn.1997.77.6.3134. [DOI] [PubMed] [Google Scholar]
- 9.Bianchi R, Wong RK. Excitatory synaptic potentials dependent on metabotropic glutamate receptor activation in guinea-pig hippocampal pyramidal cells. J Physiol (Lond) 1995;487:663–676. doi: 10.1113/jphysiol.1995.sp020908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bijak M, Misgeld U. Suppression by GABAB receptors of 4-aminopyridine-induced hyperactivity in guinea-pig dentate neurons. Neurosci Lett. 1996;205:49–52. doi: 10.1016/0304-3940(96)12368-5. [DOI] [PubMed] [Google Scholar]
- 11.Blanton MG, Kriegstein AR. Spontaneous action potential activity and synaptic currents in the embryonic turtle cerebral cortex. J Neurosci. 1991;11:3907–3923. doi: 10.1523/JNEUROSCI.11-12-03907.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blom S, Heijbel J, Bergfors PG. Incidence of epilepsy in children: a follow-up study three years after the first seizure. Epilepsia. 1978;19:343–350. doi: 10.1111/j.1528-1157.1978.tb04500.x. [DOI] [PubMed] [Google Scholar]
- 13.Burke JP, Hablitz JJ. Metabotropic glutamate receptor activation decreases epileptiform activity in rat neocortex. Neurosci Lett. 1994;174:29–33. doi: 10.1016/0304-3940(94)90111-2. [DOI] [PubMed] [Google Scholar]
- 14.Colom LV, Saggau P. Spontaneous interictal-like activity originates in multiple areas of the CA2-CA3 region of hippocampal slices. J Neurophysiol. 1994;71:1574–1585. doi: 10.1152/jn.1994.71.4.1574. [DOI] [PubMed] [Google Scholar]
- 15.Connors BW. Initiation of synchronized neuronal bursting in neocortex. Nature. 1984;310:685–687. doi: 10.1038/310685a0. [DOI] [PubMed] [Google Scholar]
- 16.Contreras D, Destexhe A, Sejnowski TJ, Steriade M. Control of spatiotemporal coherence of a thalamic oscillation by corticothalamic feedback. Science. 1996;274:771–774. doi: 10.1126/science.274.5288.771. [DOI] [PubMed] [Google Scholar]
- 17.Crair MC, Malenka RC. A critical period for long-term potentiation at thalamocortical synapses. Nature. 1995;375:325–328. doi: 10.1038/375325a0. [DOI] [PubMed] [Google Scholar]
- 18.Crick F, Koch C. Constraints on cortical and thalamic projections: the no-strong-loops hypothesis. Nature. 1998;391:245–250. doi: 10.1038/34584. [DOI] [PubMed] [Google Scholar]
- 19.Debarbieux F, Brunton J, Charpak S. Effect of bicuculline on thalamic activity: a direct blockade of IAHP in reticularis neurons. J Neurophysiol. 1998;79:2911–2918. doi: 10.1152/jn.1998.79.6.2911. [DOI] [PubMed] [Google Scholar]
- 20.Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature. 1996;381:71–75. doi: 10.1038/381071a0. [DOI] [PubMed] [Google Scholar]
- 21.Ellenberg JH, Hirtz DG, Nelson KB. Age at onset of seizures in young children. Ann Neurol. 1984;15:127–134. doi: 10.1002/ana.410150204. [DOI] [PubMed] [Google Scholar]
- 22.Forti M, Biella G, Caccia S, de Curtis M. Persistent excitability changes in the piriform cortex of the isolated guinea-pig brain after transient exposure to bicuculline. Eur J Neurosci. 1997;9:435–451. doi: 10.1111/j.1460-9568.1997.tb01621.x. [DOI] [PubMed] [Google Scholar]
- 23.Golomb D, Amitai Y. Propagating neuronal discharges in neocortical slices: computational and experimental study. J Neurophysiol. 1997;78:1199–1211. doi: 10.1152/jn.1997.78.3.1199. [DOI] [PubMed] [Google Scholar]
- 24.Golshani P, Warren RA, Jones EG. Progression of change in NMDA, non-NMDA, and metabotropic glutamate receptor function at the developing corticothalamic synapse. J Neurophysiol. 1998;80:143–154. doi: 10.1152/jn.1998.80.1.143. [DOI] [PubMed] [Google Scholar]
- 25.Gutnick MJ, Connors BW, Prince DA. Mechanisms of neocortical epileptogenesis in vitro. J Neurophysiol. 1982;48:1321–1335. doi: 10.1152/jn.1982.48.6.1321. [DOI] [PubMed] [Google Scholar]
- 26.Hablitz JJ. Picrotoxin-induced epileptiform activity in hippocampus: role of endogenous versus synaptic factors. J Neurophysiol. 1984;51:1011–1027. doi: 10.1152/jn.1984.51.5.1011. [DOI] [PubMed] [Google Scholar]
- 27.Hoffman SN, Prince DA. Epileptogenesis in immature neocortical slices induced by 4-aminopyridine. Brain Res Dev Brain Res. 1995;85:64–70. doi: 10.1016/0165-3806(94)00196-7. [DOI] [PubMed] [Google Scholar]
- 28.Hwa GG, Avoli M. The involvement of excitatory amino acids in neocortical epileptogenesis: NMDA and non-NMDA receptors. Exp Brain Res. 1991;86:248–256. doi: 10.1007/BF00228949. [DOI] [PubMed] [Google Scholar]
- 29.Isaac JT, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron. 1997;18:269–280. doi: 10.1016/s0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- 30.Ives AE, Jefferys JG. Synchronization of epileptiform bursts induced by 4-aminopyridine in the in vitro hippocampal slice preparation. Neurosci Lett. 1990;112:239–245. doi: 10.1016/0304-3940(90)90210-z. [DOI] [PubMed] [Google Scholar]
- 31.Kim HG, Fox K, Connors BW. Properties of excitatory synaptic events in neurons of primary somatosensory cortex of neonatal rats. Cereb Cortex. 1995;5:148–157. doi: 10.1093/cercor/5.2.148. [DOI] [PubMed] [Google Scholar]
- 32.Lee WL, Hablitz JJ. Initiation of epileptiform activity by excitatory amino acid receptors in the disinhibited rat neocortex. J Neurophysiol. 1991a;65:87–95. doi: 10.1152/jn.1991.65.1.87. [DOI] [PubMed] [Google Scholar]
- 33.Lee WL, Hablitz JJ. Excitatory synaptic involvement in epileptiform bursting in the immature rat neocortex. J Neurophysiol. 1991b;66:1894–1901. doi: 10.1152/jn.1991.66.6.1894. [DOI] [PubMed] [Google Scholar]
- 34.Lopantsev V, Avoli M. Participation of GABAA-mediated inhibition in ictal-like discharges in the rat entorhinal cortex. J Neurophysiol. 1998;79:352–360. doi: 10.1152/jn.1998.79.1.352. [DOI] [PubMed] [Google Scholar]
- 35.Malenka RC, Nicoll RA. Silent synapses speak up. Neuron. 1997;19:473–476. doi: 10.1016/s0896-6273(00)80362-1. [DOI] [PubMed] [Google Scholar]
- 36.Mayer ML, Westbrook GL. Permeation and block of N-methyl-d-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J Physiol (Lond) 1987;394:501–527. doi: 10.1113/jphysiol.1987.sp016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muller W, Misgeld U. Picrotoxin- and 4-aminopyridine-induced activity in hilar neurons in the guinea pig hippocampal slice. J Neurophysiol. 1991;65:141–147. doi: 10.1152/jn.1991.65.1.141. [DOI] [PubMed] [Google Scholar]
- 38.Neckelmann D, Amzica F, Steriade M. Spike-wave complexes and fast components of cortically generated seizures. III. Synchronizing mechanisms. J Neurophysiol. 1998;80:1480–1494. doi: 10.1152/jn.1998.80.3.1480. [DOI] [PubMed] [Google Scholar]
- 39.Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 40.Ojima H, Honda CN, Jones EG. Characteristics of intracellularly injected infragranular pyramidal neurons in cat primary auditory cortex. Cereb Cortex. 1992;2:197–216. doi: 10.1093/cercor/2.3.197. [DOI] [PubMed] [Google Scholar]
- 41.Psarropoulou C, Avoli M. Developmental features of 4-aminopyridine induced epileptogenesis. Brain Res Dev Brain Res. 1996;94:52–59. doi: 10.1016/0165-3806(96)00040-5. [DOI] [PubMed] [Google Scholar]
- 42.Rutecki PA, Grossman RG, Armstrong D, Irish-Loewen S. Electrophysiological connections between the hippocampus and entorhinal cortex in patients with complex partial seizures. J Neurosurg. 1989;70:667–675. doi: 10.3171/jns.1989.70.5.0667. [DOI] [PubMed] [Google Scholar]
- 43.Scharfman HE. EPSPs of dentate gyrus granule cells during epileptiform bursts of dentate hilar “mossy” cells and area CA3 pyramidal cells in disinhibited rat hippocampal slices. J Neurosci. 1994a;14:6041–6057. doi: 10.1523/JNEUROSCI.14-10-06041.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scharfman HE. Synchronization of area CA3 hippocampal pyramidal cells and non-granule cells of the dentate gyrus in bicuculline-treated rat hippocampal slices. Neuroscience. 1994b;59:245–257. doi: 10.1016/0306-4522(94)90593-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siniscalchi A, Calabresi P, Mercuri NB, Bernardi G. Epileptiform discharge induced by 4-aminopyridine in magnesium-free medium in neocortical neurons: physiological and pharmacological characterization. Neuroscience. 1997;81:189–197. doi: 10.1016/s0306-4522(97)00178-4. [DOI] [PubMed] [Google Scholar]
- 46.Steriade M, Contreras D. Spike-wave complexes and fast components of cortically generated seizures. I. Role played by neocortex and thalamus. J Neurophysiol. 1998;80:1439–1455. doi: 10.1152/jn.1998.80.3.1439. [DOI] [PubMed] [Google Scholar]
- 47.Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- 48.Steriade M, Amzica F, Neckelmann D, Timofeev I. Spike-wave complexes and fast components of cortically generated seizures. II. Extra- and intracellular patterns. J Neurophysiol. 1998;80:1456–1479. doi: 10.1152/jn.1998.80.3.1456. [DOI] [PubMed] [Google Scholar]
- 49.Sutor B, Hablitz JJ, Rucker F, ten Bruggencate G. Spread of epileptiform activity in the immature rat neocortex studied with voltage-sensitive dyes and laser scanning microscopy. J Neurophysiol. 1994;72:1756–1768. doi: 10.1152/jn.1994.72.4.1756. [DOI] [PubMed] [Google Scholar]
- 50.Timofeev I, Grenier F, Steriade M. Spike-wave complexes and fast components of cortically generated seizures. IV. Reflections of fast seizures in thalamic neurons. J Neurophysiol. 1998;80:1495–1513. doi: 10.1152/jn.1998.80.3.1495. [DOI] [PubMed] [Google Scholar]
- 51.Traub RD, Wong RK. Synaptic mechanisms underlying interictal spike initiation in a hippocampal network. Neurol. 1983;33:257–266. doi: 10.1212/wnl.33.3.257. [DOI] [PubMed] [Google Scholar]
- 52.Traub RD, Jefferys JG, Miles R. Analysis of the propagation of disinhibition-induced after-discharges along the guinea-pig hippocampal slice in vitro. J Physiol (Lond) 1993;472:267–287. doi: 10.1113/jphysiol.1993.sp019946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Traub RD, Colling SB, Jefferys JG. Cellular mechanisms of 4-aminopyridine-induced synchronized after-discharges in the rat hippocampal slice. J Physiol (Lond) 1995;489:127–140. doi: 10.1113/jphysiol.1995.sp021036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Voskuyl RA, Albus H. Spontaneous epileptiform discharges in hippocampal slices induced by 4-aminopyridine. Brain Res. 1985;342:54–66. doi: 10.1016/0006-8993(85)91352-6. [DOI] [PubMed] [Google Scholar]
- 55.Warren RA, Jones EG. Maturation of neuronal form and function in a mouse thalamocortical circuit. J Neurosci. 1997;17:277–295. doi: 10.1523/JNEUROSCI.17-01-00277.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wheeler DB, Randall A, Tsien RW. Changes in action potential duration alter reliance of excitatory synaptic transmission on multiple types of Ca2+ channels in rat hippocampus. J Neurosci. 1996;16:2226–2237. doi: 10.1523/JNEUROSCI.16-07-02226.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu G, Malinow R, Cline HT. Maturation of a central glutamatergic synapse. Science. 1996;274:972–976. doi: 10.1126/science.274.5289.972. [DOI] [PubMed] [Google Scholar]
- 58.Wu RL, Barish ME. Two pharmacologically and kinetically distinct transient potassium currents in cultured embryonic mouse hippocampal neurons. J Neurosci. 1992;12:2235–2246. doi: 10.1523/JNEUROSCI.12-06-02235.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]