

Abstract
I have previously reported that norepinephrine (NE) induces a sustained potentiation of transmitter release in the chick ciliary ganglion through a mechanism pharmacologically distinct from any known adrenergic receptors. Here I report that the adrenergic potentiation of transmitter release was enhanced by a phosphodiesterase inhibitor, 3-isobutyl-1-methylxanthine (IBMX) and by zaprinast, an inhibitor of cGMP-selective phosphodiesterase. Exogenous application of the membrane-permeable cGMP, 8-bromo-cGMP (8Br-cGMP), potentiated the quantal transmitter release, and after potentiation, the addition of NE was no longer effective. On the other hand, 8Br-cAMP neither potentiated the transmitter release nor occluded the NE-induced potentiation. The NE-induced potentiation was blocked by neither nitric oxide (NO) synthase inhibitor nor NO scavenger. The quantal transmitter release was not potentiated by NO donors, e.g., sodium nitroprusside. The NE-induced potentiation and its enhancement by IBMX was antagonized by two inhibitors of protein kinase G (PKG), Rp isomer of 8-(4-chlorophenylthio) guanosine-3′,5′-cyclic monophosphorothioate and KT5823. As with NE-induced potentiation, the effects of 8Br-cGMP on both the resting intraterminal [Ca2+] ([Ca2+]i) and the action potential-dependent increment of [Ca2+]i (ΔCa) in the presynaptic terminal were negligible. The reduction of the paired pulse ratio of EPSC is consistent with the notion that the NE- and cGMP-dependent potentiation of transmitter release was attributable mainly to an increase of the exocytotic fusion probability. These results indicate that NE binds to a novel adrenergic receptor that activates guanylyl cyclase and that accumulation of cGMP activates PKG, which may phosphorylate a target protein involved in the exocytosis of synaptic vesicles.
Keywords: adrenergic receptors, cGMP, protein kinase G, presynaptic terminal, synaptic plasticity, neurotransmitter release
Norepinephrine (NE) and epinephrine are principal neuromodulators in the central and peripheral nervous systems (Moore and Bloom, 1979; Kuba et al., 1981; Nicoll et al., 1990). The adrenergic responses are characterized according to both the receptor subtypes and the intracellular signal transduction mechanisms (Nicoll et al., 1990; Bylund et al., 1994; Goldstein, 1998). For example, the α1-adrenergic receptors usually couple with phospholipase C and use IP3 and diacylglycerol as second messengers (Nicoll et al., 1990; Bylund et al., 1994). The α2-adrenergic receptors preferentially couple with Gi-group G-proteins and downregulate adenylyl cyclase or modulate K+ channels or Ca2+channels through a membrane-delimited mechanism (Nicoll et al., 1990;Delcour and Tsien, 1993; Milligan, 1993; Bylund et al., 1994; MacKinnon et al., 1994). The β-adrenergic receptors are typical of the Gs-coupled receptors, which upregulate adenylyl cyclase (Nicoll et al., 1990; Milligan, 1993; Bylund et al., 1994). However, some adrenergic responses are resistant to conventional antagonists of any present known adrenergic receptors (Hirst et al., 1982, 1992;Benham and Tsien, 1988). In the rat arterial smooth muscle, NE causes a membrane depolarization that is resistant to α and β blockade (Hirst et al., 1982). Similarly, the NE-dependent enhancement of the L-type Ca2+ current in the arterial smooth muscle was resistant to blockers of α- and β-adrenergic receptors (Benham and Tsien, 1988). However, whether these responses are attributable to the activation of a new class of adrenergic receptors remains unknown. I have previously reported that NE induces a sustained potentiation of transmitter release in the chick ciliary ganglion through a mechanism resistant to any known adrenergic receptor antagonists (Yawo, 1996). Moreover, no receptor-selective synthetic agonists induced the potentiation of transmitter release, and only NE, adrenaline, and dopamine induced the potentiation (Yawo, 1996). Here, I report that the NE-induced presynaptic potentiation involves a nitric oxide (NO)-independent guanylyl cyclase and that an intraterminal increase of cGMP induces the potentiation of transmitter release by activating protein kinase G (PKG). It is suggested that the potentiating adrenergic receptor in the calyx-type presynaptic terminal is different from any known adrenergic receptors in terms of the intracellular signal transduction as well as pharmacological properties.
MATERIALS AND METHODS
Preparation. The methods used here are the same as those described previously (Yawo, 1996, 1999). Chick embryos (White Leghorn; Aoki Egg Farm, Nasu, Japan) were incubated at a constant temperature of 37°C. Day 14 embryos (stage 39–40; Hamburger and Hamilton, 1951) were decapitated, and the ciliary ganglion was removed with the presynaptic oculomotor nerve. A whole ganglion was mounted in a superfusing chamber (∼1 ml); the oculomotor nerve was drawn to the stimulating electrode by suction; and the collagenous envelope was enzymatically removed by focally applying a mixture of collagenase (type II, 2000 U/ml; Sigma-Aldrich, St. Louis, MO) and thermolysin (20 U/ml, Sigma-Aldrich) through a glass pipette (tip diameter, 30 μm). The ganglion was superfused with standard saline (in mm: NaCl, 132; KCl, 5; CaCl2, 1; MgCl2, 1; HEPES, 10; NaOH, 4; and glucose 11, pH adjusted to 7.4 with NaOH). All experiments were performed at room temperature (25°C).
EPSC recordings. A conventional whole-cell patch-clamp recording was made from a postsynaptic ciliary neuron (Yawo, 1996,1999) using an EPC-7 patch-clamp amplifier (List Electronic, Darmstadt-Eberstadt, Germany). Patch pipettes (2.5–3 MΩ, coated with silicon resin and fire-polished) were filled with an internal solution containing (in mm): CsCl, 130; MgCl2, 1; Na2-EGTA, 10; HEPES, 10; and MgATP, 5, pH adjusted to 7.4 with NaOH. The series resistance was usually <10 MΩ throughout the experiment. EPSC was measured at a holding potential of −60 mV. To ensure the stable recording of EPSC, the capacitative transient was minimized by electrical circuitry, and the series resistance was compensated for by 50–70%. The whole-cell currents were low-pass-filtered at 3 kHz (−3 dB, eight-pole Bessel filter, P-84P; NF Electronic Instruments, Yokohama, Japan), digitized at 10–20 kHz (ADX-98E; Canopus, Kobe, Japan), and stored in a computer (PC9801FA; NEC, Tokyo, Japan).
The quantal content (m) was estimated from the coefficient of variation (c.v.) based on Poisson statistics (Kuno and Weakly, 1972). When the occurrence of failure transmission was moderate,m calculated from the occurrence of failure was almost identical to that calculated from the c.v., indicating that the EPSC fluctuation approximately followed the Poisson statistics (Martin and Pilar, 1964a; Yawo and Chuhma, 1994). Therefore, [Ca2+]o and [Mg2+]o were adjusted so that the occurrence of transmission failure was obvious at the beginning of the experiment. Because of the infrequent occurrence of miniature EPSCs, the quantal size (q) was estimated as the mean EPSC divided by m.
Measurement of intraterminal Ca2+concentration. The method of measuring intraterminal [Ca2+] ([Ca2+]i) was almost the same as described previously (Yawo and Chuhma, 1993; Yawo, 1996, 1999). The oculomotor nerve was cut at its exit from the orbital bone in Ca2+-free saline containing 1 mm EGTA. Crystals of fura-2-conjugated dextran (fura dextran,Mr 10,000; Molecular Probes, Eugene, OR) were applied to the distal stump. After 30 min of incubation at 10°C, the ganglion was incubated at 37°C for 1.5 hr. Fura dextran was transported anterogradely and accumulated in the calyx-type nerve terminals. Because of its large molecular size, fura dextran was confined to the presynaptic axons and their terminals. A conventional epifluorescence system equipped with a water-immersion objective (40×; numerical aperture, 0.7; Olympus, Tokyo, Japan) and xenon lamp (150 W) was used. Fluorescence was excited alternately at wavelengths of 340 and 380 nm. Because the microscope was focused on the surface of the ganglion, the fluorescence from one to three terminals was measured at a single spot with a diameter of 50 μm by a photomultiplier tube (OSP-3, Olympus). Confocal pupils at both excitation and emission light paths enable a reduction in the background fluorescence behind the focal plane. The signal was integrated for 80 msec and sampled at 12.5 Hz by a computer (PC-9801RS, NEC) using software for measuring the intracellular Ca2+ (MiCa, provided by Drs. K. Furuya and K. Enomoto, National Institute of Physiological Science of Japan). Twenty records were averaged using the computer-generated stimulating pulse as a trigger. The [Ca2+]i was calculated from the ratio of fluorescence intensities at wavelengths of 340 and 380 nm (Grynkiewicz et al., 1985) using the dissociation constant determined by the manufacturer (216 nm). The minimum ratio and the maximum fluorescence at 380 nm were measured in a Ca2+-free solution containing 10 mm EGTA and 0.1 mm ionomycin (Calbiochem, La Jolla, CA). Thereafter, the solution was changed to one containing 10 mm Ca2+, and the maximum ratio and the minimum fluorescence at 380 nm were measured.
Reagents. Pharmacological agents were usually bath-applied through a surperfusing line with a constant flow rate. The solution in the chamber was completely replaced in <2 min. The agents used in this study and their sources were as follows: l-NE (Nacalai Tesque, Kyoto, Japan); phentolamine (Research Biochemicals International, Natick, MA); d,l-propranolol (Sigma-Aldrich); 3-isobutyl-1-methylxanthine (IBMX, Sigma-Aldrich); zaprinast (Sigma-Aldrich); Ro-20-1724 (Biomol Research Laboratories, Inc., Plymouth Meeting, PA); 8-bromo-cGMP (8Br-cGMP, Sigma-Aldrich); 8Br-cAMP (Sigma-Aldrich);Nω-nitro-l-arginine methyl ester (l-NAME, Sigma-Aldrich); hemoglobin (Sigma-Aldrich); sodium nitroprusside (SNP, Sigma-Aldrich); (±)-(E)-4-methyl-2-[(E)-hydroxyimino]-5-nitro-8-methoxy-3-hexenamide (NOR1; Dojin, Tabaru, Kumamoto, Japan); Rp isomer of 8-(4-chlorophenylthio) guanosine-3′,5′-cyclic monophosphorothioate (Rp-8pCPT-cGMPS; BioLog Life Science Institute, Bremen, Germany); KT5823 (Calbiochem); phorbol 12-myristate 13-acetate (PMA; Wako, Osaka, Japan); and bisindolylmaleimide I (BIS, Calbiochem). The 10 mm stock solution of NE was made with isomolar isoascorbic acid (Wako), and NE was bath-applied with 10 μmisoascorbic acid, which has no effect on the synaptic transmission. All the experiments were done under yellow fluorescent light (wavelength, >520 nm; EL40SY-F; Matsushita Electronic Co., Kadoma, Japan) to minimize the photodynamic oxidation. IBMX was dissolved as 100 mm in 0.1N NaOH. Zaprinast was dissolved as 100 mm in 0.2 mN-methyl-d-glucamine (Sigma-Aldrich). Ro-20-1724, Rp-8pCPT-cGMPS, KT-5823, PMA, and BIS were dissolved in DMSO and then diluted. The concentration of DMSO did not exceed 0.1% and by itself had no effect on the EPSC. To minimize the aberrant effects of the cyclic nucleotides on the A1-adenosine autoreceptors (Yawo and Chuhma, 1993), the antagonist 8-cyclopentyl-1,3-dimetylxanthine (10 μm, Research Biochemicals International) was present throughout the experiment.
The values in the text and figures are mean ± SEM (number of experiments). Statistically significant differences between various parameters were determined using Student’s two-tailed ttest for paired data. Otherwise, Mann–Whitney’s U test was used. Usually, p < 0.05 was considered significant.
RESULTS
Pharmacological properties of adrenergic presynaptic potentiation
The previous study (Yawo, 1996) revealed that NE increases the quantal transmitter release from calyx-type presynaptic terminals of chick ciliary ganglion with a negligible change in the postsynaptic acetylcholine sensitivity. As shown in Figure1A, NE (10 μm) potentiated the average EPSC even in the presence of both an α1- and α2-adrenergic receptor antagonist, phentolamine (10 μm), and a β-adrenergic receptor antagonist, propranolol (10 μm). A fluctuation of the EPSC amplitude was observed in extracellular solution with low Ca2+ and high Mg2+ (Fig.1B). Before the application of NE, the capacitative coupling response, which indicates the presynaptic invasion of the action potential (Yawo and Chuhma, 1994), occasionally accompanied a null EPSC response (synaptic failure). As indicated in the amplitude histogram of control EPSCs before the application of NE (Fig.1C), the occurrence of failures was 7 in 100 consecutive trials. From the c.v., m and q were estimated to be 2.7 and 8.6 pA, respectively. Based on Poisson statistics (Martin and Pilar, 1964a; Yawo and Chuhma, 1994), the expected occurrence of failure (Fig. 1C, arrow) is 7 in 100 trials, which is exactly identical to the observed occurrence. In the presence of NE, no synaptic failures were observed, whereas instead the frequency of the occurrence of large EPSCs was increased (Fig. 1D);m and q were 10.6 and 9.0 pA, respectively. Therefore, NE increased m of the EPSC by 3.9-fold of the control with a negligible change in q in the presence of both phentolamine and propranolol. In all three experiments in the presence of both phentolamine and propranolol, NE potentiated the EPSC by 1.6- to 3.6-fold of the control, which was the same as that in the absence of adrenergic antagonists (range, 1.4- to 3.5-fold of control;n = 6; p > 0.1, Mann–Whitney’sU test). Agonists selective for the α1- or β-adrenergic receptors had no effect on the EPSC, and the α2-adrenergic receptor agonist clonidine attenuated the EPSC (Yawo, 1996). Thus, the NE-dependent potentiation appears to be mediated by a mechanism pharmacologically distinct from any known adrenergic receptors.
Fig. 1.

Adrenergic presynaptic potentiation in a chick ciliary ganglion. A, Representative experimental data showing the effect of 10 μm NE on the EPSC. The extracellular solution contained 1 mm[Ca2+]o and 5 mm[Mg2+]o. Phentolamine (10 μm) and propranolol (10 μm) were present throughout the experiment. The presynaptic oculomotor nerve was stimulated at 0.5 Hz by twin pulses with an interpulse interval of 40 msec. The biphasic current between the stimulus artifact and the EPSC is the capacitative coupling response indicating the invasion of the action potential into the presynaptic terminal. Top, Average EPSC of 100 consecutive records during control. The facilitation ratio was 2.3. Bottom, Average trace of 100 consecutive records during bath application of NE. The facilitation ratio was 1.8. B, Time-dependent plots of the EPSC amplitude of the experiment shown in A. NE (10 μm) was bath-applied during the indicated period (open bar). C, EPSC amplitude histogram of 100 consecutive records before the application of NE, the average of which was shown in A (top). The quantal content (m) and the quantal size (q) were estimated from c.v. and were 2.7 and 8.6 pA, respectively. The arrow on the leftindicates the occurrence of failures expected from a Poisson distribution. D, EPSC amplitude histogram of 100 consecutive records during NE-induced potentiation, the average of which was shown in A (bottom). Them and the q were 10.6 and 9.0 pA, respectively.
Effects of phosphodiesterase inhibitors
The slow onset and the long-lasting nature of the NE-induced potentiation suggest the involvement of second messengers, e.g., cyclic nucleotides. To test this notion, the effects of a phosphodiesterase (PDE) inhibitor, IBMX, were first investigated. A moderate EPSC potentiation by 0.1 μm NE was further enhanced by IBMX (Fig. 2A), whereas IBMX alone had no effect (Fig. 2B). An increase in the dose of NE from 0.1 to 10 μm significantly increased the magnitude of potentiation (p < 0.05, Mann–Whitney’s U test), whereas the level of enhancement produced by IBMX remained virtually unchanged (p> 0.3, Mann–Whitney’s U test; Fig. 2B). This suggests that IBMX may inhibit the degradation of cyclic nucleotides, thereby potentiating the responsiveness to NE, and that this responsiveness to NE may be saturated in the presence of IBMX.
Fig. 2.

Enhancement of NE-induced potentiation by inhibitors of cyclic nucleotide PDE. A, Time-dependent plots of the average EPSC amplitude in a representative experiment. NE (0.1 μm, open bar) and IBMX (100 μm, closed bar) were bath-applied during the indicated periods. Insets, Sample records of the EPSC of control (left), in the presence of NE (middle), and in the presence of both NE and IBMX (right). The small biphasic signal preceding the EPSC is the capacitative coupling response. Calibration: 10 msec, 0.5 nA.B, Summary of the effects of IBMX (100 μm) on the NE-dependent potentiation of EPSC. EPSCs were normalized to the mean EPSC in the absence of NE and IBMX. Each column is the mean of six or seven experiments. Error bars indicate SEM. Statistical significance was evaluated as indicated by pairedt test. All differences between the groups with and without NE treatment were significant (p < 0.02, paired t test). C, Average effect of zaprinast (30 μm; n = 8) on NE-dependent potentiation of EPSC, which was normalized to that just before the application of NE (0.1 μm). Error bars indicate SEM. D, Summary (n = 7) of the effect of Ro-20-1724 (100 μm) on NE (0.1 μm)-dependent potentiation of EPSC. Eachcolumn is the mean of the value normalized to the control value in the absence of NE and Ro-20-1724; fromleft to right, the control, in the presence of NE, and in the presence of both NE and Ro-20-1724. The difference between the middle and the right columns is insignificant (p > 0.7, paired t test). Error bars indicate SEM.
The NE-dependent potentiation was also enhanced by zaprinast (Fig.2C), an inhibitor of cGMP-selective PDE (PDE5). Zaprinast enhanced the NE (0.1 μm)-dependent potentiation by 1.77 ± 0.19-fold (n = 9; p < 0.02, paired t test between raw data), whereas zaprinast alone did not potentiate the EPSC (1.10 ± 0.04 of the control;n = 6; p > 0.1, paired ttest between raw data). In contrast, Ro-20-1724, an inhibitor of cAMP-selective PDE (PDE4), had no effect on the NE-induced EPSC potentiation (Fig. 2D). Therefore, cGMP rather than cAMP appears to be involved as the second messenger of the NE-induced potentiation of transmitter release.
Effects of cyclic nucleotide analogs
Next, the effect of the membrane-permeable cGMP analog 8Br-cGMP was examined. The EPSCs fluctuated in amplitude in the solution with low Ca2+ and high Mg2+concentrations and often failed to elicit any response but, on average, were potentiated by 8Br-cGMP (30 μm) in a sustained manner (Fig.3A,B). Synaptic failure occurred in 71 of 100 trials before the application of 8Br-cGMP (Fig. 3C). Bath application of 30 μm8Br-cGMP decreased the occurrence of synaptic failures to 18 of 100 trials and increased the occurrence of large EPSCs (Fig.3B,D). Calculations based on these data (n = 6) revealed that 8Br-cGMP (30 μm) increased the mean number of quanta in a single EPSC (quantal content) by 2.23 ± 0.55-fold with a negligible change in the mean size of a single quanta (quantal size, 1.00 ± 0.14-fold), the difference being significant (p< 0.02, Mann–Whitney’s U test). As summarized in Figure3E, 8Br-cGMP potentiated the EPSC amplitude significantly, and no further potentiation was induced by the addition of NE after potentiation by 8Br-cGMP. In contrast, the same concentration of 8Br-cAMP did not potentiate the EPSC, and NE potentiated the EPSC in the presence of 8Br-cAMP to the same extent as in its absence (Fig.3E).
Fig. 3.

Effects of membrane-permeable cyclic nucleotide analogs on the transmitter release. A, Representative experimental data showing the effect of 8Br-cGMP on the EPSC. The presynaptic oculomotor nerve was stimulated at 0.33 Hz by twin pulses with an interpulse interval of 40 msec. Top, Average trace of 100 consecutive records before the application of 8Br-cGMP. The facilitation ratio was 2.1. Bottom, Average trace of 100 consecutive records after 5 min treatment with 8Br-cGMP (30 μm). The facilitation ratio was 1.5. B, Time-dependent plot of the EPSC amplitudes of the same experiments as in A. 8Br-cGMP was bath-applied during the indicated period. C, Amplitude histogram of 100 consecutive EPSCs in B just before the application of 8Br-cGMP. Them was calculated from the occurrence of failure transmissions based on the Poisson statistics and was 0.342. Theq was estimated as the mean EPSC divided bym and was 17.1 pA. D, Amplitude histogram of 100 consecutive EPSCs in B during potentiation by 8Br-cGMP. Note the reduction of the occurrence of failure responses. The m was 1.72, and the q was 14.0 pA.E, Effects of 8Br-cGMP on the EPSC and NE-induced potentiation were compared with those of 8Br-cAMP. Eachcolumn is the mean of the EPSC amplitude normalized to the mean EPSC amplitude before the application of the cyclic nucleotide analogs. Error bars indicate SEM. The three columns on the left are a summary of eight similar experiments of control, the effect of 100 μm 8Br-cGMP, and the effect of 100 μm 8Br-cGMP plus 10 μm NE. Thetwo columns on the right are a summary of six similar experiments of the effect of 100 μm 8Br-cAMP and the effect of 100 μm 8Br-cAMP plus 10 μm NE. Statistical significance was tested as indicated (paired t test). The difference between thefirst and the fourth columns was insignificant (p > 0.3, pairedt test between raw data).
The site of action of cGMP could be in either the presynaptic terminal or the postsynaptic cell. To distinguish between these possibilities, a high concentration of cGMP (0.3 mm) was injected into the postsynaptic cell through the recording electrode with an access resistance of <5 MΩ. Ten minutes after the whole-cell configuration was established, NE (10 μm) still potentiated the EPSC amplitude by 2.0- to 5.0-fold of the control (n = 4;p > 0.1, Mann–Whitney’s U test), as was the case without cGMP (Fig. 2B). Because the NE-induced potentiation is occluded by 8Br-cGMP (Fig. 3E), the site of action of cGMP appears to be presynaptic.
Contribution of NO to NE-induced potentiation
These results suggest that NE selectively activates guanylyl cyclase. Two families of guanylyl cyclases have been reported: the soluble guanylyl cyclases, which are activated by NO, and the particulate guanylyl cyclases, which are anchored to the membrane by a single transmembrane domain (Garbers, 1992). Is NO involved in the NE-induced potentiation of transmitter release? Even in the presence of NO synthase inhibitor l-NAME (100 μm, after pretreatment for 1–2 hr), NE (10 μm) again potentiated the EPSCs by 2.35 ± 0.33-fold (n = 5), which is exactly the same extent as NE alone (p > 0.5, Mann–Whitney’s U test). It is expected that extracellular NO scavengers would reduce the intracellular NO, because NO is freely permeable through the membrane (Garthwaite, 1991). However, even in the presence of 30 μm hemoglobin, NE potentiated the EPSC by 1.5- to 10.8-fold (n = 3), which was the same as in the absence of hemoglobin (p > 0.2, Mann–Whitney’s U test). Does NO itself potentiate the transmitter release? To test this, the effects of the NO donor SNP (100 μm) were examined. As exemplified in Figure4A, SNP did not potentiate the EPSC even in the presence of IBMX. The control EPSC was recorded in the presence of 100 μm IBMX, and mand q were 2.7 and 10.1 pA, respectively (Fig.4B). The addition of 100 μm SNP did not cause an obvious distortion of the amplitude histogram (Fig.4C), with m and q of 2.5 and 10.6 pA, respectively. In a total of four similar experiments in the presence of 100 μm SNP, the m was 0.93 ± 0.16 of the control (p > 0.5, paired t test between raw data), the q was 1.02 ± 0.01 of the control (p > 0.2, paired t test between raw data), and the average EPSC was 0.92 ± 0.15 of the control (p > 0.7, paired t test between raw data). Another potent NO donor, NOR1 (20 μm), failed to potentiate the EPSC even in the presence of 100 μm IBMX (0.88 ± 0.14 of the control;n = 5; p > 0.3, paired ttest between raw data).
Fig. 4.

Effects of NO donor on the transmitter release.A, Time-dependent plot of EPSC amplitudes of a representative experiment. IBMX (100 μm) was present throughout the experiments. The presynaptic oculomotor nerve was stimulated at 0.33 Hz. SNP (100 μm) was bath-applied as indicated (closed bar). B, EPSC amplitude histogram of 100 consecutive records before the application of SNP. Them and the q were estimated from c.v. and were 2.7 and 10.1 pA, respectively. The arrow on theleft indicates the occurrence of failures expected from a Poisson distribution. C, EPSC amplitude histogram of 100 consecutive records during the bath application of SNP. Them and the q were 2.5 and 10.6 pA, respectively. The arrow on the leftindicates the occurrence of failures expected from a Poisson distribution.
Involvement of PKG
Four types of molecular targets that mediate the intracellular actions of cGMP are present: cGMP-gated channels (Zagotta and Siegelbaum, 1996), a cGMP-activated cAMP PDE (PDE2) (Polson and Strada, 1996), a cGMP-inhibited PDE (PDE3) (Polson and Strada, 1996), and a cGMP-stimulated protein kinase (PKG) (Francis and Corbin, 1994). Because the effects of 8Br-cAMP on the transmitter release as well as on its potentiation by NE were negligible (Fig. 3E), the indirect regulation of cAMP by cGMP is unlikely to be the mechanism of the NE-induced potentiation. If the activation of cGMP-gated channels is involved in the potentiation of transmitter release, a change in the resting [Ca2+]i or the nerve-evoked [Ca2+]i increment (ΔCa) would be expected. This notion was tested by examining the effect of 8Br-cGMP on the Ca2+ influx into presynaptic terminals. As shown in Figure 5A, stimulation of the oculomotor nerve increased the intraterminal Ca2+ concentration ([Ca2+]i). When eight pulses were applied at 100 Hz, ΔCa was accumulated to approximately sevenfold of that by a single pulse (Fig. 5B). Calculations based on these data (n = 11) showed that the ΔCa produced by eight pulses at 100 Hz was 7.67 ± 0.08-fold of that produced by a single pulse, indicating that ΔCa was almost proportional to the number of applied pulses in this range. Similarly, the reduction of [Ca2+]o from 1 to 0.6 mm attenuated ΔCa to 0.63 ± 0.02 of the control (n = 10; Fig. 5C,D). Although [Ca2+]i transients should be much larger and faster in the vicinity of Ca2+ channel clusters (Zucker, 1996; Neher, 1998), the volume-averaged fura-2 signal accurately reflects changes in the local concentration (Sinha et al., 1997). These results indicate that this is indeed the case for the calyx-type terminal and that ΔCa appears to be nearly proportional to the Ca2+ influx into the nerve terminal in this range of [Ca2+]o (Yawo, 1999). If 8Br-cGMP increases the exocytotic fusion probability by increasing the Ca2+ influx during a presynaptic action potential, ΔCa would be expected to increase to a value above the ΔCa at 0.6 mm [Ca2+]o. Actually, 8Br-cGMP had no effect on ΔCa (Fig. 5C,D). The effect of 8Br-cGMP on the resting [Ca2+]i was also negligible (Fig.5C; 1.04 ± 0.01 of the control; n = 11; p > 0.2, paired t test between raw data).
Fig. 5.

Effects of 8Br-cGMP on the intraterminal Ca2+ concentration ([Ca2+]i) of a calyx-type presynaptic terminal of the chick ciliary ganglion. A, Sample record of the [Ca2+]i in response to a single oculomotor nerve stimulation in a solution containing 1 mm [Ca2+]o. The [Ca2+]i was 36 nmbefore stimulation. B, The same presynaptic terminal was stimulated by eight pulses at 100 Hz in the same solution. The [Ca2+]i was 46 nm before stimulation. C, Effects of 8Br-cGMP on the [Ca2+]i. The same presynaptic terminal was stimulated by eight pulses at 100 Hz in a solution containing 0.6 mm [Ca2+]o before (solid line) and after treatment with 8Br-cGMP (shaded line). During both conditions, the [Ca2+]i was 42 nm before stimulation. D, Summary of the effects of 8Br-cGMP on the nerve-evoked increment of the [Ca2+]i (ΔCa). Eachcolumn represents the mean of the ΔCa normalized to that at 1 mm [Ca2+]obefore the application of the drugs. Error bars indicate SEM. The columns are summaries of eight series of similar experiments in which the extracellular solution was changed (from left toright) from the control with 1 mm[Ca2+]o, to the 0.6 mm [Ca2+]o solution, to the 0.6 mm [Ca2+]osolution containing 100 μm 8Br-cGMP, to the 0.6 mm [Ca2+]o solution containing 10 μm NE, and to the 0.6 mm[Ca2+]o solution containing 10 μm NE plus 10 μm PA. Note that the effect of 8Br-cGMP was negligible (p > 0.8, pairedt test), as was that of NE in the presence of PA (p > 0.7, paired t test), and that the difference was insignificant (p> 0.3, paired t test).
By exclusion, PKG appears to be the most likely candidate. A previous study has demonstrated the presence of PKG in embryonic chick ciliary ganglia (Lengyel et al., 1996). Pretreatment of the ganglion with the membrane-permeable PKG inhibitor Rp-8pCPT-cGMPS (100 μm) reduced both the NE-induced potentiation and its enhancement by IBMX (Fig. 6A). Another selective PKG inhibitor, KT5823 (1 μm), which occupies the ATP-binding site of PKG (Kase et al., 1987), showed a tendency to suppress NE-induced potentiation. KT5823 evidently inhibited the enhanced adrenergic potentiation in the presence of IBMX (Fig.6A). These observations indicate that PKG-dependent phosphorylation of some presynaptic proteins must be the major mechanism of the NE-induced potentiation.
Fig. 6.
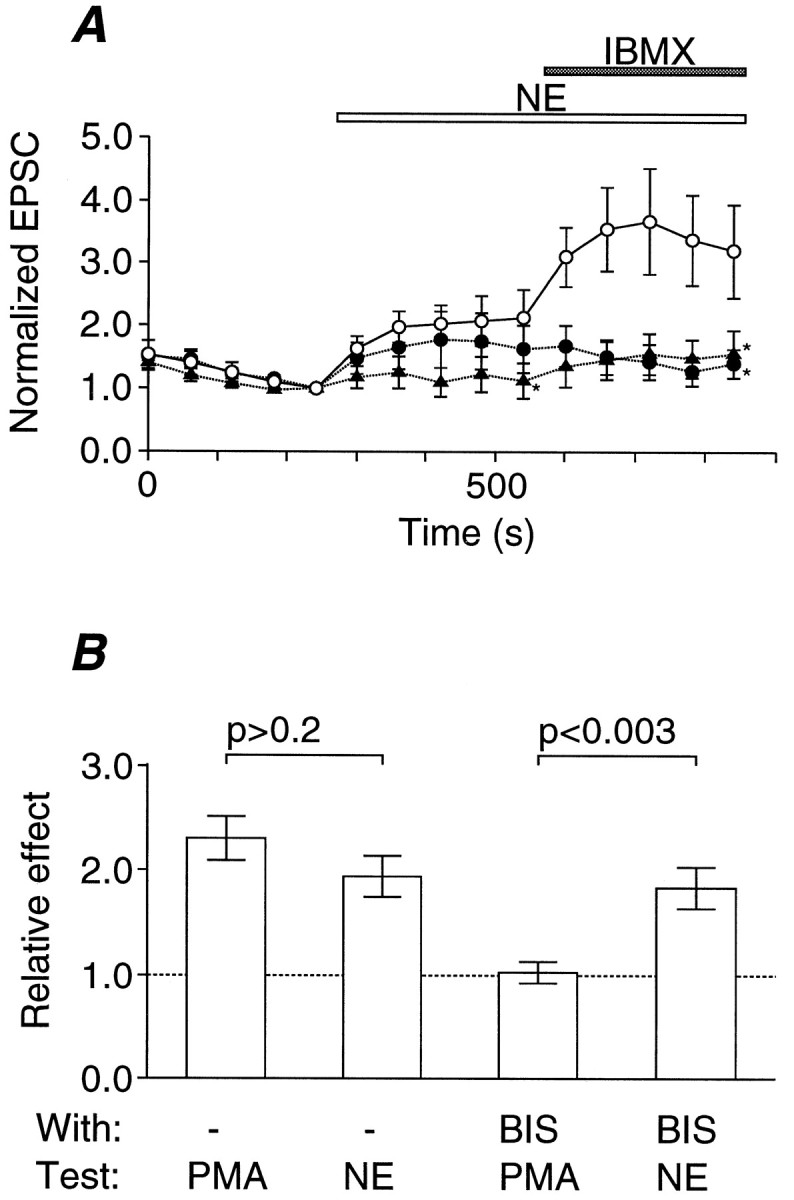
Involvement of PKG-dependent phosphorylation in the NE-dependent potentiation of transmitter release. A, Average effects of PKG inhibitors on the NE-dependent potentiation of EPSC, which is normalized to that just before the application of NE (10 μm). Error bars indicate SEM. The period of application is indicated by an open horizontal bar. The period of application of IBMX (100 μm) is also indicated by aclosed horizontal bar. Open circles, In the presence of 0.05% DMSO as control (n = 6);closed circles, in the presence of 1 μmKT5823 (n = 6); closed triangles, in the presence of 100 μm Rp-pCPT-cGMPS (n = 6). The inhibition of potentiation was statistically significant (*p < 0.05; Mann–Whitney’s U test) as indicated. B, Comparison between NE-induced potentiation and PKC-dependent potentiation. Each column shows the mean of the relative effect on the EPSC amplitude of the test substances (from left toright): the effect of a phorbol ester (PMA, 0.1 μm, n = 7), the effect of NE (10 μm; n = 8), the effect of PMA in the presence of a PKC inhibitor, BIS (10 μm;n = 7), and the effect of NE in the presence of BIS (n = 7). Error bars indicate SEM. Statistical significance was tested as indicated (Mann–Whitney’s Utest). The difference between the first and thethird columns was significant (p < 0.001, Mann–Whitney’sU test), whereas the difference between thesecond and the fourth columns was insignificant (p > 0.2, Mann–Whitney’sU test).
In this calyx-type presynaptic terminal the activation of protein kinase C (PKC) also potentiates transmitter release by upregulating the Ca2+ sensitivity of exocytosis with negligible effects on the Ca2+ dynamics (Yawo, 1999). Could NE potentiate transmitter release by activating PKC? As reported previously (Yawo, 1999), the EPSC was potentiated by a phorbol ester (PMA, 0.1 μm) to a similar extent as by NE (Fig.6B). The PMA-induced potentiation was completely suppressed by the PKC-selective inhibitor BIS (10 μm), whereas the NE-induced potentiation was not antagonized by BIS (Fig.6B). Therefore, activation of PKC does not seem to be involved in the NE-induced potentiation.
Subcellular mechanisms of cGMP-induced potentiation
As previously reported (Yawo, 1996), NE significantly reduced the ΔCa (n = 8; p < 0.02; pairedt test), and this effect was completely reversed by the α1- and α2-adrenergic receptor antagonist phentolamine (PA) (Fig. 5D) or by the α2-adrenergic receptor antagonist yohimbine. This leads to the conclusion that NE upregulates the exocytotic mechanism other than Ca2+ influx or Ca2+buffering and removal. Because 8Br-cGMP exhibited negligible effects on both the ΔCa and the resting [Ca2+]i, it might potentiate EPSCs through the same mechanism as that of NE. When the presynaptic oculomotor nerve was stimulated by twin pulses at short intervals, the second EPSC was, on average, lager than the first EPSC (Figs.1A, 3A). The mechanism of paired pulse facilitation (Martin and Pilar, 1964b; Yawo, 1999) has been attributed to the enhancement of the exocytotic fusion probability as a result of residual Ca2+ in the presynaptic terminal (Katz and Miledi, 1968; Zucker, 1996; Neher, 1998). In fact, the paired pulse facilitation was accompanied by an increase in m with a negligible change in q (H. Yawo, unpublished observation). With a pulse interval of 40 msec, the ratio of the second EPSC amplitude to the first one (paired pulse ratio) was 1.94 ± 0.12 at 1 mm [Ca2+]o and 5 mm [Mg2+]o(n = 5), whereas it was 1.07 ± 0.18 at 2 mm [Ca2+]o and 4 mm [Mg2+]o(n = 6). Because the size of the readily releasable pool of synaptic vesicles is limited, maneuvers that increase the probability of vesicular exocytosis would reduce the paired pulse ratio (Debanne et al., 1996; Schultz, 1997; Yawo, 1999). NE also reduced the facilitation ratio with a time course similar to that of EPSC potentiation (Fig. 1A). After the NE-induced potentiation, the paired pulse ratio was 1.52 ± 0.16 at 1 mm [Ca2+]o and 5 mm [Mg2+]o(n = 5; p < 0.001, paired ttest) and 0.74 ± 0.16 at 2 mm[Ca2+]o and 4 mm[Mg2+]o (n = 6;p < 0.01, paired t test). Because 8Br-cGMP also decreased the paired pulse ratio (Fig. 3A; 0.81 ± 0.06 of the control; n = 13; p < 0.005, paired t test between raw data), cGMP appears to upregulate the exocytotic fusion probability of synaptic vesicles (Debanne et al., 1996; Schultz, 1997; Yawo, 1999). Therefore, with respect to the subcellular mechanism, the 8Br-cGMP-induced potentiation was indistinguishable from the NE-induced potentiation (Yawo, 1996).
DISCUSSION
Intracellular signal transduction mechanisms of the NE-induced potentiation of transmitter release
The data presented here revealed the following five main properties of the NE-induced potentiation: (1) NE potentiated the quantal transmitter release in a manner resistant to both α1-, α2- and β-adrenergic receptors (Fig.1); (2) the NE-induced potentiation was further enhanced by IBMX, a nonspecific PDE inhibitor or zaprinast, an inhibitor of cGMP-selective PDE, whereas it was unaffected by Ro-20-1724, an inhibitor of cAMP-selective PDE (Fig. 2); (3) exogenously applied cGMP potentiated the quantal transmitter release and occluded the NE-induced potentiation, whereas the effects of the cAMP analog were negligible (Fig. 3); (4) the NE-induced potentiation was resistant to both NO synthase inhibitor and NO scavenger, and the NO donors could not potentiate the transmitter release (Fig. 4); and (5) the NE-induced potentiation as well as its enhancement by IBMX was antagonized by two PKG inhibitors with different modes of action (Fig. 6). The negligible effects of IBMX and zaprinast in the absence of NE indicate that their effect was specific to PDE. Because the NE-induced potentiation was not occluded by the intracellular injection of high cGMP into the postsynaptic cell, cGMP appears to be generated in the presynaptic terminal. The negligible effects of cAMP on the transmitter release indicate that the indirect regulation of cAMP by cGMP is unlikely to be the mechanism of the NE-induced potentiation. Because both NE-induced potentiation and cGMP-dependent potentiation were not accompanied by a change in the resting [Ca2+]i and ΔCa, the activation of cGMP-gated channels appears not to be the mechanism of the potentiation. Experiments using posthatched chicks showed that LTP of the ciliary ganglion synapse was inhibited byl-NAME (100 μm) and induced by SNP (100 μm) (Lin and Bennett, 1994). It is unclear where this difference comes from.
In summary, all the present results indicate that NE drives the following series of reactions in the calyx-type presynaptic terminals of embryonic chick ciliary ganglion: (1) NE binds to the receptor, which activates NO-insensitive guanylyl cyclases; (2) accumulation of cGMP activates PKG; and (3) PKG phosphorylates a target protein, which may be involved in the exocytosis of synaptic vesicles. Therefore, the receptor involved in the presynaptic potentiation is different from any known catecholamine receptor subtypes in terms of the signal transduction mechanisms as well as the pharmacological properties. Although this receptor pharmacologically resembles the “γ-adrenergic receptor” reported in arterial smooth muscle cells (Hirst et al., 1982; Benham and Tsien, 1988), the molecular identity is unclear. It also remains unknown how this receptor activates guanylyl cyclases.
Mechanisms involved in NE-, cGMP-, and PKG-dependent potentiation
In many synapses including the chick calyx-type synapse, the magnitude of the paired pulse ratio is negatively correlated withm of the first response (Debanne et al., 1996; Schulz, 1997). The most plausible mechanism seems to be a depletion of docked vesicles for the second release (Debanne et al., 1996; Doburunz and Stevens, 1997; O’Donovan and Rinzel, 1997; Yawo, 1999). In the present results, the NE-induced potentiation accompanied the reduction of the paired pulse ratio (Fig. 1A). It is likely that NE increased the exocytotic fusion probability in the first presynaptic release, depleting the available vesicles for the second release. The results of the paired pulse experiments are consistent with the notion that NE enhances the Ca2+sensitivity of the exocytotic fusion probability because it does not increase the Ca2+ influx (Yawo, 1996).
Because this report fills the missing link between NE and the potentiation of transmitter release, cGMP and PKG might upregulate the Ca2+ sensitivity of exocytotic fusion probability. This notion is consistent with the observation that the membrane-permeable cGMP analog 8Br-cGMP enhanced m without increasing ΔCa (Fig. 5). Therefore, the cGMP-dependent potentiation was accompanied by no detectable changes in net Ca2+influx, buffering, or removal. In addition, the reduction of the paired pulse ratio by 8Br-cGMP strongly suggests that the exocytotic fusion probability was enhanced. The same mechanism appears to be upregulated by PKC, because NE was ineffective after PKC-dependent potentiation and vice versa (H. Yawo, unpublished observation). Although the precise molecular target is unknown, this modulation might involve phosphorylation of the intracellular Ca2+ sensor molecule itself or a molecule interacting with the Ca2+ sensor (Yawo, 1999).
Footnotes
This work was supported by grants-in-aid from the Ministry of Education, Science and Culture of Japan and the Yamanouchi Foundation for Research on Metabolic Disorders. I thank S. Sai for technical support, Drs. T. Abe, K. Kawa, and M. Umemiya for comments on this manuscript, and B. Bell for reading this manuscript. A review by Dr. M. Kuno is gratefully acknowledged.
Correspondence should be addressed to Dr. Hiromu Yawo, Department of Neurophysiology, Tohoku University School of Medicine, Sendai 980-8575, Japan.
REFERENCES
- 1.Benham CD, Tsien RW. Noradrenaline modulation of calcium channels in single smooth muscle cells from rabbit ear artery. J Physiol (Lond) 1988;404:767–784. doi: 10.1113/jphysiol.1988.sp017318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bylund DA, Eikenberg DC, Hieble JP, Langer SZ, Lefkowitz RJ, Minneman KP, Molinoff PB, Ruffolo RR, Trendelenburg U. International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol Rev. 1994;46:121–136. [PubMed] [Google Scholar]
- 3.Debanne D, Guérineau NC, Gähwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol (Lond) 1996;491:163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delcour AH, Tsien RW. Altered prevalence of gating modes in neurotransmitter inhibition of N-type calcium channels. Science. 1993;259:980–984. doi: 10.1126/science.8094902. [DOI] [PubMed] [Google Scholar]
- 5.Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 6.Francis SH, Corbin JD. Structure and function of cyclic nucleotide-dependent protein kinases. Annu Rev Physiol. 1994;56:237–272. doi: 10.1146/annurev.ph.56.030194.001321. [DOI] [PubMed] [Google Scholar]
- 7.Garbers DL. Guanylyl cyclase receptors and their endocrine, paracrine, and autocrine ligands. Cell. 1992;71:1–4. doi: 10.1016/0092-8674(92)90258-e. [DOI] [PubMed] [Google Scholar]
- 8.Garthwaite J. Glutamate, nitric oxide and cell-cell signalling in the nervous system. Trends Neurosci. 1991;14:60–67. doi: 10.1016/0166-2236(91)90022-m. [DOI] [PubMed] [Google Scholar]
- 9.Goldstein DS. Catecholamine receptors and signal transduction. Adv Pharmacol. 1998;42:379–390. doi: 10.1016/s1054-3589(08)60770-x. [DOI] [PubMed] [Google Scholar]
- 10.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 11.Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. J Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- 12.Hirst GDS, Nield TO, Silverberg GD. Noradrenaline receptors on the rat basilar artery. J Physiol (Lond) 1982;328:351–360. doi: 10.1113/jphysiol.1982.sp014268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirst GDS, Bramich NJ, Edwards FR, Klemm M. Transmission at autonomic neuroeffector junctions. Trends Neurosci. 1992;15:40–46. doi: 10.1016/0166-2236(92)90024-3. [DOI] [PubMed] [Google Scholar]
- 14.Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M. K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem Biophys Res Commun. 1987;142:436–440. doi: 10.1016/0006-291x(87)90293-2. [DOI] [PubMed] [Google Scholar]
- 15.Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol (Lond) 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuba K, Kato E, Kumamoto E, Koketsu K, Hirai K. Sustained potentiation of transmitter release by adrenaline and dibutyryl cyclic AMP in sympathetic ganglia. Nature. 1981;291:654–656. doi: 10.1038/291654a0. [DOI] [PubMed] [Google Scholar]
- 17.Kuno M, Weakley JN. Quantal components of the inhibitory synaptic potential in spinal motoneurones of the cat. J Physiol (Lond) 1972;224:287–303. doi: 10.1113/jphysiol.1972.sp009895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lengyel I, Nichol KA, Sim ATR, Bennett MR, Dunkley PR, Rostas JAP. Characterization of protein kinase and phosphatase systems in chick ciliary ganglion. Neuroscience. 1996;70:577–588. doi: 10.1016/0306-4522(95)00356-8. [DOI] [PubMed] [Google Scholar]
- 19.Lin Y-Q, Bennett MR. Nitric oxide modulation of quantal secretion in chick ciliary ganglia. J Physiol (Lond) 1994;481:385–394. doi: 10.1113/jphysiol.1994.sp020447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacKinnon AC, Spedding M, Brown CM. α2-Adrenoceptors: more subtypes but fewer functional differences. Trends Pharmacol Sci. 1994;15:119–123. doi: 10.1016/0165-6147(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 21.Martin AR, Pilar G. Quantal components of the synaptic potential in the ciliary ganglion of the chick. J Physiol (Lond) 1964a;171:454–475. doi: 10.1113/jphysiol.1964.sp007499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin AR, Pilar G. Presynaptic and postsynaptic events during post-tetanic potentiation and facilitation in the avian ciliary ganglion. J Physiol (Lond) 1964b;175:17–30. doi: 10.1113/jphysiol.1964.sp007500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milligan G. Mechanisms of multifunctional signalling by G protein-linked receptors. Trends Pharmacol Sci. 1993;14:239–244. doi: 10.1016/0165-6147(93)90019-g. [DOI] [PubMed] [Google Scholar]
- 24.Moore RY, Bloom RE. Central catecholamine neuron systems: anatomy and physiology of the norepinephrine and epinephrine systems. Annu Rev Neurosci. 1979;2:113–168. doi: 10.1146/annurev.ne.02.030179.000553. [DOI] [PubMed] [Google Scholar]
- 25.Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 26.Nicoll RA, Malenka RC, Kauer JA. Functional comparison of neurotransmitter receptor subtypes in mammalian central nervous system. Physiol Rev. 1990;70:513–565. doi: 10.1152/physrev.1990.70.2.513. [DOI] [PubMed] [Google Scholar]
- 27.O’Donovan MJ, Rinzel J. Synaptic depression: a dynamic regulation of synaptic communication with varied functional roles. Trends Neurosci. 1997;20:431–433. doi: 10.1016/s0166-2236(97)01124-7. [DOI] [PubMed] [Google Scholar]
- 28.Polson JB, Strada SJ. Cyclic nucleotide phosphodiesterases and vascular smooth muscle. Annu Rev Pharmacol Toxicol. 1996;36:403–427. doi: 10.1146/annurev.pa.36.040196.002155. [DOI] [PubMed] [Google Scholar]
- 29.Schultz PE. Long-term potentiation involves increases in the probability of neurotransmitter release. Proc Natl Acad Sci USA. 1997;94:5888–5893. doi: 10.1073/pnas.94.11.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinha SR, Wu L-G, Saggau P. Presynaptic calcium dynamics and transmitter release evoked by single action potentials at mammalian central synapses. Biophys J. 1997;72:637–651. doi: 10.1016/s0006-3495(97)78702-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yawo H. Noradrenaline modulates transmitter release by enhancing the Ca2+ sensitivity of exocytosis in the chick ciliary presynaptic terminal. J Physiol (Lond) 1996;493:385–391. doi: 10.1113/jphysiol.1996.sp021390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yawo H. Protein kinase C potentiates the transmitter release from the chick ciliary presynaptic terminal by increasing the exocytotic fusion probability. J Physiol (Lond) 1999;515:169–180. doi: 10.1111/j.1469-7793.1999.169ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yawo H, Chuhma N. Preferential inhibition of ω-conotoxin-sensitive presynaptic Ca2+ channels by adenosine autoreceptors. Nature. 1993;365:256–258. doi: 10.1038/365256a0. [DOI] [PubMed] [Google Scholar]
- 34.Yawo H, Chuhma N. ω-Conotoxin-sensitive and -resistant transmitter release from the chick ciliary presynaptic terminal. J Physiol (Lond) 1994;477:437–448. doi: 10.1113/jphysiol.1994.sp020205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zagotta WN, Siegelbaum SA. Structure and function of cyclic nucleotide-gated channels. Annu Rev Neurosci. 1996;19:235–263. doi: 10.1146/annurev.ne.19.030196.001315. [DOI] [PubMed] [Google Scholar]
- 36.Zucker RS. Exocytosis: a molecular and physiological perspective. Neuron. 1996;17:1049–1055. doi: 10.1016/s0896-6273(00)80238-x. [DOI] [PubMed] [Google Scholar]