

Abstract
Long-term potentiation (LTP) is a potential cellular mechanism for learning and memory. The retrograde messenger nitric oxide (NO) is thought to induce LTP in the CA1 region of the hippocampus via activation of soluble guanylyl cyclase (sGC) and, ultimately, cGMP-dependent protein kinase (cGK). Two genes code for the isozymes cGKI and cGKII in vertebrates. The functional role of cGKs in LTP was analyzed using mice lacking the gene(s) for cGKI, cGKII, or both. LTP was not altered in the mutant mice lineages. However, LTP was reduced by inhibition of NO synthase and NMDA receptor antagonists, respectively. The reduced LTP was not recovered by the cGK-activator 8-(4 chlorophenylthio)-cGMP. Moreover, LTP was not affected by the sGC inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]-quiloxalin-1-one. In contrast, it was effectively suppressed by nicotinamide, a blocker of the ADP-ribosyltransferase. These results show that cGKs are not involved in LTP in mice and that NO induces LTP through an alternative cGMP-independent pathway, possibly ADP-ribosylation.
Keywords: synaptic plasticity, hippocampus, nitric oxide, cGMP-dependent kinase, gene targeting, mouse
Long-term potentiation (LTP) is a potential cellular mechanism underlying learning and memory. Schaffer collateral inputs to pyramidal neurons in the hippocampal CA1 region exhibit a form of LTP that critically depends on a NMDA receptor-mediated Ca2+ influx into the postsynapse (Perkel et al., 1993; Tsien et al., 1996) and is, at least partly, attributable to increased presynaptic transmitter release (Malinow, 1991; Bolshakov and Siegelbaum, 1995). Despite some controversy, convincing evidence suggests that nitric oxide (NO), generated postsynaptically by Ca2+-calmodulin-dependent NO synthase (NOS), acts as a retrograde messenger (Böhme et al., 1991; O’Dell et al., 1991;Schuman and Madison, 1991; Zhuo et al., 1993; Arancio et al., 1996;Kantor et al., 1996; Son et al., 1996; Wilson et al., 1997; but seeLum-Ragan and Gribkoff, 1993; Williams et al., 1993). However, the molecular events mediating the action of NO in the presynapse remain to be resolved unequivocally.
Soluble guanylyl cyclase (sGC) generating the intracellular second messenger cGMP is a major target of NO. Interestingly, tetanic stimulation of hippocampal preparations causes an increase of cGMP sensitive to NOS inhibitors (Chetkovich et al., 1993). Consistent with a functional role of cGMP, it has been reported that membrane-permeable dibutyryl-cGMP partially reverses reduction of LTP by an NOS inhibitor (Haley et al., 1992), and sGC inhibitors suppress LTP (Zhuo et al., 1994; Boulton et al., 1995). Cytosolic cGMP controls the activity of diverse receptor proteins, including the cGMP-dependent protein kinase (cGK). cGK has been suggested to play a role in the induction of LTP based on the following findings: cGK inhibitors block LTP, and cGK activators facilitate LTP in response to rather weak tetanic stimuli (Zhuo et al., 1994). Similar observations made at synapses between individual pyramidal neurons in hippocampal culture further support this concept (Arancio et al., 1995).
Two genes coding for cGKI and cGKII have been identified in vertebrates (Wernet et al., 1989; Ruth et al., 1991; Uhler, 1993; Jarchau et al., 1994). cGKII is expressed weakly in the hippocampus (El-Din El-Husseini et al., 1995). Expression of cGKI and the localization of both forms in the hippocampus is primarily unknown.
Conflicting with NO signaling through cGMP–cGK, others reported that cGMP fails to rescue LTP blocked by an NMDA receptor antagonist, and the protein kinase inhibitor H8 has no effect on LTP at concentrations suppressing cGK activity (Schuman et al., 1994; Selig et al., 1996). In contrast, blockers of the ADP-ribosyltransferase primarily reduced LTP, suggesting that NO, at least partly, acts via ADP-ribosylation of presynaptic proteins. This idea is substantiated further by findings that NO induces ADP-ribosylation of hippocampal proteins (Sullivan et al., 1997). ADP-ribosylation is not mimicked by cGMP but is occluded partly in hippocampal slices that received tetanic stimulation (Williams et al., 1992; Duman et al., 1993).
To gain further insight into the function of cGKs for LTP, we used mice lacking cGKI and/or cGKII. Genetic inactivation of cGKs in mice did not impair LTP. Moreover, inhibitors of NOS and ADP-ribosyltransferase suppressed LTP in cGK-deficient mice, suggesting that NO does not use the cGMP–cGK pathway during the induction of LTP.
MATERIALS AND METHODS
Field EPSP recordings in hippocampal slices.Transverse hippocampal slices (400-μm-thick) from wild-type (WT), cGKI−/− (Pfeifer et al., 1998), and cGKII−/− (Pfeifer et al., 1996) mice were prepared and maintained using standard procedures (Bliss and Lømo, 1973). After a recovery period (1.5 hr), slices were transferred into a submersion-type recording chamber perfused (1–2 ml/min) with artificial CSF (ACSF) (27.5°C) containing (in mm): glucose 10, NaCl 124, KCl 3, KH2PO4 1.25, NaHCO3 26, MgSO4 2, and CaCl2 2, bubbled with 95% O2–5% CO2, pH 7.4. Field EPSPs (fEPSPs) were recorded using an AXOCLAMP 2B amplifier (Axon Instruments, Foster City, CA), with an extracellular glass electrode (filled with 1 mm NaCl, resistance 4–8 MΩ) placed in the apical dendritic layer (stratum radiatum) of CA1 pyramidal neurons. Schaffer collaterals were stimulated using short current pulses (50 μsec) delivered through a monopolar tungsten electrode in the CA3 region. fEPSPs were low-pass filtered (1–3 kHz), digitized, stored, and analyzed using custom-made LabView software, a LabPC+ interface plus BNC-board (National Instruments München, Germany). The slope of the fEPSP was calculated and used to assess efficacy of synaptic transmission. At the beginning of each experiment, the strength of presynaptic fiber stimulation was increased stepwise until the postsynaptic response (fEPSP amplitude) saturated and then reduced to elicit an fEPSP with 40–50% of the maximal amplitude. Baseline synaptic responses evoked at 0.1–0.067 Hz were routinely recorded for 20–30 min before tetanic stimulation. LTP was induced using one of the following paradigms (same stimulus strength as for baseline recording): (1) strong tetanus (3 × 30 pulses, 100 Hz, 5 sec pause between trains); (2) weak tetanus (50 Hz for 0.5 sec); or (3) theta burst (10 × 4 pulses, 100 Hz, 200 msec pause between bursts). LTP was assessed as the increase in the slope of fEPSPs 1 hr after tetanus and expressed as the percentage of the baseline fEPSP slope (before tetanus). Paired-pulse facilitation (PPF) was examined for stimuli separated by 30, 50, 70, and 100 msec, respectively, and is expressed as the ratio between the slope of the two consecutive fEPSPs. All data shown are mean ± SEM, and statistical analysis was performed using the Student’s ttest for two independent means.
Slices were incubated with the NOS inhibitornω-nitro-l-arginine (NOArg) for 2 hr before starting an experiment. NOArg (100 μm) and the sGC inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]-quiloxalin-1-one (ODQ) (3 μm) were present throughout the experiment. 8-(4 Chlorophenylthio)-cGMP (8p-CPT-cGMP) (50 μm) was applied for 30–40 min, starting 15–20 min before tetanic stimulation.
Animals. Genotyping was routinely performed using the PCR technique (see Fig. 6). Homozygous mutants deficient in either cGKI or cGKII were F2 offspring from a cross between the chimeras (contributing 129/Sv background) and C57BL/6 mice. To minimize the possible effect of an undetermined genetic background, we primarily used littermates (in 72% of all experiments). WT offspring from heterozygous cGK-deficient lineages and C57BL/6 mice (all carrying two intact alleles ofcGKI and cGKII) matching the mutant animals in age and gender were used also as controls, because LTP was not significantly different in 129/Sv and C57BL/6 mice (data not shown). As anticipated from the fact that the two human genes encoding cGKI and cGKII are located on separate chromosomes, double-mutant mice (cGKI−/− cGKII−/−) were generated by crossing the two cGK-deficient lineages. Double mutants exhibit a combination of the phenotypes observed in mice lacking one isoform of the cGK (Pfeifer et al., 1996, 1998). Most prominent are the dwarfism and the intestinal malfunction.
Fig. 6.
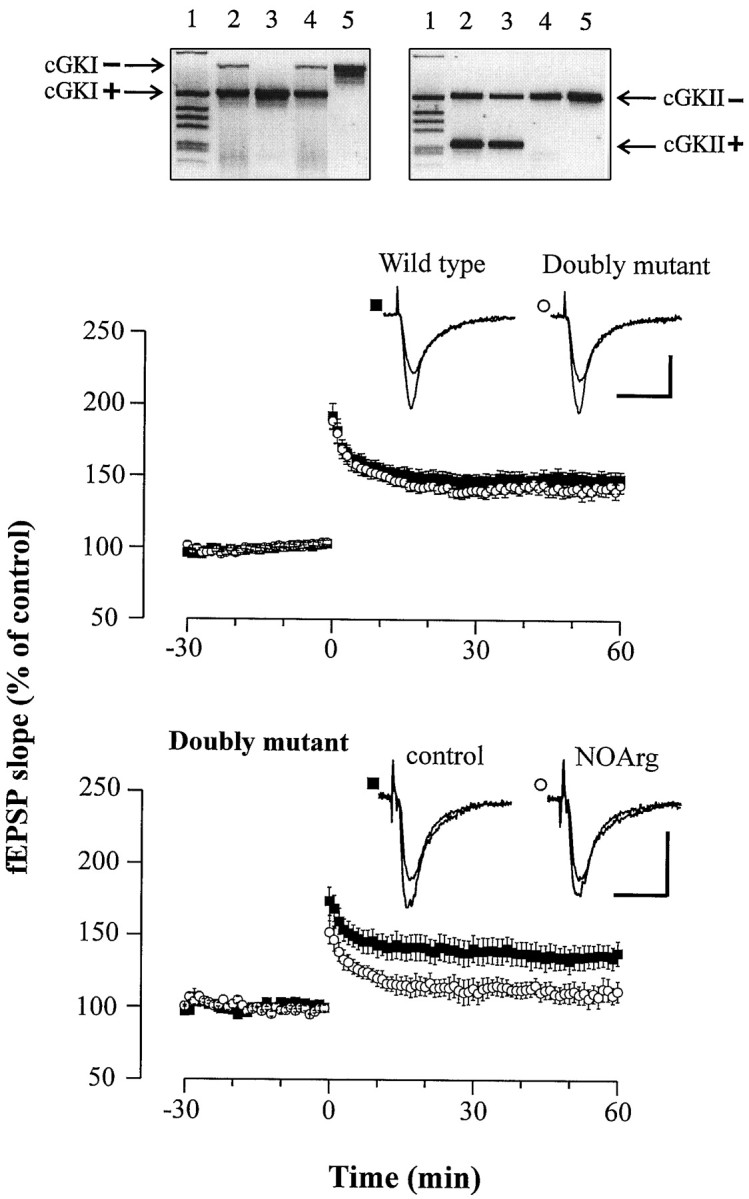
LTP in double-mutant mice (cGKI−/− cGKII−/−) is normal and reduced in the presence of the NOS inhibitor NOArg.Top, Double-mutant mice were generated by mating heterozygous cGKI+/ånd cGKII+/− mice. WT and mutant alleles of the cGKI and cGKII genes were identified by specific PCR products as illustrated for four offsprings (lanes 2–5). The DNA sample used as template for PCRs in lane 5 was derived from a double-mutant homozygous animal (cGKI−/− cGKII−/−). Lane 1, One kilobase DNA ladder (Life Technologies, Gaithersburg, MD). Middle, Average potentiation of the fEPSP slope in response to theta burst stimulation in slices from WT (▪; n = 18) and double-mutant (○;n = 15) animals. The mean baseline slope (pretetanus control) was −0.34 ± 0.02 and −0.29 ± 0.03 mV/msec in slices from WT and double-mutant animals, respectively. Representative fEPSP recordings are shown in the insets. Calibration: 20 msec, 0.5 mV. Bottom, Average potentiation of the fEPSP slope in slices from double-mutant mice bathed in normal ACSF (control) (▪;n = 7 slices) and ACSF containing 100 μm NOArg (○; n = 7 slices). The mean baseline slope (pretetanus control) was −0.46 ± 0.12 and −0.37 ± 0.10 mV/msec in control and NOArg-treated slices, respectively. Representative fEPSP recordings are shown in theinset. Calibration: 20 msec, 0.5 mV.
cGMP assay and protein kinase assay. cGMP levels in hippocampal slice preparations were determined using a commercially available immunoassay (Cayman Chemical, Ann Arbor, MI). Slices (400-μm-thick, wet weight ∼1 mg) were prepared as described above, allowed to recover in gassed ACSF at room temperature, and then preincubated with either control ACSF or ODQ (3 μm) for 15 min at 37°C before adding the NO donor 2-(N,N-diethylamino)-diazenolate-2 oxide NO (DEA-NO) (3 μm). The reaction was terminated by removing and freezing rapidly the tissue samples in liquid nitrogen. The kinase activity of hippocampal extracts was determined as described elsewhere (Ruth et al., 1991), with 10 μg of protein and 10 μm the phosphodiesterase-resistent cGMP analog 8p-CPT-cGMP (10 μm). The protein kinase A inhibitor peptide PKI(6–22) (4 μm) was added to suppress cAMP-dependent protein phosphorylation.
Immunoblotting and in situ hybridization. For Western analysis, the corresponding tissue probes were homogenized and extracted with 2× Laemmli’s buffer. Soluble proteins were then separated on 7.5% SDS-polyacrylamide gel and blotted onto nitrocellulose membranes. The blots were probed with the antibodies (Abs) B32-A3 to the COOH-terminal region of mouse cGKII and with Ab 16–14 to cGKI (Ecker et al., 1989). Bound Abs were detected using the ECL technique (Amersham, Arlington Heights, IL).
In situ hybridization analysis for cGKI and cGKII was performed adapting a protocol described previously (Pfeifer et al., 1996) to hippocampal sections. 35S-labeled hybridization probes were obtained by PCR-amplification of nt960–nt1740 of the murine cGKII sequence (Uhler, 1993) and a fragment homologous to nt1522–nt2023 of the bovine cGKI sequence (Wernet et al., 1989) from mouse brain cDNAs.
RESULTS
cGK is expressed in the hippocampus
Initially, we studied the expression of the two forms of cGK in the hippocampus of WT mice with immunoblot and in situhybridization techniques. Immunoblotting of hippocampal tissue from WT mice yielded a prominent cGKI-specific band, indicating that cGKI is highly expressed in the hippocampus (Fig.1, top). In line with this, transcripts of the cGKI gene were abundant throughout all cellular layers of the hippocampus (CA3–CA1 and dentate gyrus). Immunoblotting also demonstrated the presence of cGKII in the hippocampal tissue (Fig. 1, bottom), although significant amounts of the corresponding mRNA in pyramidal cells (CA3–CA1) were not detected by in situ hybridization. Nevertheless, the expression of the cGK proteins in the murine hippocampus supported their potential functional role in LTP.
Fig. 1.
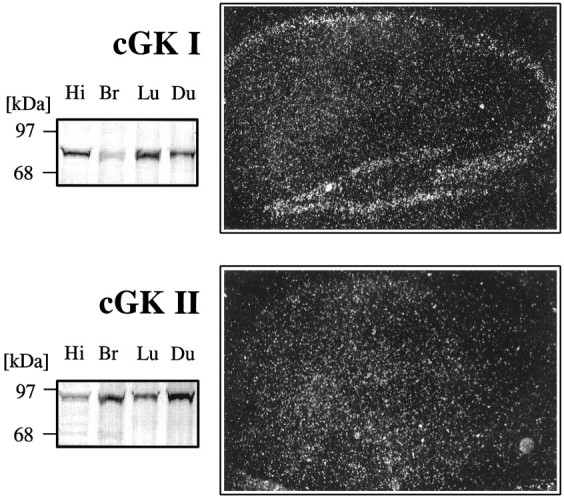
cGK is expressed in the murine hippocampus.Left, Immunoblots of tissue extracts from the hippocampus (Hi), whole brain (Br), lung (Lu), and whole duodenum (Du) with cGKI- (top) and cGKII (bottom)-specific antibodies. Right, In situ hybridization in hippocampal slices with antisense riboprobes specific for cGKI (top) and cGKII (bottom).
Mice deficient in cGKI or cGKII exhibit normal synaptic transmission and LTP in the CA1 region of the hippocampus
cGK-deficient mice had no apparent gross anatomical abnormalities in the brain. Histological analysis showed that the overall arrangement of the cellular layers in the hippocampus was normal (data not shown). To rule out possible general defects of the synaptic transmission caused by the gene deletion, we studied the dependency of the amplitude of fEPSPs on the stimulating intensity (input–output relation) and the PPF. As illustrated in Figure 2, input–output relation and PPF were similar in the WT and all types of cGK-deficient mice. Thus, the basic parameters of the synaptic transmission were normal in the mutant mice.
Fig. 2.
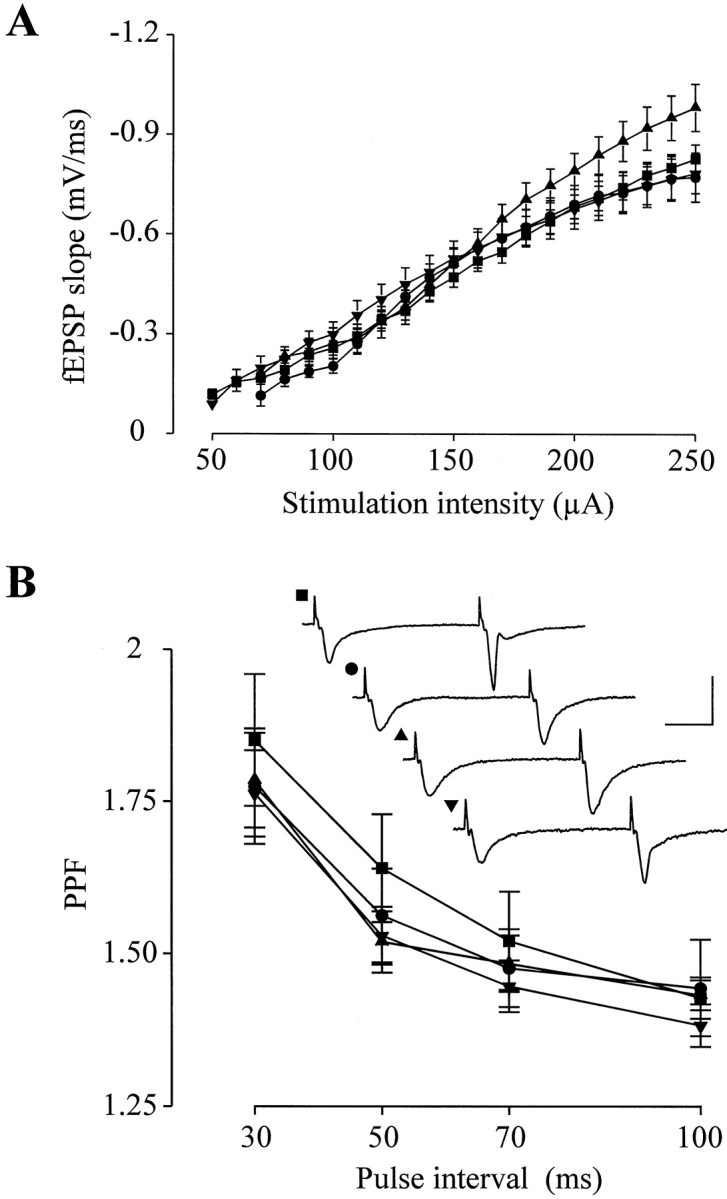
Mice deficient in cGK exhibit normal synaptic transmission. A, input–output relation for slices from WT (▪; n = 57), cGKI−/− (•;n = 24), cGKII−/− (▴; n = 22), and double-mutant (▾; n = 26) animals. The points represent the mean ± SEM for each genotype.B, PPF for WT (▪; n = 35), cGKI−/− (•; n = 30), cGKII−/− (▴;n = 27), and double-mutant (▾;n = 13) mice. The points represent mean ± SEM. Representative fEPSP recordings with an interstimulus interval of 70 msec are shown in the inset. The corresponding genotype is indicated by the label. Calibration: 20 msec, 2 mV.
The effect of the disruption of the cGK genes on hippocampal LTP was initially examined using a strong tetanic stimulus (Fig.3). Under these conditions, the fEPSP slope 1 hr after tetanus amounted to 191.6 ± 21.4% (cGKI−/−:n = 5 animals, 15 slices) versus 189.6 ± 18.4% (WT: n = 10 animals, 18 slices) and 202.7 ± 15.4% (cGKII−/−: n = 15 animals, 26 slices) versus 197.3 ± 14.8% (WT: n = 10 animals, 22 slices) of the pretetanus control. LTP might be slightly overestimated, because there was a moderate run-up of the baseline. However, LTP was not altered in cGKI−/− and cGKII−/− mice.
Fig. 3.

LTP induced by strong tetanic stimulation is normal in cGKI−/− and cGKII−/− mice. Top, Average potentiation of the fEPSP slope in slices from WT (▪;n = 18) and cGKI−/− (○; n = 15) animals. The mean baseline slope (pretetanus control) was −0.37 ± 0.04 and −0.33 ± 0.04 mV/msec in slices from WT and cGKI−/− animals, respectively. Representative fEPSP recordings for both genotypes are shown in the inset. Calibration: 20 msec, 1 mV. Bottom, Average potentiation of the fEPSP slope in slices from WT (▪; n = 22) and cGKII−/− (○; n = 26) animals. The mean baseline slope (pretetanus control) was −0.33 ± 0.03 and −0.28 ± 0.03 mV/msec in slices from WT and cGKII−/− animals, respectively. Representative fEPSP recordings for both genotypes are shown in theinset. Calibration: 20 msec, 1 mV.
Because the NO-dependent fraction of LTP might depend on the induction protocol (Lum-Ragan and Gribkoff 1993), we additionally tested a weak tetanic stimulus and a theta burst paradigm thought to approximate physiological patterns of synaptic activity in the hippocampus. These two protocols induced moderate LTP, with no significant difference between matched WT, cGKI−/−, and cGKII−/− mice (Fig.4). On average, the weak tetanus potentiated fEPSPs to 135.2 ± 8.9% (cGKI−/−: n= 5 animals, 12 slices) versus 133.2 ± 5.4% (WT:n = 5 animals, 10 slices) and 134.7 ± 6.7% (cGKII−/−: n = 8 animals, 13 slices) versus 129.5 ± 4.7% (WT: n = 4 animals, 10 slices) of the corresponding control before tetanus. After the theta burst, the slope of the fEPSPs increased to 147.2 ± 5.6% (cGKI−/−:n = 6 animals, 15 slices) versus 149.4 ± 6.8% (WT: n = 10 animals, 13 slices) and 144.5 ± 5.0% (cGKII−/−: n = 6 animals, 15 slices) versus 146.4 ± 5.9% (WT: n = 12 animals, 17 slices) of the pretetanus control, respectively.
Fig. 4.

LTP after a theta burst stimulation is normal in cGKI−/− and cGKII−/− mice. Top, Average potentiation of the fEPSP slope in slices from WT (▪; n = 13) and cGKI−/− (○; n = 15) animals. The mean baseline slope (pretetanus control) was −0.43 ± 0.05 and −0.48 ± 0.04 mV/msec in slices from WT and cGKI−/− animals, respectively. Representative fEPSP recordings for both genotypes are shown in the inset. Calibration: 20 msec, 1 mV.Bottom, Average potentiation of the fEPSP slope in slices from WT (▪; n = 17) and cGKII−/− (○;n = 15) animals. The mean baseline slope (pretetanus control) was −0.41 ± 0.04 and −0.37 ± 0.04 mV/msec in slices from WT and cGKII−/− animals, respectively. Representative fEPSP recordings for both genotypes are shown in theinset. Calibration: 20 msec, 1 mV.
Because the functional role of NO for the expression of LTP in rats is age-dependent (Williams et al., 1993), we studied LTP in young mice. Three to 4-week-old animals exhibited LTP slightly reduced compared with adult mice (8- to 12-week-old), independently of the genotype. However, there was no significant difference in LTP between matched WT and cGKI−/− mice of this age [134.8 ± 6.5% (n= 6 animals, 12 slices) vs 129.2 ± 4.4% (n = 5 animals, 20 slices)].
Inhibition of NOS attenuates LTP in the CA1 region of cGK-deficient mice
NO-dependent mechanisms involved in the induction of LTP can be blocked by inhibitors of NOS, such as NOArg. In hippocampal slices from WT mice, LTP was markedly reduced in the presence of the NOS inhibitor (Fig. 5), proving that a significant portion of LTP was NO-dependent under our experimental conditions. In slices superfused with normal ACSF and NOArg (100 μm), the fEPSP slope 1 hr after the theta burst was potentiated to 153.0 ± 9.1% (n = 9 animals, 14 slices) and 127.1 ± 7.9% (n = 9 animals, 13 slices) of the pretetanus control, respectively.
Fig. 5.
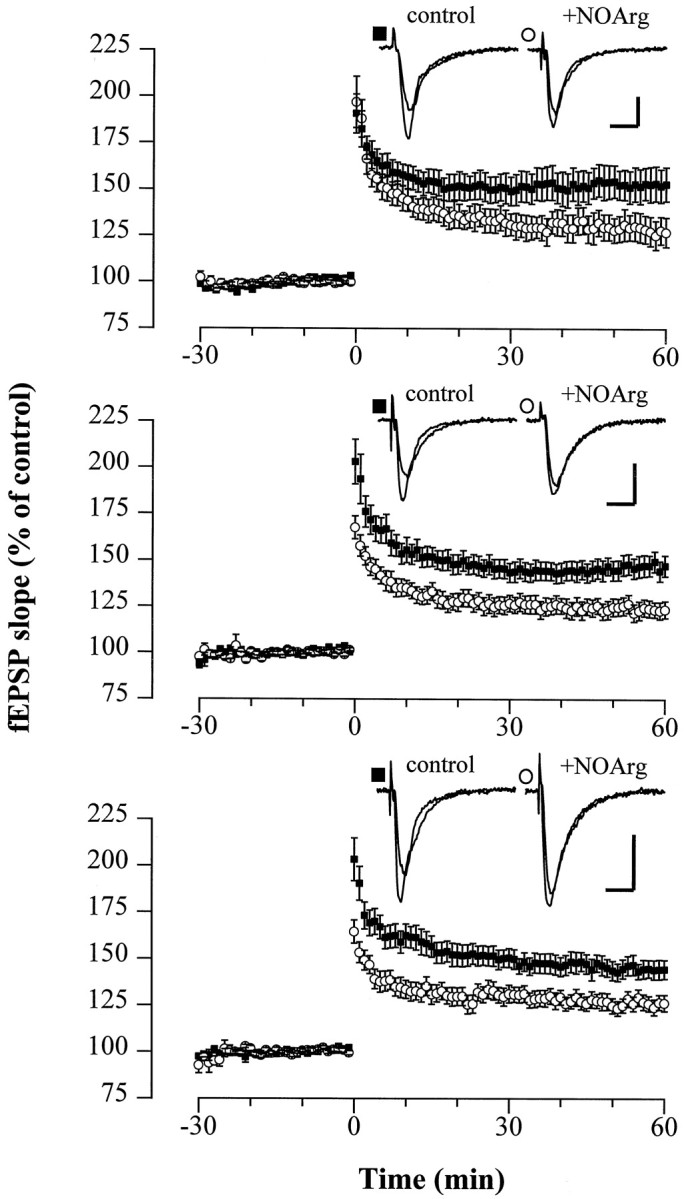
LTP is reduced in the presence of the NOS inhibitor NOArg in slices from WT, cGKI−/−, and cGKII−/− mice. Average potentiation of the fEPSP slope after theta burst stimulation in slices from WT (top), cGKI−/− (middle), and cGKII−/− (bottom) mice bathed in normal ACSF (control) (▪;n = 14, 15, and 15 slices, respectively) and ACSF containing 100 μm NOArg (○; n = 13, 11, and 13 slices, respectively). The differences between LTP in control and NOArg-treated slices were statistically significant (p < 0.05). NOArg was applied ∼2 hr before starting the experiments and was present throughout. The mean baseline slope (pretetanus control) was −0.40 ± 0.04 and −0.34 ± 0.03 mV/msec (WT), −0.48 ± 0.04 and −0.44 ± 0.05 mV/msec (cGKI−/−), and −0.29 ± 0.03 and −0.32 ± 0.03 mV/msec (cGKII−/−) in control and NOArg-treated slices.Insets show representative fEPSP recordings for the corresponding genotypes, indicated by the label. Calibration: 10 msec, 0.5 mV.
Assuming that cGK is a critical effector of NO during the induction of LTP, the NOS inhibitor NOArg should not or only marginally affect LTP in cGK-deficient mice. However, LTP in mice deficient for cGKI−/− and cGKII−/− was attenuated by NOArg (100 μm) to the same extent as in WT mice [123.2 ± 4.5% (cGKI−/−:n = 4 animals, 11 slices) and 126.4 ± 4.5% (cGKII−/−: n = 5 animals, 13 slices)] (Fig. 5). This finding further challenges the view that the expression of LTP involves a cGK-dependent step, unless both forms of cGK could functionally substitute for each other as described for the endothelial NOS (eNOS) and neuronal NOS (nNOS) (Son et al., 1996). Regarding the latter possibility, we sought to validate our observations by examining (1) LTP in double-mutant mice, (2) the effect of the NOS inhibitor NOArg on LTP in this preparation, and (3) whether activation of cGK can restore LTP suppressed by NOArg or the NMDA receptor antagonist AP-5. Double-mutant animals carrying a null mutation for both isoforms of cGK (cGKI−/− cGKII−/−) were generated by crossing heterozygotes (cGKI+/− and cGKII+/−) and were identified using the PCR technique (see Materials and Methods) (Fig. 6,top). Double mutants had no defect in LTP after the theta burst [142.8 ± 5.3% (n = 5 animals, 15 slices); WT: 148.3 ± 5.5% (n = 7 animals, 18 slices)] (Fig. 6, middle), confirming the dispensable role of cGKI and cGKII for the retrograde signaling of NO in LTP. In line with this, the NOS inhibitor NOArg reduced LTP in 3- to 4-week-old double-mutant mice, as well (138.4 ± 8.5% vs 111.8 ± 7.1%, n = 3 animals, 7 slices) (Fig. 6,bottom). The possibility that another, unidentified cGMP-dependent protein kinase could support NO-dependent LTP can also be excluded. Hippocampal extracts from the double-mutant mouse exhibited no cGMP-dependent kinase activity, because32P-incorporation into a G-kinase peptide substrate was not affected by cGMP. Accordingly, 8p-CPT-cGMP (50 μm) failed to relieve the suppression of LTP by NOArg (100 μm) or AP-5 (50 μm) in preparations from WT mice (Fig.7). The fEPSP slope 1 hr after tetanus in slices superfused with the ACSF containing NOArg plus 8p-CPT-cGMP amounted to 134.7 ± 7.2% (n = 6 animals, 10 slices) of the pretetanus control. Although no LTP was observed in the presence of AP-5 plus 8p-CPT-cGMP, robust LTP was induced by the theta burst after wash-out of the compounds (130.9 ± 12.6%,n = 2 animals, 4 slices). Furthermore, the simultaneous application of a weak tetanus and the membrane-permeable analog 8-Br-cGMP (“paired training”) did not facilitate LTP in WT mice after the weak tetanic stimulus (132.7 ± 4.6% of pretetanus control with 100 μm 8-bromo-cGMP (8-Br-cGMP),n = 5 animals, 8 slices). Together, these data clearly rule out cGMP kinases as critical determinants for the expression of LTP in the Schaffer collateral pathway of the hippocampus.
Fig. 7.

The membrane-permeable cGMP analog 8p-CPT-cGMP fails to abolish suppression of LTP by the NOS inhibitor NOArg and the NMDA receptor antagonist AP-5. A, Average potentiation of the fEPSP slope after theta burst stimulation in slices from WT mice treated with 100 μm NOArg (▪; n = 13) and 100 μm NOArg plus 50 μm 8p-CPT-cGMP (○; n = 10), respectively. For comparison, the fEPSP slope in the control (normal ACSF) 55 and 60 min after tetanus is shown (♦; n = 12). The mean baseline slope (pretetanus control) was −0.34 ± 0.03 and −0.27 ± 0.03 mV/msec in slices bathed in ACSF containing NOArg and NOArg plus 8p-CPT-cGMP, respectively. Inset shows representative fEPSP recordings. Calibration: 10 msec, 1 mV. B, Typical example of an fEPSP recording in a hippocampal slice; the theta burst stimulation (left arrow) failed to induce detectable LTP in the presence of AP-5 (50 μm) and 8-pCPT-cGMP but induced significant potentiation of fEPSPs after wash-out of the compounds (right arrow), demonstrating the functional integrity of the preparation. The mean baseline slope was −0.27 ± 0.03 mV/msec.
The sGC inhibitor ODQ failed to affect LTP but effectively suppressed cGMP production in the hippocampus
The finding that 8-pCPT-cGMP could not reverse the suppression of LTP in the presence of NOArg and AP-5 generally argues against a role of the cGMP–cGK cascade in LTP. For a more stringent test of the role of the NO–cGMP cascade, we examined the effect of ODQ, a specific inhibitor of the sGC, on hippocampal LTP in slices from WT mice. ODQ (3 μm) had no effect on LTP after the theta burst (normal ACSF: 149.5 ± 6.9%, n = 10 animals, 14 slices; ODQ: 149.2 ± 6.1%, n = 5 animals, 13 slices) (Fig. 8A). The failure of ODQ to suppress LTP was not because of insufficient inhibition of sGC; ODQ slightly reduced the basal level of cGMP (control) in hippocampal tissue and completely eliminated the increase of cGMP in the presence of the NO donor DEA-NO (3 μm) (174.2 ± 19.3% of control without ODQ vs 95.2 ± 10.5% with ODQ;n = 4) (Fig. 8B).
Fig. 8.

The sGC inhibitor ODQ fails to suppress LTP but reverses NO-stimulated cGMP increase in the hippocampus.A, Average potentiation of the fEPSP slope after theta burst stimulation in slices from WT animals bathed in normal ACSF (control) (▪; n = 14) and ACSF containing ODQ (3 μm) (○; n = 13). ODQ was present throughout the experiments. The mean baseline slope (pretetanus control) was −0.34 ± 0.03 and −0.38 ± 0.04 mV/msec in control and ODQ-treated slices, respectively. Representative fEPSP recordings are shown in the inset. Calibration: 10 msec, 1 mV. B, Concentration of cGMP in hippocampal slices (WT) treated with DEA-NO, ODQ, and both. Slices were preincubated with ODQ (3 μm) for 15 min before applying DEA-NO (3 μm). The reaction was stopped 1 min after the addition of DEA-NO. Data are presented as percentage of the cGMP concentration in control slices incubated in normal ACSF (0.528 ± 0.039 pmol/mg of wet weight; n = 6). *p < 0.01.
The ADP-ribosyltransferase inhibitor nicotinamide markedly reduced LTP in WT and cGKI−/− mice
NO-induced ADP-ribosylation of presynaptic proteins might serve as an alternative, cGMP-independent mechanism involved in the induction of LTP (Schuman et al., 1994; Sullivan et al., 1997). Therefore, we examined LTP in slices from WT, cGKI−/−, and double-mutant (cGKI−/− cGKII−/−) animals in the presence of the ADP-ribosyltransferase inhibitor nicotinamide (Rankin et al., 1989). In all three genotypes, the fEPSP potentiation induced by a theta burst stimulation was markedly suppressed by nicotinamide (10 mm) (WT: 115.7 ± 4.3%, n = 6 animals, 10 slices; cGKI−/−: 105.7 ± 5.2%, n = 3 animals, 6 slices; double-mutant: 112.2 ± 4.5%, n = 2 animals, 4 slices) (p < 0.01 compared with the corresponding controls; compare Figs. 5, 6) (Fig.9). This inhibitory effect was reversible, as demonstrated by the robust fEPSP potentiation observed after wash-out of nicotinamide (WT: 180.4 ± 7.2%,n = 6 animals, 10 slices; cGKI−/−: 156.8 ± 7.7%, n = 3 animals, 6 slices; double-mutant: 154.6 ± 12.9%, n = 2 animals, 4 slices).
Fig. 9.

The ADP-ribosyltransferase inhibitor nicotinamide suppresses LTP in the WT and cGK-deficient mice. Data from representative fEPSP recordings. Illustrated is the potentiation of the fEPSP slope in hippocampal slices from the WT (top, •), cGKI−/− (middle, •), and double-mutant (cGKI−/− cGKII−/−) (bottom, •) mice after a tetanus in the presence of nicotinamide (10 mm) (filled bars) and after wash-out. The cumulative potentiation of fEPSPs typically observed with repetitive tetanic stimulation in the control is superimposed for the WT (top, ○). Arrows indicate theta burst stimulation of the Schaffer collateral input.
DISCUSSION
The retrograde messenger NO has been suggested to promote LTP in the CA1 region of the hippocampus by stimulating the generation of cGMP and, ultimately, activating cGK (Zhuo et al., 1994; Arancio et al., 1995; Boulton et al., 1995). To shed more light onto the functional role of cGK, we combined an electrophysiological approach with a genetic approach and studied LTP in mice carrying a null mutation of the cGKI, the cGKII, or both genes. The findings described here fit well with the established role of postsynaptic NMDA receptors (Tsien et al., 1996) and the NO–NOS system (Kantor et al., 1996; Son et al., 1996; Wilson et al., 1997) for the induction of LTP. However, they are incompatible with a presynaptic NO signaling through the cGMP–cGK cascade: (1) LTP was normal in all types of cGK-deficient mice, including the double mutant. (2) The NOS inhibitor NOArg reduced LTP in cGKI−/−, cGKII−/−, and double-mutant mice to the same extent as in the WT. (3) The membrane-permeable cGMP analogs, 8p-CPT-cGMP and 8-Br-cGMP, neither reversed the suppression of LTP in WT slices treated with NOArg or the NMDA receptor antagonist AP-5, nor facilitated the expression of LTP in response to weak tetanic stimulation. (4) Finally, the sGC inhibitor ODQ failed to reduce LTP in the WT, whereas the ADP-ribosyltransferase inhibitor nicotinamide effectively suppressed LTP in WT, cGKI−/−, as well as double-mutant mice. These findings argue strongly in favor of a cGMP-independent presynaptic NO signaling.
The reason for the apparent divergence from results reported previously by Haley et al. (1992), Zhuo et al. (1994), and Boulton et al. (1995) remains unclear. It might reflect species differences between mice, rats, and guinea pigs. Mice lacking the cGK are an excellent model to prove the functional role of the enzyme, because the gene-targeting technique eliminates problems present in other studies, such as the limited specificity of pharmacological tools and the uncertainties regarding their tissue access. To minimize the possibility of false positive or negative results associated with a mixed genetic background, we used littermates in the majority of our experiments. The findings that cGK-deficient mice had no gross developmental abnormalities of the brain and the overall arrangement of the cellular layers and basic parameters of the synaptic transmission (input–output relation and PPF) in the hippocampus were normal disfavor potential nonspecific effects of the gene deletion.
LTP in the Schaffer collateral pathway is partly NO-independent (Son et al., 1996), and experimental parameters might modify the expression of the NO-dependent component (Lum-Ragan and Gribkoff, 1993; Williams et al., 1993). Defective LTP in cGK-deficient mice could, therefore, be concealed under conditions that minimize the NO-dependent fraction. We have eliminated the potential impact of the induction protocol by testing several paradigms of variable intensity causing delicate to strong LTP. To promote NO-dependent LTP, we used predominantly young animals and performed the experiments at 27.5°C (cf. Williams et al., 1993). Under these conditions, the NOS inhibitor NOArg caused a prominent reduction in LTP after the theta burst, proving the expression of a substantial NO-dependent fraction.
Normal expression of LTP and the persistence of the inhibitory effect of NOArg on LTP in mice carrying an isolated deletion of thecGKI or cGKII gene might as well reflect the regulatory overexpression of the intact gene, resulting in a functional compensation as described similarly for the eNOS and nNOS in the hippocampus (Son et al., 1996). However, this possibility can be excluded. The expression of cGKI (mRNA and protein) was not detectably altered in various tissues, including the brain, of cGKII−/− mice and vice versa (Pfeifer et al., 1996, 1998). More importantly, LTP in double-mutant mice was also not defective under conditions yielding a considerable fraction of NOArg-sensitive LTP.
Our results are in line with a previous notion that cGMP is dispensable for the NO-dependent induction of hippocampal LTP (Schuman et al., 1994; Selig et al., 1996). In our hands, addition of 8-Br-cGMP failed to facilitate the expression of LTP induced by a weak tetanus. Moreover, the membrane-permeable analog 8p-CPT-cGMP could not reverse suppression of LTP by the NOS inhibitor NOArg as well as the NMDA receptor antagonist AP-5. Likewise, in the rat hippocampus, exogenous cGMP analogs fail to rescue LTP blocked by AP-5 (Schuman et al., 1994;Selig et al., 1996). Surprisingly, inhibition of sGC by ODQ had no effect on the expression of LTP, although the compound reportedly attenuates LTP in the rat hippocampus (Boulton et al., 1995). Insufficient blockage of the sGC is unlikely to account for the failure of ODQ to inhibit LTP. It has been shown previously that 3 μm ODQ prevent NO- and NMDA-dependent accumulation of cGMP in the rat cerebellum and hippocampus (Garthwaite et al., 1995). NO-induced increase of cGMP in the murine hippocampus was completely abolished by the same concentration (see Results). Finally, the suppression of LTP in the murine hippocampus by nicotinamide confirms results of Schuman et al. (1994), strengthening the view that NO acts independent of cGMP via ADP-ribosylation of presynaptic proteins. Studies examining whether exogenous sources of NO or cGMP are capable of reinstalling LTP in the hippocampus of eNOS−/nNOS− mice or mice lacking the NMDA receptor could help to further elucidate this question.
In summary, our data demonstrate that an increase in cytosolic cGMP and activation of the cGK are neither necessary nor sufficient for the induction of LTP in the CA1 region of the murine hippocampus. Instead, it is suggested that the retrograde messenger NO acts via a cGMP–cGK-independent mechanism, possibly activation of ADP-ribosyltransferase. Direct effects of NO on other proteins, e.g., the β-subunit of a cyclic nucleotide-gated channel (Broillet and Firestein, 1997) or the ryanodine receptor (Xu et al., 1998), via nitrosylation of thiol residues might represent alternative mechanisms.
Footnotes
This research was supported by grants from Bundesministerium für Bilolung, Wissenschaft, Forschung und Technologie and Fond der Chemischen Industrie. We thank Dr. M. Korte and V. Staiger (Max-Planck-Institut für Neurobiologie, Martinsried) for their help and for providing us with the custom-made LabView software. We are thankful to K. Doerr, S. Kamm, M. Wöckner, and B. Lehnert for technical assistance.
Correspondence should be addressed to Dr. Thomas Kleppisch, Institut für Pharmakologie und Toxikologie der Technischen Universität München, Biedersteiner Strasse 29, 80802 München, Germany.
REFERENCES
- 1.Arancio O, Kandel ER, Hawkins RD. Activity-dependent long-term enhancement of transmitter release by presynaptic 3′,5′-cyclic GMP in cultured hippocampal neurons. Nature. 1995;376:74–80. doi: 10.1038/376074a0. [DOI] [PubMed] [Google Scholar]
- 2.Arancio O, Kiebler M, Lee CJ, Lev-Ram V, Tsien RY, Kandel ER, Hawkins RD. Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell. 1996;87:1025–1035. doi: 10.1016/s0092-8674(00)81797-3. [DOI] [PubMed] [Google Scholar]
- 3.Bliss TVP, Lømo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol (Lond) 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Böhme GA, Bon C, Stutzmann JM, Doble A, Blanchard JC. Possible involvement of nitric oxide in long-term potentiation. Eur J Pharmacol. 1991;199:379–381. doi: 10.1016/0014-2999(91)90505-k. [DOI] [PubMed] [Google Scholar]
- 5.Bolshakov VY, Siegelbaum SA. Regulation of hippocampal transmitter release during development and long-term potentiation. Science. 1995;269:1730–1734. doi: 10.1126/science.7569903. [DOI] [PubMed] [Google Scholar]
- 6.Boulton CL, Southam E, Garthwaite J. Nitric oxide-dependent long-term potentiation is blocked by a specific inhibitor of soluble guanylyl cyclase. Neuroscience. 1995;69:699–703. doi: 10.1016/0306-4522(95)00349-n. [DOI] [PubMed] [Google Scholar]
- 7.Broillet MC, Firestein S. β Subunits of the olfactory cyclic nucleotide-gated channel form a nitric oxide activated Ca2+ channel. Neuron. 1997;18:951–958. doi: 10.1016/s0896-6273(00)80334-7. [DOI] [PubMed] [Google Scholar]
- 8.Chetkovich DM, Klann E, Sweatt JD. Nitric oxide synthase-independent long-term potentiation in area CA1 of hippocampus. NeuroReport. 1993;4:919–922. doi: 10.1097/00001756-199307000-00020. [DOI] [PubMed] [Google Scholar]
- 9.Duman RS, Terwillinger RZ, Nestler EJ. Alteration in nitric oxide-stimulated endogenous ADP-ribosylation associated with long-term potentiation in rat hippocampus. J Neurochem. 1993;61:1542–1545. doi: 10.1111/j.1471-4159.1993.tb13652.x. [DOI] [PubMed] [Google Scholar]
- 10.Ecker T, Gobel C, Hullin R, Rettig R, Seitz G, Hofmann F. Decreased cardiac concentration of cGMP kinase in hypertensive animals. An index for cardiac vascularization? Circ Res. 1989;65:1361–1369. doi: 10.1161/01.res.65.5.1361. [DOI] [PubMed] [Google Scholar]
- 11.El-Din El-Husseini A, Bladen C, Vincent SR. Molecular characterization of type II cyclic GMP-dependent protein kinase expressed in the rat brain. J Neurochem. 1995;64:2814–2817. doi: 10.1046/j.1471-4159.1995.64062814.x. [DOI] [PubMed] [Google Scholar]
- 12.Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- 13.Haley JE, Wilcox GL, Chapman PF. The role of nitric oxide in hippocampal long-term potentiation. Neuron. 1992;8:211–216. doi: 10.1016/0896-6273(92)90288-o. [DOI] [PubMed] [Google Scholar]
- 14.Jarchau T, Häusler C, Markert T, Pöhler D, Vandekekerckhove J, De Jonge HR, Lohmann SM, Walter U. Cloning, expression, and in situ localization of rat intestinal cGMP-dependent protein kinase II. Proc Natl Acad Sci USA. 1994;91:9426–9430. doi: 10.1073/pnas.91.20.9426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kantor DB, Lanzrein M, Stary SJ, Sandoval GM, Smith WB, Sullivan BM, Davidson N, Schuman EM. A role for endothelial NO synthase in LTP revealed by adenovirus-mediated inhibition and rescue. Science. 1996;274:1744–1748. doi: 10.1126/science.274.5293.1744. [DOI] [PubMed] [Google Scholar]
- 16.Lum-Ragan JT, Gribkoff VK. The sensitivity of hippocampal long-term potentiation to nitric oxide synthase inhibitors is dependent upon the pattern of conditioning stimulation. Neuroscience. 1993;57:973–983. doi: 10.1016/0306-4522(93)90042-e. [DOI] [PubMed] [Google Scholar]
- 17.Malinow R. Transmission between pairs of hippocampal slice neurons: quantal levels, oscillations, and LTP. Science. 1991;252:722–724. doi: 10.1126/science.1850871. [DOI] [PubMed] [Google Scholar]
- 18.O’Dell TJ, Hawkins RD, Kandel ER, Arancio O. Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proc Natl Acad Sci USA. 1991;88:11285–11289. doi: 10.1073/pnas.88.24.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perkel DJ, Petrozzino JJ, Nicoll RA, Connor JA. The role of Ca2+ entry via spatially activated NMDA receptors in the induction of long-term potentiation. Neuron. 1993;11:817–823. doi: 10.1016/0896-6273(93)90111-4. [DOI] [PubMed] [Google Scholar]
- 20.Pfeifer A, Aszodi A, Seidler U, Ruth P, Hofmann F, Fässler R. Intestinal secretory defects and dwarfism in mice lacking cGMP-dependent protein kinase II. Science. 1996;274:2082–2086. doi: 10.1126/science.274.5295.2082. [DOI] [PubMed] [Google Scholar]
- 21.Pfeifer A, Klatt P, Massberg S, Ny L, Sausbier M, Hirneiβ C, Wang G-X, Korth M, Aszódi A, Andersson K-E, Krombach F, Mayerhofer A, Ruth P, Hofmann F, Fässler R. Defective smooth muscle regulation in cGMP Kinase I-deficient mice. EMBO J. 1998;17:3045–3052. doi: 10.1093/emboj/17.11.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rankin PW, Jacobson EL, Benjamin RC, Moss J, Jacobson MK. Quantitative studies of inhibitors of ADP-ribosylation in vitro and in vivo. J Biol Chem. 1989;264:4312–4317. [PubMed] [Google Scholar]
- 23.Ruth P, Landgraf W, Keilbach A, May B, Egléme C, Hofmann F. The activation of cGMP-dependent protein kinase isoenzymes Iα and Iβ is determined by the different amino termini. Eur J Biochem. 1991;202:1339–1344. doi: 10.1111/j.1432-1033.1991.tb16509.x. [DOI] [PubMed] [Google Scholar]
- 24.Schuman EM, Madison DV. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science. 1991;254:1503–1506. doi: 10.1126/science.1720572. [DOI] [PubMed] [Google Scholar]
- 25.Schuman EM, Meffert MK, Schulman H, Madison DV. An ADP-ribosyltransferase as a potential target for nitric oxide action in hippocampal long-term potentiation. Proc Natl Acad Sci USA. 1994;91:11958–11962. doi: 10.1073/pnas.91.25.11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selig DK, Segal MR, Liao D, Malenka RC, Malinow R, Nicoll RA, Lisman JE. Examination of the role of cGMP in long-term potentiation in the CA1 region of the hippocampus. Learn Mem. 1996;3:42–48. doi: 10.1101/lm.3.1.42. [DOI] [PubMed] [Google Scholar]
- 27.Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, Kandel ER. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell. 1996;87:1015–1023. doi: 10.1016/s0092-8674(00)81796-1. [DOI] [PubMed] [Google Scholar]
- 28.Sullivan BM, Wong S, Schuman EM. Modification of hippocampal synaptic proteins by nitric oxide-stimulated ADP ribosylation. Learn Mem. 1997;3:414–424. doi: 10.1101/lm.3.5.414. [DOI] [PubMed] [Google Scholar]
- 29.Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial learning. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- 30.Uhler MD. Cloning and expression of a novel cyclic GMP-dependent protein kinase from mouse brain. J Biol Chem. 1993;268:13586–13591. [PubMed] [Google Scholar]
- 31.Wernet W, Flockerzi V, Hofmann F. The cDNA of the two isoforms of bovine cGMP-dependent protein kinase. FEBS Lett. 1989;251:191–196. doi: 10.1016/0014-5793(89)81453-x. [DOI] [PubMed] [Google Scholar]
- 32.Williams JH, Li X, Gu X, Jope RS. Modulation of endogenous ADP-ribosylation in rat brain. Brain Res. 1992;592:49–56. doi: 10.1016/0006-8993(92)91657-z. [DOI] [PubMed] [Google Scholar]
- 33.Williams JH, Li YG, Nayak A, Errington ML, Murphy KPSJ, Bliss TVP. The suppression of long-term potentiation in rat hippocampus by inhibitors of nitric oxide synthase is temperature and age dependent. Neuron. 1993;11:877–884. doi: 10.1016/0896-6273(93)90117-a. [DOI] [PubMed] [Google Scholar]
- 34.Wilson RI, Yanovsky J, Gödecke A, Stevens DR, Schrader J, Haas HL. Endothelial nitric oxide synthase and LTP. Nature. 1997;386:338. doi: 10.1038/386338a0. [DOI] [PubMed] [Google Scholar]
- 35.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Nature. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 36.Zhuo M, Small SA, Kandel ER, Hawkins RD. Nitric oxide and carbon monoxide produce activity dependent long-term synaptic enhancement in hippocampus. Science. 1993;260:1946–1950. doi: 10.1126/science.8100368. [DOI] [PubMed] [Google Scholar]
- 37.Zhuo M, Hu Y, Schultz C, Kandel ER, Hawkins RD. Role of guanylyl cyclase and cGMP-dependent protein kinase in long-term potentiation. Nature. 1994;368:635–639. doi: 10.1038/368635a0. [DOI] [PubMed] [Google Scholar]