

Abstract
In this report, we provide evidence that NGF and BDNF have functionally antagonistic actions on sympathetic neuron growth and target innervation, with NGF acting via TrkA to promote growth and BDNF via p75NTR to inhibit growth. Specifically, in cultured sympathetic neurons that themselves synthesize BDNF, exogenous BDNF inhibits and function-blocking BDNF antibodies enhance process outgrowth. Both exogenous and autocrine BDNF mediate this effect via p75NTR because (1) BDNF does not inhibit growth of neurons lacking p75NTR, (2) function-blocking p75NTR antibodies enhance NGF-mediated growth, and (3) p75NTR−/− sympathetic neurons grow more robustly in response to NGF than do their wild-type counterparts. To determine the physiological relevance of this functional antagonism, we examined the pineal gland, a well defined sympathetic target organ. BDNF is present in the pineal gland during target innervation, and incoming sympathetic axons are p75NTR positive. Moreover, the pineal glands of BDNF+/− and BDNF−/−mice are hyperinnervated with sympathetic fibers, and tyrosine hydroxylase (TH) levels are elevated. Increased tyrosine hydroxylase is also observed in the BDNF+/− carotid artery, another sympathetic neuron target. Thus, BDNF, made by sympathetic neurons and/or their target organs, acts via p75NTR to antagonize NGF-mediated growth and target innervation, suggesting that sympathetic target innervation is determined by the balance of positively and negatively acting neurotrophins present in developing and potentially mature targets.
Keywords: nerve growth factor, brain-derived neurotrophic factor, sympathetic neurons, target innervation, neurotrophin receptor, TrkA, p75NTR, pineal gland
The neurotrophic factor hypothesis states that neuronal growth and survival are regulated by target-derived neurotrophic factors, such as nerve growth factor (NGF) (for review, see Thoenen and Barde, 1980; Levi-Montalcini, 1987), so that competition for limiting amounts of trophic factors match the number of innervating neurons to target cells (Oppenheim, 1991). This hypothesis is based largely on peripheral sympathetic neurons, which are absolutely dependent on NGF during the period of target competition (Levi-Montalcini, 1987). During this developmental window, target-derived NGF is thought to regulate the density of target innervation by stimulating terminal growth (Miller et al., 1994) and by serving as a discriminator that allows elimination of neurons that fail to sequester adequate target territory.
Target-derived NGF binds to two different cell surface receptors on sympathetic neurons to elicit these responses: the tyrosine kinase receptor TrkA (Cordon-Cardo et al., 1991; Kaplan et al., 1991a,b; Klein et al., 1991) and the p75 neurotrophin receptor (p75NTR) (Johnson et al., 1986; Radeke et al., 1987). In addition to these two receptors, postmitotic sympathetic neurons express low levels of another Trk family member, TrkC (Belliveau et al., 1997). Two lines of evidence indicate that NGF binding to TrkA alone is sufficient to mediate sympathetic neuron survival and growth. First, ligand-mediated activation of TrkA, but not p75NTR, supports sympathetic neuron growth and survival (Weskamp and Reichardt, 1991; Ibáñez et al., 1992; Clary et al., 1994; Belliveau et al., 1997; Bamji et al., 1998). Second, all sympathetic neurons are lost in TrkA−/− mice (Smeyne et al., 1994) as they are in NGF−/− mice (Crowley et al., 1994).
Implicit to the neurotrophic factor hypothesis is the assumption that positive signals, such as those elicited by target-derived NGF binding to TrkA, are sufficient to determine both the life and death of a developing neuron and the appropriate level of target innervation. However, we have demonstrated recently that sympathetic neuron survival is not only determined by TrkA, but it is also regulated by negatively acting neurotrophins such as BDNF, which signal though p75NTR to mediate neuronal apoptosis (Aloyz et al., 1998; Bamji et al., 1998). Specifically, when survival signals are suboptimal, BDNF-mediated activation of p75NTR causes sympathetic neuron apoptosis. Moreover, in BDNF−/− mice, sympathetic neuron number is increased, and in p75NTR−/− mice, the normal period of sympathetic neuron death is greatly delayed. This latter deficit in apoptosis is intrinsic to sympathetic neurons, because cultured p75NTR−/− neurons die much more slowly than their wild-type counterparts in the absence of NGF. Thus, naturally occurring sympathetic neuron death is regulated by positively and negatively acting neurotrophins that signal through TrkA versus p75NTR.
Because the apoptotic effect of p75NTR signaling occurs only under suboptimal survival conditions, we hypothesized that p75NTR might also inhibit other TrkA-mediated responses when survival conditions are optimal. In this report, we test this hypothesis and demonstrate that BDNF acts via p75NTR to inhibit NGF-mediated growth and target innervation. Thus, the balance of signaling mediated by the TrkA versus p75 neurotrophin receptors ultimately determines both the survival and growth of developing sympathetic neurons.
MATERIALS AND METHODS
Mass cultures of sympathetic neurons. Mass cultures of pure sympathetic neurons from the superior cervical ganglion (SCG) of postnatal day (P) 1 Sprague Dawley rats (Charles River Breeding Laboratories, St. Constant, Quebec, Canada) were prepared as described previously (Ma et al., 1992). Neurons were plated at low density (approximately one ganglion/well) in Nunclon four-well culture dishes (Life Technologies, Burlington, Ontario, Canada) coated with either rat tail collagen or poly-d-lysine and laminin (both from Collaborative Biomedical Products, Bedford, MA). Culture medium was UltraCulture (BioWhittaker, Walkersville, MD), supplemented with 3% rat serum (Harlan Bioproducts, Madison, WI), 2 mmglutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (all from BioWhittaker), and for days 2 and 3, 7 μm cytosine arabinoside (Sigma-Aldrich Canada Ltd., Oakville, Ontario, Canada).
CD-1 mouse sympathetic neurons were cultured by a modification of the method used to prepare rat neurons. Mouse cultures were essentially prepared the same way, but were dissociated in UltraCulture medium rather than in HBSS. Instead of rat serum, 3% fetal bovine serum (Life Technologies) was used, and 3.5 μm cytosine arabinoside was added to the culture medium on day 1 after plating.
NGF used in these experiments was purified from mouse salivary glands and supplied by Cedarlane Laboratories (Hornby, Ontario, Canada). The sources of recombinant human BDNF were PeproTech (Rocky Hill, NJ), for the neuritogenesis assays, and Promega Corporation (Madison, WI), for the recombinant human (rh) BDNF neutralization experiments. The p75NTR function-blocking antibody REX (Weskamp and Reichardt, 1991) was the kind gift of Dr. L. Reichardt (University of California, San Francisco, CA). REX is directed against the extracellular domain of p75NTR and was used as an antiserum at a dilution of 1:100 (Weskamp and Reichardt, 1991). Rabbit serum (Life Technologies) of the same concentration was used as the negative control for REX. Anti-human BDNF (Promega) was used at 10 μg/ml. As a negative control for anti-BDNF, nonimmune chicken IgY (Promega) was used at up to 40 μg/ml in BDNF neutralization experiments and at 10 μg/ml in neuritogenesis experiments.
BDNF neutralization. To test the capacity of anti-BDNF to neutralize BDNF, TrkB-expressing NIH-3T3 cells (the kind gift of Dr. D. Kaplan, McGill University) were cultured in DMEM. Briefly, cells were washed twice and incubated for 1 hr at 37°C in buffer, followed by a 30 min wash at 37°C in a phosphate-free buffer. Treatment consisted of incubating cells for 5 min with either 50 ng/ml BDNF (Promega) or BDNF preadsorbed for 4 hr at 4°C with increasing concentrations of the BDNF antibody (5–40 μg/ml). In addition, TrkB-3T3 cells were treated with medium only or medium plus 40 μg/ml nonimmune chicken IgY. After these treatments, cells were lysed and immunoprecipitated with anti-pan Trk (Hempstead et al., 1992), and the immunoprecipitates were analyzed for TrkB activation by Western blot analysis with phosphotyrosine antibody 4G10 (Upstate Biotechnology, Lake Placid, NY), as we have described previously (Belliveau et al., 1997; Bamji et al., 1998).
Survival assays. NGF-dependent neurons were selected by culturing sympathetic neurons for 5 d in the presence of 50 ng/ml NGF, as described previously (Ma et al., 1992; Belliveau et al., 1997;Bamji et al., 1998). Neurons were washed three times for 1 hr each in neurotrophin-free media and then fed with media containing 10 ng/ml NGF with or without 100 ng/ml BDNF, 10 μg/ml αBDNF, or a 1:100 dilution of antiserum containing the p75NTR antibody REX (Weskamp and Reichardt, 1991). Each condition was repeated in triplicate, and analysis of survival was performed 48 hr later by using nonradioactive MTT survival assays that measure mitochondrial function (Celltitre 96, Promega, Madison, WI) (Belliveau et al., 1997). Specifically, 50 μl of the MTT reagent was added to 500 μl of media in each well and incubated for 2 hr at 37°C. After aspiration of the MTT-containing media, 100 μl of a 0.065N HCL/isopropanol mixture was added to each well to lyse the cells, and colorimetric analysis was performed using an ELISA reader. For rat sympathetic neurons, 10 ng/ml NGF represents 100% survival (Bamji et al., 1998); therefore all other values were considered to be relative to 10 ng/ml NGF. For mouse neurons, 7.5 ng/ml NGF represents 100% survival and was thus considered to be the 100% survival threshold for neuritogenesis experiments.
Analysis of transgenic animals. Mice heterozygous for a targeted mutation in the BDNF gene (Ernfors et al., 1994) or homozygous for a targeted mutation in the p75NTR gene (Lee et al., 1992) were obtained from Jackson Labs (Bar Harbor, ME). The BDNF+/− mice were maintained in a C129/BALB/c background. The p75NTR−/− mice were originally generated in a C129 background (Lee et al., 1992) and were crossed back into a C129 background before purchase from Jackson Labs and then maintained as homozygotes. Progeny from BDNF heterozygote crosses were screened for the mutant allele(s) using PCR, as we have described previously (Bamji et al., 1998).
Analysis and quantification of process outgrowth.p75NTR/BDNF regulation of neuronal growth was analyzed using two different types of neuritogenesis assays. Similar results were obtained with both approaches. The first assay, which measures the number of process intersections/neuron, is described in detail in Belliveau et al. (1997) and Yang et al. (1998). Briefly, P1 rat sympathetic neurons were cultured in 50 ng/ml NGF for 2–3 d to upregulate p75NTR, whose increased expression in response to NGF occurs independently of neuronal survival (Miller et al., 1991; Ma et al., 1992). After a 1 hr washout in neurotrophin-free medium, cultures were maintained for an additional 2 d in 10 ng/ml NGF plus or minus 100 ng/ml BDNF. Fields in low-density sister cultures were randomly selected and photographed; six to eight sampling windows were used per culture. We then determined, in each field, (1) the number of visible neurite intersections and (2) the number of neuronal cell bodies. We expressed these data as the number of intersections/number of cell bodies to give us a measure of intersections/neuron. We then determined the mean number of intersections/neuron for all of the photographed fields in a given culture and used the Student’s t test to determine the statistical significance of density differences between experimental groups. Results were expressed as the mean process network density per neuron ± SEM.
The second approach allowed us to quantitate, in any given field, (1) number of neuronal cell bodies and (2) apoptotic cells, and (3) amount of area covered by neuritic processes. Specifically sympathetic neurons were labeled with terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) to visualize apoptotic cells, followed by immunolabeling with anti-α-tubulin to visualize neurites, and then staining with Hoechst 33250 to visualize nuclei. To perform these experiments, P1 SCG neurons were plated onto eight-well Nunc-Nalgene plastic Lab Tek chamber slides (Life Technologies) that were coated twice, first with a polylysine–collagen mixture followed by a second collagen coating. After the experimental treatments, cultures were washed twice with PEM (PIPES-EGTA-MgCl2) buffer and fixed for 15 min in 4% paraformaldehyde in PEM buffer containing 0.25% glutaraldehyde and 0.2% Triton X-100. After three 10 min washes in PBS, TUNEL was performed as described previously (Slack et al., 1996) (reagents from Promega) with a Streptavidin-CY3 conjugate (1:2000 in PBS; Jackson ImmunoResearch Laboratories, West Grove, PA). Cultures were then washed three times for 10 min each in PBS and incubated for 2 hr with anti-α-tubulin (1:500 in PBS, Clone DM1A; Sigma-Aldrich Canada). The tubulin immunolabel was visualized using FITC-conjugated goat anti-mouse IgG (1:800 in PBS; Jackson ImmunoResearch). Finally, cultures were incubated with Hoechst 33250 (2 μg/ml in PBS; ICN Biomedicals, Costa Mesa, CA) for 1 min to label cell nuclei and washed an additional three times for 10 min each in PBS. Slides were then coverslipped using Sigma Mounting Medium (Sigma Diagnostics, St. Louis, MO) and viewed by epifluorescence microscopy.
To analyze these cultures, images were captured using a Sony XC-75CE CCD video camera module attached to an Axioskop microscope (Carl ZeissCanada) and a 16× plan-neofluar lens. The Northern Eclipse image analysis system (Empix Imaging, Mississauga, Ontario, Canada) was used to analyze these images as follows. Images of low-density neuronal fields were captured, and the area labeled with tubulin was measured and expressed as a percentage of the total field area. The numbers of Hoechst-positive and TUNEL-positive cell bodies were quantitated by viewing the same field with the appropriate filters and analyzed using the same software package. For each culture, seven to eight independent, randomly chosen fields were analyzed. These data were expressed as average percentage tubulin immunoreactive area per live cell, and Student’s t test was used to determine the statistical significance of differences between experimental groups.
Immunocytochemistry and analysis of sympathetic innervation density. For quantitative analysis of sympathetic innervation density, P13–P15 BDNF+/+, +/−, and−/− mice were deeply anesthetized with isoflurane and decapitated. Pineal glands were removed immediately, immersion-fixed in 4% paraformaldehyde in phosphate buffer, pH 7.4, overnight, and subsequently cryoprotected in graded sucrose solutions. Pineal glands taken from mice of each genotype were cut on a cryostat (12 μm sections), and the entire pineal gland was serially thaw-mounted onto three Superfrost slides (Fisher Scientific, Houston, TX). Thus, each pineal gland was completely represented on each of three slides, and an entire pineal gland of each genotype could be analyzed, after immunostaining, to obtain a quantitative measure of innervation density. These sections were then post-fixed in 4% paraformaldehyde for 10 min at room temperature and washed for 10 min in PBS, pH 7.4. After nonspecific blocking with 4% goat serum and 4% rat serum (both from Jackson ImmunoResearch) plus 0.2% Triton X-100 in PBS, pH 7.4, the sections were incubated overnight at 4°C with a commercially available polyclonal antibody directed against tyrosine hydroxylase (TH) (1:400; Chemicon International, Temecula, CA) in blocking solution. Slides were then washed three times for 10 min each in PBS, and incubated for 2 hr in blocking solution containing a CY3-conjugated secondary antibody (goat anti-rabbit IgG, 1:2000, Jackson ImmunoResearch). After three 10 min washes in PBS, slides were coverslipped using Sigma Mounting Medium and viewed by epifluorescence microscopy.
To quantitatively analyze sympathetic innervation density, images were captured and analyzed using the Northern Eclipse Imaging System. For a given slide, the percentage area covered by TH immunolabeling was measured for every section on that slide, and the mean percentage area covered by TH immunolabeling was determined from all of these sections, thereby avoiding errors attributable to potential differences in distribution of sympathetic fibers in pineal glands of different genotypes. Comparisons were only made between slides that were processed together.
To directly compare the pattern of p75NTR immunoreactivity with that of TH immunoreactivity, two slides containing serial sections obtained from the same P13 BDNF+/− pineal gland were immunostained as described above with either anti-TH (Chemicon) or anti-human p75NTR (Promega, 1:500) antibodies. The secondary antibody used was a CY3-conjugated goat anti-rabbit IgG (1:2000 in blocking solution; Jackson ImmunoResearch).
Western blot analysis. For biochemistry, groups of P13–P15 pineal glands from +/+, +/−, or−/− BDNF mice, or portions of +/+ or+/− common carotid artery (dissected at the point of bifurcation into the internal and external branches), or adult rat pineal glands or cortex were homogenized in Tris buffered saline (TBS) containing 137 mm NaCl, 20 mm Tris, pH 8.0, 1% v/v NP-40, 0.1% SDS, 10% glycerol, and the protease inhibitors phenylmethyl sulfonyl fluoride (1 mm), aprotinin (10 μg/ml), leupeptin (0.2 μg/ml), and sodium vanadate (1.5 mm). The tissue was rocked for 10 min at 4°C, and after a 10 min centrifugation at 4°C, the supernatant was collected and lysates were normalized for protein concentration using a BCA Protein Assay kit (Pierce Chemical Co., Rockford, IL). For Western blot analysis, equal amounts of pineal, carotid, or cortical protein were boiled in sample buffer for 5 min and separated by 7.5 or 15% SDS-PAGE (7.5% gel for TH, p75NTR, α-tubulin, and ERK1; 15% gel for BDNF). rhBDNF (20 ng) (Amgen, Thousand Oaks, CA) was also run on a 15% SDS-PAGE gel. After electrophoresis, proteins were transferred to 0.2 μm nitrocellulose membranes for 1.5 hr at 0.6 amps, and washed three times for 10 min each with either PBS (for BDNF) or TBS (for all other proteins). After a 1.5 hr block in blotto (3% nonfat milk in PBST for BDNF, or TBST for all other proteins) at room temperature, membranes were incubated overnight at 4°C in blocking solution containing either anti-BDNF (1:3000; Santa Cruz Biotechnology, Santa Cruz, CA), anti-recombinant human p75NTR (1:10,000; Promega), anti-TH (1:5000; Chemicon), anti-α-tubulin (1:5000; Calbiochem/Oncogene Research Products, Cambridge, MA), or anti-ERK1 (1:10,000; Santa Cruz Biotechnology). The membranes were washed four times for 10 min each with either PBST (for BDNF) or TBST (for all other proteins) and then incubated with secondary antibody [1:10,000 goat anti-rabbit HRP (Boehringer Mannheim, Laval, Quebec, Canada) for anti-BDNF, anti-p75NTR, anti-ERK1, and anti-TH, or 1:10,000 goat anti-mouse HRP (also from Boehringer) for anti-α-tubulin] for 1.5 hr at room temperature. After three 10 min washes in PBST (for BDNF protein) or TBST (for all other proteins), detection was performed using enhanced chemiluminescence (ECL Western blotting detection reagent; Amersham Canada Ltd., Oakville, Ontario, Canada) and XAR x-ray film (Eastman Kodak Co., Rochester, NY).
RESULTS
BDNF-mediated activation of p75NTR inhibits NGF-induced growth of cultured sympathetic neurons.
BDNF-mediated activation of p75NTR antagonizes TrkA-mediated sympathetic neuron survival when NGF levels are suboptimal, but has no effect on survival at higher levels of NGF (Aloyz et al., 1998; Bamji et al., 1998). To determine whether p75NTR activation also antagonized other TrkA-mediated biological responses, we focused on sympathetic neuron growth. Specifically, we cultured sympathetic neurons in NGF to maintain their survival and then activated p75NTR using BDNF. For rat sympathetic neurons, 10 ng/ml NGF mediates 100% sympathetic neuron survival but elicits limited morphological growth and TrkA activation relative to higher concentrations of NGF (Ma et al., 1992; Belliveau et al., 1997), whereas 100 ng/ml BDNF is sufficient to activate p75NTR in apoptosis experiments (Bamji et al., 1998) but does not bind to the two Trk receptors, TrkA and TrkC, that are present on sympathetic neurons (Belliveau et al., 1997).
Initially, we confirmed, as we have reported previously (Bamji et al., 1998), that the addition of 100 ng/ml BDNF in the presence of 10 ng/ml NGF had no negative effects on sympathetic neuron survival (Fig.1A). Sympathetic neurons were cultured for 5 d in 50 ng/ml NGF, washed free of neurotrophin, and then switched into 10 ng/ml NGF plus or minus 100 ng/ml BDNF. Two days later, neuronal survival was measured using MTT assays, which measure mitochondrial function (Belliveau et al., 1997;Bamji et al., 1998). As shown previously (Bamji et al., 1998), the addition of 100 ng/ml BDNF had no effect on sympathetic neuron survival in 10 ng/ml NGF (Fig. 1A).
Fig. 1.

BDNF decreases the growth of sympathetic neuronsin vitro without affecting their survival.A, Results of colorimetric MTT assays to measure mitochondrial function and cell survival. Neonatal sympathetic neurons were cultured in 50 ng/ml NGF for 5 d, washed free of neurotrophin-containing medium, and then switched for 2 d to NGF or NGF plus BDNF. The data derive from a representative survival assay that was performed in triplicate. In these assays, absolute values are normalized so that the value obtained with 0 neurotrophin is 0% survival, whereas that obtained with 10 ng/ml NGF is considered 100% survival. Error bars represent SEM. The values obtained for NGF versus NGF plus BDNF were not significantly different (p > 0.05). B–D, Quantitative analysis of neurite process density in sympathetic neuron cultures grown in the presence of NGF or NGF plus BDNF. B, Six separate experiments were performed to determine the effect of BDNF on NGF-mediated process density in sympathetic neurons. Sympathetic neurons were plated at low density on collagen or poly-d-lysine and laminin in 50 ng/ml NGF for 2–3 d and were then switched to 10 ng/ml NGF plus or minus 100 ng/ml BDNF for 2 d. In all six experiments, significantly fewer neurite intersections were observed after exposure to NGF + BDNF versus NGF alone (*p < 0.05).C, The experiments shown in B were normalized so that the neurite density at 10 ng/ml NGF is 100, and then averaged to provide an index of the relative neurite density.D, Dose–response curve to determine the effect of different concentrations of BDNF on NGF-mediated process density. Experiments were performed as in B. No significant different in process density was observed when 50 ng/ml BDNF was added, but statistically significant differences were seen with both 100 and 200 ng/ml BDNF (*p < 0.05). The data derive from one neurite outgrowth assay, where each condition was sampled four to five times. E, Results of TUNEL to measure apoptotic neurons. Neurons were treated as in A, but were TUNEL-labeled to measure apoptotic cells and then stained with Hoechst 33250 to quantitate total neuronal nuclei. Seven to eight fields of cells were analyzed per treatment in each experiment, and results are expressed as the percentage of TUNEL-labeled nuclei/total neuronal nuclei. Results represent the mean ± SEM. Note that BDNF had no effect on the number of TUNEL-labeled neurons in these experiments. F, G, Quantitative analysis of the area covered by tubulin immunoreactive neurites in sympathetic neuron cultures grown in 10 ng/ml NGF with or without BDNF. The same fields of cells shown in E were analyzed for the percentage of area within a given field that was covered by tubulin-immunoreactive processes. These data were normalized for the total number of neuronal cell bodies in the same field (as indicated by Hoechst staining) and then used to determine the relative tubulin-immunolabeled area per neuron. Results represent the mean ± SEM; in both experiments, BDNF caused a statistically significant decrease in tubulin-immunoreactive processes (*p < 0.05), with no effect on neuronal apoptosis (E).G, The experiments shown in F were normalized so that the tubulin immunolabeled area at 10 ng/ml NGF was 100 and then averaged to provide an index of the mean relative tubulin immunolabel.
We next determined whether p75NTR activation by BDNF affected neuronal growth by measuring the level of neurite extension that occurs in response to 10 ng/ml NGF with or without BDNF. For these experiments, neurons were cultured for 2–3 d in 50 ng/ml NGF and switched to 10 ng/ml NGF plus or minus 100 ng/ml BDNF, and the density of neuritic processes was determined 2 d later by quantitating the number of neurite intersections per neuron (Figs. 1B,C,2A,B). Results from six separate experiments indicated that BDNF reduced the process network density from 22 to 52%, for an average decrease of 40% (Figs.1B,C, 2A,B). Having determined that BDNF reduced neurite density, we then performed a dose–response curve with 50, 100, and 200 ng/ml BDNF, using the same experimental approach. This analysis revealed that 50 ng/ml BDNF produced a small decrease in process density that was not statistically significant, whereas both 100 and 200 ng/ml BDNF produced similar, significant decreases (Fig.1D). This dose–response is similar to that observed for the apoptotic effects of BDNF on the same neurons (Bamji et al., 1998).
Fig. 2.

Exogenous BDNF inhibits and anti-BDNF and anti-p75NTR enhance NGF-promoted growth of sympathetic neuronsin vitro. Phase-contrast micrographs of cultured neonatal rat sympathetic neurons maintained in 50 ng/ml NGF for 2–3 d, and then switched to (A) 10 ng/ml NGF, (B) 10 ng/ml NGF plus 100 ng/ml BDNF, (C) 10 ng/ml NGF plus 10 μg/ml anti-BDNF, or (D) 10 ng/ml NGF plus anti-p75NTR (REX). Exogenous BDNF inhibited and BDNF or p75NTR antibodies enhanced process outgrowth. Scale bar, 65 μm.
To confirm that the observed decrease in neurite intersections per neuron reflected a decrease in the total amount of growth in these cultures, we used a second approach. As in the previous experiments, neurons were first grown for 2–3 d in 50 ng/ml NGF and then switched to 10 ng/ml NGF plus or minus 100 ng/ml BDNF for 2 additional days. As a control, neurons were withdrawn from NGF for these final 2 d. We triple-labeled these neurons (Fig. 3) by (1) using TUNEL to assess the number of apoptotic neurons (pink/red nuclei in Fig. 3), (2) using anti-tubulin to visualize the neuritic network (green in Fig. 3), and (3) using Hoechst 33250 to visualize neuronal nuclei (blue in Fig. 3). We then selected random fields in each sister culture and used image analysis to quantitate the amount of area covered by tubulin-immunoreactive processes per Hoechst-labeled neuron and to determine the number of apoptotic neurons per field. This analysis confirmed that, as indicated by the MTT assay, the number of dying cells was similar in cultures maintained in 10 ng/ml NGF alone versus those in 10 ng/ml NGF plus 100 ng/ml BDNF (Fig.1E). Moreover, the relative amount of tubulin-immunoreactive processes per neuron was decreased ∼40% in the neurons treated with BDNF (Fig. 1F,G), a decrease similar to that seen when the number of neuritic intersections per neuron was determined (Fig. 1B,C). Thus, these two approaches indicate that under these conditions, BDNF was able to antagonize NGF-promoted sympathetic neuron growth with no perturbation of neuronal survival.
Fig. 3.
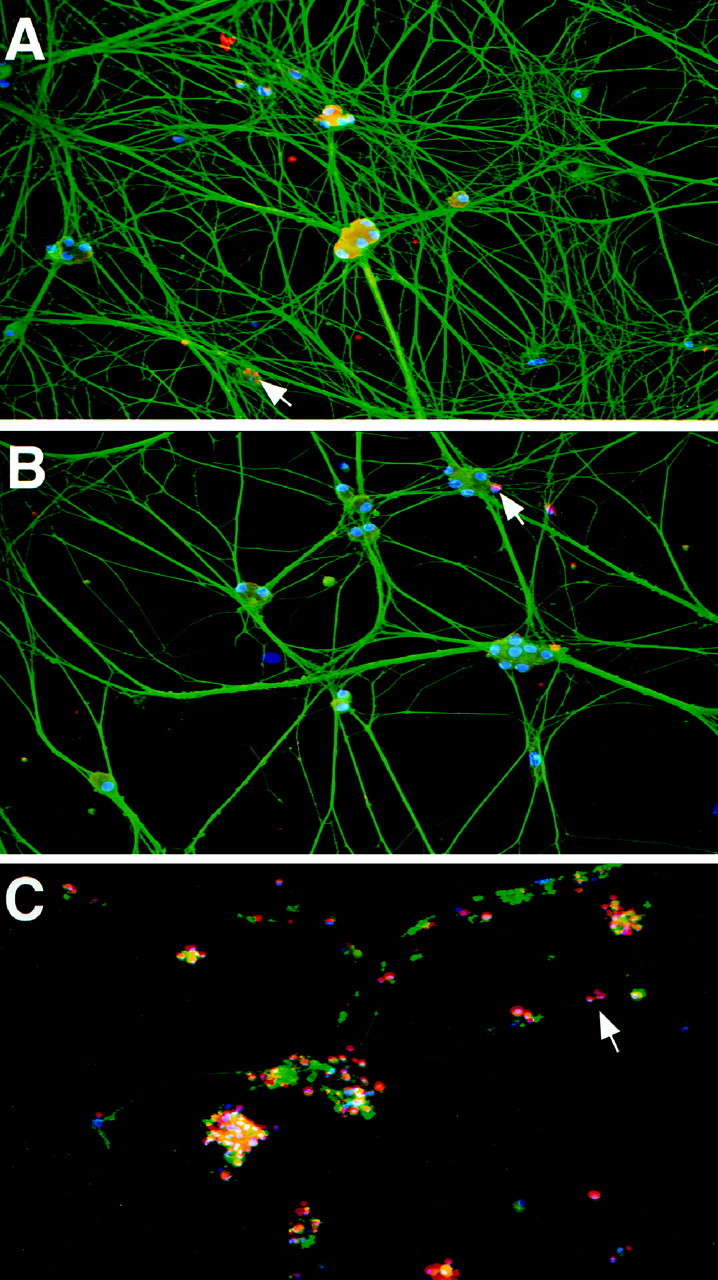
BDNF decreases neurite outgrowth in cultured neonatal rat sympathetic neurons without decreasing their survival. Digitized micrographs are of postnatal day 1 sympathetic neuron cultures triple-labeled to visualize neurite outgrowth (α-tubulin,green), apoptotic cells (TUNEL-labeled,red/pink), and total number of cells in the culture (Hoechst nuclear stain, blue). Cultures were grown in 50 ng/ml NGF for 2 d and then switched to either (A) 10 ng/ml NGF or (B) 10 ng/ml NGF plus 100 ng/ml BDNF, or (C) withdrawn from NGF. After 2 additional days, the cultures were labeled as indicated above. Arrows point to nuclei of apoptotic cells. Note that the addition of BDNF to the cultures did not increase the degree of TUNEL over that observed in cultures treated with NGF alone, whereas cultures in which NGF was withdrawn exhibit a high number of TUNEL-labeled neurons and a complete disintegration of neurites. Magnification, 160×.
Autocrine BDNF, acting through p75NTR, decreases the growth of cultured sympathetic neurons
These data suggested that exogenous BDNF is able to activate p75NTR and negatively influence TrkA-mediated neuritogenesis. However, because sympathetic neurons themselves synthesize BDNF (Causing et al., 1997), which can be detected in conditioned medium obtained from cultured SCG neurons (C. G. Causing, R. Aloyz, and F. D. Miller, unpublished observations), we hypothesized that autocrine BDNF might play a role in negatively regulating levels of sympathetic neuron growth through a BDNF/p75NTR autocrine loop. To test this hypothesis, we used a function-blocking BDNF antibody.
Initially, to ensure that this anti-BDNF was capable of neutralizing BDNF, we incubated TrkB-expressing NIH-3T3 cells for 5 min with 50 ng/ml BDNF plus or minus 5–40 μg/ml of anti-BDNF. TrkB protein was then immunoprecipitated using anti-panTrk, and the immunoprecipitates were analyzed by Western blot analysis with anti-phosphotyrosine. As controls, cells were incubated either with culture medium or medium with BDNF plus 40 μg/ml nonimmune IgY. This analysis revealed that BDNF caused a robust increase in tyrosine phosphorylation of TrkB and that anti-BDNF inhibited BDNF-stimulated TrkB phosphorylation at concentrations of 10 μg/ml or higher (data not shown). In contrast, the control IgY had no effect on BDNF-mediated TrkB activation (data not shown).
We then used this function blocking anti-BDNF to test the role of autocrine BDNF in sympathetic neuron growth. Initially, we determined whether anti-BDNF had any effect on sympathetic neuron survival under the conditions of our growth experiments; neurons were cultured for 5 d in 50 ng/ml NGF and then were switched to 10 ng/ml NGF with or without 10 μg/ml anti-BDNF. Measurement of neuronal survival using MTT assays 2 d later revealed that anti-BDNF had no effect on sympathetic neuron survival (Fig.4A). We then determined whether anti-BDNF affected neuronal growth under these same conditions; neurons were grown in 50 ng/ml NGF for 2–3 d and then switched into 10 ng/ml NGF plus or minus 10 μg/ml anti-BDNF. Measurement of neurite process density revealed that in three separate experiments, cultures exposed to anti-BDNF exhibited an average increase in neuritogenesis of 80% relative to 10 ng/ml NGF alone (Figs. 2A,C,4B,C). Nonimmune IgY had no effect on NGF-mediated growth (Fig. 4B), demonstrating the specificity of the effect. Thus, autocrine BDNF inhibits TrkA-mediated neuritogenesis, at least in vitro.
Fig. 4.

Function-blocking antibodies directed against BDNF or p75NTR enhance growth of sympathetic neurons without affecting their survival. A, Results of colorimetric MTT assays to measure mitochondrial function and cell survival. Neonatal sympathetic neurons were cultured in 50 ng/ml NGF for 5 d, washed free of neurotrophin-containing medium, and then switched for 2 d to 10 ng/ml NGF or 10 ng/ml NGF plus 10 μg/ml anti-BDNF (α-BDNF). These data represent the values obtained in a representative survival assay that was performed in triplicate. In these assays, absolute values are normalized so that the value obtained with 0 neurotrophin is 0% survival, whereas that obtained with 10 ng/ml NGF is considered 100% survival. Error bars represent SEM. These two values were not significantly different (p = 0.3710). B–H, Quantitative analysis of neuritic process density in sympathetic neuron cultures grown in the presence of NGF, NGF plus anti-BDNF (B, C), NGF plus REX (D, E), increased NGF (F), or (G, H) NGF and REX plus or minus BDNF. B, Three separate experiments were performed to determine the effect of anti-BDNF on NGF-promoted neurite process growth in sympathetic neurons. Sympathetic neurons were plated at low density in 50 ng/ml NGF on collagen, or poly-d-lysine and laminin for 2–3 d, and then switched for an additional 2 d to 10 ng/ml NGF plus or minus 10 μg/ml anti-BDNF (α-BDNF) or, as a control, 10 μg/ml nonimmune IgY. In all three experiments, significantly more neurite intersections were observed after exposure to anti-BDNF relative to NGF alone (*p < 0.05). C, The experiments shown in B were normalized so that the neuritic density at 10 ng/ml NGF is 100 and then averaged to provide an index of the relative neurite density. D, Three separate experiments were performed to determine the effect of anti-p75NTR on NGF-promoted neurite process growth in sympathetic neurons. Sympathetic neurons were treated as in B, except that they were treated with a 1:100 dilution of p75NTR antiserum (REX) or, as a control, with the same dilution of nonimmune rabbit serum. In all three experiments, significantly more neurite intersections were observed after exposure to anti-p75NTR than after NGF alone (*p < 0.05). Serum itself had no effect (p > 0.05).E, The experiments shown in D were normalized so that the neuritic density at 10 ng/ml NGF is 100 and then averaged to provide an index of the relative neurite density.F, The increase in relative neurite process density in 40 ng/ml NGF relative to 10 ng/ml NGF. These data represent the results from one experiment and are shown here for comparison only. We have previously documented the reproducibility and significance of this increase in Belliveau et al. (1997). G, Three separate experiments were performed to determine whether BDNF could antagonize the p75NTR-mediated increase in sympathetic neuron growth. Sympathetic neurons were treated as in B, except that they were treated with a 1:100 dilution of p75NTR antiserum (REX) plus or minus 100 ng/ml BDNF. In all three experiments, exogenous BDNF significantly inhibited the REX-induced increase in process density (*p < 0.05). H, The experiments shown in G were normalized so that the neuritic density at 10 ng/ml NGF is 100 and then averaged to provide an index of the relative neurite density.
If autocrine BDNF is mediating these effects via p75NTR, then we would predict a similar increase in TrkA-mediated neuritogenesis if we blocked p75NTR. To test this prediction, we performed neuritogenesis experiments using the function-blocking p75NTR antibody REX. As before, cultures were initially grown for 2 d in 50 ng/ml NGF and then incubated for 2 additional days with 10 ng/ml NGF with or without REX (Figs. 2A,D, 4D,E). As a control, sister cultures were incubated with rabbit serum at the same volume as the REX antiserum. These experiments revealed that when p75NTR was blocked by REX, neuritogenesis was enhanced almost twofold relative to NGF alone (Figs. 2A,D, 4D,E), an effect that was not observed with rabbit nonimmune serum (Fig.4D) and was similar to the response elicited by anti-BDNF (Fig. 4B,C). The increased neuritogenesis observed with both REX and anti-BDNF is similar to the 2- to 2.5-fold increase that occurs when NGF is increased from 10 to 40 ng/ml NGF (Fig. 4F) (Belliveau et al., 1997), a treatment that causes increased TrkA activation (Belliveau et al., 1997), supporting the idea that a BDNF/p75NTR autocrine loop antagonizes TrkA-mediated sympathetic neuron growth.
In a final experiment, we tested whether exogenous BDNF could reverse the effect of REX on neuritogenesis, as it should if REX is acting by disrupting a BDNF/p75NTR loop. Neurons were cultured for 2–3 d in 50 ng/ml NGF and then switched to 10 ng/ml NGF with REX plus or minus 100 ng/ml BDNF for an additional 2 d; in experiments with REX and BDNF, cultures were preincubated with REX for 2 hr before the addition of BDNF. These experiments revealed that exogenous BDNF blocked the ability of REX to increase sympathetic neuron growth (Fig.4G,H). This antagonism between REX and exogenous BDNF is similar to results we have obtained previously when examining BDNF-induced apoptosis of sympathetic neurons (Bamji et al., 1998) and supports the idea of an inhibitory BDNF/p75NTR growth loop.
p75NTR−/− sympathetic neurons show enhanced neuritogenesis in response to NGF and do not respond to exogenous BDNF
Although our data strongly suggest that BDNF acts through p75NTR to antagonize TrkA, they do not conclusively demonstrate the necessity of p75NTR for BDNF’s effects. To examine this more directly, we cultured neurons from both p75NTR−/− and wild-type control mice and repeated the neuritogenesis assays. Because mouse sympathetic neurons have been reported to be more sensitive to NGF than rat sympathetic neurons, we initially performed survival assays to determine an appropriate NGF concentration. Neurons were maintained for 5 d in 50 ng/ml NGF and were switched to concentrations of NGF ranging from 0.1 to 10 ng/ml NGF for 3 d, and survival was then measured using MTT assays. These experiments revealed that 5, 7.5, and 10 ng/ml NGF were all able to mediate maximal mouse sympathetic neuron survival (Fig.5A). To ensure that BDNF had no apoptotic effect under these survival conditions, we performed similar experiments with 7.5 or 10 ng/ml NGF plus or minus 100 ng/ml BDNF. MTT assays revealed that, as for rat neurons (Fig.1A), BDNF did not affect mouse sympathetic neuron survival in the presence of optimal concentrations of NGF (Fig.5B). On the basis of these data, we selected 7.5 ng/ml NGF for our experiments, a concentration that was optimal for survival and in which BDNF had no significant effect on survival.
Fig. 5.

Analysis of neurite outgrowth in response to NGF or NGF plus BDNF in p75NTR−/− versus p75NTR+/+ sympathetic neurons. A, B, Results of colorimetric MTT assays to measure mitochondrial function and survival of murine sympathetic neurons in response to NGF or NGF plus BDNF. A, Neonatal murine sympathetic neurons were cultured in 50 ng/ml NGF for 5 d, washed free of neurotrophin-containing medium, and switched for 2 d to various concentrations of NGF as indicated on the x-axis. Results from one representative experiment performed in triplicate are shown. In these assays, absolute values are normalized so that the value obtained with 0 neurotrophin is 0% survival, whereas that obtained with 10 ng/ml NGF is considered 100% survival. Error bars represent SEM. Note that 5 ng/ml NGF is capable of eliciting maximal survival of murine sympathetic neurons. B, Neonatal murine sympathetic neurons were cultured as in A and were then switched into various concentrations of NGF plus 100 ng/ml BDNF, as denoted on the x-axis. These data represent values from one representative experiment performed in triplicate. Values are normalized as in A; error bars represent SEM. As with rat sympathetic neurons, BDNF does not affect survival in optimal concentrations of NGF. C, D, Quantitative analysis of neuritic process density in p75NTR−/−versus wild-type murine sympathetic neurons in response to NGF or NGF plus BDNF. In the two separate experiments shown here, p75NTR−/− versus wild-type neonatal sympathetic neurons were cultured as sister cultures for 3 d in 50 ng/ml NGF and were then switched for an additional 2 d to 7.5 ng/ml NGF plus or minus 100 ng/ml BDNF. * denotes values that were significantly different in the comparison between NGF versus NGF plus BDNF (*p < 0.05); ** denotes those values that were significantly different in the comparison between p75NTR−/− versus wild-type (WT) neurons in response to 7.5 ng/ml NGF (**p < 0.05). Note that p75NTR−/− neurons grow more robustly than wild-type neurons in response to the same concentration of NGF. Note also that BDNF significantly reduces the NGF-mediated growth of wild-type (p < 0.05) but not p75NTR−/− (in both experiments,p > 0.38) neurons. D, The experiments shown in C were normalized so that the neuritic density of wild-type neurons at 10 ng/ml NGF was 100 and then averaged to provide an index of the relative neurite density.
We then examined sympathetic neurons from p75NTR−/− mice to determine first whether the lack of p75NTR imparted to p75NTR−/− neurons an intrinsic ability to extend more neuritic processes, and second, whether BDNF was acting through p75NTR to inhibit TrkA-mediated growth. To perform these experiments, sympathetic neurons were cultured from p75NTR−/− versus control mice on the same day, were maintained for 3 d in 50 ng/ml NGF, and were subsequently switched to 7.5 ng/ml NGF plus or minus 100 ng/ml BDNF. Measurement of neuritic process density revealed that p75NTR−/−neurons exhibited an almost twofold increase in neurite outgrowth relative to their wild-type counterparts (Figs. 5C,D, Fig.6A,C), a result similar to that observed with the REX and anti-BDNF antibodies in experiments using rat sympathetic neuron cultures (Fig. 4). Moreover, although BDNF decreased the degree of neuritogenesis in wild-type mouse cultures by an average of 35% (Figs. 5C,D, 6A,B), a result similar to that observed with rat neurons (Fig.1B,C), exogenous BDNF had no effect on growth of p75NTR−/− neurons (Figs. 5C,D,6C,D). Thus p75NTR is required for BDNF to inhibit NGF-mediated sympathetic neuron growth, and NGF is more effective at eliciting growth in the absence of p75NTR.
Fig. 6.

p75NTR−/− sympathetic neurons show enhanced neuritogenesis in response to NGF and do not respond to exogenous BDNF. Phase-contrast micrographs of cultured Coomassie blue-stained (A, B) wild-type and (C, D) p75NTR−/− murine sympathetic neurons maintained in 50 ng/ml NGF for 2 d and then switched to (A, C) 7.5 ng/ml NGF or (B, D) 7.5 ng/ml NGF plus 100 ng/ml BDNF. As observed in cultured rat sympathetic neurons (Fig.2B), exogenous BDNF inhibited NGF-promoted process outgrowth in wild-type but not p75NTR−/−murine sympathetic neurons. Note that the degree of process outgrowth is greatly enhanced in p75NTR−/− neurons, relative to their wild-type counterparts. Scale bar, 65 μm.
BDNF is present in sympathetic target organs and p75NTR is present in sympathetic neuron axons during the period of target innervation
If these culture results are relevant to the process of target innervation in vivo, then we would predict that BDNF (derived from incoming sympathetic fibers or target tissue or both) would be present in sympathetic neuron targets during the developmental period of target competition. To test this prediction, we focused on the pineal gland, a sympathetic target organ that (1) is bilaterally innervated by neurons from the SCG (Kappers, 1960; Owman, 1964), (2) does not receive any other peripheral innervation from sensory or motor neurons (Stanley et al., 1987), and (3) is innervated postnatally. Ingrowth of sympathetic fibers to the pineal gland begins during the first week of postnatal life, reaching adult levels after 3–4 weeks (Hakanson et al., 1967). To perform this experiment, lysates of pineal glands from adult rats were separated by PAGE, transferred to nitrocellulose, and probed for the presence of BDNF using a BDNF antibody that we have previously characterized extensively for specificity (Causing et al., 1997; Fawcett et al., 1997, 1998). This analysis revealed a BDNF-immunoreactive band in the pineal gland that is the same size as BDNF in the rat cortex and human recombinant BDNF (Fig. 7A,B). To confirm the identity of this band, we analyzed the pineal gland from mice in which the BDNF gene was mutated by homologous recombination (Ernfors et al., 1994). Western blot analysis revealed that the BDNF-immunoreactive band was completely lost in the pineal glands of BDNF−/− mice (Fig. 7C), as we have observed previously for this BDNF band in other BDNF−/− tissues (Causing et al., 1997; Fawcett et al., 1998).
Fig. 7.

BDNF is present in the pineal gland during the period of sympathetic target innervation, as detected by Western blot analysis. A, Western blot analysis for BDNF in the adult rat pineal gland. Tissue lysates were separated on polyacrylamide gels, transferred to nitrocellulose, and probed with an antibody to BDNF that we have previously extensively characterized (Causing et al., 1997;Fawcett et al., 1997, 1998). B, Pineal gland BDNF is the same size as recombinant human BDNF. Western blot analysis of tissue lysates revealed that the band seen in the adult pineal gland (Pineal) is similar in size to recombinant human BDNF (rhBDNF) and to BDNF in the adult rat cortex (Cortex). Lysates from the brain and pineal contain equal amounts of protein. C, Western blot analysis of BDNF in the pineal gland of BDNF−/− and BDNF+/+ littermates at P13–P15. Note that the BDNF-immunoreactive band is not present in the BDNF−/− pineal gland.
We next determined whether incoming sympathetic axons were positive for p75NTR over this same time frame. Immunocytochemical analysis of the rat pineal gland using the anti-p75NTR antibody MC192 revealed the presence of numerous p75NTR-positive fibers throughout the pineal gland at P6 (data not shown), in a pattern similar to that observed previously for tyrosine hydroxylase-positive sympathetic fibers (Kuchel, 1993). To confirm that this immunostaining corresponded to bona fide p75NTR, we also performed Western blot analysis, which demonstrated that p75NTR was present in both the rat (data not shown) and mouse (see Fig. 9) pineal gland during the first few postnatal weeks. Thus, both BDNF and p75NTR are present in the pineal gland at the time of sympathetic target competition.
Fig. 9.

Levels of sympathetic axon markers are increased in sympathetic targets in BDNF+/− and BDNF−/− mice. Western blot analysis of tyrosine hydroxylase (TH), p75NTR (P75), α-tubulin (Tubulin), and ERK1 (Erk1) in equal amounts of protein from the pineal glands and carotid arteries of BDNF+/+, BDNF+/−, and BDNF−/− littermates at P13–P15. Note that the blots shown for ERK1 are reprobes of the same blots shown for p75NTR, in the case of the pineal gland, and for α-tubulin, in the case of the carotid artery.
The pineal gland, a sympathetic neuron target organ, is hyperinnervated in BDNF+/− and −/−mice
Together, our in vivo and in vitro data predict that when BDNF levels are lowered, sympathetic neuron target innervation should increase. To test this prediction, we examined the level of sympathetic innervation to the pineal gland of BDNF+/− and BDNF−/− mice at P13–P15. Initially, we analyzed the density of sympathetic fibers immunocytochemically, using an antibody against TH, a protein that is a marker for sympathetic axons. To perform this analysis, we serially sectioned the pineal gland from BDNF+/+,+/−, and −/− littermates and performed TH immunostaining on every third serial section from pineal glands of each genotype. We then used an image analysis system to quantitate the percentage area covered by TH-positive fibers on each of these sections and averaged the percentage area obtained from sections throughout each pineal gland, thereby ensuring that sampling errors were not incurred as a result of differences in the pattern of sympathetic innervation to the pineal gland in animals of different genotypes. This analysis revealed that thick, TH-positive fibers were interspersed throughout the entire pineal gland of BDNF+/− and BDNF−/− mice, whereas fibers in the pineal gland of BDNF+/+ littermates were less abundant and appeared qualitatively thinner (Fig.8A–C). This qualitative difference was reflected in the numbers obtained using image analysis; in each of four sets of littermates, the TH-positive innervation density was increased approximately two- to threefold in the pineal glands of BDNF+/− versus BDNF+/+ animals, and the level of innervation was similar in the BDNF+/− and BDNF−/− pineal glands (Fig.8F).
Fig. 8.
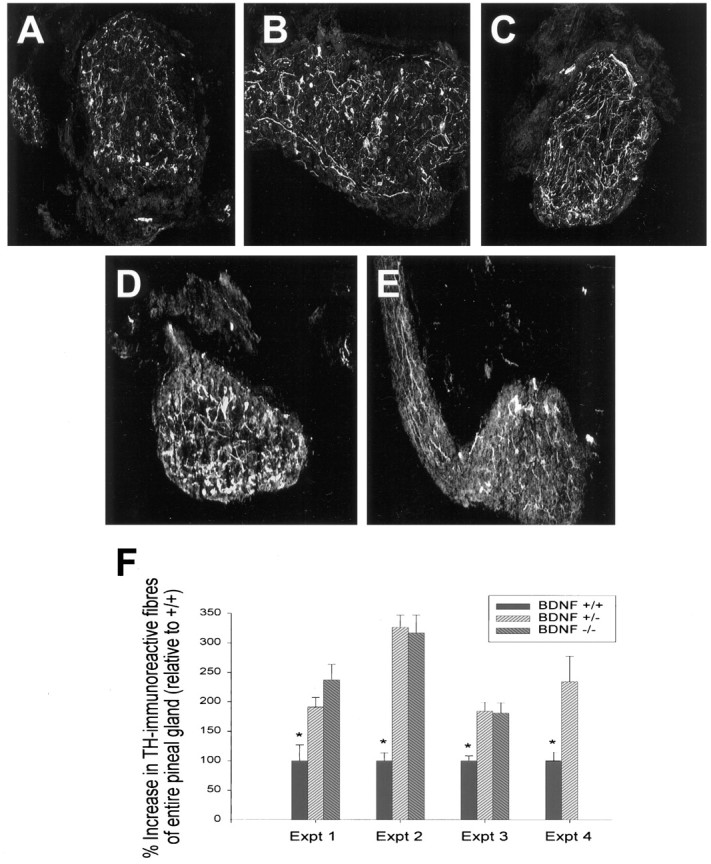
The pineal gland is hyperinnervated with sympathetic fibers in BDNF+/− and BDNF−/− mice at P13. A–C, Immunocytochemical analysis of tyrosine hydroxylase, a specific marker for sympathetic axons, in sections of the pineal gland from (A) BDNF+/+, (B) BDNF+/−, and (C) BDNF−/− littermates. Note that the density of TH-positive fibers is increased in both the BDNF+/− and −/− sections relative to the section from the control littermate. D, E, Immunocytochemical analysis of TH (D) and p75NTR (E) in sections from the same P13 BDNF+/− pineal gland. Note that although the p75NTR-immunoreactivity is somewhat fainter, the pattern of immunoreactivity is similar to that seen with anti-TH . F, Quantitative analysis of the relative amount of pineal gland area covered by TH-immunoreactive fibers in BDNF+/+, +/−, and −/−animals, obtained using sections similar to those shown inA–C. For details of the analysis, see Results and Materials and Methods. Each experiment represents the results obtained from the pineal glands of one set of littermates of different genotypes. Note that in all four experiments, the amount of TH-positive innervation in the BDNF+/+ pineal gland was significantly lower than that seen in either the BDNF+/− or BDNF−/− pineal glands (*p < 0.05). A–E, Magnification, 160×.
To confirm the sympathetic hyperinnervation in the BDNF+/− and −/− pineal glands, we also measured the level of TH biochemically. Western blot analysis of equal amounts of protein from P13–P15 pineal glands revealed that TH levels were increased in BDNF+/− and BDNF−/− tissue relative to controls (Fig.9), consistent with the immunocytochemical results (Fig. 8). We also used the same approach to quantitate the levels of p75NTR and α-tubulin, the former of which is present in incoming sympathetic afferents (Fig.8D,E), and the latter of which is enriched but not specific to all axons. Western blot analysis revealed that like TH, levels of both of these proteins were increased in quantity in the pineal glands of BDNF+/− and −/− mice relative to their wild-type littermates (Fig. 9). In contrast, levels of ERK1, a signaling protein that is present in all cells, were similar in all of the samples (Fig. 9).
To determine whether this increase in sympathetic innervation was limited to the pineal gland or whether it generalized to other sympathetic targets such as blood vessels, we also analyzed the common carotid artery of BDNF+/− versus BDNF+/+ littermates. Western blot analysis of equal amounts of protein revealed that, as observed for the pineal gland, levels of TH, p75NTR, and tubulin were all increased in the+/− carotid artery (Fig. 9). In contrast, levels of ERK1 were similar (Fig. 9). Thus, when BDNF levels are reduced as they are in the BDNF+/− mice (Fawcett et al., 1998), sympathetic innervation to at least two peripheral targets is increased.
DISCUSSION
Data presented in this paper demonstrate that BDNF-mediated activation of p75NTR antagonizes NGF-mediated growth of sympathetic neurons, thereby playing an essential role in the establishment of appropriate target innervation in vivo. Specifically, these experiments indicate that when sympathetic neurons are cultured under optimal survival conditions, exogenous BDNF and autocrine BDNF made by sympathetic neurons themselves inhibit NGF-promoted neuronal growth. BDNF mediates this inhibition via p75NTR because (1) function-blocking p75NTR antibodies enhance NGF-promoted growth, (2) BDNF cannot inhibit the growth of p75NTR−/− neurons, and (3) p75NTR−/− neurons grow more robustly in response to NGF than do their wild-type counterparts. The physiological relevance of these culture findings is demonstrated by the pineal gland studies presented in this paper. Specifically, BDNF is present in the pineal gland, and p75NTR is localized to sympathetic neuron fibers at the time of developmental target innervation. When BDNF is reduced or absent, as in BDNF+/− or BDNF−/− mice, the pineal gland is hyperinnervated with sympathetic fibers, and tyrosine hydroxylase levels are increased in two sympathetic targets, the pineal gland and the carotid artery. Together, these data indicate that BDNF, made by sympathetic neurons and/or their target organs, acts via p75NTR to antagonize NGF-mediated growth and target innervation, suggesting that sympathetic target innervation is determined by the balance of positively and negatively acting neurotrophins present in developing target organs.
What is the biological rationale for having two neurotrophin receptors, one of which, TrkA, mediates survival and growth, and one of which, p75NTR, acts antagonistically to cause apoptosis and inhibit growth? With regard to neuronal survival, we have previously proposed that p75NTR provides a molecular mechanism for ensuring rapid and active apoptosis when a neuron is unsuccessful in competing for adequate amounts of the appropriate neurotrophin (Aloyz et al., 1998; Bamji et al., 1998; Miller and Kaplan, 1998). We propose that the antagonistic effects of TrkA and p75NTR on growth, as described here, are part of the same biological mechanism, as exemplified in the following three situations. First, if a sympathetic neuron is late-arriving and/or reaches an inappropriate target, then TrkA would be only weakly induced as a consequence of the lack of NGF, and p75NTR would be robustly activated by neurotrophins such as BDNF, leading to the rapid apoptotic elimination of that neuron. Second, if a sympathetic neuron extends only one main axon collateral and that collateral reaches an appropriate target and sequesters NGF, then TrkA would be robustly activated, allowing for survival of that neuron. The third possibility would occur when a sympathetic neuron extends several axon collaterals or terminal branches. If one of those collaterals reaches an appropriate target and sequesters NGF, then the subsequent TrkA activation would retrogradely mediate survival of the neuron and locally promote terminal growth of that axon. If the second collateral arrives at a target that is already innervated or is inappropriate, then the low level of available NGF would cause only weak TrkA activation, and p75NTR would be robustly activated by neurotrophins such as BDNF. In this final case, p75NTR activation would not affect neuronal survival, because survival would be maintained by TrkA activation from the other collateral. Instead, the major effect would be on target innervation itself, with p75NTR acting locally to attenuate the growth of that specific axon branch. The net result would be selection of one collateral over the other. In this way, p75NTR would act as a “fine-tuning” mechanism that would allow both the selection of those neurons that reach appropriate targets at an appropriate time, and the selection and maintenance of axon collaterals or terminal branches that meet the same criteria. Disruption of such a mechanism could well explain the perturbations in sympathetic neuron number (Bamji et al., 1998) and sympathetic innervation (Lee et al., 1994) observed previously in the p75NTR−/− mice. In the absence of p75NTR, sympathetic target organs that are innervated late, such as the pineal gland, are never appropriately innervated (Lee et al., 1994), suggesting that the disruption of appropriate competition for innervation of early sympathetic targets completely disrupts the later period of sympathetic target innervation.
Data presented here also indicate that BDNF is one p75NTR ligand that is responsible for regulating sympathetic neuron growth. Specifically, we have demonstrated that when BDNF in the pineal gland is reduced or absent, both the density of sympathetic innervation and the levels of tyrosine hydroxylase are increased. We believe that this increased pineal innervation is directly attributable to the loss of BDNF for the following reasons. First, the pineal gland does not receive peripheral sensory or motor innervation, making it unlikely that the observed effects are caused by a BDNF-dependent loss of, for example, sensory innervation. Second, although sympathetic neuron number is increased ∼30% in BDNF−/− mice (Bamji et al., 1998), the increases in innervation level and tyrosine hydroxylase are two- to threefold in the pineal gland, indicating that the degree of hyperinnervation is greater than the increase in cell number. Third, and perhaps most importantly, increased innervation to the pineal gland is also seen in BDNF+/− mice, which are not delayed developmentally and have normal sympathetic and sensory neuron numbers. There are several precedents for such a BDNF gene dosage effect in which the absence of one BDNF allele is sufficient to significantly perturb nervous system structure and/or function (Korte et al., 1995;Carroll et al., 1998; Fawcett et al., 1998), strongly suggesting that even minor alterations in the ratios of the neurotrophins are of major physiological importance. In this regard, we believe that the most likely explanation for our findings is that the BDNF present in the pineal gland inhibits NGF-mediated target innervation and that this inhibition is lost when BDNF is reduced or absent.
What is the source of BDNF in vivo? Although the CNS is the most abundant postnatal source of BDNF (Ernfors et al., 1990; Hofer et al., 1990; Phillips et al., 1990), it is clear that many peripheral targets, including dermal mesenchyme, mandible, and whisker pad (Schecterson and Bothwell, 1992), as well as muscle, heart, and lung (Maisonpierre et al., 1990), synthesize BDNF. Moreover, these same targets synthesize NGF (Levi-Montalcini, 1987; Schecterson and Bothwell, 1992), suggesting that targets themselves determine the precise neurotrophin cohort encountered by arriving axons. However, sympathetic neurons also synthesize BDNF (Schecterson and Bothwell, 1992; Causing et al., 1997), and cultured neonatal sympathetic neurons secrete processed BDNF into the media (Causing, Aloyz, and Miller, unpublished observations). Moreover, our data indicate that this autocrine BDNF is sufficient to inhibit NGF-mediated growth, at least in culture. However, it is not possible, on the basis of the data presented here, to assess the relative contribution of target-derived versus sympathetic neuron-derived BDNF in vivo. Nonetheless, it is tempting to speculate that if BDNF is anterogradely trafficked into sympathetic axons as it is into sensory (Zhou and Rush, 1996) and central (von Bartheld et al., 1996; Altar et al., 1997; Conner et al., 1997; Fawcett et al., 1998) axons, then secretion from sympathetic terminals may well provide a cellular mechanism whereby “successful” terminals could eliminate and/or make the environment unfavorable for later-arriving axon collaterals. A precedent for a related mechanism derives from the neuromuscular junction, where active synaptic sites apparently destabilize inactive synapses in their vicinity (Colman et al., 1997).
How does p75NTR inhibit TrkA-mediated neuronal growth? One potential mechanism involves p75NTR-mediated generation of intracellular ceramide (Dobrowsky et al., 1994, 1995). Recently, Posse de Chaves et al. (1997)demonstrated that elevation of intracellular ceramide in distal sympathetic neurites locally inhibited NGF-promoted sympathetic axon growth. Thus, as proposed by the authors, activation of p75NTR could well attenuate neurite growth via increased ceramide. A similar and potentially additive attenuation would occur if activated p75NTR directly downregulated TrkA-mediated growth signals by serine–threonine phosphorylation of the TrkA receptor via ceramide-activated kinases (MacPhee and Barker, 1997). The net outcome of these ceramide-driven mechanisms would be inhibition of growth and potential axonal retraction. This receptor cross-talk is also likely to be bidirectional. Robust TrkA activation would likely silence a p75NTR-mediated ceramide flux, as previously demonstrated in PC12 cells (Dobrowsky et al., 1995), thereby further favoring axonal growth. Such negative feedback between these two receptors provides a mechanism for biasing the cell to one of two outcomes, growth or no growth, thereby ensuring that axon collaterals that are only minimally successful in terms of sequestering target territory do not remain to compete during this critical developmental period.
Does this functional antagonism between TrkA and p75NTR generalize to neurons other than sympathetic neurons? The most compelling case that it does, to at least some degree, derives from studies on basal forebrain cholinergic neurons. In p75NTR−/− mice, the number of basal forebrain cholinergic neurons increases (Van der Zee et al., 1996) and the hippocampus is hyperinnervated (Yeo et al., 1997), two phenotypes reminiscent of sympathetic neurons in p75NTR−/− and BDNF−/− mice (Lee et al., 1994; Bamji et al., 1998; data presented here). Such functional antagonism may also occur, in at least some situations, for TrkA-positive sensory neurons. For example, the local NGF-promoted growth of adult sensory neurons is inhibited by BDNF (Kimpinski et al., 1997), cultured dorsal root ganglia neurons showed a decrease in growth cone turning toward NGF-coated beads in the presence of BDNF (Gallo et al., 1997), and functional ablation of p75NTR using antisense oligonucleotides enhanced the survival of cultured postnatal sensory neurons (Barrett and Bartlett, 1994). Moreover, one recent study indicates that this functional antagonism between p75NTR and TrkA may not only regulate the innervation of appropriate targets, but may also allow axons to distinguish between permissive and nonpermissive growth substrates. In particular, when NGF is overexpressed in astrocytes of transgenic mice, absence of p75NTR leads to robust sympathetic axon growth on myelinated tracts in the mature CNS, indicating that p75NTR plays a significant role in making CNS myelin an inhibitory growth environment, at least for sympathetic axons (Walsh et al., 1999). However, it is also clear that, like other members of the p75NTR family, the outcome of p75NTR-mediated signal transduction is a function of cellular context. For example, depending on the cellular environment, p75NTR regulates cell migration (Anton et al., 1994) and gene expression (Itoh et al., 1995) and can positively modulate TrkA signaling (Barker and Shooter, 1994; Verdi et al., 1994).
In summary, our studies provide evidence for a mechanism whereby two receptors that are coexpressed in sympathetic neurons, TrkA and p75NTR, can functionally interact to regulate process outgrowth during the time of target innervation, thereby ensuring appropriate matching of neurons and their target territory. Thus, not only do neurotrophins regulate neuronal survival and growth, depending on the particular receptors that they activate, but they may also provide a mechanism whereby neurons can recognize whether they are exposed to the “right” versus the “wrong” targets. Such functional antagonism, mediated by TrkA and p75NTR, appears to be essential for appropriate sympathetic neuron target innervation, and similar mechanisms may well play an essential role in the matching of neurons with their targets throughout the nervous system.
Footnotes
This study was supported by grants from the Canadian Medical Research Council and NeuroSciences Network (F.D.M.). F.D.M. is a Killam Scholar, and during the course of this work, R. Aloyz was a NeuroSciences Network fellow. We thank Christine Pozniak and Marta Majdan (both of McGill University) for their help with the BDNF+/−and p75NTR−/− colonies, and other members of the Miller laboratory for their advice and input throughout the course of this work.
Correspondence should be addressed to Dr. Freda D. Miller, Centre for Neuronal Survival, Montréal Neurological Institute, 3801 rue University, Montréal, Québec, Canada, H3A 2B4.
REFERENCES
- 1.Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, Miller FD. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol. 1998;143:1691–1703. doi: 10.1083/jcb.143.6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay R, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- 3.Anton ES, Weskamp G, Reichardt LF, Matthew WD. Nerve growth factor and its low affinity receptor promote schwann cell migration. Proc Natl Acad Sci USA. 1994;91:2795–2799. doi: 10.1073/pnas.91.7.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. 1998;140:911–923. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barker PA, Shooter EM. Disruption of NGF binding to the low affinity neurotrophin receptor p75LNTR reduces NGF binding to TrkA on PC12 cells. Neuron. 1994;13:203–215. doi: 10.1016/0896-6273(94)90470-7. [DOI] [PubMed] [Google Scholar]
- 6.Barrett GL, Bartlett PF. The p75 nerve growth factor receptor mediates survival or death depending on the stage of sensory neuron development. Proc Natl Acad Sci USA. 1994;91:6501–6505. doi: 10.1073/pnas.91.14.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belliveau DJ, Krivko I, Kohn J, Lachance C, Pozniak C, Rusakov D, Kaplan D, Miller FD. NGF and neurotrophin-3 both activate TrkA on sympathetic neurons but differentially regulate survival and neuritogenesis. J Cell Biol. 1997;136:375–388. doi: 10.1083/jcb.136.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carroll P, Lewin GR, Koltzenburg M, Toyka KV, Thoenen H. A role for BDNF in mechanosensation. Nat Neurosci. 1998;1:42–46. doi: 10.1038/242. [DOI] [PubMed] [Google Scholar]
- 9.Causing CG, Gloster A, Aloyz R, Bamji SX, Chang E, Fawcett J, Kuchel G, Miller FD. Synaptic innervation density is regulated by neuron-derived BDNF. Neuron. 1997;18:257–267. doi: 10.1016/s0896-6273(00)80266-4. [DOI] [PubMed] [Google Scholar]
- 10.Clary DO, Weskamp G, Austin LR, Reichart LF. TrkA cross-linking mimics neuronal responses to nerve growth factor. Mol Biol Cell. 1994;5:549–563. doi: 10.1091/mbc.5.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colman H, Nabekura J, Lichtman JW. Alterations in synaptic strength preceding axon withdrawal. Science. 1997;275:356–361. doi: 10.1126/science.275.5298.356. [DOI] [PubMed] [Google Scholar]
- 12.Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cordon-Cardo C, Tapley P, Jing SQ, Nanduri V, O’Rourke E, Lamballe F, Kovary K, Klein R, Jones KR, Reichardt LF, Barbacid M. The trk tyrosine protein kinase mediates the mitogenic properties of nerve growth factor and neurotrophin-3. Cell. 1991;66:173–183. doi: 10.1016/0092-8674(91)90149-s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts-Meek S, Armanini MP, Ling LH, McMahon SB, Shelton DL, Levinson AD, Phillips HS. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons, yet develop basal forebrain cholinergic neurons. Cell. 1994;76:1–20. doi: 10.1016/0092-8674(94)90378-6. [DOI] [PubMed] [Google Scholar]
- 15.Dobrowsky RT, Werner MH, Castellino AM, Chao MV, Hannun YA. Activation of the sphingomyelin cycle through the low-affinity neurotrophin receptor. Science. 1994;265:1596–1599. doi: 10.1126/science.8079174. [DOI] [PubMed] [Google Scholar]
- 16.Dobrowsky RT, Jenkins GM, Hannun YA. Neurotrophins induce sphingomyelin hydrolysis: modulation by co-expression of p75 with Trk receptors. J Biol Chem. 1995;270:22135–22142. doi: 10.1074/jbc.270.38.22135. [DOI] [PubMed] [Google Scholar]
- 17.Ernfors P, Wetmore C, Olson L, Persson L. Identification of cells in rat brain and peripheral tissues expressing mRNA for members of the nerve growth factor family. Neuron. 1990;5:511–526. doi: 10.1016/0896-6273(90)90090-3. [DOI] [PubMed] [Google Scholar]
- 18.Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- 19.Fawcett JP, Aloyz R, McLean JH, Pareek S, Miller FD, McPherson PS, Murphy RA. Detection of brain-derived neurotrophic factor in a vesicular fraction of brain synaptosomes. J Biol Chem. 1997;272:8837–8840. doi: 10.1074/jbc.272.14.8837. [DOI] [PubMed] [Google Scholar]
- 20.Fawcett JP, Bamji SX, Causing CG, Aloyz R, Ase AR, Reader TA, McLean JH, Miller FD. Functional evidence that BDNF is an anterograde neuronal trophic factor in the CNS. J Neurosci. 1998;18:2808–2821. doi: 10.1523/JNEUROSCI.18-08-02808.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallo G, Lefcort FB, Letourneau PC. The TrkA receptor mediates growth cone turning toward a localized source of nerve growth factor. J Neurosci. 1997;17:5445–5454. doi: 10.1523/JNEUROSCI.17-14-05445.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hakanson R, Lombard Des Gouttes M-N, Owman C. Activities of tryptophan hydroxylase, dopa decarboxylase and monoamine oxidases correlated with the appearance of monoamines in the developing rat pineal gland. Life Sci. 1967;6:2577–2585. doi: 10.1016/0024-3205(67)90107-5. [DOI] [PubMed] [Google Scholar]
- 23.Hempstead BL, Rabin SJ, Kaplan L, Reid S, Parada LF, Kaplan DR. Overexpression of the trk tyrosine kinase rapidly accelerates nerve growth factor-induced differentiation. Neuron. 1992;9:883–896. doi: 10.1016/0896-6273(92)90241-5. [DOI] [PubMed] [Google Scholar]
- 24.Hofer M, Pagliusi SR, Hohn A, Leibrock J, Barde Y-A. Regional distribution of brain-derived neurotrophic factor mRNA in adult mouse brain. EMBO J. 1990;9:2459–2464. doi: 10.1002/j.1460-2075.1990.tb07423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ibáñez CF, Ebendal T, Barbany G, Murray-Rust J, Blundell TL, Persson H. Disruption of the low affinity receptor binding site in NGF allows for neuronal survival and differentiation by binding to the trk gene product. Cell. 1992;69:329–341. doi: 10.1016/0092-8674(92)90413-7. [DOI] [PubMed] [Google Scholar]
- 26.Itoh K, Brackenbury R, Akeson RA. Induction of L1 mRNA in PC12 cells by NGF is modulated by cell–cell contact and does not require the high-affinity NGF receptor. J Neurosci. 1995;15:2504–2512. doi: 10.1523/JNEUROSCI.15-03-02504.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson D, Lanahan A, Buck CR, Sehgal A, Morgan C, Mercer E, Bothwell M, Chao MV. Expression and structure of the human NGF receptor. Cell. 1986;47:545–554. doi: 10.1016/0092-8674(86)90619-7. [DOI] [PubMed] [Google Scholar]
- 28.Kaplan DR, Hempstead BL, Martin-Zanca D, Chao MV, Parada LF. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991a;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- 29.Kaplan DR, Martin-Zanca D, Parada LF. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature. 1991b;350:158–160. doi: 10.1038/350158a0. [DOI] [PubMed] [Google Scholar]
- 30.Kappers JA. The development, topographical relations and innervation of the epiphysis cerebri in the albino rat. Z Zellforsch Mikrosk Anat. 1960;52:163–215. doi: 10.1007/BF00338980. [DOI] [PubMed] [Google Scholar]
- 31.Kimpinski K, Campenot RB, Mearow K. Effects of the neurotrophins nerve growth factor, neurotrophin-3, and brain-derived neurotrophic factor (BDNF) on neurite growth from adult sensory neurons in compartmented cultures. J Neurobiol. 1997;33:395–410. doi: 10.1002/(sici)1097-4695(199710)33:4<395::aid-neu5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 32.Klein R, Jing S, Nanduri V, O’Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65:189–197. doi: 10.1016/0092-8674(91)90419-y. [DOI] [PubMed] [Google Scholar]
- 33.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuchel GA. Alterations in target innervation and collateral sprouting in the aging sympathetic nervous system. Exp Neurol. 1993;124:381–386. doi: 10.1006/exnr.1993.1210. [DOI] [PubMed] [Google Scholar]
- 35.Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R. Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell. 1992;69:737–749. doi: 10.1016/0092-8674(92)90286-l. [DOI] [PubMed] [Google Scholar]
- 36.Lee KF, Bachman K, Landis S, Jaenisch R. Dependence on p75 for innervation of some sympathetic targets. Science. 1994;263:1447–1449. doi: 10.1126/science.8128229. [DOI] [PubMed] [Google Scholar]
- 37.Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- 38.Ma Y, Campenot RB, Miller FD. Concentration-dependent regulation of neuronal gene expression by nerve growth factor. J Cell Biol. 1992;117:135–141. doi: 10.1083/jcb.117.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacPhee IJ, Barker PA. Brain-derived neurotrophic factor binding to the p75 neurotrophin receptor reduced TrkA signalling while increasing serine phosphorylation in the TrkA intracellular domain. J Biol Chem. 1997;272:23547–23551. doi: 10.1074/jbc.272.38.23547. [DOI] [PubMed] [Google Scholar]
- 40.Maisonpierre PC, Belluscio L, Squinto S, Ip NY, Firth ME, Lindsay RM, Yancopoulos GD. Neurotrophin-3: a new neurotrophic factor related to NGF and BDNF. Science. 1990;247:1446–1451. doi: 10.1126/science.247.4949.1446. [DOI] [PubMed] [Google Scholar]
- 41.Miller FD, Kaplan DR. Life and death decisions: a biological role for the p75 neurotrophin receptor. Cell Death Differ. 1998;5:343–345. doi: 10.1038/sj.cdd.4400385. [DOI] [PubMed] [Google Scholar]
- 42.Miller FD, Mathew TC, Toma JG. Regulation of nerve growth factor receptor gene expression by NGF in the developing peripheral nervous system. J Cell Biol. 1991;112:303–312. doi: 10.1083/jcb.112.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller FD, Speelman A, Mathew TC, Fabian J, Chang E, Pozniak C, Toma JG. Nerve growth factor derived from terminals selectively increases the ratio of p75 to trkA NGF receptors on mature sympathetic neurons. Dev Biol. 1994;161:206–217. doi: 10.1006/dbio.1994.1021. [DOI] [PubMed] [Google Scholar]
- 44.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 45.Owman C. Sympathetic nerves probably storing two types of monoamines in the rat pineal gland. Int J Neuropharmacol. 1964;2:105–112. doi: 10.1016/0028-3908(64)90052-8. [DOI] [PubMed] [Google Scholar]
- 46.Phillips HS, Hains JM, Laramee GR, Rosenthal A, Winslow JW. Widespread expression of BDNF but not NT3 by target areas of basal forebrain cholinergic neurons. Science. 1990;250:290–294. doi: 10.1126/science.1688328. [DOI] [PubMed] [Google Scholar]
- 47.Posse de Chaves EI, Bussière M, Vance DE, Campenot RB, Vance JE. Elevation of ceramide within distal neurites inhibits neurite growth in cultured rat sympathetic neurons. J Biol Chem. 1997;272:3028–3035. doi: 10.1074/jbc.272.5.3028. [DOI] [PubMed] [Google Scholar]
- 48.Radeke MJ, Misko TP, Hsu C, Herzenberg LA, Shooter EM. Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature. 1987;325:593–596. doi: 10.1038/325593a0. [DOI] [PubMed] [Google Scholar]
- 49.Schecterson LC, Bothwell M. Novel roles for neurotrophins are suggested by BDNF and NT-3 mRNA expression in developing neurons. Neuron. 1992;9:449–463. doi: 10.1016/0896-6273(92)90183-e. [DOI] [PubMed] [Google Scholar]
- 50.Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Lochmuller H, Aloyz R, Haghighi A, Lach B, Seth P, Cooper E, Miller FD. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J Cell Biol. 1996;135:1–12. doi: 10.1083/jcb.135.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira S, Barbacid M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature. 1994;368:246–249. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- 52.Stanley LC, Horikawa K, Powell EW. Innervation of the superficial pineal of the rat using retrograde tracing methods. Am J Anat. 1987;180:249–254. doi: 10.1002/aja.1001800306. [DOI] [PubMed] [Google Scholar]
- 53.Thoenen H, Barde Y-A. Physiology of nerve growth factor. Physiol Rev. 1980;60:1284–1335. doi: 10.1152/physrev.1980.60.4.1284. [DOI] [PubMed] [Google Scholar]
- 54.Van der Zee CEEM, Ross GM, Riopelle RJ, Hagg T. Survival of cholinergic forebrain neurons in developing p75NFGR-deficient mice. Science. 1996;274:1729–1732. doi: 10.1126/science.274.5293.1729. [DOI] [PubMed] [Google Scholar]
- 55.Verdi JM, Birren SJ, Ibáñez CF, Persson H, Kaplan DR, Benedetti M, Chao MV, Anderson DJ. p75(NGFR) regulates trk signal transduction and NGF-induced differentiation in MAH cells. Neuron. 1994;12:733–745. doi: 10.1016/0896-6273(94)90327-1. [DOI] [PubMed] [Google Scholar]
- 56.von Bartheld CS, Byers MR, Williams R, Bothwell M. Anterograde transport of neurotrophins and axodendritic transfer in the developing visual system. Nature. 1996;379:830–833. doi: 10.1038/379830a0. [DOI] [PubMed] [Google Scholar]
- 57.Walsh GS, Krol KM, Crutcher KA, Kawaja MD. Enhanced neurotrophin-induced axon growth in myelinated portions of the central nervous system in mice lacking the p75 neurotrophin receptor. J Neurosci. 1999;19:4155–4168. doi: 10.1523/JNEUROSCI.19-10-04155.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weskamp G, Reichardt LF. Evidence that biological activity of NGF is mediated through a novel subclass of high affinity receptors. Neuron. 1991;6:649–663. doi: 10.1016/0896-6273(91)90067-a. [DOI] [PubMed] [Google Scholar]
- 59.Yang X-M, Toma JG, Bamji SX, Belliveau DJ, Kohn J, Park M, Miller FD. Autocrine hepatocyte growth factor provides a local mechanism for promoting axonal growth. J Neurosci. 1998;18:8369–8381. doi: 10.1523/JNEUROSCI.18-20-08369.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yeo TT, Chua-Couzens J, Butcher LL, Bredesen DE, Cooper JD, Valletta JS, Mobley WC, Longo FM. Absence of p75NTR causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. J Neurosci. 1997;17:7594–7605. doi: 10.1523/JNEUROSCI.17-20-07594.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou XF, Rush RA. Endogenous brain-derived neurotrophic factor is anterogradely transported in primary sensory neurons. Neuroscience. 1996;74:945–951. doi: 10.1016/0306-4522(96)00237-0. [DOI] [PubMed] [Google Scholar]