

Abstract
Most early onset cases of familial Alzheimer’s disease (AD) are caused by mutations in presenilin-1 (PS1) and presenilin-2 (PS2). These mutations lead to increased β-amyloid formation and may induce apoptosis in some model systems. Using primary cultured hippocampal neurons (HNs) and rat pheochromocytoma (PC12) cells transiently transfected with replication-defective recombinant adenoviral vectors expressing wild-type or mutant PS1, we demonstrate that mutant PS1s induce apoptosis, downregulate the survival factor Akt/PKB, and affect several Akt/PKB downstream targets, including glycogen synthase kinase-3β and β-catenin. Expression of a constitutively active Akt/PKB rescues HNs from mutant PS1-induced neuronal cell death, suggesting a potential therapeutic target for AD. Downregulation of Akt/PKB may be a mechanism by which mutant PS1 induces apoptosis and may play a role in the pathogenesis of familial AD.
Keywords: presenilin, Alzheimer’s disease, apoptosis, Akt/PKB, adenoviral vectors, hippocampal neurons
Mutations in the presenilin genes PS1 and PS2 cause most early onset familial Alzheimer’s disease (AD) (Price and Sisodia, 1998). These genes encode integral membrane proteins of unknown function that are predicted to have six to eight membrane-spanning domains (Price and Sisodia, 1998). The presenilins facilitate β-amyloid processing and are believed to play a role in Notch signaling (Levitan et al., 1996; Scheuner et al., 1996). Several lines of evidence suggest that PS1 may also be involved in the Wnt signaling pathway (Zhou et al., 1997; Murayama et al., 1998; Takashima et al., 1998; Yu et al., 1998; Zhang et al., 1998; Nishimura et al., 1999). Using yeast two-hybrid analysis, Zhou et al. (1997) identified δ-catenin, an armadillo repeat protein, as a putative partner with PS1. Other studies have demonstrated that PS1 interacts in vivo with β-catenin and its regulatory kinase glycogen synthase kinase-3β (GSK-3β) (Murayama et al., 1998; Takashima et al., 1998;Yu et al., 1998; Zhang et al., 1998; Nishimura et al., 1999). PS1 and PS2 have also been reported to modulate apoptosis. Vito et al. (1996)demonstrated that Alg-3, a cDNA with homology to PS2, rescues T lymphocytes from apoptotic death. In addition, Wolozin et al. (1996)showed that mutant PS2 transfection of rat pheochromocytoma (PC12) cells enhanced basal levels of apoptosis over that seen after PS2-wild-type (WT) transfection. Furthermore, PC12 cells that stably express mutant PS1-L286V show an increased sensitivity to apoptosis after trophic factor withdrawal and β-amyloid administration (Guo et al., 1996). However, other studies have failed to demonstrate that mutant PS1 results in increased sensitivity to apoptotic stimuli (Bursztajn et al., 1998).
Studies of mutant PS-induced apoptotic cell death have involved stably transfected non-neuronal and neuroblastoma cell lines (Guo et al., 1996; Wolozin et al., 1996). We investigated hippocampal neurons (HNs) and PC12 cells transiently expressing PS1-WT or mutant PS1 using adenoviral vectors (AdVs) to identify the effects of mutant PS1 on cell viability and apoptosis. The advantages of this system are that the cells studied, HNs, are prominently affected by AD, and that the use of transient expression avoids selection of cell lines that may have undergone mutation to avoid deleterious effects of mutant PS1 overexpression. Because PS1 may play a role in Wnt signal transduction, and PS1 mutations may induce apoptosis, we questioned whether PS1 may act at the point of convergence between these two signaling pathways. We specifically investigated the effect of PS1 on Akt/PKB, a serine/threonine kinase, and its downstream targets because Akt/PKB is associated with antiapoptotic signaling (Dudek et al., 1997; Kennedy et al., 1997) and is capable of inactivating GSK-3β (Cross et al., 1995), a key component in Wnt signaling.
MATERIALS AND METHODS
Cell culture and reagents. PC12 cells were maintained in DMEM supplemented with 10% bovine calf serum and 10 μg/ml penicillin-streptomycin (Sigma, St. Louis, MO). HNs were prepared from the hippocampi of fetal rats at 17 d of gestation, as previously described (Ghadge et al., 1997). LY294002, a gift from Dr. Clive Palfrey (University of Chicago), was diluted in DMSO and used at 10 μm.
Construction of AdVs and bicistronic expression plasmids.AdPS1-WT, AdPS1-A246E, and AdPS1-C410Y were previously described (Weihl et al., 1999). Bicistronic expression plasmids containing PS1-WT, PS1-A246E, or PS1-C410Y cDNAs upstream of a LacZ reporter gene were constructed as follows: the encephalomyocarditis virus internal ribosomal entry site and β-galactosidase cDNA (LacZ) were digested from plasmid p1704LacZ and blunt end-ligated into an EcoRV restriction site of pAdCMV containing PS1-WT or mutant PS1.
Plasmid pAdKNmAkt/PKB contains the Akt/PKB cDNA fused to a myristoylation sequence allowing for plasma membrane translocation. To construct AdmAkt/PKB, the plasmid pAdKNmAkt/PKB was linearized withNheI and cotransfected, using the calcium phosphate precipitation method, with XbaI- andClaI-digested adenovirus 5 (sub360) DNA into HEK293 cells, atrans-complementing cell line for E1 function. The AdVs were purified by CsCl isopycnic ultracentrifugation and then dialyzed against HEPES-buffered saline to produce a high-titered virus stock. AdV transgene expression was determined via Western analysis.
Recombinant virus infections and transfections. PC12 cells were infected for 2 hr with an aliquot of purified virus to achieve a multiplicity of infection (MOI) of 40 or 200 pfu/cell in a sufficient volume of postinfection media (culture media containing 2% serum) to cover the cells, washed once, and then incubated with postinfection media. In the case of HNs, coverslips were removed from the glial feeder layer, and HNs were washed once in glial conditioned media and then infected at an MOI of 1000 (except for AdmAkt when the MOI was 250) in conditioned media. Two hours later HNs were washed and replaced inverted over the glial feeder layer.
DNA transfections were performed as previously described. In brief, PC12 cells were plated in six-well culture plates, and 2 μg of the respective expression plasmid was diluted in 100 μl of 0.15m NaCl and 0.6 μl of 0.1 m polyethylenimine (Aldrich, St. Louis, MO) and applied to wells containing 1.5 ml of serum-free DMEM. The plates were spun in a clinical centrifuge for 10 min at 1000 rpm and then incubated for 2 hr, after which media were replaced with DMEM containing 5% serum. After 48 and 96 hr, the plates were rinsed in PBS, fixed in 4% paraformaldehyde, and incubated for 4 hr at 37°C in 5-bromo-4-chloro-3-indolyl-β-d-galactopyranosidase (X-gal) staining solution (50 mm Tris-Cl, pH 7.5, 15 mm NaCl, 2 mm MgCl2, 0.5 mg/ml X-gal, and 2.5 mm ferriferrocyanide).
Immunohistochemistry and cell death assay. Coverslips were rinsed in PBS, pH 7.4, fixed with ice-cold methanol for 10 min, and blocked overnight in PBS containing 2% BSA and 0.1% Tween 20. Rat monoclonal anti-PS1 antibody (Chemicon, Temecula, CA) diluted 1:200 in blocking buffer was applied 1 hr at room temperature and rinsed three times in blocking buffer. FITC-conjugated anti-rat IgG diluted 1:200 in blocking buffer was then applied 1 hr at room temperature and rinsed three times in blocking buffer, and immunostaining was detected via fluorescent microscopy. For nuclear staining, coverslips were rinsed in PBS, pH 7.4, fixed 10 min in 4% paraformaldehyde, and then stained with 50 μg/ml Hoescht 33342 (Molecular Probes, Eugene, OR). The percentage of fragmented nuclei was determined using five experiments and >10 fields per experiment.
MTT assay was performed as follows. One hundred microliters of 5 mg/ml MTT (Sigma) in PBS were incubated per milliliter of culture media for 3 hr at 37°C. Precipitated MTT was resuspended in culture media with the addition of 1 volume of acid isopropanol (0.04N HCl) and read on a spectrophotometer at 570 nm.
Immunoprecipitation and Western blot. The monolayer was rinsed in PBS, pH 7.4, scraped, and centrifuged. The cell pellet was resuspended in Laemmli buffer with 100 μg/ml phenylmethylsulfonyl fluoride (PMSF), 1 μg/ml aprotinin, and 1 μg/ml leupeptin and sonicated on ice for 5 sec to shear chromosomal DNA. For the preparation of cytosolic extracts, the cell pellet was resuspended in lysis buffer (10 mm HEPES, pH 7.5, 10 mm KCl, 1 mm dithithreitol, and 1% NP-40) and centrifuged for 15 min to remove cell debris. For immunoprecipitation, these lysates were resuspended in PBS, precleared for 1 hr with protein A-Sepharose (Pharmacia, Piscataway, NJ) and incubated overnight at 4.0°C with primary antibody. The antigen–antibody complex was captured with protein A-Sepharose and released by boiling in Laemmli buffer. Sonicated or immunoprecipitated material was electrophoresed on a 10% Tris-glycine-buffered SDS-polyacrylamide gel. The gel was electroblotted to a 0.2 μm polyvinylidene difluoride membrane (Schleicher & Schuell, Keene, NH) for 2 hr. The blot was blocked overnight in PBS containing 0.1% Tween 20 and 5.0% milk and then incubated at room temperature for 3 hr with primary antibody, rinsed three times for 15 min, and incubated for 1 hr in peroxidase-conjugated secondary antibody diluted in PBS containing 0.05% Tween 20. Detection was performed using ECL Plus and Hyperfilm ECL (Amersham, Arlington Heights, IL).
The following antibodies were used: anti-PS1loop (Thinakaran et al., 1996), anti-GSK-3β (QCB, Hopkinton, MA), anti-GSK-3β(Ser9) (QCB), anti-GSK-β(Tyr216) (Upstate Biotechnology, Lake Placid, NY), anti-p85 (Upstate), anti-phosphotyrosine (Upstate), and anti-β-catenin (Transduction Laboratories, Lexington, KY). Phospho-Akt(Ser473), phospho-stress-activated protein kinase (SAPK)/c-Jun N-terminal protein kinase (JNK)(Thr183/Tyr185), phospho-p38 MAP kinase(Thr180/Tyr182), phospho-p44/42 extracellular signal-regulated kinase (ERK1/2) kinase(Thr202/Tyr204), and phospho-p70S6 kinase(Ser411) antibody kits are from New England Biolabs (Beverly, MA). All dilutions were according to manufacturers’ instructions.
Akt/PKB kinase assay. Cells from transfected 100 mm plates were lysed in buffer A (50 mm Tris-Cl, pH 7.5, 1 mm EDTA, 1 mm EGTA, 0.5 mmNa3VO4, 0.1% β-mercaptoethanol, 1% Triton X-100, 50 mm NaF, 5 mm sodium pyrophospate, 10 mm sodium β-glycerophosphate, 0.1 mm PMSF, and 1 μg/ml aprotinin and leupeptin) and immunoprecipitated with 4 μg of anti-Akt/PKB PH domain antibody (Upstate) prebound to protein A/G-agarose (Santa Cruz Biotechnology, Santa Cruz, CA) for 90 min at 4.0°C. Immunoprecipitated material was washed three times in buffer A containing 0.5 m NaCl and two times in buffer B (100 mm 4-morpholinepropanesulfonic acid, pH 7.2, 125 mm β-glycerophosphate, 25 mm EGTA, 5 mmNa3VO4, and 5 mm DTT). The immune complex was incubated in 50 μl of buffer B, with 10 μm inhibitor peptide (Upstate), 100 μmAkt/PKB-specific substrate (Upstate), and 10 μCi of [γ-33P]ATP at room temperature for 10 min. The reaction was stopped with addition of 40% TCA for 5 min, and 40 μl of the reaction mix was then transfered to P81 phosphocellulose paper and washed three times with 50 ml of 0.75% phosphoric acid. Samples were transfered to scintillation tubes with 5 ml of scintillation mixture and counted in a scintillation counter.
RESULTS
Mutant PS1 expression induces apoptosis in HNs and PC12 cells
HNs were transduced with AdVs that expressed PS1-WT or one of two mutants associated with familial AD, PS1-A246E and PS1-C410Y. Using a sensitive monoclonal antibody directed against the N terminus of human PS1, we were able to specifically immunostain HNs that expressed human PS1, because the antibody does not cross-react with rodent PS1. Immunofluorescent studies showed that ∼50% of HNs carried the human PS1 transgene 24 hr after transduction. Human PS1 localized in a perinuclear distribution and in both axonal and dendritic processes (Fig. 1a), indicating that overexpression of PS1 results in a localization similar to that described for endogenous PS1 (Cook et al., 1996; Lah et al., 1997). Confocal microscopy showed that PS1-WT and mutant PS1 had a similar localization (data not shown). The intensity of the immunofluorescent staining as well as Western analysis of lysates from adenovirally transduced cells 72 hr after transduction demonstrated that PS1 protein expression levels of PS1-WT- and mutant PS1-expressing cells were similar (Fig. 1b). Of note is a 14 kDa PS1-immunoreactive fragment present in lysates from PS1-A246E- and PS1-C410Y-expressing cells (Fig. 1b). This fragment coincides with a previously reported putative PS1 caspase-cleaved fragment (Loetscher et al., 1997;Weihl et al., 1999).
Fig. 1.

AdV epression of PS1 in neurons.a, Immunohistochemistry of HNs with a rat monoclonal antibody specific for human PS1 24 hr after transduction with PS1-WT or the indicated PS1-mutant. b, Western analysis using anti-PS1loop antibody on lysates of PC12 cells 36 hr after transduction. The AdVs used in the transduction of cells processed for this and subsequent Western blots are noted above the lanes. The full-length holoprotein (HP), processed C-terminal fragment (CTF) and caspase-cleaved CTF (CTFcasp) are noted.
We used Hoescht 33342 fluorescent staining to determine whether mutant PS1 induced apoptosis of HNs. The staining demonstrated that the number of fragmented apoptotic nuclei was greatly increased in HNs expressing mutant PS1s when compared with those expressing PS1-WT or a control gene (LacZ) or when compared with mock-transduced neurons (Fig. 2a). The number of fragmented nuclei peaked at 5 d after transduction (Fig.2b). Additional staining for human PS1 expression showed that it was the mutant PS1-expressing HNs that exhibited fragmented nuclei, whereas HNs expressing PS1-WT had intact nuclei (Fig.2c). Similar results were obtained after PC12 cell transduction with mutant PS1s using Hoescht 33342 nuclear staining (data not shown). To quantify this data by an alternative method, we used a colormetric assay based on the tetrazolium salt MTT, which measures only living cells (Fig. 2d).
Fig. 2.
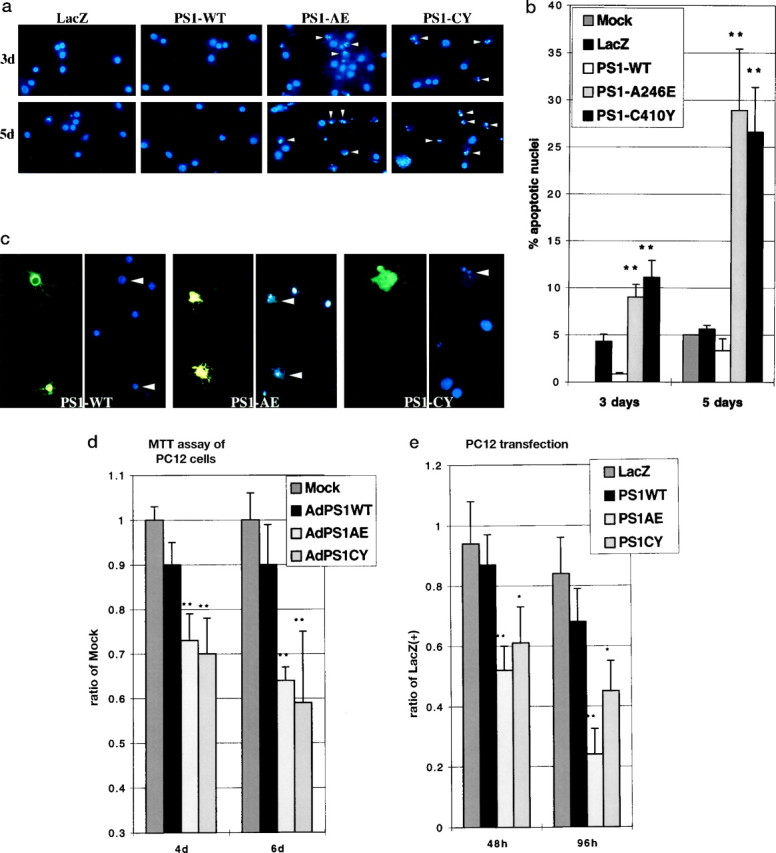
Mutant PS1 expression induces apoptosis in HNs and PC12 cells. a, Hoescht 33342 nuclear staining of HNs 3 d (top panel) or 5 d (bottom panel) after transduction with AdLacZ, AdPS1-WT, or the indicated AdPS1-mutant. Arrowheads show fragmented nuclei. b, Percentage of fragmented nuclei from HNs 3 and 5 d after transduction with mock, LacZ, PS1-WT, or mutant PS1. **p < 0.005 for PS1-mutant when compared with control transduced HNs from five separate experiments.c, Double fluorescent staining of HNs 4 d after transduction with PS1-WT or the indicated PS1-mutant using a rat monoclonal antibody for human PS1 and Hoescht 33342 for nuclear morphology. d, Ratio of MTT production from PC12 cells 4 or 6 d after transduction with AdPS1-WT or the indicated AdPS1-mutant compared with mock controls at each time point, which was arbitrarily set to 1. **p < 0.005 for PS1-mutant when compared with control transduced PC12 cells from three separate experiments. e, Ratio of the number of LacZ-positive cells 48 or 96 hr after transient transfection of a bicistronic expression vector containing LacZ in the second cistron preceded by PS1-WT or PS1-mutant cDNAs in the first cistron compared with the number LacZ-positive cells 24 hr after transfection of a monocistronic LacZ construct. *p < 0.05; **p< 0.005 for PS1 mutant when compared with monocistronic LacZ-transfected PC12 cells from four separate experiments.
To confirm that mutant PS1-associated cell death was unrelated to the use of adenoviral vectors, we transiently transfected PC12 cells with bicistronic expression vectors expressing LacZ downstream of PS1-WT, PS1-A246E, or PS1-C410Y cDNAs. Forty-eight and 96 hr after transfection the number of LacZ-positive cells was counted. PS1-mutant-transfected cells demonstrated significantly fewer LacZ-positive cells at both time points when compared with PS1-WT or a LacZ monocistronic plasmid control, suggesting that transient transfection of mutant PS1 decreases cell survival (Fig. 2e). These studies confirm that mutant PS1 expression induced the nuclear condensation and neuronal apoptosis.
Downregulation of Akt/PKB activity is an early step in PS1-mutant-associated apoptosis
Lysates of HNs and naive (undifferentiated) PC12 cells 36 hr after PS1-WT or mutant PS1 transduction were Western blotted using an antibody specific for the phosphorylated, active form of Akt/PKB. No morphological signs of apoptosis or cell death were apparent with mutant PS1-transduced HNs and PC12 cells at this time point, suggesting that any change in Akt/PKB activity would be an early step in mutant PS1-induced neuronal death. HNs and naive PC12 cells transduced with mutant PS1s demonstrated a 25–50% decrease in basal levels of Akt/PKB phosphorylation (Fig. 3a–c) when compared with lysates from HNs or PC12 cells expressing PS1-WT or a control [wild-type superoxide dismutase (SOD); (Ghadge et al., 1997)] or with lysates from mock-transduced cells. A similar decrease was seen in naive PC12 cells that were serum starved for 1 hr and stimulated with nerve growth factor (NGF) for 10 min (Fig.3a,c). In this experiment and all subsequent experiments, NGF stimulation refers to a brief 10 min application of NGF to serum-starved naive PC12 cells before cell lysis. The increase in Akt/PKB phosphorylation after SOD expression is a result of AdV infection, because transduction with an AdV expressing LacZdemonstrated a similar increase (data not shown).
Fig. 3.

Mutant PS1 expression downregulates Akt/PKB. a, Western analysis using anti-Akt/PKB (top panel) or anti-phospho-Akt/PKB (Ser473) antibody (AKTpS) (bottom panel) on lysates of PC12 cells 36 hr after transduction with (right panel) and without (left panel) 10 min NGF stimulation. b, Western analysis using anti-Akt/PKB (top panel) or anti-phospho-Akt/PKB(Ser473) antibody (bottom panel) on lysates of HNs 36 hr after transduction.c, Densitometric analysis of the ratio of phospho-Akt/ PKB(Ser473) to total Akt/PKB from four separate experiments. **p < 0.005 for PS1 mutant when compared with controls. In calculating this ratio and ratios in subsequent figures, the mock was arbitrarily set to 1 for each condition. d, In vitro kinase assay of PC12 cell lysates 36 hr after transduction with and without 10 min NGF stimulation. *p < 0.05; **p < 0.005 for PS1-mutants when compared with PS1-WT are from more than four separate experiments.
Although the phosphorylation state of Akt/PKB is a good indicator of its activity, we chose to further confirm downregulation of Akt/PKB after mutant PS1 expression using an in vitro kinase assay. Immunoprecipitated Akt/PKB from PC12 cells expressing PS1-WT, PS1-A246E, or PS1-C410Y was assayed for the transfer of radiolabeled ATP to an Akt/PKB-specific substrate peptide. Figure 3dshows that Akt/PKB activity was reduced by 40% in PS1-A246E-expressing cell lysates and by 65% in PS1-C410Y-expressing cell lysates when compared with PS1-WT-expressing cell lysates. NGF stimulation for 10 min modestly increased Akt/PKB activity in both PS1-WT- and PS1-mutant-expressing cells.
Mutant PS1-associated downregulation of Akt/PKB is not associated with a perturbation of known upstream kinase pathways
Akt/PKB is phosphorylated through a cascade of kinases that are stimulated by NGF. More specifically, NGF stimulation leads to TrkA receptor autophosphorylation, which in turn recruits the p85 regulatory subunit of phosphoinositide-3 kinase (PI3K) to the plasma membrane. At the plasma membrane, p85 is phosphorylated by both TrkA and p110, the catalytic subunit of PI3K, which in turn phosphorylates lipid messengers that signal for Akt/PKB activation (Coffer et al., 1998). To determine whether NGF and TrkA signaling was perturbed in mutant PS1-expressing neurons and contributed to the observed decrease in basal and NGF-stimulated Akt/PKB activity, we examined other kinase pathways associated with this trophic factor signaling. Western blots of PC12 cells 36 hr after transduction with mutant PS1s showed no difference, when compared with control lysates, in basal or NGF-stimulated phosphorylation of TrkA or in global tyrosine phosphorylation (Fig. 4a). There blots and subsequent blots are representative of at least three experiments; hence minor inconsistencies present in the representative blots were not seen with all experiments. When possible, we have densitometrically quantified the results of separate experiments and the data are shown when relevant. We assessed the activation of PI3K by immunoprecipitating similar NGF-stimulated PC12 lysates with an antibody specific for phosphorylated tyrosines and subsequently immunoblotting with an antibody directed to the p85 subunit of PI3K. The phosphorylation of p85 was unchanged after mutant PS1 expression compared with lysates from cells expressing PS1-WT or controls, suggesting that TrkA activation of PI3K is unaffected (Fig.4b). As a negative control we immunoprecipitated unstimulated PC12 cell lysates [Fig. 4b,mock(−NGF)]. In addition, TrkA activation of MAP kinases ERK1 and ERK2 was unaffected after mutant PS1 expression (Fig. 4c). These results indicate that the trophic factor signaling necessary for Akt/PKB phosphorylation and concomitant activation is intact and unchanged in mutant PS1-expressing neurons.
Fig. 4.

Mutant PS1 expression does not affect other kinase pathways. All blots are representative of at least three independent experiments. a, Western analysis using anti-phosphotyrosine antibody on lysates of PC12 cells 36 hr after transduction with (right panel) and without (left panel) 10 min NGF stimulation.b, Densitometric analysis of the amount of tyrosine-phosphorylated p85 from three western blots of transduced PC12 cell lysates 36 hr after transduction following a 10 min NGF stimulation (which were immunoprecipitated with anti-phosphotyrosine antibody before electrophoresis) immunostained with anti-p85 antibody.Mock(−NGF), Negative control for NGF stimulation. c, Western analysis using anti-p44/42 ERK1/2 kinase antibody (top panel) or anti-phospho-p44/42 ERK1/2 kinase(Thr202/Tyr204) antibody (bottom panel) on lysates of PC12 cells 36 hr after transduction with (right panel) and without (left panel) 10 min NGF stimulation. TheMock lane is overexposed to allow a comparison between the PS1-WT and mutant PS1 lanes.d, Western analysis using anti-SAPK/JNK antibody (top panel) or anti-phospho-SAPK/JNK(Thr183/Tyr185) antibody (bottom panel) on lysates of PC12 cells 36 hr after transduction with (right panel) and without (left panel) 10 min NGF stimulation. e, Western analysis using anti-p38 MAP kinase antibody (top panel) or anti-phospho-p38 MAP kinase(Thr180/Tyr182) antibody (bottom panel) on lysates of PC12 cells 36 hr after transduction with (right panel) and without (left panel) 10 min NGF stimulation.
Mutant PS1-induced apoptosis is not related to stress-activated kinase pathways
We wondered whether other kinase pathways unassociated with the direct regulation of Akt/PKB activity mediated changes in neurons induced by mutant PS1 expression. The SAPKs, for example, have been shown to be activated by a variety of cellular stresses and apoptotic stimuli and may indirectly regulate Akt/PKB activity (Berra et al., 1998; Zundel and Giaccia, 1998). We found no differences in the phosphorylation states of SAPK/JNK or p38/high-osmolarity glycerol response (HOG) stress-related kinases in Western blots of lysates of PC12 cells after transduction with mutant PS1s compared with PS1-WT (Fig. 4d,e). The increase in phospho-SAPK/JNK seen in all groups after AdV transduction is consistent with results from previous reports using AdV infection (See and Shi, 1998). These results indicate that the observed differences in apoptosis or Akt/PKB activity after mutant PS1 expression are not a result of cell signaling through stress-activated kinase pathways.
Mutant PS1-associated apoptosis affects the ribosomal S6 kinase
Akt/PKB affects several aspects of cellular metabolism, in particular, protein synthesis, glycogen metabolism, cell cycle regulation, cell differentiation, and cell survival (Coffer et al., 1998). We examined several of these putative downstream targets in PC12 cells expressing PS1-WT or mutant PS1. The ribosomal S6 kinase (p70S6k), which changes the pattern of protein synthesis after mitogenic stimulation of cells, lies downstream of Akt/PKB and is phosphorylated by a number of kinases (Coffer et al., 1998). Western analysis of lysates from PC12 cells transduced with mutant PS1s demonstrated a decrease in active p70S6k when compared with lysates from PC12 cells expressing PS1-WT or SOD or with lysates from mock-transduced PC12 cells, consistent with its activation through an Akt/PKB signaling pathway (Fig.5a,b). NGF stimulation of mutant PS1-transduced cells boosted phosphorylated p70S6k to mock levels (Fig. 5a,b), suggesting that basal levels of p70S6k activity may be mediated through Akt/PKB and that the other signaling pathways necessary to activate p70S6k via NGF are intact. It is interesting to note that NGF-stimulated activation of p70S6k occurs through a wortmannin-sensitive PI3K pathway and that p70S6k is phosphorylated by phosphoinositide-dependent protein kinase 1 (PDK1), the same kinase responsible for Akt/PKB activation (Alessi et al., 1998; Pullen et al., 1998). Because NGF-stimulated p70S6k phosphorylation is normal in mutant PS1-expressing cells, and because p70S6k uses a pathway identical to that involved in NGF-stimulated Akt/PKB activity, we suggest that mutant PS1-associated basal and NGF-stimulated downregulation of Akt/PKB in HNs is a specific defect and not secondary to a more general abnormality in this pathway.
Fig. 5.

Mutant PS1-induced apoptosis is associated with pathways downstream of Akt/PKB. a, Western analysis using anti-p70S6k (top panel) or anti-phospho-p70S6k(Ser411) antibody (bottom panel) on lysates of PC12 cells 36 hr after transduction with (right panel) and without (left panel) 10 min NGF stimulation. b,Densitometric analysis of the ratio of phospho-p70S6k(Ser411) to total p70S6k from four separate experiments. **p < 0.005 for PS1 mutant when compared with controls.
Mutant PS1-associated apoptosis affects GSK-3β and β-catenin
Considerable attention has recently been focused on the role of PS1 in the Wnt signaling pathway. It has been shown that PS1 associates with β-catenin and its regulatory kinase, GSK-3β (Zhou et al., 1997; Murayama et al., 1998; Takashima et al., 1998; Yu et al., 1998;Zhang et al., 1998). Wnt signaling occurs through activation of the Frz receptor, which signals the inactivation of GSK-3β, resulting in increased β-catenin stability (Cadigan and Nusse, 1997). This allows for β-catenin to enter the nucleus and activate the T-cell factor/lymphoid-enhancing factor-1 (Tcf/LEF-1) family of transcription factors (Cadigan and Nusse, 1997). A complementary pathway involves trophic factor stimulation of Akt/PKB, resulting in phosphorylation of GSK-3β on serine residue 9, leading to its inactivation (Cross et al., 1995). We surmised that a decrease in Akt/PKB activity associated with mutant PS1 expression in neurons would lead to decreased inactivation (and enhanced activity) of GSK-3β. Immunoprecipitation of GSK-3β and subsequent immunostaining with an antibody specific for phophoserine-9 of GSK-3β demonstrated that mutant PS1-expressing PC12 cells failed to phosphorylate GSK-3β as efficiently as cells expressing PS1-WT or SOD or mock-transduced cells (Fig.6a). In agreement with these data, immunoblots of the same lysates with an antibody against phosphotyrosine-216 of (activated) GSK-3β demonstrated that mutant PS1-expressing neurons had an increase in phosphotyrosine-216 GSK-3β immunoreactivity when compared with PS1-WT-, SOD-, or mock-transduced control cells (Fig. 6b). NGF stimulation for 10 min did not significantly alter the phosphorylation of GSK-3β in PS1-mutant expressing cells (data not shown). These data confirm that the mutant PS1-associated decrease in Akt/PKB activity results in decreased inactivation of GSK-3β with a subsequent enhancement in activity.
Fig. 6.

Mutant PS1-associated apoptosis modulates components of Wnt signaling. a,Immunoprecipitation with anti-GSK-3β followed by Western analysis with anti-GSK-3β (top panel) or anti-phospho-GSK-3β(Ser9) antibody (bottom panel) on lysates of PC12 cells 36 hr after transduction. b, Western analysis using anti-GSK-3β (top panel) or anti-phospho-GSK-3β(Tyr216) antibody (bottom panel) on lysates of PC12 cells 36 hr after transduction. c, Western analysis from two separate experiments using anti-β-catenin antibody on cytosolic lysates of PC12 cells 36 hr after transduction. Loading controls show immunoblotted anti-Akt/PKB antibody from the same lysates.d, Densitometry of β-catenin normalized to total protein concentration from three separate experiments. **p < 0.005 for PS1 mutant when compared with controls.
GSK-3β has many targets, including glycogen synthase, tau, and β-catenin (Plyte et al., 1992). To examine the physiological consequence of increased GSK-3β activity, we assessed the levels of soluble β-catenin, because phosphorylation of β-catenin targets it for proteosomal degradation (Plyte et al., 1992). Immunoblots of PC12 cytosolic lysates with an antibody for β-catenin demonstrated a decrease in the amount of β-catenin in cells expressing PS1-WT and mutant PS1s when compared with SOD- or mock-transduced cells (Fig.6c,d). The decrease in mutant PS1-transduced cells was greater than that seen in PS1-WT-transduced cells, suggesting that mutant PS1-associated upregulation of GSK-3β results in a decrease in β-catenin levels.
Mutant PS1-induced apoptosis is sensitive to PI3K inhibition and is rescued by a constitutively active Akt/PKB
If mutant PS1-induced apoptosis is mediated through Akt/PKB signaling, we reasoned that inhibiting Akt/PKB phosphorylation should induce apoptosis in HNs and increase the apoptosis associated with mutant PS1 expression. To test this, we used LY294002, a potent PI3K inhibitor that has been shown to decrease Akt/PKB phosphorylation and activity in vivo (Coffer et al., 1998). After treatment with LY294002, the levels of apoptosis were elevated in PS1-WT- and SOD-expressing HNs and highest after mutant PS1 expression (Fig.7a). Similarly, we tested whether expression of a constitutively active Akt/PKB could rescue HNs from mutant PS1-induced apoptosis. To express a constitutively active Akt/PKB, we prepared an AdV that expresses a myristoylated form of Akt/PKB (AdmAkt/PKB), because Akt/PKB must move to the plasma membrane before it is phosphorylated and becomes active (Alessi and Cohen, 1998). Figure 7b demonstrates the increase in total as well as phosphorylated Akt/PKB after transduction of PC12 cells with AdmAkt/PKB compared with mock and AdSODWT controls. Co-transduction of HNs with both mAkt/PKB and mutant PS1s resulted in complete rescue from the PS1-mutant-induced HN apoptosis at 3 d after transduction and partial rescue at 5 d after transduction (Fig. 7c).
Fig. 7.

Mutant PS1-associated apoptosis is increased by inhibition of Akt/PKB and rescued by constitutively active Akt/PKB. a, c, Percentage of fragmented nuclei in HNs 3 d after transduction with mock, SOD, PS1-WT, or mutant PS1 along with co-treatment with LY294002 (a) or co-transduction with AdmAkt/PKB (c), and 5 d after co-transduction with mutant PS1 along with AdmAkt/PKB (c). b, Western analysis using anti-Akt/PKB or anti-phospho-Akt/PKB(Ser473) antibody on lysates of PC12 cells 36 hr after transduction.
DISCUSSION
In summary, we have demonstrated that expression of familial AD-associated mutant PS1s in HNs and PC12 cells results in apoptotic cell death. Our results are the first to show a direct proapoptotic effect of mutant PS1 in HNs, the target cell of AD pathogenesis. This death is accompanied by an early decrease in survival signaling through the Akt/PKB pathway. The decrease in Akt/PKB activity affects several proteins associated with cell metabolism and cell survival: the basal activity of the translational regulator p70S6k is reduced; Akt/PKB-mediated inactivation of GSK-3β is reduced, resulting in an increase in the activity of GSK-3β; and the levels of soluble β-catenin are decreased. Mutant PS1-expressing HNs have increased cell death after Akt/PKB inhibition, whereas expression of a constitutively active form of Akt/PKB rescues HNs from mutant PS1-induced cell death. The latter observations may have therapeutic implications in AD.
We considered whether downregulation of Akt/PKB after expression of mutant PS1 is a direct effect or merely a consequence of the accompanying apoptosis. Recent evidence suggests that downregulation of Akt/PKB activity can occur through mechanisms associated with some forms of apoptosis. For example, ceramide-induced apoptosis in some cells has been shown to be associated with a decrease in Akt/PKB activity (Zhou et al., 1998; Zundel and Giaccia, 1998). In addition, an increase in phosphatase activity present during apoptosis can result in an increase in Akt/PKB inactivation (Meier et al., 1998).
Despite these reports, we doubt that the decrease in Akt/PKB activity, as well as the increase in GSK-3β activity and the decrease in β-catenin levels, are merely a result of apoptosis. Of importance is the fact that these biochemical changes occur 36 hr after transduction, well before any morphological signs of apoptosis, which peak at 5 d after transduction. In addition, we found that PS1-mutant-expressing stably transfected neuroblastoma cells, which do not show an enhanced level of basal apoptosis, show a decrease in Akt/PKB activity (data not shown), demonstrating that the decline in Akt/PKB is not a result of apoptosis. Our data are supported by several reports demonstrating an increase in apoptosis after PS1-mutant expression (Guo et al., 1996;Wolozin et al., 1998). In addition, other groups have found that mutant PS1-expressing cells have a decrease in the stability or trafficking of β-catenin and an increase in GSK-3β activity in cells that were not undergoing apoptosis (Murayama et al., 1998; Takashima et al., 1998;Zhang et al., 1998; Nishimura et al., 1999), suggesting that our findings are not an artifact of apoptosis.
The question remains how PS1, an integral membrane and endoplasmic reticulum- and Golgi-associated protein, mediates the activity of Akt/PKB. Recent evidence suggests that PS1 associates with GSK-3β (Takashima et al., 1998) and β-catenin (Zhou et al., 1997; Murayama et al., 1998; Yu et al., 1998; Zhang et al., 1998; Nishimura et al., 1999). Because GSK-3β is known to associate with Akt/PKB (van Weeren et al., 1998), it may be that PS1 associates with GSK-3β, β-catenin, and Akt/PKB in a large complex. It is intriguing to note that Akt/PKB must move from an intracellular location to the plasma membrane before becoming activated by PDK1 (Alessi and Cohen, 1998) and that constitutively active mutants of Akt/PKB target the membrane facilitating this transit (Alessi and Cohen, 1998). Perhaps PS1 is responsible for this trafficking step, whereas mutant PS1 disrupts the trafficking, resulting in a decreased activation of Akt/PKB.
Mutant PS1 could result in an increased sensitivity to apoptotic stimuli by several mechanisms, including a decrease in Akt/PKB survival signaling, upregulation of GSK-3β, or a decrease in β-catenin-associated transcriptional activation. Akt/PKB has a clear role in some cell survival paradigms, because expression of dominant-negative constructs leads to an increase in apoptotic cell death, whereas overexpression of constitutively active Akt/PKB rescues some cells from a variety of apoptotic stimuli (Kennedy et al., 1997). Upregulation of GSK-3β may also mediate apoptosis. Its constitutive expression results in increased basal levels of apoptosis in PC12 cells (Pap and Cooper, 1998). Finally, a recent report demonstrates that expression of a dominant negative β-catenin mediates apoptosis in cortical neurons (Zhang et al., 1998).
Although compelling evidence links PS mutations to abnormal amyloid precursor processing and β-amyloid deposition (Scheuner et al., 1996), little is known about how PS mutations result in the hyperphosphorylation of microtuble-associated protein tau, a pathological hallmark of AD. Interestingly, GSK-3β phosphorylates tau as well as β-catenin (Lovestone et al., 1994). Therefore, the increased activity of GSK-3β that we found after mutant PS1 expression may lead to tau hyperphosphorylation. Our observations are consistent with reports that AD brain tissues have increased levels of GSK-3β and that inhibition of GSK-3β reduces tau phosphorylation in neurons (Hong et al., 1997; Pei et al., 1997). Therefore, the decrease in Akt/PKB-mediated inactivation of GSK-3β associated with mutant PS1 expression may play a central and critical role in AD pathogenesis by increasing the hyperphosphorylation of tau, increasing neuronal apoptosis, and altering amyloid precursor protein processing.
Footnotes
C.C.W. was supported by US Public Health Service Grant F30MH11697. We thank Drs. S. Sisodia and G. Thinakaran for reagents and advice in the preparation of this manuscript, Dr. J. Schaak for AdLacZ, and C. P. Mauer for culturing hippocampal neurons. Special thanks to Dr. V. P. Bindokas for help with the cover illustration.
Correspondence should be addressed to Dr. Raymond P. Roos, University of Chicago Medical Center, Department of Neurology MC2030, 5841 S. Ellis Avenue, Chicago, Illinois 60637.
REFERENCES
- 1.Alessi DR, Cohen P. Mechanism of activation and function of protein kinase B. Curr Opin Genet Dev. 1998;8:55–62. doi: 10.1016/s0959-437x(98)80062-2. [DOI] [PubMed] [Google Scholar]
- 2.Alessi DR, Kozlowski MT, Weng QP, Morrice N, Avruch J. 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr Biol. 1998;8:69–81. doi: 10.1016/s0960-9822(98)70037-5. [DOI] [PubMed] [Google Scholar]
- 3.Berra E, Diaz-Meco MT, Moscat J. The activation of p38 and apoptosis by the inhibition of Erk is antagonized by the phosphoinositide 3-kinase/Akt pathway. J Biol Chem. 1998;273:10792–10797. doi: 10.1074/jbc.273.17.10792. [DOI] [PubMed] [Google Scholar]
- 4.Bursztajn S, DeSouza R, McPhie DL, Berman SA, Shioi J, Robakis NK, Neve RL. Overexpression in neurons of human presenilin-1 or a presenilin-1 familial alzheimer disease mutant does not enhance apoptosis. J Neurosci. 1998;18:9790–9799. doi: 10.1523/JNEUROSCI.18-23-09790.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11:3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- 6.Coffer PJ, Jin J, Woodgett JR. Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem J. 1998;335:1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cook DG, Sung JC, Golde TE, Felsenstein KM, Wojczyk BS, Tanzi RE, Trojanowski JQ, Lee VM, Doms RW. Expression and analysis of presenilin 1 in a human neuronal system: localization in cell bodies and dendrites. Proc Natl Acad Sci USA. 1996;93:9223–9228. doi: 10.1073/pnas.93.17.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 9.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 10.Ghadge GD, Lee JP, Bindokas VP, Jordan J, Ma L, Miller RJ, Roos RP. Mutant superoxide dismutase-1-linked familial amyotrophic lateral sclerosis: molecular mechanisms of neuronal death and protection. J Neurosci. 1997;17:8756–8766. doi: 10.1523/JNEUROSCI.17-22-08756.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo Q, Furukawa K, Sopher BL, Pham DG, Xie J, Robinson N, Martin GM, Mattson MP. Alzheimer’s PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid beta-peptide. NeuroReport. 1996;8:379–383. doi: 10.1097/00001756-199612200-00074. [DOI] [PubMed] [Google Scholar]
- 12.Hong M, Chen DC, Klein PS, Lee VM. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem. 1997;272:25326–25332. doi: 10.1074/jbc.272.40.25326. [DOI] [PubMed] [Google Scholar]
- 13.Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- 14.Lah JJ, Heilman CJ, Nash NR, Rees HD, Yi H, Counts SE, Levey AI. Light and electron microscopic localization of presenilin-1 in primate brain. J Neurosci. 1997;17:1971–1980. doi: 10.1523/JNEUROSCI.17-06-01971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levitan D, Doyle TG, Brousseau D, Lee MK, Thinakaran G, Slunt HH, Sisodia SS, Greenwald I. Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc Natl Acad Sci USA. 1996;93:14940–14944. doi: 10.1073/pnas.93.25.14940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loetscher H, Deuschle U, Brockhaus M, Reinhardt D, Nelboeck P, Mous J, Grunberg J, Haass C, Jacobsen H. Presenilins are processed by caspase-type proteases. J Biol Chem. 1997;272:20655–20659. doi: 10.1074/jbc.272.33.20655. [DOI] [PubMed] [Google Scholar]
- 17.Lovestone S, Reynolds CH, Latimer D, Davis DR, Anderton BH, Gallo JM, Hanger D, Mulot S, Marquardt B, Stabel S, Woodget JR, Miller CCJ. Alzheimer’s disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr Biol. 1994;4:1077–1086. doi: 10.1016/s0960-9822(00)00246-3. [DOI] [PubMed] [Google Scholar]
- 18.Meier R, Thelen M, Hemmings BA. Inactivation and dephosphorylation of protein kinase B alpha (PKBalpha) promoted by hyperosmotic stress. EMBO J. 1998;17:7294–7303. doi: 10.1093/emboj/17.24.7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murayama M, Tanaka S, Palacino J, Murayama O, Honda T, Sun X, Yasutake K, Nihonmatsu N, Wolozin B, Takashima A. Direct association of presenilin-1 with beta-catenin. FEBS Lett. 1998;433:73–77. doi: 10.1016/s0014-5793(98)00886-2. [DOI] [PubMed] [Google Scholar]
- 20.Nishimura M, Yu G, Levesque G, Zhang DM, Ruel L, Chen F, Milman P, Holmes E, Liang Y, Kawarai T, Jo E, Supala A, Rogaeva E, Xu DM, Janus C, Levesque L, Bi Q, Duthie M, Rozmahel R, Mattila K, Lannfelt L, Westaway D, Mount HT, Woodgett J, Fraser PE, St George-Hyslop P. Presenilin mutations associated with Alzheimer disease cause defective intracellular trafficking of beta-catenin, a component of the presenilin protein complex. Nat Med. 1999;5:164–169. doi: 10.1038/5526. [DOI] [PubMed] [Google Scholar]
- 21.Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- 22.Pei JJ, Tanaka T, Tung YC, Braak E, Iqbal K, Grundke-Iqbal I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J Neuropathol Exp Neurol. 1997;56:70–78. doi: 10.1097/00005072-199701000-00007. [DOI] [PubMed] [Google Scholar]
- 23.Plyte SE, Hughes K, Nikolakaki E, Pulverer BJ, Woodgett JR. Glycogen synthase kinase-3: functions in oncogenesis and development. Biochim Biophys Acta. 1992;1114:147–162. doi: 10.1016/0304-419x(92)90012-n. [DOI] [PubMed] [Google Scholar]
- 24.Price DL, Sisodia SS. Mutant genes in familial Alzheimer’s disease and transgenic models. Annu Rev Neurosci. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- 25.Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, Thomas G. Phosphorylation and activation of p70s6k by PDK1. Science. 1998;279:707–710. doi: 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
- 26.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 27.See RH, Shi Y. Adenovirus E1B 19,000-molecular-weight protein activates c-Jun N- terminal kinase and c-Jun-mediated transcription. Mol Cell Biol. 1998;18:4012–4022. doi: 10.1128/mcb.18.7.4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takashima A, Murayama M, Murayama O, Kohno T, Honda T, Yasutake K, Nihonmatsu N, Mercken M, Yamaguchi H, Sugihara S, Wolozin B. Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc Natl Acad Sci USA. 1998;95:9637–9641. doi: 10.1073/pnas.95.16.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 30.van Weeren PC, de Bruyn KM, de Vries-Smits AM, van Lint J, Burgering BM. Essential role for protein kinase B (PKB) in insulin-induced glycogen synthase kinase 3 inactivation. Characterization of dominant-negative mutant of PKB. J Biol Chem. 1998;273:13150–13156. doi: 10.1074/jbc.273.21.13150. [DOI] [PubMed] [Google Scholar]
- 31.Vito P, Wolozin B, Ganjei JK, Iwasaki K, Lacana E, D’Adamio L. Requirement of the familial Alzheimer’s disease gene PS2 for apoptosis. Opposing effect of ALG-3. J Biol Chem. 1996;271:31025–31028. doi: 10.1074/jbc.271.49.31025. [DOI] [PubMed] [Google Scholar]
- 32.Weihl CC, Ghadge GD, Miller RJ, Roos RP. Processing of wild-type and mutant familial Alzheimer’s disease-associated presenilin-1 in cultured neurons. J Neurochem. 1999;73:31–40. doi: 10.1046/j.1471-4159.1999.0730031.x. [DOI] [PubMed] [Google Scholar]
- 33.Wolozin B, Iwasaki K, Vito P, Ganjei JK, Lacana E, Sunderland T, Zhao B, Kusiak JW, Wasco W, D’Adamio L. Participation of presenilin 2 in apoptosis: enhanced basal activity conferred by an Alzheimer mutation. Science. 1996;274:1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- 34.Wolozin B, Alexander P, Palacino J. Regulation of apoptosis by presenilin 1. Neurobiol Aging. 1998;19:S23–S27. doi: 10.1016/s0197-4580(98)00041-4. [DOI] [PubMed] [Google Scholar]
- 35.Yu G, Chen F, Levesque G, Nishimura M, Zhang DM, Levesque L, Rogaeva E, Xu D, Liang Y, Duthie M, St George-Hyslop PH, Fraser PE. The presenilin 1 protein is a component of a high molecular weight intracellular complex that contains beta-catenin. J Biol Chem. 1998;273:16470–16475. doi: 10.1074/jbc.273.26.16470. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Sommer B, van de Wetering M, Clevers H, Saftig P, De Strooper B, He X, Yankner BA. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998;395:698–702. doi: 10.1038/27208. [DOI] [PubMed] [Google Scholar]
- 37.Zhou H, Summers SA, Birnbaum MJ, Pittman RN. Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis. J Biol Chem. 1998;273:16568–16575. doi: 10.1074/jbc.273.26.16568. [DOI] [PubMed] [Google Scholar]
- 38.Zhou J, Liyanage U, Medina M, Ho C, Simmons AD, Lovett M, Kosik KS. Presenilin 1 interaction in the brain with a novel member of the Armadillo family. NeuroReport. 1997;8:2085–2090. doi: 10.1097/00001756-199705260-00054. [DOI] [PubMed] [Google Scholar]
- 39.Zundel W, Giaccia A. Inhibition of the anti-apoptotic PI(3)K/Akt/Bad pathway by stress. Genes Dev. 1998;12:1941–1946. doi: 10.1101/gad.12.13.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]