

Abstract
Marijuana and related drugs (cannabinoids) have been proposed as treatments for a widening spectrum of medical disorders.R(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-yl]-(1-naphthalenyl)methanone mesylate (R(+)-WIN 55212-2), a synthetic cannabinoid agonist, decreased hippocampal neuronal loss after transient global cerebral ischemia and reduced infarct volume after permanent focal cerebral ischemia induced by middle cerebral artery occlusion in rats. The less active enantiomerS(−)-WIN 55212-3 was ineffective, and the protective effect of R(+)-WIN 55212-2 was blocked by the specific central cannabinoid (CB1) cannabinoid receptor antagonistN-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide-hydrochloride.R(+)-WIN 55212-2 also protected cultured cerebral cortical neurons from in vitro hypoxia and glucose deprivation, but in contrast to the receptor-mediated neuroprotection observed in vivo, this in vitro effect was not stereoselective and was insensitive to CB1 and CB2 receptor antagonists. Cannabinoids may have therapeutic potential in disorders resulting from cerebral ischemia, including stroke, and may protect neurons from injury through a variety of mechanisms.
Keywords: cannabinoid, ischemia, stroke, glutamate, excitotoxicity, infarct, neuronal culture
Cannabis, the marijuana plant, has been used since antiquity for its medicinal and psychoactive properties (Snyder, 1971). Both its principal active ingredient, Δ9-tetrahydrocannabinol (THC), and synthetic analogs thereof (cannabinoids) have been proposed as therapy for a variety of medical conditions, including glaucoma, cancer chemotherapy-induced nausea and vomiting, acquired immunodeficiency syndrome, inflammatory disorders, and epilepsy (Jack, 1997). This has contributed to efforts to legalize marijuana use for therapeutic purposes (Annas, 1997; Kassirer, 1997). However, concern exists about the safety of cannabinoids, including their possible role in infertility (Schmid et al., 1997) and the extent to which they share effects with narcotics (Rodríguez de Fonseca et al., 1997; Tanda et al., 1997). This controversy persists despite major advances regarding the basic molecular and cellular mechanisms of cannabinoid action (for review, see Felder and Glass, 1998), including the discovery of endogenous cannabinoids (Devane et al., 1992; Stella et al., 1997; Randall and Kendall, 1998), mechanisms for their synthesis and termination of action (Di Marzo et al., 1994; Beltramo et al., 1997), cannabinoid receptors (Matsuda et al., 1990; Kuster et al., 1993; Howlett, 1995), receptor–effector coupling pathways (Mackie and Hille, 1992;Derkinderen et al., 1996), and synthetic cannabinoid agonist and antagonist drugs (Compton et al., 1992; Rinaldi-Carmona et al., 1994).
Central cannabinoid (CB1) receptors are coupled to several signal transduction pathways, including G-proteins that inhibit N-type voltage-gated calcium channels involved in the release of neurotransmitters (Mackie and Hille, 1992). These channels participate in release of the excitatory transmitter l-glutamate, which has been implicated in the death of neurons from stroke, hypoxia, hypoglycemia, and epilepsy. In hippocampal cultures, cannabinoid agonists acting through CB1 receptors and G-proteins inhibit glutamate release (Shen et al., 1996), suggesting that they might reduce glutamate-mediated neuronal injury. Therefore, we examined the effect of cannabinoids on neuronal death in the selectively vulnerable CA1 region of rat hippocampus after transient global cerebral ischemia induced by four-vessel occlusion, on infarct volume after permanent occlusion of the middle cerebral artery (MCA) and on the viability of cultured cerebral cortical neurons deprived of oxygen and glucose in vitro.
MATERIALS AND METHODS
Animals. Animal experiments were approved by local committee review and were conducted according to policies on the use of animals of the Society for Neuroscience. Male Sprague Dawley rats weighing 300–330 gm (global ischemia studies) or 280–310 gm (focal ischemia studies) were used. Anesthesia was induced with 4% isoflurane, 66% N2O, and 30% O2 and, after intubation, maintained with 1.5% isoflurane, 68.5% N2O, and 30% O2. The left femoral artery was cannulated to monitor arterial blood pressure, blood gases, and blood glucose concentration. Rectal temperature was monitored continuously and maintained at 37.0–37.5°C using a heating pad. In global ischemia studies, brain temperature was monitored with a 29 gauge thermocouple implanted in the right striatum and was maintained at 36–37°C with a temperature-regulated heating lamp. In focal ischemia studies, the temperature of the temporalis muscle contralateral to MCA occlusion was monitored and maintained at 37.0–37.5°C in the same manner.
Global cerebral ischemia. Global cerebral ischemia lasting 15 min, followed by reperfusion, was induced by four-vessel occlusion in anesthetized rats (Pulsinelli et al., 1982). Animals were placed in a Kopf stereotactic frame, and the vertebral arteries were coagulated and transected at the junction of the C1 and C2 vertebrae under microscopic guidance. The common carotid arteries (CCAs) were then exposed, the external carotid arteries (ECAs) were ligated, and administration of isoflurane was discontinued. Three minutes later, the CCAs were occluded reversibly for 15 min with microvascular clips, and perfusion was then restored. The electroencephalogram was monitored to ensure isoelectricity during the period of ischemia. Temperature was monitored from the time of intubation until ∼30 min after the onset of reperfusion, for a total of ∼50–70 min.
Focal cerebral ischemia. Permanent focal ischemia was induced by intraluminal occlusion of the MCA with a suture (Longa et al., 1989). Under a microscope, the left ECA was ligated with a 6-0 silk suture and dissected distally, and the left internal carotid artery (ICA) was isolated and separated from the vagus nerve. The extracranial branch of the left ICA was ligated close to its origin with a 6-0 silk suture. A 3-0 surgical monofilament nylon suture with a rounded tip was introduced into the left ICA lumen through the stump of the left ECA and advanced 20–21 mm past the CCA bifurcation. The suture was left in place until rats were killed 24 hr after the onset of ischemia. Temperature was monitored from the time of intubation until ∼30 min after the onset of ischemia, for a total of ∼30–45 min.
Drug administration. R(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-yl]-(1-naphthalenyl)methanone mesylate (R(+)-WIN 55212-2 mesylate) andS(−)-WIN 55212-3 mesylate were purchased from Research Biochemicals (Natick, MA) andN-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamidehydrochloride (SR141716A) was obtained from the National Institute on Drug Abuse. Drugs were dissolved in dimethylsulfoxide (DMSO) and given as 300 μl injections by the intraperitoneal route, 40 min before occlusion of the CCAs (global ischemia) or 30 min before or 30, 60, or 120 min after MCA occlusion (focal ischemia). R(+)-WIN 55212-2 produced dose-dependent behavioral effects consisting of slight drowsiness (1 mg/kg), more marked drowsiness with limb rigidity and hypokinesia in ∼50% of animals (3 mg/kg), and more severe rigidity and hypokinesia in 100% of animals (10 mg/kg).
Quantification of neuronal loss. Three days after exposure to global ischemia, animals were perfused transcardially with 200 ml of saline and then 300 ml of 4% paraformaldehyde in 0.1 mphosphate buffer, pH 7.4, and killed by decapitation. The brains were removed and post-fixed in the same paraformaldehyde solution for 5 d and then embedded in paraffin, and 6 μm sections through the dorsal hippocampus (anteroposterior coordinate, bregma − 3.0 mm) were cut on a microtome and processed for staining with cresyl violet. Neuronal counts in a predesignated region of CA1 were obtained from six to eight animals per condition.
Quantification of infarct volume. Rats subjected to focal ischemia were anesthetized with an overdose of chloral hydrate and decapitated. The brains were removed and sectioned coronally at 2 mm intervals. Sections were immersed in 2% 2,3,5-triphenyltetrazolium hydrochloride (TTC) in saline for 20 min at 37°C and then fixed for 30 min in 4% paraformaldehyde (Isayama et al., 1991). Six sections per brain were analyzed for infarct size using a computerized image analysis system (MCID, St. Catharine’s, Ontario, Canada). Infarct area in each section was calculated by subtracting the residual uninfarcted, TTC-stained area of the ischemic (left) hemisphere from the total area of the nonischemic (right) hemisphere (Swanson et al., 1990). Infarct volume at 24 hr measured in this manner is equivalent to infarct volume determined from hematoxylin- and eosin-stained sections (Isayama et al., 1991).
Neuronal cell culture. Neuronal cultures were prepared from 16–17 d Sprague Dawley rat embryos (Yu et al., 1986). Cerebral hemispheres were removed aseptically, freed of meninges, olfactory bulbs, basal ganglia, and hippocampi, and incubated at 37°C in Ca2+- and Mg2+-free Earle’s balanced salt solution containing 0.01% trypsin (1:250). After 30 min, 10% horse serum (HS) was added. Cells were placed in 2 ml of fresh MEM, triturated, and resuspended in Eagle’s MEM prepared without glutamine and with twice the usual concentration of other amino acids and four times the usual concentration of vitamins (MEM-Pak; Cell Culture Facility, University of California, San Francisco, CA); this medium had been supplemented on the day of plating with glucose (final concentration, 30 mm), 2 mm glutamine, and 15 mm HEPES, pH 7.4. Cell suspensions were filtered through a 70 μm Falcon nylon cell strainer, supplemented with 10% HS and 10% fetal bovine serum (FBS), and seeded at 3 × 105 cells per well on 24-well Corning (Corning, NY) tissue culture dishes coated with 100 μg/ml poly-d-lysine. Cultures were incubated for 20 min at 37°C in humidified 95% air and 5% CO2, and one-half of the medium was replaced with medium containing 5% HS and 5% FBS. Cytosine arabinoside (AraC, 10 μm) was added on the sixth day in vitro (DIV). At 7 DIV, two-thirds of the medium was replaced with medium lacking AraC; thereafter, one-half of the medium was replaced with fresh medium twice weekly. Experiments were conducted at 18 DIV, when cultures consisted primarily of neurons (92 ± 1% MAP2-immunoreactive cells, 6 ± 1% GFAP-immunoreactive cells;n = 12).
In vitro model of ischemia. To model neuronal ischemia in vitro, cultures were exposed to combined hypoxia and glucose deprivation (Koretz et al., 1994). Two-thirds of the medium was replaced three times with serum-free medium, with or without 30 mm glucose. Glucose-containing cultures were then incubated for 24 hr at 37°C in humidified 95% air and 5% CO2. Glucose-deprived cultures were placed in a Billups-Rothenberg (Del Mar, CA) modular incubator chamber, which was flushed for 5 min with 95% N2 and 5% CO2, sealed for 15 min, flushed again for 30 sec with 5% N2 and 5% CO2, and resealed. The chamber was placed in a water-jacketed incubator at 37°C for 8 hr and then returned to 95% air and 5% CO2 and glucose-containing medium for 16 hr. In some experiments, glucose-containing, normoxic cultures were treated for 8 hr with and then 16 hr without 1 mm NMDA or 1 mm AMPA in the presence or absence of the NMDA antagonist (5R,10S)-(+)-5-methyl-10,11-dihydro-5H-benzo[a,d]cyclohepten-5,10-imine hydrogen maleate (MK-801) or the AMPA antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (all from Research Biochemicals, Natick, MA). Stock solutions of cannabinoid receptor agonists and antagonists were prepared in DMSO and diluted to a final concentration not exceeding 7.36 μm DMSO in the drug-treated cultures. At concentrations at least 10-fold higher than this, DMSO had no effect on neuronal viability in either normoxic or hypoxic cultures.
Cytotoxicity assays. Fluorescence of Alamar blue (Accumed International, Westlake, OH), an indicator that changes color from blue to red and fluoresces when reduced by cellular metabolic activity, was used to measure the viability of cultured neurons. One-half of the culture medium was replaced with MEM-Pak containing 10% (v/v) Alamar blue, and cultures were incubated for 3 hr at 37°C in humidified 95% air and 5% CO2. Fluorescence was determined in a Millipore (Bedford, MA) CytoFluor 2300 automated plate-reading fluorometer, with excitation at 530 nm and emission at 590 nm. As reported previously, Alamar blue fluorescence in these cultures varies linearly with cell number, decreases with exposure to hypoxia or excitotoxins, and correlates with the extent of cellular injury determined by lactate dehydrogenase release (White et al., 1996).
Detection of DNA damage. DNA polymerase I-mediated biotin-dATP nick translation (PANT) labeling was used to detect DNA single-strand breaks (Didier et al., 1996). Cultures were fixed for 15 min at room temperature by adding 20% paraformaldehyde in PBS, pH 7.4, to a final concentration of 4%, washed three times with PBS, and permeabilized for 10 min with 0.5% Triton X-100 in PBS. H2O2 (1%) was added for 15 min at room temperature to neutralize endogenous peroxidase, after which cultures were again washed three times with PBS. Cells were then incubated for 60 min at 37°C in PBS, pH 7.6, containing 5 mmMgCl2, 10 mm 2-mercaptoethanol, 50 μg/ml bovine serum albumin (BSA), 20 μm dCTP, dGTP, dTTP, and biotinylated-dATP, and 50 U/ml Escherichia coliDNA polymerase I (Sigma, St. Louis, MO). Cells were washed three times with ice-cold PBS and once with 5 mg/ml BSA in PBS (PBS-BSA), incubated for 1 hr at room temperature with streptavidin-horseradish peroxidase (HRP) in PBS-BSA (Vector Laboratories, Burlingame, CA), and washed four times in PBS. The HRP complex was detected after incubation for 5 min at room temperature in PBS containing 0.5 mg/ml diaminobenzidine, 0.03% H2O2, and nickel (added according to the manufacturer’s instructions). Cells with dense nuclear staining were considered PANT-positive and were counted at 200× magnification in at least three randomly selected fields per well. Data were expressed as a percentage of all cells in the same fields, with reactions conducted in the absence of DNA polymerase I serving as controls for nonspecific labeling.
Statistical analysis. Results are reported as mean values ± SE. The significance of differences between means was assessed by Student’s t test (single comparisons) or by ANOVA and post hoc t tests (multiple comparisons), with p < 0.05 considered statistically significant.
RESULTS
Global cerebral ischemia
In control animals that underwent sham CCA occlusions, ∼300 neurons could be counted in the designated region of CA1, whereas only zero to five neurons were seen after global ischemia (Figs.1, 2).R(+)-WIN 55212-2, a synthetic aminoalkylindole cannabimimetic compound, competes with THC for binding to cannabinoid receptor sites in brain (Kuster et al., 1993) and reproduces pharmacological and behavioral effects of THC (Compton et al., 1992). When animals were given R(+)-WIN 55212-2 mesylate intraperitoneally 40 min before the induction of global cerebral ischemia, there was a dose-dependent increase in neuronal survival that reached maximal levels (56% of sham-operated controls) at 1 mg/kg. At higher doses, neuronal survival declined. The doses that protected against ischemic neuronal death were similar to those associated with behavioral actions such as locomotor inhibition, antinociception, and THC-like discriminative effects (Compton et al., 1992). Neuroprotection by R(+)-WIN 55212-2 (1 mg/kg) was not associated with significant changes before, during, or after the induction of ischemia in mean arterial blood pressure, PaO2, PaCO2, pH, or blood glucose concentration compared with values in animals that received DMSO vehicle alone (Table1).
Fig. 1.
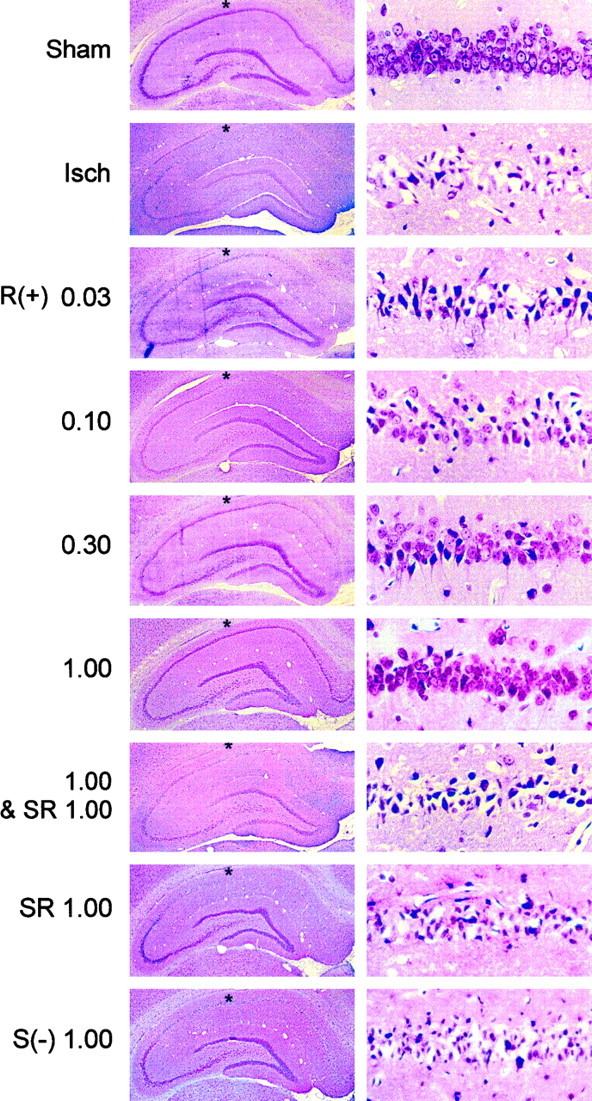
Histological evidence for cannabinoid-mediated protection of CA1 hippocampal neurons in global cerebral ischemia. Rats underwent sham surgery or 15 min of cerebral ischemia induced by four-vessel occlusion, with or without the administration of drugs 40 min before the onset of ischemia. Animals were killed 3 d later and paraffin-embedded brain sections were stained with cresyl violet. Sections are shown at low (left, 25×) and high (right, 400×) power, after sham surgery (Sham) or after the induction of ischemia without (Isch) or with previous administration ofR(+)-WIN 55212-2 [R(+)],S(−)-WIN 55212-3 [S(−)], or SR 141716A (SR) at the indicated doses (milligrams per kilogram, i.p.). Asterisks at leftindicate the region within CA1 from which the fields shown atright were taken.
Fig. 2.

Neuron counts in CA1 after global cerebral ischemia. Animals were treated as described in the legend to Figure 1, undergoing sham surgery (Sham) or global cerebral ischemia without drugs (Isch) or with previous administration of R(+)-WIN 55212-2 [R(+)], S(−)-WIN 55212-3 [S(−)], or SR 141716A (SR) at the indicated doses (milligrams per kilogram, i.p.). Neurons were counted in cresyl violet-stained sections. Data shown are mean values ± SEs from six to eight animals per condition. *p < 0.05 (ANOVA with post hoc t tests) relative to neuron counts in animals subjected to ischemia without drug treatment (Isch).
Table 1.
Blood pressure, arterial blood gases, and serum glucose in transient global cerebral ischemia
| Time relative to ischemia | Drug treatment | MABP | PaO2 | PaCO2 | pH | Glucose |
|---|---|---|---|---|---|---|
| Before | Vehicle | 102 ± 3 | 151 ± 21 | 38 ± 1 | 7.42 ± 0.01 | 116 ± 4 |
| R(+)-WIN (0.03 mg/kg) | 102 ± 6 | 143 ± 10 | 38 ± 1 | 7.41 ± 0.01 | 115 ± 8 | |
| R(+)-WIN (0.1 mg/kg) | 105 ± 5 | 142 ± 13 | 40 ± 2 | 7.40 ± 0.01 | 114 ± 12 | |
| R(+)-WIN (0.3 mg/kg) | 108 ± 5 | 147 ± 3 | 38 ± 2 | 7.42 ± 0.00 | 111 ± 13 | |
| R(+)-WIN (1 mg/kg) | 111 ± 8 | 141 ± 3 | 38 ± 3 | 7.41 ± 0.01 | 105 ± 15 | |
| R(+)-WIN (1 mg/kg) + SR (1 mg/kg) | 107 ± 8 | 152 ± 8 | 37 ± 2 | 7.41 ± 0.01 | 119 ± 19 | |
| SR (1 mg/kg) | 104 ± 10 | 156 ± 12 | 39 ± 1 | 7.41 ± 0.01 | 114 ± 21 | |
| S(−)-WIN (1 mg/kg) | 104 ± 6 | 143 ± 6 | 38 ± 1 | 7.41 ± 0.00 | 117 ± 9 | |
| During | Vehicle | 132 ± 2 | 150 ± 9 | 38 ± 2 | 7.41 ± 0.01 | 110 ± 2 |
| R(+)-WIN (0.03 mg/kg) | 135 ± 6 | 144 ± 6 | 39 ± 2 | 7.42 ± 0.01 | 116 ± 15 | |
| R(+)-WIN (0.1 mg/kg) | 135 ± 9 | 148 ± 20 | 40 ± 2 | 7.40 ± 0.01 | 125 ± 22 | |
| R(+)-WIN (0.3 mg/kg) | 137 ± 4 | 156 ± 7 | 39 ± 0 | 7.41 ± 0.00 | 121 ± 15 | |
| R(+)-WIN (1 mg/kg) | 137 ± 5 | 155 ± 1 | 39 ± 0 | 7.41 ± 0.00 | 126 ± 23 | |
| R(+)-WIN (1 mg/kg) + SR (1 mg/kg) | 140 ± 8 | 136 ± 10 | 38 ± 1 | 7.42 ± 0.01 | 116 ± 7 | |
| SR (1 mg/kg) | 139 ± 11 | 141 ± 14 | 39 ± 1 | 7.41 ± 0.02 | 117 ± 18 | |
| S(−)-WIN (1 mg/kg) | 138 ± 8 | 146 ± 15 | 39 ± 1 | 7.41 ± 0.02 | 120 ± 17 | |
| After | Vehicle | 121 ± 9 | 151 ± 17 | 38 ± 1 | 7.42 ± 0.01 | 122 ± 5 |
| R(+)-WIN (0.03 mg/kg) | 119 ± 12 | 149 ± 9 | 39 ± 1 | 7.41 ± 0.02 | 112 ± 17 | |
| R(+)-WIN (0.1 mg/kg) | 122 ± 8 | 148 ± 15 | 39 ± 1 | 7.40 ± 0.01 | 114 ± 21 | |
| R(+)-WIN (0.3 mg/kg) | 116 ± 11 | 143 ± 8 | 41 ± 2 | 7.41 ± 0.00 | 121 ± 8 | |
| R(+)-WIN (1 mg/kg) | 124 ± 6 | 143 ± 7 | 38 ± 2 | 7.40 ± 0.02 | 119 ± 23 | |
| R(+)-WIN (1 mg/kg) + SR (1 mg/kg) | 113 ± 11 | 140 ± 17 | 38 ± 1 | 7.41 ± 0.01 | 129 ± 15 | |
| SR (1 mg/kg) | 122 ± 8 | 141 ± 20 | 39 ± 1 | 7.41 ± 0.02 | 125 ± 12 | |
| S(−)-WIN (1 mg/kg) | 116 ± 10 | 147 ± 19 | 39 ± 1 | 7.41 ± 0.01 | 120 ± 18 |
Data shown are mean values ± SEs from six to eight animals per condition. MABP, Mean arterial blood pressure (mm Hg); Glucose, plasma concentration of glucose (mg/dl); R(+)-WIN,R(+)-WIN 55212-2; SR, SR141716A; S(−)-WIN,S(−)-WIN 55212-3.
R(+)-WIN 55212-2 is one of a pair of enantiomers that exhibit stereoselectivity in cannabinoid receptor radioligand binding assays and behavioral studies. R(+)-WIN 55212-2 has ∼2500-fold greater affinity than its enantiomer, S(−)-WIN 55212-3, for CB1 receptor binding sites on rat cerebellar membranes (Kuster et al., 1993), and the S(−) enantiomer is inactive in behavioral assays of cannabimimetic activity that show potent effects of R(+)-WIN 55212-2 (Compton et al., 1992). When animals were treated with S(−)-WIN 55212-3 before ischemia, neuronal counts in CA1 were not significantly different from counts obtained after ischemia alone and were significantly lower than counts in animals given R(+)-WIN 55212-2.
SR141716A is a selective, competitive antagonist that inhibits radioligand binding to CB1 cannabinoid receptors in rat brain synaptosomal membranes, cannabinoid-induced inhibition of smooth muscle contraction in mouse vas deferens, and behavioral effects ofR(+)-WIN 55212-2, including locomotor inhibition and antinociception (Rinaldi-Carmona et al., 1994). When administered alone before ischemia, SR141716A failed to alter neuronal counts. However, when 1 mg/kg SR141716A was given together with 1 mg/kgR(+)-WIN 55212-2, the neuroprotective effect of the latter was reduced by 80%.
Focal cerebral ischemia
MCA occlusion produced ipsilateral cerebral infarcts averaging ∼210 mm3 in volume, which could be detected after 24 hr by the loss of TTC staining (Figs.3, 4). Pretreatment with 1 mg/kg R(+)-WIN 55212-2 given 30 min before the onset of ischemia reduced infarct size by ∼30%. The drug was similarly effective when given 30 min after the onset of ischemia, but its effect was lost when administration was delayed by 60–120 min. Cerebral salvage occurred primarily in the periphery of the ischemic region, leading to enhanced survival of penumbral cortical tissue, without affecting the ischemic core in the striatum. Reduction of infarct size by R(+)-WIN 55212-2 (1 mg/kg) was not accompanied by alterations in mean arterial blood pressure, PaO2, PaCO2, pH, or blood glucose concentration, measured 10 min after the onset of ischemia, compared with values in control animals given DMSO vehicle alone (Table 2).
Fig. 3.
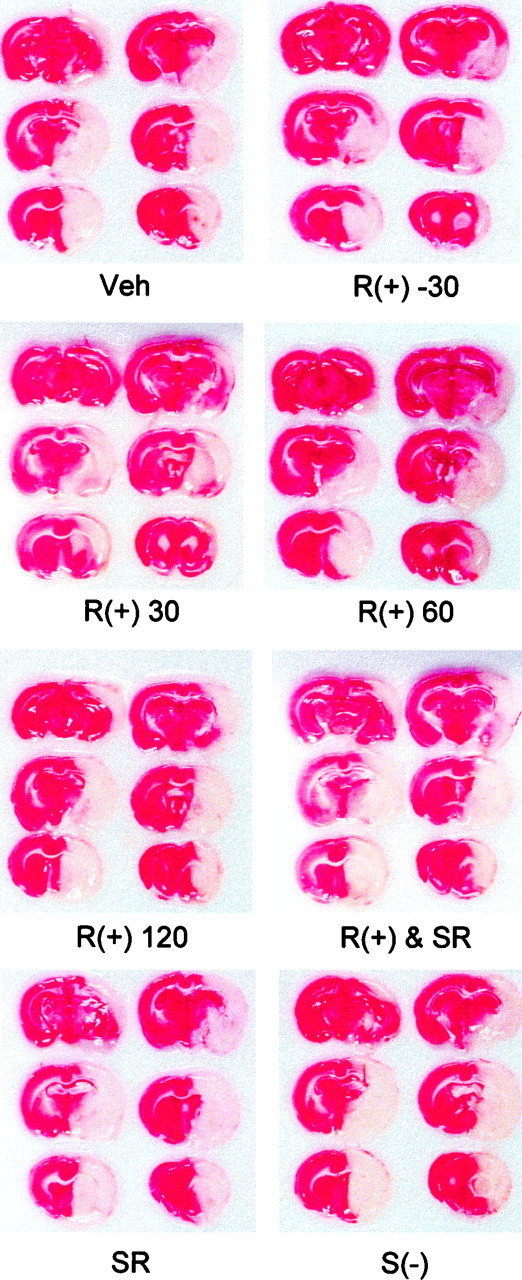
Histological evidence for cannabinoid-mediated reduction in infarct size after permanent MCA occlusion. The left MCA (on the viewer’s right) was occluded with a nylon suture as described in Materials and Methods. Animals were given DMSO vehicle 30 min before occlusion (Veh); 1 mg/kgR(+)-WIN 55212-2 30 min before [R(+) −30], or 30 [R(+) 30], 60 [R(+) 60], or 120 [R(+) 120] min after occlusion; or 1mg/kg R(+)-WIN 55212-2 and 1 mg/kg SR141716A [R(+) & SR], 1 mg/kg SR141716A alone (SR), or 1 mg/kg S(−)-WIN 55212-3 [S(−)], all 30 min before occlusion. Drugs were administered intraperitoneally. Animals were killed 24 hr after the onset of occlusion, and brain sections were treated with TTC (red), which stains viable tissue red but does not stain infarcted tissue. The brains shown are representative of six animals per condition. S(−)-WIN 55212-3 [S(−)], or 10 μm THC. After an additional 16 hr under control conditions, cell viability was measured by Alamar blue fluorescence and expressed as a percentage of fluorescence above background in control cultures. Data shown are mean values ± SEs from 4–12 cultures per condition. *p < 0.05 (ANOVA with post hoc t tests) relative to fluorescence in cultures exposed to hypoxia and glucose deprivation in the presence of 10 nmR(+)-WIN 55212-2. C, Concentration dependence of neuroprotection induced by R(+)-WIN 55212-2. Experiments were performed as described in Babove, except that different concentrations of R(+)-WIN 55212-2 were used, and results were expressed as a percentage of the maximal drug-induced increase in viability above that in cultures exposed to hypoxia and glucose deprivation without drugs. Data shown are mean values ± SEs from 4–12 cultures per condition.
Fig. 4.

Infarct volumes after focal cerebral ischemia. Animals were treated as described in the legend to Figure 3, undergoing permanent MCA occlusion without drugs (Veh) or with intraperitoneal administration of 1 mg/kg R(+)-WIN 55212-2 given 30 min before (−30) or 30, 60, or 120 min after the onset of ischemia; 1 mg/kg R(+)-WIN 55212-2 and 1 mg/kg SR141716A, both given 30 min before ischemia; 1 mg/kg SR141716A alone given 30 min before ischemia; or 1 mg/kgS(−)-WIN 55212-3. Data shown are mean values ± SEs from six animals per condition. *p < 0.05 (ANOVA with post hoc t tests) relative to infarct volumes in animals subjected to ischemia and treated with vehicle only (Veh).
Table 2.
Blood pressure, arterial blood gases, and serum glucose 10 min after the onset of permanent focal cerebral ischemia
| Drug treatment | MABP | PaO2 | PaCO2 | pH | Glucose |
|---|---|---|---|---|---|
| Vehicle | 120 ± 11 | 124 ± 9 | 32 ± 1 | 7.40 ± 0.01 | 117 ± 6 |
| R(+)-WIN, −30 min | 124 ± 8 | 118 ± 11 | 33 ± 1 | 7.39 ± 0.01 | 117 ± 6 |
| R(+)-WIN, +30 min | 125 ± 8 | 115 ± 4 | 37 ± 4 | 7.38 ± 0.03 | 113 ± 12 |
| R(+)-WIN, +60 min | 122 ± 14 | 113 ± 3 | 37 ± 5 | 7.39 ± 0.02 | 117 ± 6 |
| R(+)-WIN, +120 min | 118 ± 16 | 112 ± 6 | 35 ± 4 | 7.39 ± 0.01 | 110 ± 10 |
| R(+)-WIN + SR | 124 ± 12 | 122 ± 15 | 36 ± 9 | 7.41 ± 0.03 | 109 ± 14 |
| SR | 117 ± 7 | 115 ± 4 | 36 ± 2 | 7.40 ± 0.02 | 113 ± 13 |
| S(−)-WIN | 121 ± 6 | 115 ± 10 | 33 ± 3 | 7.39 ± 0.04 | 107 ± 6 |
Data shown are mean values ± SEs from six animals per condition. MABP, Mean arterial blood pressure (mm Hg); Glucose, plasma concentration of glucose (mg/dl); R(+)-WIN,R(+)-WIN 55212-2; SR, SR141716A; S(−)-WIN,S(−)-WIN 55212-3.
The CB1 cannabinoid receptor antagonist SR141716A had no effect on infarct size when administered alone but reversed the protective effect of R(+)-WIN 55212-2 given 30 min before the onset of ischemia. The less active agonist isomer,S(−)-WIN 55212-3, failed to alter infarct volume.
In vitro hypoxia and glucose deprivation
Both normoxic and hypoxic cortical cultures expressed CB1 receptor immunoreactivity on Western blots probed with a polyclonal rabbit IgG (Calbiochem, La Jolla, CA) raised against the N-terminal domain of the human CB1 receptor (Fig.5A). Exposure of cortical cultures to hypoxia and glucose deprivation reduced neuronal viability (Alamar blue fluorescence), with approximately half-maximal toxicity at 8 hr and maximal toxicity by 24 hr. The NMDA antagonist MK-801 reduced toxicity, as did R(+)-WIN 55212-2 and the endogenous cannabinoid anandamide (Fig. 5B). Exposure for 8 hr to 1 mm NMDA or AMPA decreased neuronal viability to an extent similar to that seen with hypoxia and glucose deprivation, but although MK-801 blocked NMDA toxicity, and the AMPA antagonist CNQX blocked AMPA toxicity under these conditions, R(+)-WIN 55212-2 was ineffective (data not shown). PANT staining, indicative of cells with DNA single-strand breaks (Chen et al., 1997), also increased after hypoxia and glucose deprivation, and this increase was attenuated by both MK-801 and R(+)-WIN 55212-2 (Fig.6).
Fig. 5.
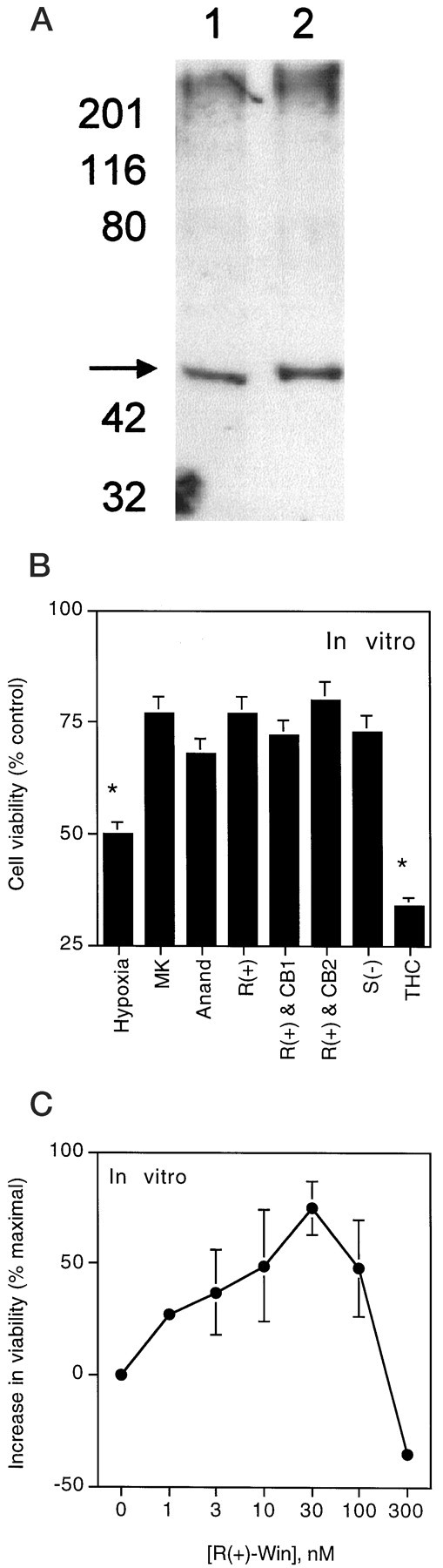
Pharmacological characterization of cannabinoid-mediated neuroprotection from hypoxia and glucose deprivation in vitro. Cultured cerebral cortical neurons were exposed for 24 hr to normoxia and glucose (control), or to 8 hr of hypoxia and glucose deprivation followed by 16 hr of recovery (hypoxic). A, Western analysis of control (lane 1) and hypoxic (lane 2) cultures probed with an antibody against the CB1 cannabinoid receptor (arrow). B, Pharmacological features of cannabinoid-mediated neuroprotection. Cultures were exposed to hypoxia and glucose deprivation in the absence (Hypoxia) or presence of 10 μm MK-801 (MK), 100 nm anandamide (Anand), 10 nmR(+)-WIN 55212-2 (alone [R(+)] or together with the CB1 antagonist SR141617A [R(+) & CB1] or the CB2 antagonist SR144528 [R(+) & CB2], each at 300 nm–1 μm), 10 nm
Fig. 6.
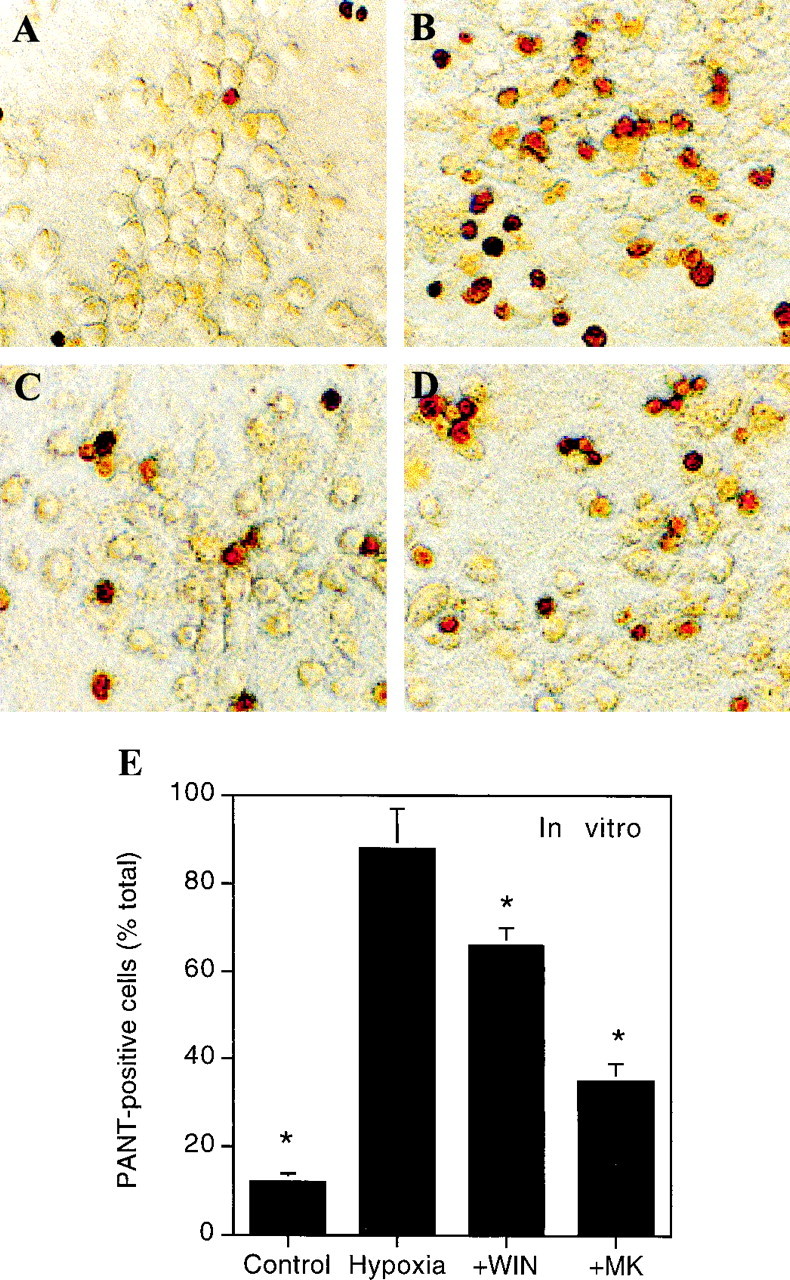
PANT staining for DNA single-strand breaks afterin vitro hypoxia and glucose deprivation. Cultures were maintained for 8 hr under control conditions (A) or exposed for 8 hr to hypoxia and glucose deprivation in the absence (B) or presence of 10 μm MK-801 (C) or 10 nmR(+)-WIN 55212-2 (D). After an additional 16 hr under control conditions, cultures were fixed and processed for PANT staining as described in Materials and Methods. Cells with dense nuclear staining were considered PANT-positive and were counted at 200× magnification in at least three randomly selected fields per well.E, Quantitative data were expressed as a percentage of all cells in the same fields and are shown are mean values ± SEs from 6–12 cultures per condition. *p < 0.05 (ANOVA with post hoc t tests) relative to values in cultures exposed to hypoxia and glucose deprivation in the absence of drugs (Hypoxia).
To test whether the same CB1 receptor-mediated mechanism involved in protection from in vivo ischemia was also responsible for in vitro neuroprotection byR(+)-WIN 55212-2, we examined the effects of the less-active enantiomer S(−)-WIN 55212-3, the CB1 agonists anandamide and THC, the CB1 antagonist SR141716A, and the CB2 antagonistN-[(1S)-endo-1,3,3-trimethyl bicyclo[2.2.1]heptan-2-yl]-5-(4-chloro-3-methylphenyl)-1-(4-methylbenzyl)-pyrazole-3-carboxamide (SR144528; obtained from the National Institute on Drug Abuse) (Rinaldi-Carmona et al., 1998). As shown in Figure 5B, protection from hypoxia and glucose deprivation in our cultures did not have the pharmacological features of a CB1 or CB2 receptor-mediated process. Thus, although the endogenous cannabinoid anandamide reduced hypoxic injury, THC did not. Moreover, neuroprotection lacked stereoselectivity and was insensitive to CB1 and CB2 antagonists. As was also observed in our studies on global ischemia, the protective effect ofR(+)-WIN 55212-2 was lost at higher concentrations (Fig.5C).
DISCUSSION
The major finding we report is that the synthetic cannabinoidR(+)-WIN 55212-2, an analog of the psychoactive constituents of marijuana, protects brain tissue against ischemic injury. This effect appears to be mediated through CB1 cannabinoid receptors, because it is stereospecific and is blocked by a selective CB1 receptor antagonist, SR141716A. We observed neuroprotection by R(+)-WIN 55212-2 in both global and focal ischemia and, in the latter case, with drug administration either before or up to 30 min after the onset of ischemia. Cannabinoid-mediated protection from global ischemia was seen in the CA1 region of hippocampus, which is especially vulnerable to ischemic injury, and inhibition of glutamate release by cannabinoids has been demonstrated in cells cultured from this region (Shen et al., 1996). Cannabinoid receptor levels in hippocampus, including CA1, and in cerebral cortex, which was the major site of cannabinoid-mediated neuroprotection in our focal ischemia model, are among the highest in the brain (Matsuda et al., 1990; Kuster et al., 1993; Adams et al., 1998).
In principle, the neuroprotective action of R(+)-WIN 55212-2 could be exerted in a variety of ways. For example, the ability of cannabinoids to inhibit ion flux through calcium channels (Mackie and Hille, 1992) and thereby reduce glutamate release (Shen et al., 1996) suggests that these effects could be responsible for neuroprotection. However, cannabinoid receptor activation has been shown to affect a variety of second mesenger systems, including the cAMP and phospholipase A2–cyclooxygenase signal transduction pathways (Chan et al., 1998), so reduction of calcium influx may not necessarily be responsible for cannabinoid-mediated neuroprotection. The vasoactive properties of cannabinoids (Randall and Kendall, 1998) raise the possibility that they may alter blood flow in the ischemic brain, but although it would be premature to conclude that such alterations have no role in the neuroprotective effect of cannabinoids, a major contribution of altered blood flow seems unlikely. Protection was seen not only in focal ischemia, in which blood flow might be redistributed from nonischemic to ischemic brain regions, but also in global cerebral ischemia, which affects the brain diffusely. Moreover, isoelectricity of the electroencephalogram, reflecting ischemia severe enough to interrupt brain function, was present during global ischemia in both cannabinoid-treated and untreated animals, arguing against cannabinoid-induced preservation of cerebral blood flow. Cannabinoids can also promote hypothermia, which is neuroprotective in some settings. Although rectal and brain temperature were held constant during the early stages of our global and focal ischemia studies, a contributory effect of late hypothermia on outcome cannot be excluded. Finally, cannabinoids exert anti-inflammatory effects, at least in part by inhibiting the proliferation of lymphocytes and inducing their death by apoptosis (Schwarz et al., 1994), and inflammation has been implicated in focal ischemic brain injury (Nogawa et al., 1997). However, inflammation appears to be a less prominent feature of transient global cerebral ischemia (Dirnagl et al., 1994), in which we found neuroprotection by cannabinoids similarly effective.
We also observed neuronal protection in an in vitro cell culture model of neuronal hypoxia and glucose deprivation, but the mechanism appeared to differ from that observed in vivo. In particular, neuroprotection in vitro lacked stereoselectivity for R(+)-WIN 55212-2 overS(−)-WIN 55212-3 and was insensitive to inhibition by cannabinoid receptor antagonists. This resembles in some respects the effect reported by Hampson et al. (1998b), who found that comparatively high concentrations of THC reduced excitotoxicity in cultured cortical neurons by a receptor-independent mechanism. In contrast to those investigators, however, we found no effect of THC and no protection against directly applied excitatory amino acids or the nitric oxide donor sodium nitroprusside (data not shown), arguing against an antioxidant effect such as that they proposed. Whether the nonreceptor-mediated neuroprotective effect of cannabinoids that we observed in vitro also operates in vivo is unclear, although there is precedent for the coexistence of parallel, receptor-mediated and non-receptor-mediated effects of cannabinoids on neurotransmission and signal transduction (Felder et al., 1992; Hampson et al., 1998a). Therefore, it would not be altogether surprising if similarly parallel neuroprotective mechanisms were found.
Two other in vitro studies of cannabinoid effects have appeared recently and require comment. One described a toxiceffect of THC on cultured hippocampal neurons (Chan et al., 1998), and the other showed a protective effect of R(+)-WIN 55212-2 on cultured hippocampal neurons exposed to excitotoxicity induced by Mg2+ depletion (Shen and Thayer, 1998). Both effects appeared to be CB1 receptor-mediated. Thus, neurotoxic and neuroprotective—as well as receptor-mediated and non-receptor-mediated—effects of cannabinoids can be observed in vitro. These differences are likely to depend on a variety of influences, apparently including the nature of the toxic insult, the source of cells, and the particular cannabinoid used. In light of our finding that cannabinoids afford receptor-mediated neuroprotection against global and focal cerebral ischemia in vivo(occurring in hippocampus and cerebral cortex, respectively), the discrepant results of our and other in vitro studies accent the importance of using in vivo models to establish how potential therapeutic agents are likely to affect intact organisms.
Whether the in vivo protective effect of cannabinoids that we observed is permanent or only delays ischemic death beyond the time frames examined remains to be shown, as does the relationship between histological and functional improvement. Nevertheless, the ability of cannabinoids to improve histological outcome after both global and focal cerebral ischemia in rats indicates that further investigation of its potential therapeutic role in cerebral ischemia, such as occurs in stroke and after cardiac arrest, may be warranted.
Footnotes
This work was supported by National Institutes of Health Grants NS24728 and NS35965. We thank Wei Pei and Marie Rose for help with biochemical assays.
Correspondence should be addressed to Dr. David A. Greenberg, Department of Neurology, University of Pittsburgh, S-526 Biomedical Science Tower, 3500 Terrace Street, Pittsburgh, PA 15213.
REFERENCES
- 1.Adams IB, Compton DR, Martin BR. Assessment of anandamide interaction with the cannabinoid brain receptor: SR 141716A antagonism studies in mice and autoradiographic analysis of receptor binding in rat brain. J Pharmacol Exp Ther. 1998;284:1209–1217. [PubMed] [Google Scholar]
- 2.Annas GJ. Reefer madness—the federal response to California’s medical-marijuana law. N Engl J Med. 1997;337:435–439. doi: 10.1056/NEJM199708073370621. [DOI] [PubMed] [Google Scholar]
- 3.Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–1097. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- 4.Chan GC-K, Hinds TR, Impey S, Storm DR. Hippocampal neurotoxicity of Δ9-tetrahydrocannabinol. J Neurosci. 1998;18:5322–5332. doi: 10.1523/JNEUROSCI.18-14-05322.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Jin K, Chen M, Pei W, Kawaguchi K, Greenberg DA, Simon RP. Early detection of DNA strand breaks in the brain after transient focal ischemia: implications for the role of DNA damage in apoptosis and neuronal cell death. J Neurochem. 1997;69:232–245. doi: 10.1046/j.1471-4159.1997.69010232.x. [DOI] [PubMed] [Google Scholar]
- 6.Compton DR, Gold LH, Ward SJ, Balster RL, Martin BR. Aminoalkylindole analogs: cannabimimetic activity of a class of compounds structurally distinct from Δ9-tetrahydrocannabinol. J Pharmacol Exp Ther. 1992;263:1118–1126. [PubMed] [Google Scholar]
- 7.Derkinderen P, Toutant M, Burgaya F, Le Bert M, Siciliano JC, de Franciscis V, Gelman M, Girault J-A. Regulation of a neuronal form of focal adhesion kinase by anandamide. Science. 1996;273:1719–1722. doi: 10.1126/science.273.5282.1719. [DOI] [PubMed] [Google Scholar]
- 8.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 9.Didier M, Bursztajn S, Adamec E, Passani L, Nixon RA, Coyle JT, Wei JY, Berman SA. DNA strand breaks induced by sustained glutamate excitotoxicity in primary neuronal cultures. J Neurosci. 1996;16:2238–2250. doi: 10.1523/JNEUROSCI.16-07-02238.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz J-C, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- 11.Dirnagl U, Niwa K, Sixt G, Villringer A. Cortical hypoperfusion after global forebrain ischemia in rats is not caused by microvascular leukocyte plugging. Stroke. 1994;25:1028–1038. doi: 10.1161/01.str.25.5.1028. [DOI] [PubMed] [Google Scholar]
- 12.Felder CC, Glass M. Cannabinoid receptors and their endogenous agonists. Annu Rev Pharmacol Toxicol. 1998;38:179–200. doi: 10.1146/annurev.pharmtox.38.1.179. [DOI] [PubMed] [Google Scholar]
- 13.Felder CC, Veluz JS, Williams HL, Briley EM, Matsuda LA. Cannabinoid agonists stimulate both receptor- and non-receptor-mediated signal transduction pathways in cells transfected with and expressing cannabinoid receptor clones. Mol Pharmacol. 1992;42:838–845. [PubMed] [Google Scholar]
- 14.Hampson AJ, Bornheim LM, Scanziani M, Yost CS, Gray AT, Hansen BM, Leonoudakis DJ, Bickler PE. Dual effects of anandamide on NMDA receptor-mediated responses and neurotransmission. J Neurochem. 1998a;70:671–676. doi: 10.1046/j.1471-4159.1998.70020671.x. [DOI] [PubMed] [Google Scholar]
- 15.Hampson AJ, Grimaldi M, Axelrod J, Wink D. Cannabidiol and (−)Δ9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci USA. 1998b;95:8268–8273. doi: 10.1073/pnas.95.14.8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Howlett AC. Pharmacology of cannabinoid receptors. Annu Rev Pharmacol Toxicol. 1995;35:607–634. doi: 10.1146/annurev.pa.35.040195.003135. [DOI] [PubMed] [Google Scholar]
- 17.Isayama K, Pitts LH, Nishimura MC. Evaluation of 2,3,5-triphenyltetrazolium chloride staining to delineate rat brain infarcts. Stroke. 1991;22:1394–1398. doi: 10.1161/01.str.22.11.1394. [DOI] [PubMed] [Google Scholar]
- 18.Jack DB. Wider use of cannabinoids likely soon? Drug News Perspect. 1997;10:440–442. [Google Scholar]
- 19.Kassirer JP. Federal foolishness and marijuana. N Engl J Med. 1997;336:366–367. doi: 10.1056/NEJM199701303360509. [DOI] [PubMed] [Google Scholar]
- 20.Koretz B, Ahern KvB, Lustig HS, Greenberg DA. Pre- and postsynaptic modulators of excitatory neurotransmission: comparative effects on hypoxia/hypoglycemia in cortical cultures. Brain Res. 1994;643:334–337. doi: 10.1016/0006-8993(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 21.Kuster JE, Stevenson JI, Ward SJ, D’Ambra TE, Haycock DA. Aminoalkylindole binding in rat cerebellum: selective displacement by natural and synthetic cannabinoids. J Pharmacol Exp Ther. 1993;264:1352–1363. [PubMed] [Google Scholar]
- 22.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 23.Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci USA. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 25.Nogawa S, Zhang F, Ross ME, Iadecola C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci. 1997;17:2746–2755. doi: 10.1523/JNEUROSCI.17-08-02746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 27.Randall MD, Kendall DA. Endocannabinoids: a new class of vasoactive substances. Trends Pharmacol Sci. 1998;19:55–58. doi: 10.1016/s0165-6147(97)01161-9. [DOI] [PubMed] [Google Scholar]
- 28.Rinaldi-Carmona M, Barth F, Héaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Néliat G, Caput D, Ferrara P, Soubrié P, Brelière JC, Le Fur G. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350:240–244. doi: 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- 29.Rinaldi-Carmona M, Barth F, Millan J, Derocq J-M, Casellas P, Congy C, Oustric D, Sarran M, Bouaboula M, Calandra B, Portier M, Shire D, Brelière JC, Le Fur G. SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J Pharmacol Exp Ther. 1998;284:644–650. [PubMed] [Google Scholar]
- 30.Rodríguez de Fonseca F, Carrera MRA, Navarro M, Koob GF, Weiss F. Activation of corticotropin-releasing factor in the limbic system during cannabinoid withdrawal. Science. 1997;276:2050–2054. doi: 10.1126/science.276.5321.2050. [DOI] [PubMed] [Google Scholar]
- 31.Schmid PC, Paria BC, Krebsbach RJ, Schmid HHO, Dey SK. Changes in anandamide levels in mouse uterus are associated with uterine receptivity for embryo implantation. Proc Natl Acad Sci USA. 1997;94:4188–4192. doi: 10.1073/pnas.94.8.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwarz H, Blanco FJ, Lotz M. Anandamide, an endogenous cannabinoid receptor agonist, inhibits lymphocyte proliferation and induces apoptosis. J Neuroimmunol. 1994;55:107–115. doi: 10.1016/0165-5728(94)90152-x. [DOI] [PubMed] [Google Scholar]
- 33.Shen M, Thayer SA. Cannabinoid receptor agonists protect cultured rat hippocampal neurons from excitotoxicity. Mol Pharmacol. 1998;54:459–462. doi: 10.1124/mol.54.3.459. [DOI] [PubMed] [Google Scholar]
- 34.Shen M, Piser TM, Seybold VS, Thayer SA. Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J Neurosci. 1996;16:4322–4334. doi: 10.1523/JNEUROSCI.16-14-04322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Snyder SH. Uses of marijuana. Oxford UP; New York: 1971. [Google Scholar]
- 36.Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 37.Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 38.Tanda G, Pontieri FE, Di Chiara G. Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common μ1 opioid receptor mechanism. Science. 1997;276:2048–2050. doi: 10.1126/science.276.5321.2048. [DOI] [PubMed] [Google Scholar]
- 39.White MJ, DiCaprio MJ, Greenberg DA. Assessment of neuronal viability with Alamar blue in cortical and granule cell cultures. J Neurosci Methods. 1996;70:195–200. doi: 10.1016/s0165-0270(96)00118-5. [DOI] [PubMed] [Google Scholar]
- 40.Yu ACH, Chan PH, Fishman RA. Effects of arachidonic acid on glutamate and γ-aminobutyric acid uptake in primary cultures of rat cerebral cortical astrocytes and neurons. J Neurochem. 1986;47:1181–1189. doi: 10.1111/j.1471-4159.1986.tb00738.x. [DOI] [PubMed] [Google Scholar]