

Abstract
Previous studies indicate that a variety of pharmacological agents interfere with the prepulse inhibition of the acoustic startle (PPI) response including phencyclidine (PCP), 8-hydroxy-2-(di-n-propylamino)tetralin (8-OH-DPAT), amphetamine, and apomorphine. Strain differences have been observed in the ability of apomorphine to disrupt PPI, although the degree to which these strain differences occur after administration of nondopaminergic drugs or the degree to which differences can be observed in other models of dopamine (DA) receptor activation has not been elucidated. The present study tested the effects of apomorphine, amphetamine, 8-OH-DPAT, and PCP on PPI in the Sprague Dawley and Wistar rat strains. Because apomorphine disrupts PPI via activation of DA receptors in the nucleus accumbens, apomorphine-induced hyperlocomotion, also a behavioral model of nucleus accumbens DA receptor activation, was measured in both rat strains. Administration of PCP or 8-OH-DPAT attenuated PPI in both strains, whereas apomorphine and amphetamine only attenuated PPI in Wistar rats. The ability of apomorphine to increase motor activity in the absence of a startle-eliciting stimulus was similar in the two strains, as was apomorphine-induced hyperlocomotion. A time course analysis of the effects of apomorphine on startle response in Sprague Dawley rats found that changes in the magnitude of PPI followed changes in basic startle amplitude. Similarly, no apomorphine-induced attenuation of PPI was observed in Sprague Dawley rats after 6-OHDA–induced DA receptor supersensitivity in the nucleus accumbens. These data suggest a dissociation between the effects of DA receptor agonists in PPI and other behavioral models of DA receptor activation.
Keywords: amphetamine, apomorphine, phencyclidine, 8-OH-DPAT, prepulse inhibition, dopamine, motor behavior, schizophrenia, rats, startle
The acoustic startle response is an easily quantified, reflexive movement after a loud acoustic stimulus. Prepulse inhibition (PPI) occurs when the startle response is reduced because of the previous presentation of a less intense sensory stimulus (Hoffman and Searle, 1965). PPI can be attenuated by administration of dopamine (DA) agonists such as apomorphine (APO) and amphetamine (AMPH), and this effect is reversed by dopamine receptor antagonists such as haloperidol and clozapine (Mansbach et al., 1988; Swerdlow et al., 1991, 1998). As such, attenuation of PPI by DA receptor agonists has been proposed as an animal model of the deficits in sensory-gating processes observed in schizophrenia (Braff and Geyer, 1990).
Evidence has accumulated suggesting that different strains of rats can respond diversely in the PPI paradigm (Rigdon, 1990; Varty and Higgins, 1994; Swerdlow et al., 1998). For example, several reports have documented strain differences in APO-induced attenuation of PPI. Using identical testing conditions for both strains of rat, Rigdon (1990)reported that systemic injections of APO in Wistar but not CD rats attenuated acoustic PPI. More recently, Swerdlow et al. (1998)demonstrated that Sprague Dawley rats are more sensitive to disruption of PPI by APO than are Wistar rats and that clozapine can reverse this APO-induced attenuation at lower concentrations in Sprague Dawley rats than in Wistar rats.
Because studies that have found strain-related differences in the ability of compounds to attenuate PPI have been restricted to APO-induced PPI attenuation (Rigdon, 1990; Swerdlow et al., 1998) and not that of other compounds [e.g., phencyclidine (Varty and Higgins, 1994)], the question of whether strain differences were restricted to compounds acting primarily via dopaminergic mechanisms remains. The present study, which followed from our observation that PPI was not robustly attenuated by APO in Sprague Dawley rats in contrast to previously published literature, bears directly on this issue. Specifically, the present study examined the effect of APO, the DA-releasing agent amphetamine, the use-dependent NMDA antagonist phencyclidine (PCP), and the 5-HT1A receptor agonist 8-hydroxy-2-(di-n-propylamino)tetralin (8-OH-DPAT) on two rat strains (Sprague Dawley and Wistar) in the PPI paradigm. These compounds have been reported previously to attenuate PPI (Mansbach and Geyer, 1989; Rigdon and Weatherspoon, 1992).
It is thought that the effects of APO on PPI act via DA receptors in the nucleus accumbens. This idea is supported by the fact that intra-accumbens injection of dopamine results in an attenuation of PPI similar to that observed with systemic APO or AMPH administration (Swerdlow et al., 1990b, 1992) and that lesion of nucleus accumbens DA innervation potentiates the effects of systemic APO administration but abolishes the effects of systemic AMPH administration (Swerdlow et al., 1986, 1990a). Because similar experiments suggest that APO-induced hyperlocomotion may also be mediated at the level of the nucleus accumbens (Kelly et al., 1975; for review, see Koob and Swerdlow, 1988), the hyperlocomotor response to APO in the Wistar and Sprague Dawley strains was also examined in the present study. This latter test was performed, in part, to determine whether strain differences in the response to APO were restricted to the ability of this compound to attenuate PPI or were generalized to other behavioral models of DA receptor activation.
MATERIALS AND METHODS
Subjects. Male Sprague Dawley and Wistar rats (Bantin-Kingman, Hull, UK), weighing between 250 and 400 gm, were used for these experiments. The animals were housed in groups of three or six, according to weight, in a separate colony room on a 12:12 hr light/dark cycle (lights on at 8:00 A.M.). The animals were allowed access to food and water ad libitum before testing.
Acoustic startle studies. Six SR-LAB (San Diego Instruments, San Diego, CA) stabilimeter chambers were contained in a testing room separate from the colony room. The animal-testing chamber consisted of a Plexiglas cylinder 8.8 cm in diameter attached to a 12.5 cm × 20.5 cm Plexiglas base. This was contained in a ventilated, sound-attenuating chamber. Any movement of a test animal resulted in the displacement of a piezoelectric cartridge secured to the bottom of the Plexiglas base. The cartridge generated voltage that was proportional to the amount of displacement. Startle amplitude, measured as mean displacement during a 100 msec period beginning at the onset of the startle-eliciting stimulus, was digitized and recorded using a commercially available program (SR-LABS). In addition, the SR-LABS program, via a microcomputer and interface, controlled the delivery of all acoustic stimuli to a speaker located 24 cm above the animal.
Before the first session in each experiment, all testing chambers were calibrated both for movement, using equipment provided by SR-LABS, and for sound levels, using a Realistic brand Tandy sound level meter (catalog #33-2050).
In each session, batches of six animals were randomly assigned to a drug condition. Ten minutes after subjects received their appropriate injections, they were placed in the testing apparatus and given a 5 min acclimation period during which the background noise (i.e., 65 dB white noise) was present. Treatment conditions were always counterbalanced between the six startle boxes. At the conclusion of the acclimation period, a series of 10 40 msec 118 dB tones was presented at a 30 sec interstimulus interval to habituate the animals partially to the startle-eliciting stimulus (see Davis, 1988). After these 10 tones, the test session, which consisted of 15 repetitions of trials, began.
Because the ability of drugs to disrupt PPI is influenced by the salience of the prepulse stimuli (Davis et al., 1990; Keith et al., 1991), conditions in Sprague Dawley and Wistar rats were matched to produce similar degrees of PPI in the two strains. Preliminary experiments confirmed previous reports that the salience of prepulse stimuli (and the magnitude of prepulse inhibition) could be altered by changing either the intensity or duration of the prepulse. In these experiments, a 70 dB, 10 msec duration prepulse stimulus reproducibly reduced startle amplitude by ∼50% in Sprague Dawley rats. These parameters are similar to those used in previous studies (Davis et al., 1990; Mansbach, 1991; Swerdlow et al., 1992), which typically use a prepulse stimulus 10–15 dB above background and produce a 50% reduction in startle amplitude. When these same parameters were used in Wistar rats, a 77% reduction in startle amplitude was observed (data not shown); thus prepulse duration was reduced to 5 msec for experiments in the Wistar strain.
Four different trial types were presented during the startle session: (1) a 10 or 5 msec 70 dB prepulse stimulus alone (prepulse-alone), (2) the same prepulse stimulus followed by a 40 msec 118 dB burst of white noise (prepulse-pulse) that began 100 msec after the beginning of the prepulse stimulus, (3) the 118 dB stimulus alone (pulse-alone), and (4) a period in which no stimulus was presented (no-stimulus). The stimuli were presented in random order with interstimulus intervals averaging 15 sec. At the conclusion of each session, the animals in that session were removed, the testing chambers were cleaned, and six new animals were brought in for the next session.
Locomotor activity studies. Rats were placed in individual activity cages (300 × 200 × 300 mm) containing five infrared beams. Two beams at the base of the cage recorded cage crossings as consecutive beam breaks. Animals were habituated to the boxes for 2 hr before injection with APO. Cage crossings were recorded in 10 or 20 min time blocks for 120 min after injection with APO.
6-OHDA lesions of nucleus accumbens. Thirty-six rats were randomly assigned to two groups to receive either 6-OHDA lesions or sham procedures. Rats were anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and placed into a stereotaxic frame with the tooth bar placed 5.0 mm above the interaural line (Pellegrino et al., 1981). 6-OHDA HBr (Sigma, St. Louis, MO) was freshly prepared at a concentration of 4 μg/μl (expressed as base) in isotonic saline containing 0.1% ascorbic acid. Bilateral infusions of either 6-OHDA or vehicle were then made into the nucleus accumbens (anteroposterior, +3.2; lateral, ±1.7; and dorsoventral, −7.8). An injection volume of 2 μl was administered via an infusion pump at a constant rate over an 8 min period. The cannula was left in place for 2 min after infusion of 6-OHDA or vehicle to allow time for drug diffusion. At the completion of surgery, the burr hole in the skull was sealed, and the wound was closed with wound clips. Animals were monitored closely until they recovered from anesthesia and were kept in a recovery room with soft bedding overnight. The following day they were returned to the colony room and housed in groups of three.
One week after surgery, one-half of the animals in each group (lesioned or sham) were randomly assigned to receive an injection of APO (0.6 mg/kg, s.c.) or vehicle (0.1% ascorbic acid). Ten minutes after the injection, the animals were placed in the acoustic startle–testing apparatus and tested as described above. The following week, treatment conditions were reversed, and the procedure was repeated. Five days after the second startle-testing session, the locomotor response to APO was assessed in these animals. Rats were randomly assigned to receive either 0.3 or 1.0 mg/kg APO, placed in individual activity cages, and tested as described above. The doses used in this study were chosen to “bracket” the dose of APO used in the startle study because APO induces stereotyped behavior at higher doses, which could interfere with the expression of locomotor activity. Because the primary purpose of this experiment was to compare the effect of APO in sham and lesioned animals, no saline controls were used.
One day after the locomotor testing, rats were killed by decapitation, and the nucleus accumbens and caudate nucleus were dissected on ice. Samples were frozen immediately on dry ice and stored at −70°C. Samples were homogenized for analysis in 10 vol of homogenizing medium (0.4 m perchloric acid containing 0.1% sodium metabisulphite, 0.01% EDTA, and 0.1% cysteine) and centrifuged at 3000 × g for 10 min. Aliquots of the supernatant were analyzed for dopamine content by HPLC as described previously by Hutson et al. (1991).
Drugs. All drugs were prepared freshly before the beginning of the first session in each experiment. Apomorphine HCl, 8-OH-DPAT hydrobromide (Research Biochemicals, Natick, MA), and phencyclidine HCl (Ultrafine Chemicals) were dissolved into an isotonic saline solution, which contained 0.1% ascorbic acid in the case of apomorphine. d-Amphetamine sulfate (Research Biochemicals) was dissolved into double distilled water. All injections were given subcutaneously, at the region near the nape of the neck. Doses of APO, PCP, and 8-OH-DPAT are expressed as the weight of the salt, whereas doses of AMPH are expressed in terms of the free base. The animals were put in the startle boxes 10 min after injection.
Statistical analyses. Mean startle amplitude for each type of stimulus (i.e., no-stimulus, prepulse-alone, prepulse-pulse, and pulse-alone) was calculated for each subject. These data sets were analyzed using repeated measures ANOVA [BMDP, programs 4V and 7D (Dixon et al., 1981)]. In the first analysis, a block ANOVA (BMDP 7D) was performed on startle amplitude to each type of stimulus, with Tukey’s post hoc t test if significant effects were found. In addition, a repeated measures ANOVA (BMDP 4V) was performed on pulse-alone and prepulse-pulse startle amplitudes, using drug as a between-subjects variable and stimulus as a within-subjects variable. A significant drug × stimulus interaction was interpreted as a significant alteration in prepulse inhibition. Significant effects in this analysis were interpreted using a simple effects ANOVA forpost hoc analysis (BMDP 4V). Finally, levels of prepulse inhibition in each rat were determined by expressing the prepulse-pulse startle amplitude as a percentage decrease from pulse-alone startle amplitude [i.e., 100 × (pulse-alone − prepulse-pulse)/pulse-alone]. This transformation was performed because it reduces statistical variability attributable to differences between animals and is a direct measure of the level of prepulse inhibition. Significant effects in this analysis were examined using the Tukey post hoc t test. This transformation was only performed in cases in which no significant effect of drug was observed on pulse-alone startle amplitude. Locomotor scores were analyzed as the number of cage crosses in each 10 or 20 min period using repeated measures ANOVA (BMDP 2V). In the case of the 6-OHDA lesion study, drug effects on locomotion followed a quadratic curve. Thus, in this instance, ANOVA results are reported from the second orthogonal comparison.
RESULTS
Prepulse inhibition: apomorphine and amphetamine
Sprague Dawley: apomorphine
As can be seen in Figure 1,top, APO administration produced a significant increase in the startle response to the pulse-alone stimulus [F(3,44) = 5.36; p< 0.005]. Specifically, the pulse-alone startle amplitude after a 1.0 mg/kg dose was significantly increased from that of both the vehicle control and the 0.1 mg/kg doses (p < 0.05). Thus, the data were not transformed to a percentage of pulse-alone trials. Analysis of prepulse-pulse and pulse-alone data failed to reveal a significant drug × stimulus interaction [F(3,44) = 2.16; p= 0.11], indicating that APO did not influence prepulse-pulse trials independently of pulse-alone trials. Correspondingly, analyses of simple main effects indicated robust levels of PPI at all doses (p < 0.001 in all cases).
Fig. 1.
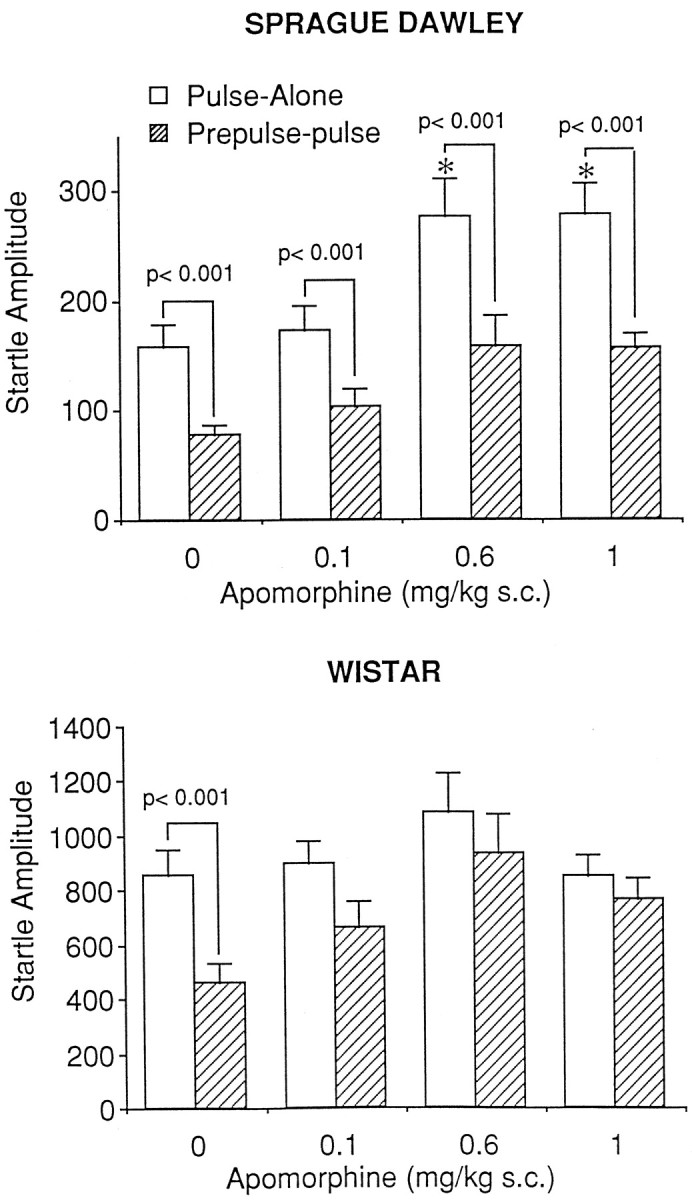
The effects of apomorphine administration on prepulse inhibition in the Sprague Dawley (top) and Wistar (bottom) strains of rat. Administration of apomorphine had no effect on prepulse inhibition in Sprague Dawley rats (n = 12/group) but attenuated prepulse inhibition in Wistar rats (n = 10–11/group). Anasterisk indicates significantly different from pulse-alone response in vehicle-treated animals (p < 0.05).
Wistar: apomorphine
The effect of APO on PPI for the Wistar strain of rat is shown in Figure 1, bottom. Administration of APO significantly influenced PPI as revealed by a significant drug × stimulus interaction [F(3,38) = 3.65;p < 0.05]. Further analyses of simple main effects demonstrated significant levels of PPI after vehicle administration only (p < 0.001). Because there was no significant effect of APO on pulse-alone startle levels [F(3,38) = 2.65; p= 0.063], prepulse-pulse startle amplitude was expressed as a percentage decrease from that of pulse-alone. Analysis of the transformed data indicated a significant effect of APO [F(3,38) = 5.37; p< 0.005]. Tukey pairwise comparisons indicated significant attenuation of PPI at the 0.6 and 1.0 mg/kg doses (p < 0.05).
Sprague Dawley: amphetamine
Figure 2, top, represents the effect of amphetamine on PPI in Sprague Dawley rats. Amphetamine did not influence PPI as revealed by the lack of a drug × stimulus interaction [F(3,32) = 0.12; p = 0.95]. Correspondingly, analyses of simple main effects demonstrated a significant difference between pulse-alone startle amplitude and prepulse-pulse startle amplitude (significant levels of PPI) at each dose of amphetamine (p < 0.001 in each case). Because amphetamine did not influence pulse-alone startle amplitudes [F(3,32) = 0.51; p= 0.679], the data were analyzed as a percentage decrease from that of pulse-alone. When the transformed data were analyzed, a nonsignificant effect of dose [F(3,32) = 1.91;p = 0.148] confirmed that amphetamine had no significant influence on PPI.
Fig. 2.
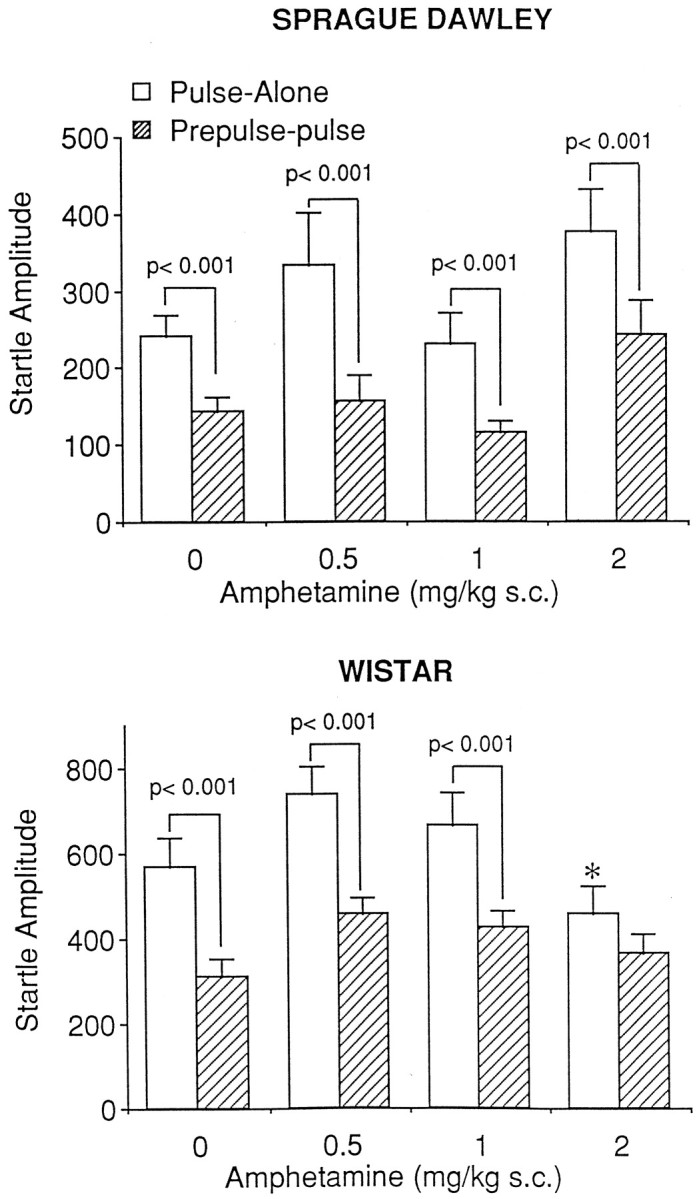
The effects of amphetamine administration on prepulse inhibition in the Sprague Dawley (top) and Wistar (bottom) strains of rat. Administration of amphetamine had no effect on prepulse inhibition in the Sprague Dawley strain (n = 9/group) but attenuated prepulse inhibition in the Wistar strain of rats (n = 10/group). An asterisk indicates significantly different from pulse-alone response in vehicle-treated animals (p < 0.05).
Wistar: amphetamine
The effects of amphetamine on PPI in Wistar rats are illustrated in Figure 2, bottom. Amphetamine significantly attenuated PPI as demonstrated by a significant drug × stimulus interaction [F(3,36) = 2.90; p< 0.05]. Analyses of simple main effects showed a significant difference between pulse-alone and prepulse-pulse startle levels in animals receiving vehicle and 0.5 and 1.0 mg/kg amphetamine (p < 0.001 in each case), whereas there was no significant difference between pulse-alone and prepulse-pulse startle amplitudes after administration of amphetamine at 2.0 mg/kg. Because amphetamine administration had a significant effect on the response to the pulse-alone stimulus [F(3,36) = 3.26; p < 0.05], the data were not expressed as a percentage decrease from that of pulse-alone. Startle response to the pulse-alone stimulus after administration of 2 mg/kg amphetamine was significantly different from that of vehicle (p< 0.05).
Prepulse inhibition: phencyclidine and 8-OH-DPAT
Sprague Dawley: PCP
Figure 3, top, demonstrates the effect of PCP on PPI in Sprague Dawley rats. PCP significantly influenced PPI as revealed by a significant drug × stimulus interaction [F(3,32) = 5.24; p < 0.005]. Further analyses of simple main effects demonstrated a significant difference between pulse-alone and prepulse-pulse startle amplitudes at the saline (p < 0.001) and 0.1 mg/kg (p < 0.005) but not the 3.0 or 5.0 mg/kg doses. Administration of PCP had no significant effect on the pulse-alone stimulus [F(3,32) = 0.36;p = 0.78], allowing data to be analyzed as a percentage decrease from that of pulse-alone. Analysis of the transformed data indicated a significant effect of PCP [F(3,32) = 12.63; p< 0.001]. Tukey pairwise comparisons indicated that there was significant attenuation of PPI at all doses of PCP tested (p < 0.01).
Fig. 3.
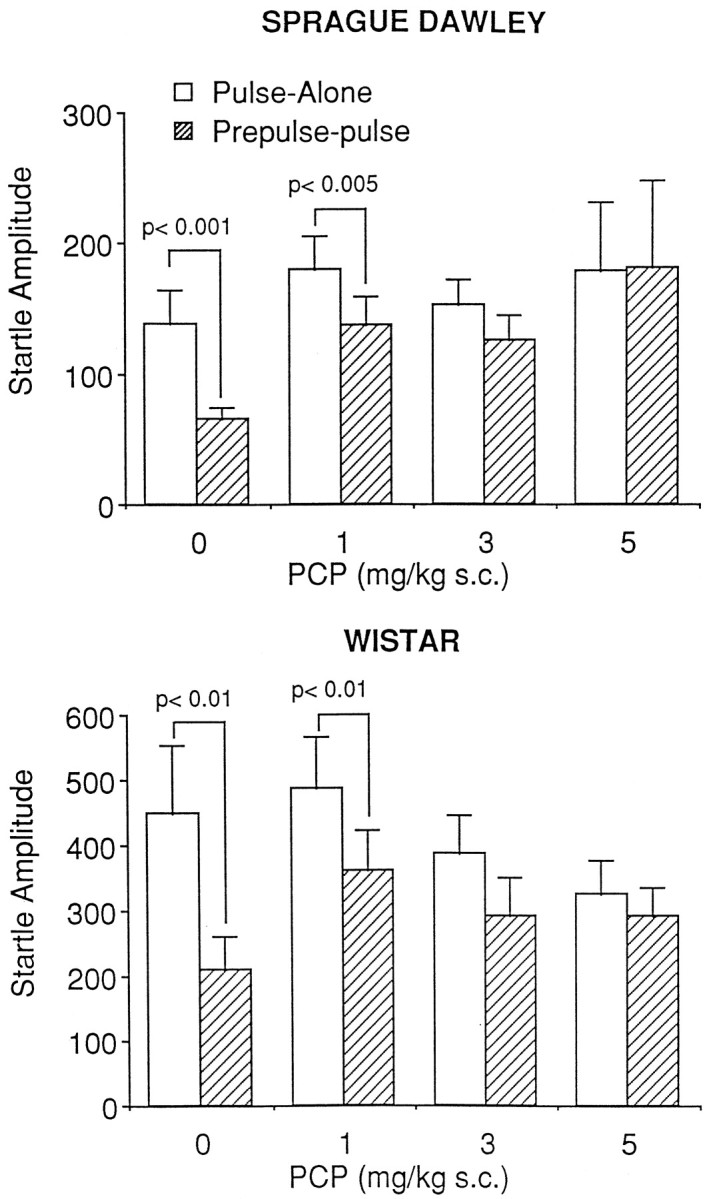
The effects of PCP administration on prepulse inhibition in the Sprague Dawley (top) and Wistar (bottom) strains of rat. Administration of PCP significantly attenuated prepulse inhibition in both rodent strains (Sprague Dawley, n = 9/group; Wistar,n = 10/group).
Wistar: PCP
Figure 3, bottom, represents the effects of PCP on PPI in Wistar rats. PCP had a significant influence on PPI as revealed by a significant drug × stimulus interaction [F(3,36) = 3.37; p< 0.05]. Further analysis of simple main effects demonstrated a significant difference between pulse-alone and prepulse-pulse startle amplitudes in the saline- and 1 mg/kg– treated groups (p < 0.01 in each case) but not at the 3.0 or 5.0 mg/kg doses. Administration of PCP had no effect on pulse-alone trials [F(3,36) = 0.96;p = 0.42]; accordingly data could be analyzed as a percentage decrease from that of pulse-alone. Analysis of the transformed data indicated a significant influence of PCP on PPI [F(3,36) = 8.0; p< 0.001], with PPI after 1, 3, and 5 mg/kg PCP significantly decreased from control (p < 0.05).
Sprague Dawley: 8-OH-DPAT
Figure 4, top, represents the effects of 8-OH-DPAT on mean startle amplitude in the Sprague Dawley rat. 8-OH-DPAT administration produced a significant increase in baseline startle levels [F(3,32) = 6.01; p < 0.005]. Specifically, administration of 1.0 mg/kg 8-OH-DPAT produced a significant increase in pulse-alone startle amplitude compared with that in saline (p < 0.05). A significant drug × stimulus interaction [F(3,32) = 4.63; p= 0.008] indicated attenuation of PPI by 8-OH-DPAT. Further analyses of simple main effects suggested significant levels of PPI in vehicle-treated rats (p = 0.005) and the rats given 0.1 mg/kg (p < 0.001) and no significant difference between pulse-alone and prepulse-pulse startle amplitudes after administration of 0.3 and 1.0 mg/kg 8-OH-DPAT.
Fig. 4.
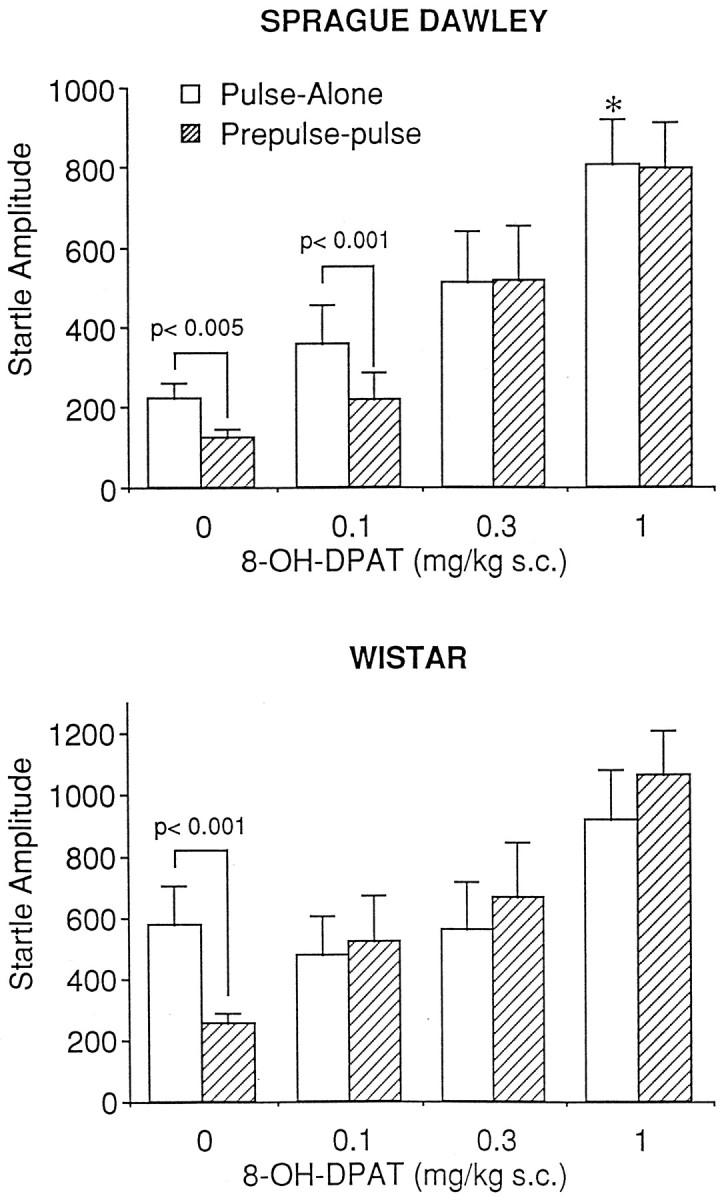
Effects of 8-OH-DPAT administration on prepulse inhibition in the Sprague Dawley (top) and Wistar (bottom) strains of rat. Administration of 8-OH-DPAT significantly attenuated prepulse inhibition in both strains of rodent (Sprague Dawley, n = 9/group; Wistar,n = 10/group). An asterisk indicates significantly different from pulse-alone response in vehicle-treated rats (p < 0.05).
Wistar: 8-OH-DPAT
Figure 4, bottom, represents the effect of 8-OH-DPAT on startle amplitude in Wistar rats. Administration of 8-OH-DPAT had a significant influence on PPI as revealed by a significant drug × stimulus interaction [F(3,36) = 5.33; p < 0.05]. Analyses of simple main effects confirmed this, in that there was a significant difference between pulse-alone and prepulse-pulse startle amplitude in vehicle-treated animals only (p < 0.001), indicating attenuation of PPI at all doses of 8-OH-DPAT tested. Administration of 8-OH-DPAT had no significant effect on pulse-alone startle amplitude [F(3,36) = 1.83; p= 0.15], which allowed the data to be calculated as a percentage decrease from that of pulse-alone. Analysis of the transformed data revealed a significant effect of drug [F(3,36) = 15.12; p< 0.0001]. Tukey post hoc comparisons indicated that PPI after administration of 8-OH-DPAT at the 0.1, 0.3, and 1.0 mg/kg doses all differed significantly from control (p < 0.01 in all cases).
Motor activity in the absence of a startle-eliciting stimulus
In the preceding experiments, one-fourth of the trials presented to the animals consisted of a no-stimulus presentation trial. In this trial, the mean cage displacement was recorded during a 100 msec period during which no startle-eliciting acoustic stimulus was presented. Drug effects on this measure have been reported previously (Svensson and Ahlenius, 1983; Mansbach et al., 1988; Rigdon, 1990) and reflect an increase in the level of motor activity of the animals within the startle boxes.
APO administration produced a significant increase in stabilimeter output in the no-stimulus trial in both the Sprague Dawley and Wistar strains [F(3,82) = 23.44;p < 0.001]. Specifically, stabilimeter output was increased after administration of APO at 0.6 or 1 mg/kg (p < 0.05) in both strains (Fig.5). There was no difference between the Sprague Dawley and Wistar strains on this measure [F(1,82) = 0.12; p= 0.71].
Fig. 5.

The effects of apomorphine administration on cage displacement in the startle boxes in the absence of a startle-eliciting stimulus (no-stimulus trial) in the Sprague Dawley (filled circles; n = 12/group) and Wistar (open circles;n = 10/group) rat strains. The effects of apomorphine on this measure were identical in the two rodent strains. An asterisk indicates significantly different from vehicle (p < 0.05).
APO was the only drug tested that produced a robust and reliable alteration in stabilimeter output during the no-stimulus trial. Although significant effects on the no-stimulus trial type were observed after administration of amphetamine in Sprague Dawley [F(3,32) = 3.18; p= 0.037] and Wistar [F(3,36) = 2.17; p < 0.05] rats, no statistical differences were observed using Tukey’s post hoc test at a criterion ofp < 0.05. Similarly, there was an increase in stabilimeter output after administration of 8-OH-DPAT in Sprague Dawley [F(3,32) = 3.10; p= 0.04] and Wistar [F(3,36) = 3.1;p < 0.05] rats, although no statistical differences were observed using Tukey’s post hoc t test. Administration of PCP had no effect on stabilimeter output during the no-stimulus trial in Sprague Dawley [F(3,32) = 1.32; p= 0.28] or Wistar [F(3,36) = 1.79;p = 0.166] rats.
Effects of apomorphine on locomotor activity in Sprague Dawley and Wistar rats
Figure 6 illustrates total cage crosses produced in response to APO or vehicle administration in Sprague Dawley and Wistar rats. Analysis using repeated measures ANOVA indicated a significant effect of drug [F(2,42) = 4.40; p< 0.05] but no significant effect of strain [F(1,42) = 1.36; p= 0.24] and no drug × strain interaction [F(2,42) = 0.35; p= 0.70].
Fig. 6.

The effects of apomorphine administration (top, vehicle; middle, 0.3 mg/kg;bottom, 0.6 mg/kg) on locomotor activity in the Sprague Dawley (filled circles) and Wistar (open circles) strains of rat. The locomotor response to apomorphine administration was identical in the two rat strains. Cage crossings were defined as two consecutive beam breaks in photocell beams along the bottom of the cage floor. Error bars represent SEM (n = 8/group).
Time course of apomorphine effects on prepulse inhibition in Sprague Dawley rats
In agreement with previous reports (Davis and Aghajanian, 1976), we found that APO increased the basic startle response in Sprague Dawley rats (see Fig. 1). This effect has been shown to be biphasically time dependent. Thus, APO increases startle amplitude during the first 40 min after drug injection but decreases startle amplitude from 50 to 80 min after drug injection (Davis and Aghajanian, 1976). In contrast, the effects of APO on PPI are not time dependent and can be observed up to 90 min after drug injection (Young et al., 1991). Thus, we examined the time course of the effect of APO on PPI in the Sprague Dawley rat to determine whether an effect of APO on PPI could be observed after the effect of APO on the basic startle amplitude had disappeared.
As shown in Figure 7, APO had a time-dependent influence on startle reactivity. Repeated measures ANOVA using drug as a between-subject variable and stimulus type and time as within-subject variables indicated a significant time × drug interaction [F(3,48) = 6.58;p = 0.0008]. However APO influenced startle scores to the pulse-alone and prepulse-pulse stimuli to the same degree, as revealed by the lack of a drug by stimulus interaction [F(1,16) = 1.68; p= 0.213] and no drug × time × stimulus interaction [F(3,48) = 1.32; p= 0.277]. Matched t test comparisons indicated robust levels of PPI at all time points in APO-treated and vehicle-treated animals (p values < 0.01), confirming a lack of effect of APO on PPI. Analyses of simple effects were performed to examine the effects of APO on startle. These analyses indicated that APO had a significant influence on basic startle scores during the first [20–40 min; F(1,16) = 4.55;p = 0.049] and last [80–100 min;F(1,16) = 5.93; p = 0.027] time points compared with that in saline controls. In addition, analyses of simple main effects using all four stimuli types indicated that there was no effect of time in saline-treated animals [F(3,48) = 1.35; p= 0.27] but there was a significant effect of time in APO-treated animals [F(3,48) = 17.93;p < 0.0001]. The time effect in APO-treated animals likely reflects the overall difference between the first two (20–40 and 40–60 min) and the last two (60–80 and 80–100 min) blocks of time in APO-treated rats. Specifically, the pulse-alone startle amplitude at the third (60–80 min) and fourth (80–100 min) time blocks in APO-treated rats was significantly different from that at the first time block (20–40 min; p < 0.05, Tukey’s post hoc t test), and the responses recorded during the prepulse-pulse, prepulse-alone, and no-stimulus trial types during the third and fourth time blocks were each significantly different from the response to each of these stimuli during the first and second time blocks (p values < 0.05, Tukey’s post hoc t test).
Fig. 7.

Time course of the effects of apomorphine on prepulse inhibition in the Sprague Dawley rat. Apomorphine administration (0.6 mg/kg, s.c.) had a biphasic influence on the startle response, increasing startle amplitudes during the first 20 min sampling period (20–40 min) and decreasing startle amplitudes during the last 20 min sampling period (80–100 min). Apomorphine administration did not influence prepulse inhibition, because it had a parallel influence on pulse-alone and prepulse-pulse startle amplitudes. An asterisk indicates significantly different from saline treatment (p < 0.05;n = 9/group).
6-OHDA nucleus accumbens lesions in Sprague Dawley rats: prepulse inhibition and locomotor activity
The effects of DA receptor agonists on PPI are thought to be mediated, in part, by activation of DA receptors in the nucleus accumbens (Swerdlow et al., 1986, 1990a,b; Wan and Swerdlow, 1996). However, DA infusions into the nucleus accumbens do not influence baseline startle amplitude (Swerdlow et al., 1990b); thus, the effect of APO on basic startle amplitude per se is unlikely to be mediated via the nucleus accumbens. One possible explanation for the difference between Sprague Dawley and Wistar rats in the PPI tests described above may be a differential sensitivity of nucleus accumbens DA receptors in the two strains, possibly attributable to differences in receptor density and/or level of receptor reserve. To test this hypothesis, an experiment was undertaken to examine whether an attenuation of PPI could be observed in Sprague Dawley rats with supersensitive nucleus accumbens DA receptors achieved by 6-OHDA lesion of this brain area. To verify the induction of DA receptor supersensitivity by 6-OHDA, the locomotor response to APO was also examined in these animals.
Neurochemical verification of lesions
The tissue concentration of DA in the nucleus accumbens and caudate nucleus in 6-OHDA– and sham-lesioned rats is presented in Table 1. Two rats in the 6-OHDA lesion group that appeared to have unsuccessful lesions (nucleus accumbens DA level decreases of 33 and 54% from control) were excluded from all further analysis. DA levels in the nucleus accumbens of 6-OHDA–lesioned rats were significantly decreased from control (p < 0.0001, unpaired t test), whereas DA levels in the caudate nucleus were not significantly different in sham and lesioned rats (p = 0.202).
Table 1.
Tissue dopamine concentrations in rats with 6-OHDA lesions of the nucleus accumbens and in sham-lesioned animals
| Nucleus accumbens | Caudate nucleus | |
|---|---|---|
| Sham | 4.99 ± 0.21 | 8.35 ± 0.87 |
| Lesion | 0.48 ± 0.11*** | 6.88 ± 0.68 |
Dopamine concentration is expressed in picograms per milligram wet weight of tissue ± SEM. Dopamine levels in lesioned rats were significantly decreased in the nucleus accumbens but not in the caudate nucleus. Asterisks indicate significantly different from the sham control (p < 0.0001, unpaired ttest).
Acoustic startle response
The effect of nucleus accumbens 6-OHDA lesions on PPI in response to APO (0.6 mg/kg) is depicted in Figure8. Because the design of this experiment involved repeated testing in two separate startle sessions, the first statistical analysis used lesion and session number as between-subject variables and drug and stimulus as within-subject variables. Only pulse-alone and prepulse-pulse scores were included in this analysis. There was no significant difference between week 1 and week 2 [F(1,30) = 1.06; p= 0.318] and no significant interaction between session number and drug or stimulus or lesion. As such, data from both weeks were combined for further analysis. Analysis of these data revealed no significant interaction between stimulus and drug [F(1,32) = 1.31; p= 0.26], indicating that APO administration did not influence PPI. Moreover, there was no three-way interaction between stimulus, lesion, and drug [F(1,32) = 1.25;p = 0.27], indicating that the lesions did not influence the effects of APO on the startle response. There was a significant effect of stimulus [F(1,32) = 145.6; p< 0.00001], and robust levels of PPI were observed in each treatment group (i.e., differences between pulse-alone and prepulse-pulse startle amplitudes were p < 0.00005 in each case). However, a significant effect of drug [F(1,31)= 25.16; p < 0.0001] indicated that APO significantly influenced startle scores. Because there was no significant effect of lesion on startle amplitude [F(1,32) = 0.6; p = 0.44], data were collapsed across lesion type to investigate further the effects of APO. Subsequent analysis again revealed a significant effect of drug [F(1,33) = 23.89;p < 0.001] but no drug × stimulus interaction [F(1,33) = 1.16; p= 0.29], indicating that APO administration produced an increase in startle amplitude in response to both pulse-alone and prepulse-pulse startle stimuli. Subsequent analyses on each of the four stimuli types using lesion and stimulus type as block variables (BMDP 7D) confirmed the finding that APO administration had a significant effect on startle scores [F(1,64) = 18.98;p < 0.0001), producing an overall increase in each of the four stimuli types (no-stimulus, prepulse-alone, prepulse-pulse, and pulse-alone; p values < 0.001, ANOVA of simple effects in each case).
Fig. 8.

Effects of apomorphine (0.6 mg/kg, s.c.;filled symbols) or vehicle (Veh; (open symbols) administration on prepulse inhibition in animals with sham (circles) or 6-OHDA (squares) lesions of the nucleus accumbens. Apomorphine administration increased startle reactivity to a similar degree in sham and lesioned animals but had no effect on prepulse inhibition, because the effects on pulse-alone and prepulse-pulse startle amplitudes were similar in all groups. See text for statistical comparisons (n = 9/group).
The effects of APO on startle amplitude in the absence of a startle-eliciting stimulus (no-stimulus trial type) were also examined in a separate ANOVA that used lesion as a between-subject variable and drug as a within-subject variable. APO increased startle amplitudes in the no-stimulus condition from 2.3 ± 1.4 to 4.7 ± 1.4 and 2.3 ± 1.3 to 6.1 ± 2.5 in sham and lesioned rats, respectively. Thus, APO significantly increased the stabilimeter output in the no-stimulus trials [F(1,32)= 45.05; p < 0.0001], but there was no significant effect of lesion [F(1,32) = 3.49;p = 0.07] and no drug × lesion interaction [F(1,32) = 2.35; p= 0.13].
Locomotor activity
The effects of nucleus accumbens 6-OHDA lesions on locomotor activity are depicted in Figure 9. Because a Box–Cox diagnostic test on the raw data scores revealed unequal variances among groups (slope of regression line = 0.69), the data were subjected to square root transformation. Analysis of the transformed data revealed a significant time × drug interaction [F(1,30) = 7.83; p= 0.0089], a significant time × lesion interaction [F(1,30) = 19.94; p= 0.0001], and a significant three-way interaction between time, drug, and lesion [F(1,30) = 5.03;p = 0.0324]. Locomotor activity in 6-OHDA–lesioned rats after administration of 0.3 mg/kg APO was significantly increased over that in sham rats treated with the same dose during the first 10 min time period and from 30 to 90 min after APO administration (p < 0.05, t test). In 6-OHDA–lesioned rats treated with 1 mg/kg APO, locomotor activity was increased from 70 to 110 min after drug administration (compared with the same dose in sham rats). Analysis of data collapsed across time confirmed that the APO-induced increases in locomotor activity were significantly greater in rats with 6-OHDA lesions of the nucleus accumbens (mean cage crosses = 11.2 ± 3.7 and 9.3 ± 2.0 in animals treated with 0.3 and 1.0 mg/kg, respectively) than in rats with sham lesions [mean cage crosses = 1.9 ± 0.5 and 2.8 ± 0.9, respectively;F(1,30) = 37.08; p< 0.0001]. A Newman–Keuls multiple range test revealed that locomotor scores in lesioned rats were significantly different from locomotor scores in sham rats at each dose of APO (p < 0.01).
Fig. 9.

Effects of apomorphine administration [0.3 mg/kg (open symbols) and 1.0 mg/kg (filled symbols), s.c.] on locomotor activity in animals with sham (circles) or 6-OHDA (squares) lesions of the nucleus accumbens. The effects of apomorphine administration were significantly enhanced in animals with 6-OHDA lesions of the nucleus accumbens. Asterisks indicate p < 0.05 compared with same-dose sham controls (matched ttest; n = 9/group).
DISCUSSION
The first series of experiments reported here examined the effects of drugs that act primarily via the DA system on PPI in the Sprague Dawley and Wistar strains of rat. Both the direct DA agonist APO and the indirect DA agonist AMPH were found to disrupt PPI in Wistar rats but did not influence PPI in Sprague Dawley rats. AMPH disrupted PPI in Wistar rats only at the highest dose tested (2 mg/kg). Thus, the possibility remains that higher doses of AMPH may have disrupted PPI in both strains of rats. At a minimum, however, the Wistar strain of rat is more sensitive to the disruptive effects of AMPH in the present conditions. It appears that the present findings reflect a difference in the ability of DA agonists to influence PPI in these two strains and are not an artifact of the experimental conditions used. Thus, the observation that both PCP and 8-OH-DPAT administration produced a marked attenuation of PPI in the Sprague Dawley rat indicates that our experimental conditions were conducive to pharmacological manipulation of PPI in the Sprague Dawley strain. Indeed, there was a marked similarity between the two strains in both the effective dose range and the magnitude of the effects of 8-OH-DPAT and PCP. With specific regard to APO, it is also unlikely that alterations in drug metabolism, distribution, or bioavailability could account for the observed strain differences. Although strain differences were observed in the ability of APO to influence PPI, no strain difference was observed in the ability of APO to increase motor activity in the absence of a startle-eliciting stimulus. Because these two measures were obtained simultaneously, this finding suggests that the differential actions of APO on PPI in Wistar and Sprague Dawley rats cannot be explained solely by pharmacokinetic differences between the two rat strains. This argument is further strengthened by the observation that hyperlocomotion induced by APO was similar in Sprague Dawley and Wistar rats. The similarity in the response of Sprague Dawley and Wistar rats in this model suggests a broad similarity in the brain penetration, distribution, and metabolism of APO in the two strains.
The present results also demonstrate a differential sensitivity to the excitatory effects of APO on baseline startle amplitudes between Sprague Dawley and Wistar rat strains. Thus, APO increased the pulse-alone scores of Sprague Dawley rats but did not produce a significant increase in this measure in the Wistar rat. This finding led to the hypothesis that the excitatory effect of APO on the basic startle response could theoretically mask an effect of the drug on PPI in Sprague Dawley rats. The latter hypothesis was tested by examining the time course of effects of APO on PPI. In agreement with a previous report (Davis and Aghajanian, 1976), we found that APO had a biphasic effect on the startle response. Thus, APO increased startle amplitudes during the first 40 min but decreased startle amplitudes from 80 to 100 min after drug administration. APO also had a time-dependent effect on motor activity during periods when no startle-eliciting stimulus was presented. However, APO did not influence PPI as reflected in the observation that the effects of APO on pulse-alone scores were mirrored by similar changes on the prepulse-pulse startle amplitude at all time points. This suggests that a failure to observe PPI in Sprague Dawley animals was not caused by any “response competition” between the effects of APO on startle and the effect of APO on PPI. In agreement with this conclusion, previous examinations of the effect of compounds in the PPI model indicate that PPI can be manipulated independently of baseline startle levels. Thus, in Sprague Dawley and Wistar rats, administration of 8-OH-DPAT produces an attenuation of PPI despite the fact that it produces significant increases in the basic startle response (Rigdon and Weatherspoon, 1992; present results). In contrast, PCP attenuates PPI in both strains with no influence on the basic startle response (Mansbach and Geyer, 1989; Keith et al., 1991; present results).
The present findings reinforce previous indications of differences in the mechanism of action of dopamine receptor agonists and PCP in the PPI model. Thus, although administration of PCP causes an increase in DA turnover in the nucleus accumbens (Deutch et al., 1987), this effect does not seem to be the mechanism for the effect of PCP on PPI, because the effect of PCP cannot be attenuated by administration of haloperidol (Keith et al., 1991). The fact that PCP attenuated PPI in both Wistar and Sprague Dawley rats, although there were strain differences in the effects of dopamine receptor agonists, is consistent with the hypothesis that PCP does not disrupt PPI via its actions on brain DA systems. The results similarly indicate differences in the mechanism of action between 8-OH-DPAT and dopamine receptor agonists. However, the effects of 8-OH-DPAT on PPI have been shown to be attenuated partially by haloperidol administration (Rigdon and Weatherspoon, 1992), suggesting that further studies are needed to address the role of the DA system in the effects of 5-HT1A receptor agonists.
Current evidence suggests that DA receptors in the nucleus accumbens are an integral component of the normal PPI response (Swerdlow et al., 1986, 1990a,b, 1992; Wan and Swerdlow, 1996). In an effort to examine nucleus accumbens DA receptor sensitivity in Sprague Dawley rats further, 6-OHDA lesions of the nucleus accumbens were used to supersensitize this receptor population. Our protocol was based on that used by Swerdlow et al. (1986), in which 6-OHDA lesions of the nucleus accumbens enhanced the effects of APO on PPI in Wistar rats. Neurochemical measurements of DA levels in the nucleus accumbens and caudate nucleus of the rats used in our studies were consistent with an effective DA depletion in the nucleus accumbens. 6-OHDA lesions reduced nucleus accumbens DA by 90.4% [compare with 93% depletion bySwerdlow et al. (1986)]. These lesions also appeared primarily to spare the caudate nucleus (17% DA depletion), whereas Swerdlow et al. (1986) reported a 57% depletion of the anterior caudate DA and a 16% depletion in the posterior caudate. In agreement with the findings ofKelly et al. (1975), the locomotor response to APO administration was significantly enhanced in Sprague Dawley rats with 6-OHDA lesions of the nucleus accumbens, providing behavioral confirmation of the ability of these lesions to induce supersensitivity in nucleus accumbens DA receptors. However, the effect of APO administration on the startle response was similar in lesioned and sham-lesioned animals. Thus, APO did not influence PPI in Sprague Dawley rats with supersensitive DA receptors in the nucleus accumbens.
These results stress the importance of using caution when selecting a strain of rat for use in studies with the PPI paradigm. Previous work using the Sprague Dawley strain has indicated that the doses of APO needed to attenuate PPI can vary between laboratories (Mansbach et al., 1988; Davis et al., 1990). Populations of rats in both Sprague Dawley and Wistar strains can be identified with differences in markers of DA turnover and sensitivity to APO (Cools et al., 1990; Pradhan et al., 1990; Hooks et al., 1992). These findings suggest that strain or “substrain” differences may, in part, explain differences between laboratories in this model. In our studies, the effects of APO in Wistar rats is very reproducible, with an ED50 of ∼0.1 mg/kg subcutaneously, similar to the effects observed when APO is tested in other laboratories using Wistar rats (Rigdon, 1990). In contrast, Swerdlow et al. (1998) reported less PPI attenuation relative to that in Sprague Dawley rats at 0.5 or 2.0 mg/kg subcutaneous APO; however the APO-induced PPI attenuation in Wistar rats was significant. Thus, existing studies collectively suggest that the Wistar strain is more consistent for disruption of PPI by APO across laboratories than is the Sprague Dawley strain.
The factors underlying the observed strain differences in this model remain to be determined. The observation of genetic differences in the D2 receptor locus of Sprague Dawley and Wistar rats (Luedtke et al., 1992) may be implicated in our observations. Alternatively, there could be anatomical or biochemical differences in the intrinsic level of dopaminergic modulation of the neuronal networks involved in startle. Because the DA receptor subtype influencing PPI in these rats has not yet been clearly identified (D2, D3, or D4), there may also be a role for a mixed population of DA receptors and/or glutamatergic systems in the effects reported in this paper. In support of this, Wan and Swerdlow (1996) have demonstrated that interactions of dopaminergic and glutamatergic systems within the nucleus accumbens likely modulate PPI. The two behavioral models used in this study (PPI and locomotor activity) have both been associated with increased DA function in the nucleus accumbens. In the present experiments, the effects of APO in these models could be dissociated; APO administration induced hyperlocomotion in the Sprague Dawley rat but did not attenuate PPI in this strain. The possibility that these two behavioral models may reflect activation of different populations of DA receptor could therefore be considered.
In summary, the present study found that administration of PCP or 8-OH-DPAT attenuated PPI in both Wistar and Sprague Dawley rats whereas APO and AMPH only attenuated PPI in Wistar rats. The ability of APO to increase motor activity in the absence of a startle-eliciting stimulus and APO-induced hyperlocomotion were similar in both strains. Furthermore, no APO-induced attenuation of PPI was observed in Sprague Dawley rats after 6-OHDA–induced DA receptor supersensitivity in the nucleus accumbens. Collectively, these data suggest a dissociation between the effects of DA receptor agonists, especially the direct DA agonist APO, in PPI and other behavioral models of DA receptor activation.
Footnotes
We thank Dr. Gerry Dawson for constructive discussions and statistical assistance.
Correspondence should be addressed to Dr. Gene G. Kinney, Bristol-Myers Squibb Pharmaceutical Research Institute, Neuroscience Drug Discovery, Department Number 404, 5 Research Parkway, Wallingford, CT 06492-7660.
Dr. Wilkinson’s present address: Pfizer Central Research, Groton, CT 06340-1596.
Dr. Saywell’s present address: Roche Products Limited, Broadwater Road, Welwyn Garden City, Hertfordshire, AL7 3AY United Kingdom.
Dr. Tricklebank’s present address: Novartis Pharma AG, Nervous System Research, S-368.7.26, CH-4002 Basel, Switzerland.
REFERENCES
- 1.Braff DL, Geyer MA. Sensorimotor gating and schizophrenia: human and animal studies. Arch Gen Psychiatry. 1990;47:181–188. doi: 10.1001/archpsyc.1990.01810140081011. [DOI] [PubMed] [Google Scholar]
- 2.Cools AR, Brachten R, Heeren D, Willemen A, Ellenbroek B. Search after neurobiological profile of individual-specific features of Wistar rats. Brain Res Bull. 1990;24:49–69. doi: 10.1016/0361-9230(90)90288-b. [DOI] [PubMed] [Google Scholar]
- 3.Davis M. Apomorphine, d-amphetamine, strychnine and yohimbine do not alter prepulse inhibition of the acoustic startle reflex. Psychopharmacology (Berl) 1988;95:151–156. doi: 10.1007/BF00174500. [DOI] [PubMed] [Google Scholar]
- 4.Davis M, Aghajanian GK. Effects of apomorphine and haloperidol on the acoustic startle response in rats. Psychopharmacology (Berl) 1976;47:217–223. doi: 10.1007/BF00427605. [DOI] [PubMed] [Google Scholar]
- 5.Davis M, Mansbach RS, Swerdlow NR, Campeau S, Braff DL, Geyer MA. Apomorphine disrupts the inhibition of acoustic startle induced by weak prepulses in rats. Psychopharmacology (Berl) 1990;102:1–4. doi: 10.1007/BF02245735. [DOI] [PubMed] [Google Scholar]
- 6.Deutch AY, Tam S-Y, Freeman AS, Bowers MB, Jr, Roth RH. Mesolimbic and mesocortical dopamine activation induced by phencyclidine: contrasting pattern to striatal response. Eur J Pharmacol. 1987;134:257–264. doi: 10.1016/0014-2999(87)90356-6. [DOI] [PubMed] [Google Scholar]
- 7.Dixon WJ, Brown MB, Engleman L, France JW, Hill MA, Jennrich RI, Toparek JD. BMDP statistical software. University of California; Berkeley, CA: 1981. [Google Scholar]
- 8.Hoffman HS, Searle JL. Acoustic variables in the modification of startle reaction in the rat. J Comp Physiol Psychol. 1965;60:53–58. doi: 10.1037/h0022325. [DOI] [PubMed] [Google Scholar]
- 9.Hooks MS, Colvin AC, Juncos JL, Justice JB. Individual differences in basal and cocaine-stimulated extracellular dopamine in the nucleus accumbens using quantitative microdialysis. Brain Res. 1992;587:306–312. doi: 10.1016/0006-8993(92)91012-4. [DOI] [PubMed] [Google Scholar]
- 10.Hutson PH, Bristow LJ, Thorn L, Tricklebank MD. R-(+)-HA-966, a glycine/NMDA receptor antagonist, selectively blocks the activation of the mesolimbic dopamine system by amphetamine. Br J Pharmacol. 1991;103:2037–2044. doi: 10.1111/j.1476-5381.1991.tb12372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keith VA, Mansbach RS, Geyer MA. Failure of haloperidol to block the effects of phencyclidine and dizocilpine on prepulse inhibition of startle. Biol Psychiatry. 1991;30:557–566. doi: 10.1016/0006-3223(91)90025-h. [DOI] [PubMed] [Google Scholar]
- 12.Kelly PH, Seviour PW, Iversen SD. Amphetamine and apomorphine responses in the rat following 6-OHDA lesions of the nucleus accumbens septi and corpus striatum. Brain Res. 1975;94:507–522. doi: 10.1016/0006-8993(75)90233-4. [DOI] [PubMed] [Google Scholar]
- 13.Koob GF, Swerdlow NR. The functional output of the mesolimbic dopamine system. In: Kalivas PW, Nemeroff CB, editors. Annals of the New York Academy of Sciences, Vol 537, The mesocorticolimbic dopamine system. New York Academy of Sciences; New York: 1988. pp. 216–227. [DOI] [PubMed] [Google Scholar]
- 14.Luedtke RR, Artymyshyn RP, Monks BR, Molinoff PB. Comparison of the expression, transcription and genomic organization of D2 dopamine receptors in outbred and inbred strains of rat. Brain Res. 1992;584:45–54. doi: 10.1016/0006-8993(92)90876-b. [DOI] [PubMed] [Google Scholar]
- 15.Mansbach RS. Effects of NMDA receptor ligands on sensorimotor gating in the rat. Eur J Pharmacol. 1991;202:61–66. doi: 10.1016/0014-2999(91)90253-m. [DOI] [PubMed] [Google Scholar]
- 16.Mansbach RS, Geyer MA. Effects of phencyclidine and phencyclidine biologs on sensorimotor gating in the rat. Neuropsychopharmacology. 1989;2:299–308. doi: 10.1016/0893-133x(89)90035-3. [DOI] [PubMed] [Google Scholar]
- 17.Mansbach RS, Geyer MA, Braff DL. Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacology (Berl) 1988;94:507–514. doi: 10.1007/BF00212846. [DOI] [PubMed] [Google Scholar]
- 18.Pellegrino LJ, Pellegrino AS, Cushman AJ. A stereotaxic atlas of the rat brain, 2nd Edition. Plenum; New York: 1981. [Google Scholar]
- 19.Pradhan N, Arunasmitha S, Udaya HB. Behavioral and neurochemical differences in an inbred strain of rats. Physiol Behav. 1990;47:705–708. doi: 10.1016/0031-9384(90)90081-e. [DOI] [PubMed] [Google Scholar]
- 20.Rigdon GC. Differential effects of apomorphine on prepulse inhibition of acoustic startle reflex in two rat strains. Psychopharmacology (Berl) 1990;102:419–421. doi: 10.1007/BF02244115. [DOI] [PubMed] [Google Scholar]
- 21.Rigdon GC, Weatherspoon JK. 5-Hydroxytryptamine1A receptor agonists block prepulse inhibition of acoustic startle reflex. J Pharmacol Exp Ther. 1992;263:486–493. [PubMed] [Google Scholar]
- 22.Svensson L, Ahlenius S. Enhancement by the putative 5-HT receptor agonist 8-OH-2-(di-n-propylamino)tetralin of the acoustic startle response in the rat. Psychopharmacology (Berl) 1983;79:104–107. doi: 10.1007/BF00427793. [DOI] [PubMed] [Google Scholar]
- 23.Swerdlow NR, Braff DL, Geyer MA, Koob GF. Central dopamine hyperactivity in rats mimics abnormal acoustic startle in schizophrenics. Biol Psychiatry. 1986;21:23–33. doi: 10.1016/0006-3223(86)90005-3. [DOI] [PubMed] [Google Scholar]
- 24.Swerdlow NR, Mansbach RS, Geyer MA, Pulvirenti L, Koob GF, Braff DL. Amphetamine disruption of prepulse inhibition of acoustic startle is reversed by depletion of mesolimbic dopamine. Psychopharmacology (Berl) 1990a;100:413–416. doi: 10.1007/BF02244616. [DOI] [PubMed] [Google Scholar]
- 25.Swerdlow NR, Braff DL, Masten VL, Geyer MA. Schizophrenic-like sensorimotor gating abnormalities in rats following dopamine infusion into the nucleus accumbens. Psychopharmacology (Berl) 1990b;101:414–420. doi: 10.1007/BF02244063. [DOI] [PubMed] [Google Scholar]
- 26.Swerdlow NR, Keith VA, Braff DL, Geyer MA. Effects of spiperone, raclopride, SCH 23390 and clozapine on apomorphine inhibition of sensorimotor gating of the startle response in the rat. J Pharmacol Exp Ther. 1991;256:530–536. [PubMed] [Google Scholar]
- 27.Swerdlow NR, Caine SB, Geyer MA. Regionally selective effects of intracerebral dopamine infusion on sensorimotor gating of the startle reflex in rats. Psychopharmacology (Berl) 1992;108:189–195. doi: 10.1007/BF02245306. [DOI] [PubMed] [Google Scholar]
- 28.Swerdlow NR, Varty GB, Geyer MA. Discrepant findings of clozapine effects on prepulse inhibition of startle: is it the route or the rat? Neuropsychopharmacology. 1998;18:50–56. doi: 10.1016/S0893-133X(97)00110-3. [DOI] [PubMed] [Google Scholar]
- 29.Varty GB, Higgins GA. Differences between three rat strains in sensitivity to prepulse inhibition of an acoustic startle response: influence of apomorphine and phencyclidine pretreatment. J Psychopharmacol (Oxf) 1994;8:148–156. doi: 10.1177/026988119400800302. [DOI] [PubMed] [Google Scholar]
- 30.Wan FJ, Swerdlow NR. Sensorimotor gating in rats is regulated by different dopamine-glutamate interactions in the nucleus accumbens core and shell subregions. Brain Res. 1996;722:168–176. doi: 10.1016/0006-8993(96)00209-0. [DOI] [PubMed] [Google Scholar]
- 31.Young KA, Randall PK, Wilcox RE. Dose and time response analysis of apomorphine’s effect on prepulse inhibition of acoustic startle. Behav Brain Res. 1991;42:43–48. doi: 10.1016/s0166-4328(05)80038-5. [DOI] [PubMed] [Google Scholar]