

Abstract
Neurons are polarized cells, the activity of which relies on the morphological and functional differences between their axonal and somatodendritic domains. One mechanism for establishing and maintaining neuronal polarity is via the selective targeting of proteins to these domains. The endocytic pathway plays a major role in the generation and maintenance of cellular polarity by selectively sorting and recycling endocytosed plasma membrane proteins. In this study we first show that endogenous syntaxin 13 localizes to tubulovesicular organelles that are present in the somatodendritic and axonal domains of neurons. These organelles contain and actively recycle transferrin receptor and are sensitive to brefeldin A, suggesting that they are analogous to the tubulovesicular recycling endosomes in non-neuronal cells. We next use a syntaxin 13–GFP fusion protein transiently expressed in hippocampal neurons, together with time-lapse microscopy, to study the dynamics of the endosomal system in neurons. The analysis revealed the presence of two distinct classes of syntaxin 13-labeled endosomes: round–oval stationary organelles and highly mobile tubulovesicular structures. The dynamic population of tubulovesicular endosomes travels in both directions along microtubules in dendrites and axons. The mobile organelles appear to fuse with and bud from the stationary endosomes, possibly as a means of delivering and picking up their cargo.
Keywords: vesicular transport, endosomes, protein recycling, membrane trafficking, syntaxin, microtubules, hippocampal neurons
Neurons are highly polarized cells in which cellular morphology and accurate protein distribution lay the basis for their function: the ability to receive, process, and transmit information. To generate and maintain their polarity, neurons have to target proteins selectively to their axonal or somatodendritic domains. In addition to the direct sorting of newly synthesized proteins in distinct trafficking vesicles in the trans-Golgi network (TGN) (Matter and Mellman, 1994), the endocytic pathway also plays a major role in the generation and maintenance of cellular polarity by selectively sorting and recycling endocytosed plasma membrane proteins (Mellman, 1996; Robinson et al., 1996). The endocytic system comprises a series of heterogeneous organelles, the morphological and functional identities of which are becoming more defined (Mellman, 1996; Robinson et al., 1996). Early endosomes (EE) (Helenius et al., 1983; Mayor et al., 1993), late endosomes (LE), and recycling endosomes (RE) (Hopkins and Trowbridge, 1983; Gruenberg and Maxfield, 1995) are part of a system that correctly direct internalized proteins and lipids to the recycling and degradative pathways.
In neurons, although much effort has focused on elucidating the events underlying the exocytotic and endocytotic steps of the synaptic vesicle life cycle, less is known about the general endosomal system. Electron microscopy studies have shown the presence of extensive networks of tubular endosomes in dendrites and cell bodies whereas, in axons, early endosomes were found exclusively in the presynaptic terminals and in varicosities (Parton et al., 1992).
In the past few years a set of proteins, collectively named solubleN-ethylmaleimide-sensitive factor (NSF) attachment protein receptors (SNAREs), has emerged for which the function is to mediate and regulate membrane fusion events (Bennett and Scheller, 1993;Sollner et al., 1993). The prototypic interaction between SNAREs has been investigated extensively in the nerve terminal. The synaptic vesicle-associated membrane protein (VAMP) and the plasma membrane proteins syntaxin and synaptosomal-associated protein of 25 kDa (SNAP-25) form a four helix bundle structure that is thought to bring the two membranes into close apposition and directly drive the fusion event (Hanson et al., 1997; Lin and Scheller, 1997; Sutton et al., 1998). The interaction between SNAREs from donor and acceptor compartments throughout the cellular exocytotic and endocytotic pathways may be at the basis, at least in part, of the specificity of all vesicular trafficking steps in the cell.
Two members of the syntaxin family, syntaxin 7 (Wang et al., 1997; Wong et al., 1998) and syntaxin 13 (Prekeris et al., 1998), so far have been implicated in the endosomal pathway in non-neuronal cells. In nonpolarized cells syntaxin 13 is found primarily in tubular early and recycling endosomes, where it colocalizes with transferrin receptor (TfR) (Prekeris et al., 1998). Anti-syntaxin 13 antibodies inhibit TfR recycling in permeabilized PC12 cells, establishing a function for syntaxin 13 in the recycling of internalized proteins to the plasma membrane (Prekeris et al., 1998).
In this study we used a syntaxin 13–green fluorescent protein (GFP) fusion protein transiently expressed in hippocampal cultures, together with time-lapse microscopy, to gain insights into the dynamics of the endosomal system in neurons. Here we describe two distinct classes of syntaxin 13-labeled endosomes: round–oval stationary organelles and highly mobile tubulovesicular structures. The behavior of these organelles suggests the presence of a dynamic population of endosomes that travel in both directions along microtubules in dendrites and axons, possibly fusing with and budding from the stationary endosomes as a means of delivering and picking up their cargo.
MATERIALS AND METHODS
Antibodies and reagents. Rabbit polyclonal antibodies directed against syntaxin 13 have been described previously (Prekeris et al., 1998). The following mouse monoclonal antibodies were used: anti-MAP2 from Transduction Laboratories (Lexington, KY), anti-β-tubulin from Boehringer Mannheim (Indianapolis, IN), and anti-transferrin receptor from Zymed Laboratories (South San Francisco, CA). Secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA) and included fluorescein isothiocyanate (FITC)- and Texas Red (TxR)-conjugated AffiniPure goat anti-mouse IgG as well as FITC- and TxR-conjugated AffiniPure goat anti-rabbit IgG. FITC-conjugated transferrin (Tf), used at 80 μg/ml, was obtained from Molecular Probes (Eugene, OR). Brefeldin A (5 mg/ml stock solution in ethanol, used at 5 μg/ml), nocodazole (10 mg/ml stock solution in DMSO, used at 5 μg/ml), and taxol (paclitaxel, 10 mmstock solution in DMSO, used at 10 nm) were purchased from Calbiochem (La Jolla, CA). Equine serum was obtained from HyClone (Logan, UT). Unless otherwise stated, all other reagents were obtained from Sigma (St. Louis, MO) or Life Technologies (Grand Island, NY).
Hippocampal cultures and transfections. Primary cultures of hippocampal neurons were prepared from the hippocampi of 18-d-old fetal rats, as described previously (Banker and Cowan, 1977; Hazuka et al., 1999). Cultures were transfected at 8–10 d in vitro (DIV) with the calcium phosphate method primarily as described (Xia et al., 1996; Dudek et al., 1997). The glutamate receptor inhibitors kynurenate and d-2-amino-5-phosphonovaleric acid (d-APV) were not included in the transfection. Optimization of the volume of calcium phosphate/DNA precipitate, 120 μl per 60-mm-diameter dish, and of the duration of the incubation, typically 45 min, resulted in low levels of toxicity and transfection efficiencies of 5% or more. Cultures were used for immunocytochemistry or imaging experiments 2 d after transfection.
Immunocytochemistry. The cells were fixed in 4% formaldehyde and 120 mm sucrose in PBS for 20 min at room temperature (RT). After the formaldehyde was quenched with 0.1m glycine in PBS, the cells were permeabilized, and nonspecific sites were blocked in PBS containing 0.4% saponin, 2% normal goat serum, and 1% bovine serum albumin (permeabilization/blocking buffer). Primary antibodies, diluted in permeabilization/blocking buffer, were applied for 1 hr at RT. After the cells were rinsed five times for 5 min with PBS, secondary antibodies were applied for 1 hr at RT. Finally, the cells were washed five times for 5 min and mounted onto slides with Vectashield (Vector Laboratories, Burlingame, CA) as a mounting medium. Microscopy was performed with a Molecular Dynamics (Sunnyvale, CA) laser confocal imaging system (Stanford University, Cell Sciences Imaging Facility). To measure the overlap between two fluorophores in double-stained samples, we analyzed confocal images with the Molecular Dynamics MultiProbe 2010 image analysis software. The data shown are the means ± SE of at least three separate images.
Construction of the syntaxin 13–GFP fusion protein. The coding region of syntaxin 13 was amplified by using PCR. The amplicon was digested with the restriction enzymes EcoRI andXhoI (Promega, Madison, WI) and cloned in frame in the mammalian expression vector pEGFP-N3 (Clontech, Palo Alto, CA). The resulting construct was verified by DNA sequencing. Before use for transfections experiments in hippocampal neurons, the syntaxin 13–GFP construct was transfected into normal rat kidney (NRK) cells, and the expression of the fusion protein was analyzed by fluorescence microscopy as described previously (Chao et al., 1999).
Time-lapse microscopy. Hippocampal neurons on glass coverslips were mounted into an imaging chamber (Warner Instruments, Hamden, CT) containing culture medium. Cells were visualized with an Olympus IX70 inverted microscope and a 60× magnification oil immersion objective. The imaging chamber and the microscope stage were kept at 37°C. Sequential images were acquired with a CCD camera and Deltavision software. Exposure times were between 0.5 and 1 sec, the time lapse was either 2.5 or 3 sec, and 100–200 images were collected for each experiment. Recordings were analyzed on a Silicon Graphics Octane computer and Deltavision image analysis software. Selected sequential frames were processed for publication with Adobe Photoshop.
Fluorescence recovery after photobleaching (FRAP) analysis.Hippocampal neurons were transfected with the syntaxin 13–GFP construct and mounted into an imaging chamber as described above. Cells were visualized with a Molecular Dynamics laser confocal imaging system. The imaging chamber and the microscope stage were kept at 37°C. Portions of selected axons were bleached by scanning the area six times with the laser at 100% power. These conditions were sufficient to reduce the fluorescence of syntaxin 13–GFP by >95%. Sequential images were taken at different time points, using the laser at 10% power. These conditions did not result in significant additional bleaching (data not shown). The data are expressed as a percentage of the prebleaching value. Where indicated, LineScan analysis was performed by using the Measure 2-D 3.2 software (Molecular Dynamics).
RESULTS
Subcellular localization of syntaxin 13 in cultured hippocampal neurons
We previously reported that syntaxin 13 is localized to tubular extensions of the sorting endosomes as well as to tubulovesicular recycling endosomes in nonpolarized cells (Prekeris et al., 1998). To determine the subcellular localization of syntaxin 13 in neuronal cells, we stained embryonic and postnatal cultured hippocampal neurons with affinity-purified polyclonal antibodies raised against syntaxin 13. In both neuronal cultures syntaxin 13 was detected in small puncta throughout the cell, including soma and processes. Costaining of embryonic hippocampal cultures with antibodies against syntaxin 13 and MAP2 as a dendritic marker established the presence of syntaxin 13 in both the somatodendritic and axonal domains of the neurons (Fig.1). Previously, we have demonstrated that in non-neuronal cells syntaxin 13 is present in TfR-positive endosomes, where it mediates TfR recycling (Prekeris et al., 1998). To determine whether syntaxin 13 might be involved in a similar pathway in neurons, we costained hippocampal neurons for TfR and syntaxin 13 (Fig.2A–C). As in nonpolarized cells, 98.9 ± 0.3% of syntaxin 13 colocalized with TfR, and 99.0 ± 0.2% of TfR colocalized with syntaxin 13, strongly indicating that both proteins are located on the same organelles (Fig. 2A–C). The dynamics of membrane proteins in the presence of the fungal toxin brefeldin A (BFA) can reveal features of their native localization and life cycle. The addition of BFA to a wide variety of established cell lines results in the disassembly of the Golgi apparatus and redistribution of its components into the endoplasmic reticulum (ER) via the formation of an extensive network of tubulovesicular structures extending from the Golgi apparatus along microtubules (Lippincott-Schwartz et al., 1991;Reaves and Banting, 1992; Mundigl et al., 1993). The drug also affects the endosomal system and TGN (Lippincott-Schwartz et al., 1991; Reaves and Banting, 1992). Analogous to the mixing of the Golgi with the ER, the TGN mixes with the recycling endosomal system. To determine whether syntaxin 13-positive endosomes recycle via BFA-sensitive compartments, we treated differentiated hippocampal neurons with BFA before costaining them for TfR and syntaxin 13. In agreement with earlier studies in non-neuronal cells, BFA treatment caused the tubulation of syntaxin 13-containing endosomes that also costained for TfR (Fig.2D–F). The presence of the tubules was not restricted to the soma and extended toward the distal ends of the dendrites (Fig. 2D–F), indicating the presence of recycling endosomes throughout the dendrites as an extension of the endosomal network located in the soma. To confirm further the identity of the syntaxin 13-positive organelles as recycling endosomes, we incubated hippocampal neurons with TxR-labeled Tf. Cells fixed immediately after the incubation showed that the syntaxin 13-positive organelles were loaded effectively with TxR–Tf (Fig. 2G–I). As expected for recycling endosomes, a chase of 45 min in the absence of labeled Tf resulted in the complete TxR–Tf unloading of the syntaxin 13-positive organelles (Fig.2J–L).
Fig. 1.

Syntaxin 13 is localized to tubulovesicular structures present in the somatodendritic and axonal domains of mature cultured hippocampal neurons. Embryonic hippocampal neurons 14 DIV were fixed and stained with antibodies against syntaxin 13 (A, E) and MAP-2 (B, F). C, Merged image of A and B. D, G, DIC images. E–G, Higher magnification of part of the field in A–D, showing the presence of syntaxin 13-labeled organelles in both dendrites and axons.Largearrowheads point to dendrites;small arrows point to axons. Scale bars:A–D, 10 μm; E–G, 2 μm.
Fig. 2.

Syntaxin 13 in dendrites localizes to organelles that contain TfR, tubulate in the presence of BFA, and actively recycle labeled Tf. A–C, Embryonic hippocampal neurons 11 DIV were fixed and stained with antibodies against syntaxin 13 (A) and TfR (B),C, Shown is the merge of the two images.D–F, Hippocampal neurons were treated with 5 μg/ml BFA for 15 min before being fixed and stained for syntaxin 13 (D) and TfR (E).F, Shown is the merged image. G–L, Hippocampal neurons were loaded with 80 μg/ml TxR–Tf for 30 min and either fixed immediately (G–I) or chased for 45 min in medium without labeled Tf (J–L) before fixation. Then the cultures were stained for syntaxin 13 (G, J) and imaged to detect TxR–Tf (H, K); I and L show the respective merged images. Scale bars: A–C, 2 μm;D–F, 5 μm; G–L, 20 μm.
Although in dendrites syntaxin 13 colocalized with TfR, syntaxin 13-positive endosomes in axons did not stain for TfR, nor could they be loaded with TxR–Tf (data not shown), consistent with the reports that TfR is absent from axons (Parton et al., 1992; Mundigl et al., 1993). The presence of punctate syntaxin 13 staining in axons raised the possibility that syntaxin 13 might be involved in the formation and/or recycling of synaptic vesicles. To test these possibilities, we costained hippocampal neurons for syntaxin 13 and known SV markers such as SV2a (Fig. 3) and synaptotagmin (data not shown). In embryonic hippocampal cultures of 11 DIV the axons, which by running along dendrites established mature synaptic contacts, did not show a restricted or enriched localization of syntaxin 13 at the synapses (Fig. 3, compare A–C). In the same cultures the axons developing in isolation did not possess mature synapses. Staining with SV2a showed a punctate labeling pattern that likely represents the well characterized mobile clusters of synaptic vesicles (Kraszewski et al., 1995) (Fig. 3E). The syntaxin 13 staining pattern in isolated axons also appeared punctate (Fig.3D,F), with 5.0 ± 1.5% of SV2a colocalized with syntaxin 13. Likewise, 4.4 ± 1.9% of axonal syntaxin 13 colocalized with SV2a. As previously reported (Mundigl et al., 1993), in these cultures some neurons have axons that, at variable distance from the cell body, become very large and flat. These giant axons, with a width of 40 μm or more, offer a better spatial resolution for the localization of subcellular organelles at the light microscopy level. Taking advantage of their presence, we again performed double staining with antibodies against syntaxin 13 and SV2a. As in immature axons, syntaxin 13 showed only 1.6 ± 1.3% overlap with SV2a, and SV2a showed 1.4 ± 1.2% overlap with syntaxin 13 (Fig.3G–I). At least some of the syntaxin 13-positive organelles are probably early endosomes, because they could be loaded with HRP, a bulk endocytotic marker (Fig. 3J–L, arrows). Taken together, these studies established the presence of syntaxin 13 in the early/recycling endosomal compartments in neurons analogous to its localization in non-neuronal cells. Moreover, the data suggest that syntaxin 13 likely does not play a role in the recycling of SV proteins in mature nerve terminals despite the fact that the presence of sorting endosomes in synaptic terminals has been reported (Parton et al., 1992). Additionally, the overlap of syntaxin 13 staining and HRP indicates the presence of syntaxin 13 in early endosomes in the axon.
Fig. 3.

Syntaxin 13 is not enriched at mature synapses. Embryonic hippocampal neurons 11 DIV were fixed and stained for syntaxin 13 (A, D, G) and SV2a (B, E, H); the respective merged images are shown in C, F, and I, with areas of overlap inyellow. A–C, Portion of an axon running along a dendrite. Note that mature synapses, marked by the SV2a staining, do not show enrichment of syntaxin 13. D–F, Isolated immature axon that does not contact any dendrites. The SV2a staining likely represents mobile clusters of synaptic vesicles.G–I, Giant axon. J–L, Giant axon of embryonic hippocampal neuron 11 DIV that was loaded first with 10 mg/ml HRP–biotin for 30 min and then was fixed and stained for syntaxin 13 (J). HRP–biotin was detected by using streptavidin–TxR (K); L shows the merged image, with areas of overlap in yellow. Scale bars: A–C, 1 μm; D–I, 2 μm;J–L, 10 μm.
Time-lapse microscopy analysis of endocytic trafficking in dendrites
Using syntaxin 13 as a marker for recycling endosomes, we sought to gain insights into the dynamics of the endocytic pathway in living neurons. To this end we constructed a fusion protein between syntaxin 13 and the GFP (Chao et al., 1999), transfected hippocampal neurons in culture, and imaged the labeled endosomes by time-lapse microscopy. Syntaxin 13 is a type II integral membrane protein, and linking GFP to its C terminus results in a fluorescent tag on the luminal side of the endosomes. This topology minimizes the risk of interference between GFP and the cytoplasmically located functional domains of syntaxin 13. Our laboratory has reported previously that syntaxin 13–GFP transfected in NRK cells is targeted appropriately and inserted into the endosomal membrane, with the GFP tag oriented in the lumen of the endocytic organelle (Chao et al., 1999). In transfected hippocampal cultures syntaxin 13–GFP showed a staining pattern very similar to that obtained by staining untransfected cells for the endogenous protein (compare Fig. 4B,E for syntaxin 13–GFP with 1A and 2A for the endogenous protein). To analyze further the localization of syntaxin 13–GFP, we also stained transfected neurons for TfR. As in nontransfected neurons a majority (90.7%) of syntaxin 13-containing organelles also contained TfR, indicating that syntaxin 13–GFP is localized predominantly to tubulovesicular recycling endosomes (Fig.4A–C). The correct localization of the fusion protein was also confirmed by the tubulation of these organelles in response to BFA treatment (data not shown). In addition, syntaxin 13–GFP-containing organelles could, like the structures containing the endogenous protein, be loaded and unloaded with Tf–TxR (Fig.4D–I). These results indicate that syntaxin 13–GFP is targeted to the correct endosomal compartment and that its expression does not cause malfunctioning of the endosomal system. Therefore syntaxin 13–GFP appears to be a suitable marker to examine the endocytic pathway in neurons.
Fig. 4.

Syntaxin 13–GFP displays a subcellular localization equivalent to that of the endogenous protein.A–C, Embryonic hippocampal neurons 11 DIV transfected with syntaxin 13–GFP were fixed and stained with antibodies against TfR (A) and were imaged to detect syntaxin 13–GFP (B); C shows the merge of the two images, with areas of overlap in yellow.D–I, Hippocampal neurons expressing syntaxin 13–GFP were loaded with 80 μg/ml TxR–Tf for 30 min and either fixed immediately (D–F) or chased for 45 min in medium without labeled Tf before fixation (G–I). Then the cultures were imaged to detect syntaxin 13–GFP (E, H) and TxR–Tf (D, G); F andI show the respective merged images. Scale bars, 5 μm.
We first sought to gain insights into the dynamics of syntaxin 13-labeled endosomes in dendrites. Hippocampal neurons in culture were transfected with syntaxin 13–GFP and imaged by time-lapse microscopy, recording the localization and movement of the syntaxin 13–GFP-labeled endosomes. The analysis revealed the presence of two classes of labeled organelles (Fig. 5). Some of the endosomes had a round–oval morphology, variable size of ∼1 by 0.5 μm, and appeared to be mostly stationary (Fig. 5, large arrowheads). The second class of endosomes had a tubulovesicular morphology, variable length of ∼1–2 μm, and displayed fast movement with the appearance of an almost continuous flow both in the retrograde and anterograde direction (Fig. 5, small arrows). Although because of the resolution limitations imposed by the light microscopic analysis we cannot discount the possibility that the organelles simply are aggregating and separating, our observations are suggestive of fusion and budding events between the mobile tubulovesicular organelles and the stationary organelles (Fig. 5,asterisk). So, while the function of these mobile tubular endosomes remains to be elucidated, we suggest that they are involved in the bidirectional movement of cargo from and to the soma, probably using the larger stationary endosomes as relay stations to pick up or deliver their cargo. Indeed, we observe similar mobile structures in hippocampal neurons preloaded with TxR–Tf (data not shown).
Fig. 5.
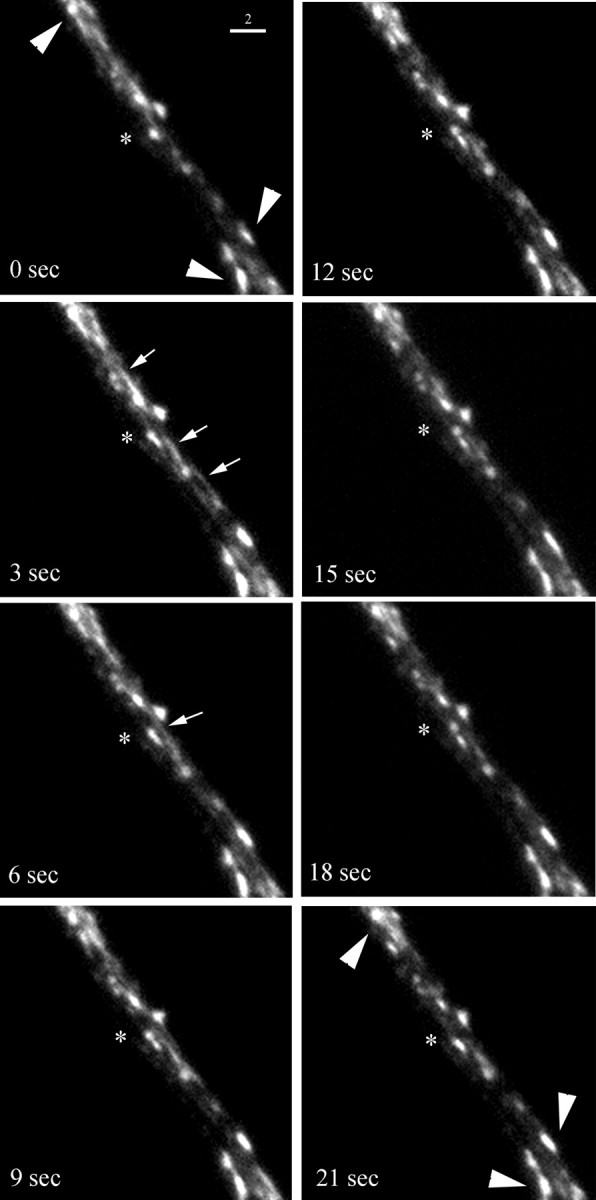
Time-lapse microscopy analysis of syntaxin 13–GFP-labeled endosomes in dendrites. Embryonic hippocampal neurons 11 DIV were transfected with syntaxin 13–GFP and imaged by time-lapse microscopy (eight sequential frames from a 5 min recording are shown).Large arrowheads point to stationary endosomes.Small arrows point to highly mobile tubular endosomes. The asterisk marks putative fusion and budding events. Scale bar, 2 μm.
Time-lapse microscopy analysis of endocytic trafficking in axons
To analyze the kinetics of single endosomal organelles, we imaged their movement in axons, taking advantage of the presence of fewer endosomes in thinner processes as compared with dendrites. As in dendrites, axons contained round–oval stationary endosomes (Figs.6A,C, 7A, large arrowheads) as well as highly mobile tubulovesicular endosomes (Figs. 6A–C, 7A,B, small arrows). Although a slight preference for retrograde movement was observed (67.7 ± 4%; n = 26 organelles from three separate axons), the tubular endosomes clearly moved bidirectionally (Fig. 7B; the two organelles first moved apart in opposite directions and then reversed their movement for few seconds before changing direction again). In most cases the tubular organelles seemed to pass by stationary endosomes, approaching them on one side of the axonal process and reappearing on the other side in consecutive frames (Fig. 6A,B, small arrows). As in dendrites, although acknowledging the possibility that the organelles might simply be aggregating and separating, we observed putative fusion and budding events. We detected tubular endosomes moving toward stationary endosomes, coalescing with them, and not reappearing in consecutive frames (Fig. 6C). In other cases we observed mobile organelles that appeared to be budding from and fusing with stationary endosomes (Fig. 7A, asterisk). When followed over the length of the recording, the tubular endosomes exhibited a saltatory behavior, with periods of movement interrupted by stationary phases. The resulting apparent speed of the organelles (the total distance traveled divided by time) varied greatly from 0.2 to 0.53 μm/sec (Fig. 8A, Table 1). This difference in speed was, however, mostly attributable to the different amount of time that the organelles spent in the stationary periods, because the actual speed (calculated after subtracting the stationary phases) for all of the examined organelles was not significantly different (Table 1). These rates are comparable with those reported for the fast axonal transport (Allen et al., 1982; Parton et al., 1992).
Fig. 6.
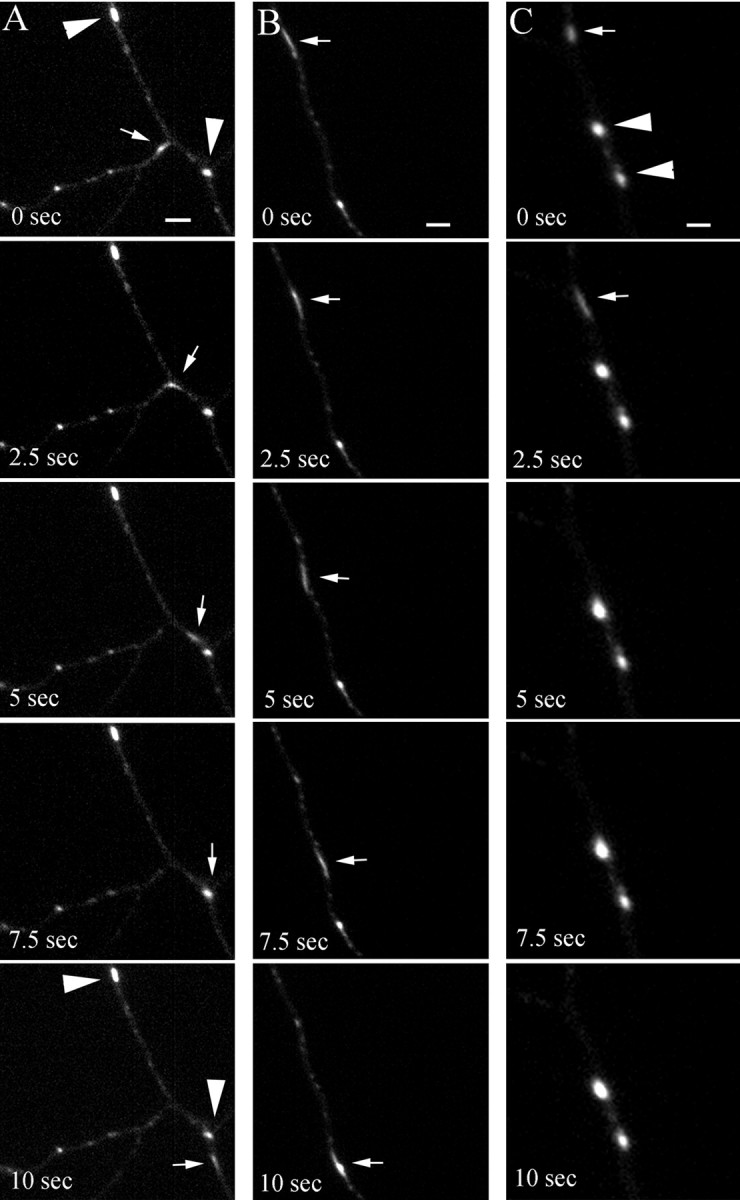
Time-lapse microscopy analysis of syntaxin 13–GFP-labeled endosomes in axons. Embryonic hippocampal neurons 11 DIV were transfected with syntaxin 13–GFP and imaged by time-lapse microscopy (selected sequential frames from 4 min recordings are shown). Large arrowheads point to stationary endosomes;small arrows point to highly mobile tubular endosomes. Scale bar, 2 μm.
Fig. 7.
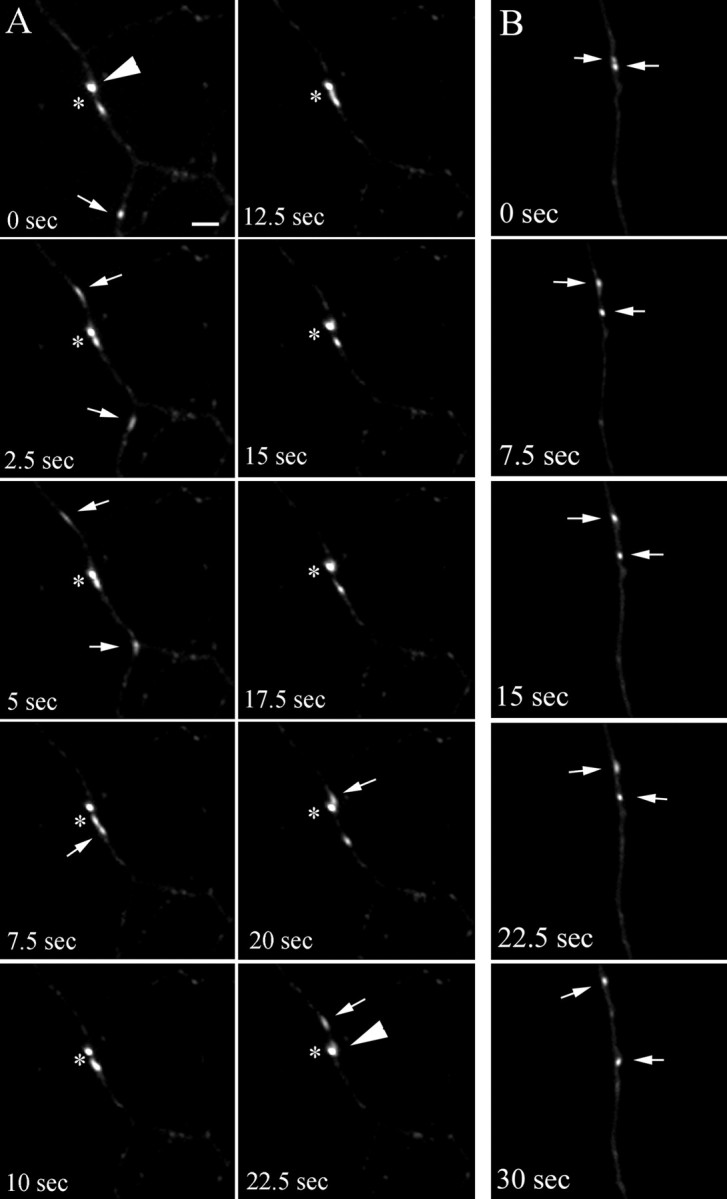
Time-lapse microscopy analysis of syntaxin 1–GFP-labeled endosomes in axons. Embryonic hippocampal neurons 11 DIV were transfected with syntaxin 13–GFP and imaged by time-lapse microscopy (selected sequential frames from 4 min recordings are shown). Large arrowheads point to stationary endosomes;small arrows point to highly mobile tubular endosomes that appear to be budding from the stationary endosomes (A) or that appear to move bidirectionally in the same axon (B). The asterisk inA marks putative fusion and budding events. Scale bar, 2 μm.
Fig. 8.
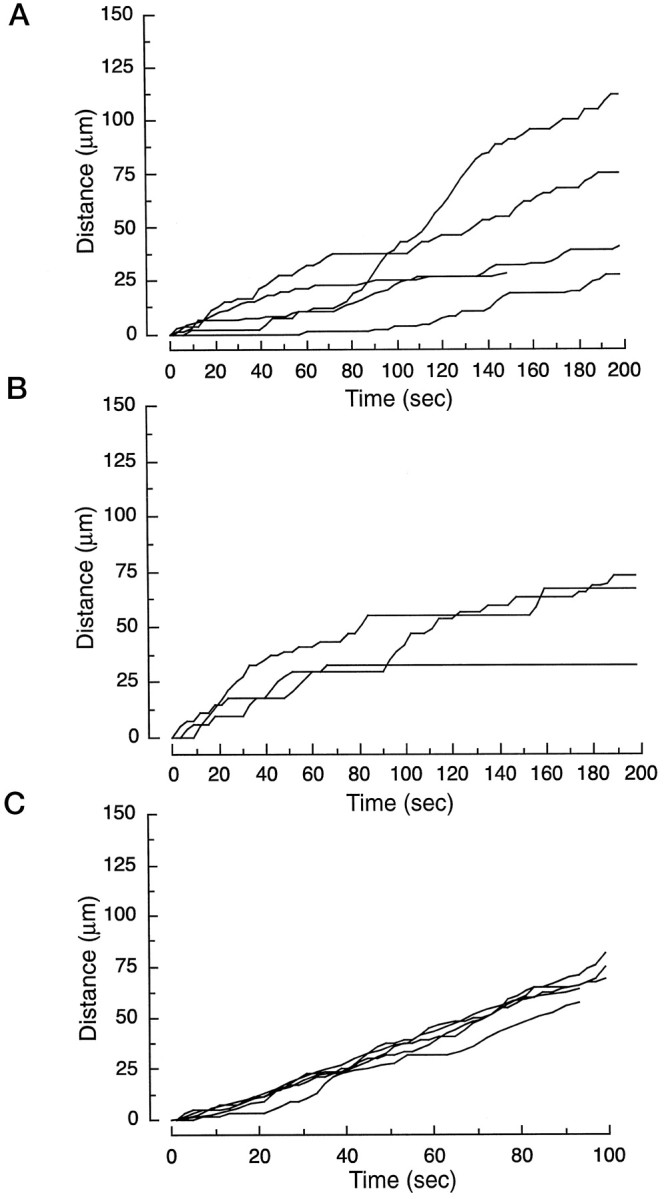
Kinetic analysis of syntaxin 13–GFP-labeled endosomes in axons. Embryonic hippocampal neurons 11 DIV were transfected with syntaxin 13–GFP and imaged by time-lapse microscopy. The movement of single organelles was traced and plotted versus time.A, Traces of five randomly selected organelles from three untreated neurons. B, Traces of three randomly selected organelles from three different neurons treated with 5 μg/ml nocodazole; cultures on the microscope stage were perfused with the drug at time 0. C, Traces of five randomly selected organelles from three different neurons pretreated with 10 nm taxol for 30 min.
Table 1.
Effect of microtubule stability on endosomal dynamics in hippocampal neurons
| Treatment organelles | Apparent speed (μm/sec) | Actual speed (μm/sec) | On/off ratio | Stationary (% total) |
|---|---|---|---|---|
| Untreated | 0.34 ± 0.11 | 0.85 ± 0.1 | 0.42 ± 0.06 | 60.6 ± 6.1 |
| Nocodazole (5 μg/ml) | 0.22 ± 0.05 | 1.09 ± 0.02 | 0.2 ± 0.05 | 91.8 ± 1.2 |
| Taxol (10 nM) | 0.72 ± 0.03 | 0.91 ± 0.1 | 0.81 ± 0.06 | 53.3 ± 2.3 |
The data shown are the means of five traces ± SEM for untreated and taxol-treated axons and means of three traces ± SEM for nocodazole-treated axons. In all cases data were collected from three different neurons. The apparent speed represents the total distance traveled by the organelles divided by time. The actual speed represents the speed of the organelles after subtracting the stationary periods. The On/off ratio is the ratio between the mobile time and total measured time. The fraction of stationary organelles was determined from either four (untreated and taxol-treated) or three (nocodazole-treated) axons.
Effect of microtubule stability on axonal and dendritic endosomal trafficking
It is widely accepted that microtubule-dependent movement is a predominant means of axonal and dendritic transport. Depolymerization of microtubules (MT) results in the inhibition of protein as well as phospholipid transport from the cell soma to the axonal and dendritic processes (Zakharenko and Popov, 1998). To determine whether an intact MT network is also necessary for endosomal trafficking in differentiated neurons, we used nocodazole to depolymerize MT in living hippocampal neurons. Treatment of hippocampal cultures for 30 min with 5 μg/ml nocodazole resulted in the depolymerization of MT, as clearly shown in the giant axons of neurons stained with antibodies against β-tubulin (Fig. 9A,B). The effect of nocodazole was reversible, because 2 hr after the drug was removed the MT network had fully re-formed (Fig. 9C). To analyze the effect of MT depolymerization on axonal endosomal trafficking in living cells, we again used syntaxin 13–GFP-transfected hippocampal neurons. Because a substantial number of axonal endosomes are stationary even in untreated cells, we analyzed the effect of MT depolymerization on the mobile tubular endosomes. Cultures on the microscope stage were perfused with nocodazole, and the recordings were started immediately, allowing us to follow the full course of the drug effects. The presence of nocodazole resulted in almost complete cessation of endosomal movement (see Fig. 8B). As expected, nocodazole decreased the apparent speed of the endosomes in axons, an effect that was attributable mainly to an increase in the stationary periods rather than to a decrease in the actual speed (see Fig. 8B, Table 1). Indeed, whereas in the first phase after the addition of the drug the organelles traveled with an actual speed similar to that observed in untreated cells, with increasing depolymerization of MT their stationary periods increased up to a full stop (see Fig. 8, Table 1). A similar effect of MT depolymerization was observed on the endosomes in dendrites, where nocodazole treatment resulted in the inhibition of endosomal movement (Fig. 9G). The most striking and reversible morphological effect of the drug was the formation of large accumulations of syntaxin 13 staining toward the distal end of the dendrites (Fig. 9D–F, arrow inG, I). These structures, which also costained for TfR (Fig. 9D–F), seemed to be generated by a partial retraction of the dendritic tip and left behind an area largely devoid of staining (Fig. 9I) in contrast to untreated neurons, in which syntaxin 13-labeled endosomes were present all the way to the tip of the dendrite (Fig.9H). At the light microscopy level we could not determine whether these accumulations were endosomal aggregates or whether they actually represented enlarged organelles resulting from the fusion between multiple endosomes. These data suggest that the efficiency of endosomal trafficking depends on the stability of the MT network. The local depolymerization of MT would result in the dissociation of organelles from tracks, increasing the duration and frequency of the stationary periods. To look further into this possibility, we analyzed the movement of syntaxin 13-containing endosomes in hippocampal neurons treated with 10 nm taxol. Taxol, a drug isolated from the bark of the yew tree, binds tubulin and suppresses the dynamic instability of MT. Thus, taxol treatment should stabilize the MT network and influence the role of local MT depolymerization on endosomal trafficking. Indeed, taxol treatment resulted in a dramatic increase in the apparent speed of the endosomes as a consequence of a decrease in the duration and frequency of the stationary periods (see Fig. 8C, Table 1). The actual speed was not affected significantly by taxol (Table 1).
Fig. 9.
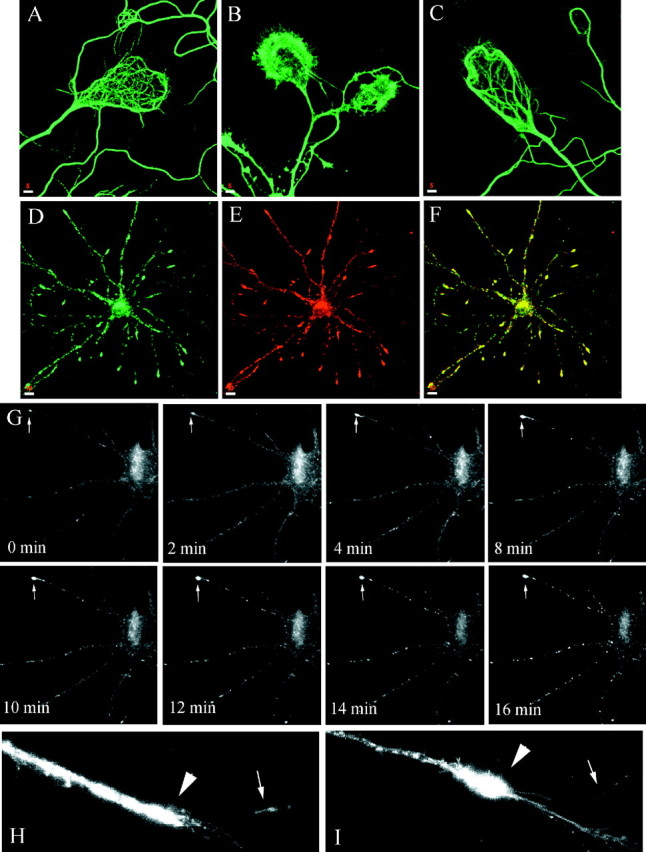
Nocodazole effect on localization and dynamics of syntaxin 13-containing endosomes in dendrites. A–C, Embryonic hippocampal neurons 11 DIV were fixed and stained with antibodies against β-tubulin before (A) and after treatment with 5 μg/ml nocodazole for 30 min (B); the MT network clearly visible in the growth cones was disrupted completely by the drug. Then 2 hr after the drug was removed, the MT had re-formed (C).D–F, Hippocampal neurons were treated with 5 μg/ml nocodazole for 30 min and then were fixed and stained for syntaxin 13 (D) and TfR (E).F, Shown is a merged image of D andE, with areas of overlap in yellow.G, Hippocampal neurons 11 DIV were transfected with syntaxin 13–GFP and imaged by video microscopy. Cultures on the microscope stage were perfused with 5 μg/ml nocodazole at time0. H, I, Higher magnification of the distal portion of a dendrite in an untreated cell (H) or after nocodazole treatment (I). Scale bars:A–C, 5 μm; D–F, 10 μm.
The time-lapse microscopy data suggest the presence, both in dendrites and axons, of two classes of syntaxin 13-labeled endosomes: round–oval stationary and mobile tubulovesicular organelles. However, we cannot discount completely the possibility that the stationary organelles are simply “stalled” mobile organelles. The apparent difference in the shape of mobile and stationary organelles could be caused by the movement itself. Indeed, changes in shape in relation to the movement have been observed in several other GFP-labeled organelles (Hirschberg et al., 1998). It is possible that the round–oval organelles stretch into tubules when they begin to move and that the tubular organelles collapse into a round–oval shape when they stop moving. To address this issue, we determined the relative number of stationary organelles in untreated as well as nocodazole- and taxol-treated axons. Although nocodazole substantially increased the relative number of stationary organelles (as the mobile structures were coming to a stop because of MT depolymerization), taxol did not have any effect on the ratio between mobile and stationary organelles (Table 1). Thus, these data are consistent with the existence of two distinct classes of syntaxin 13–GFP-positive organelles.
To characterize further their dynamics and relationship to each other, we used FRAP to bleach a selected area in the axonal tree of syntaxin 13–GFP-transfected hippocampal neurons. After bleaching, we quantitated the recovery of the GFP signal in the bleached area. As shown in Figure 5 for dendrites and in Figures 6 and 7 for axons, the greatest portion of the fluorescent signal in any given field is contributed by the large stationary endosomes. In untreated cells the signal recovered to 60 ± 4% of its original intensity (before bleaching) after 5 min (Fig. 10). This time course and the lack of mobility of the stationary endosomes argue against a simple “moving in” of this class of endosomes in the bleached field. The data are more consistent with the movement of unbleached tubulovesicular endosomes into the bleached area and their fusion with the stationary endosomes. After several such fusion events unbleached syntaxin 13–GFP would have exchanged for the bleached fusion protein in the stationary endosomes, leading to the recovery of their fluorescent signal. Indeed, we observed recovery of the fluorescent signal in stationary endosomes that seemed to be at the same position before bleaching and at the end of the recovery time (data not shown). As expected, the treatment of cultures with 5 μg/ml nocodazole for 30 min almost completely abolished the extent of the recovery (Fig. 10). Interestingly, hippocampal neurons treated with 10 nm taxol for 30 min also were unable to recover the fluorescent signal (Fig. 10). As we showed in Figure 8 and Table 1, taxol does not inhibit the movement of the tubulovesicular endosomes. Rather it decreases the frequency and length of the stationary periods. Although the mechanism of taxol inhibition on fluorescence recovery remains unclear, one possibility is that in the presence of the drug the mobile tubulovesicular organelles could not disengage from the MT and therefore were unable to fuse with other compartments in the axons, impairing the efficient delivery of unbleached syntaxin 13–GFP.
Fig. 10.

Recovery of syntaxin 13–GFP fluorescent signal after bleaching. Embryonic hippocampal neurons 11 DIV were transfected with syntaxin 13–GFP. Selected areas of the axonal network of transfected cells were bleached, and the recovery of the fluorescent signal was followed over a time course of 5 min. The quantitation of the fluorescent signal is expressed as a percentage of the value before bleaching for untreated cells (○), cells treated with 5 μg/ml nocodazole for 30 min (●), and cells treated with 10 nmtaxol for 30 min (▿). The data are the means ± SEM of four independent experiments for untreated and nocodazole-treated cells and the means ± SEM of eight independent experiments for taxol-treated cells.
To analyze further the syntaxin 13–GFP fluorescence recovery and to address the question of possible preferential movement of organelles from the proximal or distal end into the bleached segment, we performed a LineScan analysis on axons before and after photobleaching as well as during fluorescence recovery. The fluorescence was measured along the selected fragment of the axon, starting at the distal end of the axon, and was plotted as a function of the distance (data not shown). The fluorescence recovery appeared to progress from both the proximal (somatodendritic) and distal ends of the bleached area in agreement with the bidirectional movement of the syntaxin 13-labeled organelles. Interestingly, small fluorescence peaks appeared in the bleached area within the first minute of the recovery, whereas large peaks started to appear only after 5 min and did not reach their original intensity until after 10 min of the recovery. We interpret these results as an indication that, after photobleaching, small mobile organelles rapidly moved in the bleached area and started exchanging unbleached syntaxin 13–GFP with the large stationary organelles that contained bleached protein, gradually restoring their fluorescence signal.
DISCUSSION
Although in recent years much has been learned about the mechanisms underlying the exocytosis and recycling of synaptic vesicles (Cameron et al., 1993; Kraszewski et al., 1995), less is known about the dynamics and function of the general endosomal/lysosomal system in neuronal cells. This is partly because the boundaries between the different endocytic compartments remain difficult to define. Indeed, the existence of several distinct endosomal/lysosomal compartments still remains controversial, and the characterization of these compartments very often is based entirely on their morphological description in fixed cells. Furthermore, the endocytic organelles are highly dynamic and their structure is remodeled continuously, making it difficult to appreciate their three-dimensional organization. Finally, the asynchronous internalization of typical endocytic markers does not allow discrimination of the multiple endocytic compartments. The spatial separation of subcellular domains within the dendrites and axons of neurons makes embryonic hippocampal neurons in culture an excellent system to investigate the pathways of endosomal recycling. In this study we used syntaxin 13–GFP as a fluorescent marker for tubulovesicular recycling endosomes (Prekeris et al., 1998; Chao et al., 1999), in combination with time-lapse microscopy, to analyze the dynamics of endosomal trafficking in living neurons.
In dendrites, the syntaxin 13-positive endosomes could be clearly divided in two morphologically different types of organelles. We observed round–oval organelles with an apparent size of ∼1 by 0.5 μm and tubulovesicular organelles with a variable length of ∼1–2 μm. The round–oval endosomes were mostly stationary, whereas the tubular endosomes exhibited a high degree of mobility. We observed membrane dynamics suggestive of putative fusion and budding events between the two types of organelles consistent with the hypothesis that the mobile tubulovesicular endosomes might shuttle cargo from and to the stationary endosomes. The stationary endosomes may be analogs of the vacuolar-sorting endosomes that have been described in several nonpolarized cell lines (Helenius et al., 1983; Mayor et al., 1993). Our data are in agreement with earlier studies that suggested the presence of an extensive tubulovesicular endosomal network in dendrites (Parton et al., 1992; Mundigl et al., 1993). The high degree of subcellular colocalization of syntaxin 13 and TfR-positive organelles suggests that endocytosed Tf and syntaxin 13 are present in the same tubulovesicular recycling endosomes. Indeed, our laboratory (data not shown) and other groups (Cameron et al., 1993) have shown the existence of mobile tubulovesicular endosomes that can be labeled efficiently with exogenously applied Tf–TxR or Tf–HRP. Thus, we suggest that syntaxin 13-positive tubular endosomes are involved in the trafficking of endocytosed membrane proteins throughout the dendrites. It remains to be determined whether syntaxin 13 mediates the endosomal trafficking in dendrites or whether it is merely a cargo protein. Nevertheless, in the light of recent studies of syntaxin 13 role in Tf recycling (Prekeris et al., 1998), it is tempting to speculate that syntaxin 13 might be the SNARE involved in endosome–endosome fusion within the tubulovesicular endosomal network.
Our studies additionally revealed the presence of an extensive endosomal network in axons that also consists of stationary endosomes and mobile tubulovesicular endosomes. After photobleaching the syntaxin 13–GFP-positive endosomes in segments of axons, we observed a rapid microtubule-dependent recovery of the fluorescent signal in the stationary endosomes, perhaps as a result of the transport of unbleached syntaxin 13–GFP from both the proximal (somatodendritic) and distal ends of the bleached area. The existence of a continuous tubulovesicular compartment has been described in other polarized cells, including Madin-Darby canine kidney (MDCK) cells (Gibson et al., 1998). Different exogenous tracers, including Tf, EGF, and Ig, appeared to accumulate in endosomal tubules from which they later were sorted and delivered to the different subcellular domains (Gibson et al., 1998). Thus, it is tempting to speculate that neurons also might possess a similar continuous tubulovesicular network that is responsible for the sorting and polarized delivery of endocytosed plasma membrane proteins.
Most of the protein synthesis and degradation compartments are centrally located in the cell body. Thus, to maintain the ongoing protein and lipid turnover in the cell periphery, neurons must have an efficient long-range organelle transport system. Our observations suggest that syntaxin 13-positive tubular endosomes move predominantly along microtubules. These results are consistent with earlier studies in nonpolarized cells, in which recycling has been shown to be dependent on an intact microtubule network and the exit of recycling proteins from the early endosomes is blocked by nocodazole, a MT-depolymerizing agent (Yamashiro et al., 1984). MT-dependent transport is achieved via molecular motors belonging to the kinesin or dynein family of proteins (Hirokawa et al., 1998). Kinesins are plus end-directed motor proteins (Vale et al., 1985; Vale and Fletterick, 1997), whereas dyneins move cargo toward the MT minus end (Hirokawa et al., 1990). It remains to be established which of the molecular motors are involved in transport of tubular endosomes. Nevertheless, because endosomal movement is bidirectional, members from both protein families are expected to mediate the trafficking of tubular endosomes. This bidirectional movement is likely to be especially important for endosomal trafficking in axons because, unlike dendrites, axons have a uniform MT polarity. Interestingly, in axons we rarely observed single organelles reversing their movement. Thus, we speculate that the association of tubular endosomes with specific motor proteins might determine the type and direction of their movement. However, the mechanisms that determine the specificity of the interactions between motor proteins and different endosomes remain to be determined.
Kinesins remain bound to MT while undergoing multiple rounds of activity (Vale and Fletterick, 1997). The average distance traveled by one kinesin molecule after binding to a MT is ∼600 nm, as determined by in vitro motility studies (Vale et al., 1996). In living cells, single organelles likely are associated with multiple kinesin molecules, so that the traveled distance is much longer than 600 nm. The length of a single MT, however, is much shorter than the length of a dendrite or axon. Thus, the “trafficking endosome” would be expected to “fall off” its track and stop moving once it reaches the end of the microtubule. To resume movement, the “trafficking endosome” then would need to reattach to another MT. Indeed, our data clearly show that endosomes move along microtubules in a staggered way, with periods of steady movement interrupted by stationary phases of various lengths. The stationary periods presumably represent times when the endosomes are dissociated from the MT. Supporting this model, depolymerization of the MT network with nocodazole resulted in the inhibition of the endosomal movement, mainly by increasing the length and frequency of the stationary periods. Moreover, taxol, a drug that inhibits the dynamic instability of microtubules, significantly increased the apparent speed of the tubular endosomes by decreasing the length and frequency of the stationary periods. Interestingly, the inhibition of the dynamic microtubule instability prevented recovery of the syntaxin 13–GFP fluorescence in the FRAP assay. A similar effect of taxol has been described for the delivery of cell body-derived vesicles to the growth cone region. Although the movement of the vesicles was not affected, their fusion with the plasma membrane was inhibited dramatically (Zakharenko and Popov, 1998). Thus, we propose that the association of the tubular endosomes with the microtubules inhibits their ability to fuse with stationary endosomes. Although the mechanism of such inhibition remains unclear, one possible explanation is that the tethering of organelles to MT imposes physical constraints on their ability to interact with other endosomes. Alternatively, the molecular motor proteins could inhibit the fusion directly by binding to proteins that are necessary for the endosome–endosome fusion event.
Our work suggests the existence of a dynamic tubulovesicular recycling endosomal network in axons and dendrites, the function of which is regulated via the microtubule network. After dissociating from the MT tracks, the endosomes can fuse with other compartments or associate with another MT. The probability of an organelle associating with a MT will depend on the availability of MT tracks. The intrinsic instability of the MT network is regulated in the cell extensively. In vivo, the turnover of microtubules is much more rapid as compared with that of MT formed from purified tubulin (Belmont et al., 1990;Verde et al., 1992). This is attributable in large part to the increase in the frequency of the transitions from the polymerization phase to the depolymerization phase, known as the frequency of catastrophe (Belmont et al., 1990; Verde et al., 1992). This high frequency of catastrophe in vivo is believed to result from the action of numerous cellular proteins that disassemble the stabilizing caps at the ends of the MT strands. Several microtubule-associated proteins (MAPs) such as MAP2, tau, and MAP4 have been identified (Hirokawa, 1994;Mandelkow and Mandelkow, 1995). It will be interesting to see whether these proteins affect the efficiency of endosomal trafficking. Moreover, the differential localization of MAPs within mature neurons (Binder et al., 1985) raises the intriguing possibility that neurons might have different mechanisms of regulating endosomal trafficking in dendrites and axons.
Footnotes
We acknowledge Dr. Christopher D. Hazuka for help in culturing embryonic hippocampal neurons. We thank Dr. Susan L. Palmieri, Dr. Stephen J. Smith, and Daniel S. Chao for assistance with confocal and time-lapse microscopy, as well as Susanne Ahmari for help with FRAP studies. We also thank Kelly C. Lee for the critical reading of this manuscript.
R.P. and D.L.F. contributed equally to this work.
Correspondence should be addressed to Dr. Richard H. Scheller at the above address. E-mail: scheller@cmgm.stanford.edu.
REFERENCES
- 1.Allen RD, Metuzals J, Tasaki I, Brady ST, Gilbert SP. Fast axonal transport in squid axon. Science. 1982;218:1127–1128. doi: 10.1126/science.6183744. [DOI] [PubMed] [Google Scholar]
- 2.Banker GA, Cowan MW. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–425. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- 3.Belmont LD, Hyman AA, Sawin KE, Mitchison TJ. Real-time visualization of cell cycle-dependent changes in microtubule dynamics in cytoplasmic extracts. Cell. 1990;62:579–589. doi: 10.1016/0092-8674(90)90022-7. [DOI] [PubMed] [Google Scholar]
- 4.Bennett MK, Scheller RH. The molecular machinery for secretion is conserved from yeast to neurons. Proc Natl Acad Sci USA. 1993;90:2559–2563. doi: 10.1073/pnas.90.7.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Binder LI, Frankfurter A, Rebhum LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cameron PL, Mundigl O, De Camilli P. Traffic of synaptic vesicle proteins in polarized and nonpolarized cells. J Cell Sci Suppl. 1993;17:93–100. doi: 10.1242/jcs.1993.supplement_17.14. [DOI] [PubMed] [Google Scholar]
- 7.Chao DS, Hay JC, Winnick S, Prekeris R, Klumperman J, Scheller RH. SNARE membrane trafficking dynamics in vivo. J Cell Biol. 1999;144:869–881. doi: 10.1083/jcb.144.5.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 9.Gibson A, Futter CE, Maxwell S, Allchin EH, Shipman M, Kraehenbuhl JP, Domingo D, Odorizzi G, Trowbridge IS, Hopkins CR. Sorting mechanisms regulating membrane protein traffic in the apical transcytosis pathway of polarized MDCK cells. J Cell Biol. 1998;143:81–94. doi: 10.1083/jcb.143.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gruenberg J, Maxfield FR. Membrane transport in the endocytic pathway. Curr Opin Cell Biol. 1995;7:552–563. doi: 10.1016/0955-0674(95)80013-1. [DOI] [PubMed] [Google Scholar]
- 11.Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell. 1997;90:523–535. doi: 10.1016/s0092-8674(00)80512-7. [DOI] [PubMed] [Google Scholar]
- 12.Hazuka CD, Foletti DL, Hsu SC, Kee Y, Hopf W, Scheller RH. The Sec6/8 complex is located at neurite outgrowth and axonal synapse–assembly domains. J Neurosci. 1999;19:1324–1334. doi: 10.1523/JNEUROSCI.19-04-01324.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helenius A, Mellman I, Wall D, Hubbart A. Endosomes. Trends Biochem Sci. 1983;8:245–250. [Google Scholar]
- 14.Hirokawa N. Microtubule organization and dynamics dependent on microtubule-associated proteins. Curr Opin Cell Biol. 1994;6:74–81. doi: 10.1016/0955-0674(94)90119-8. [DOI] [PubMed] [Google Scholar]
- 15.Hirokawa N, Yoshida Y, Sato-Yoshitake R. Brain dynein (MAP1C) localizes on both anterogradely and retrogradely transported organelles in vivo. J Cell Biol. 1990;111:1027–1037. doi: 10.1083/jcb.111.3.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirokawa N, Noda Y, Okada Y. Kinesin and dynein superfamily proteins in organelle transport and cell division. Curr Opin Cell Biol. 1998;10:60–73. doi: 10.1016/s0955-0674(98)80087-2. [DOI] [PubMed] [Google Scholar]
- 17.Hirschberg K, Miller CM, Ellenberg J, Presley JF, Siggia ED, Phair RD, Lippincott-Schwartz J. Kinetic analysis of secretory protein traffic and characterization of Golgi to plasma membrane transport intermediates in living cells. J Cell Biol. 1998;143:1485–1503. doi: 10.1083/jcb.143.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hopkins CR, Trowbridge IS. Internalization and processing of transferrin and transferrin receptor in human carcinoma A431 cells. J Cell Biol. 1983;97:508–521. doi: 10.1083/jcb.97.2.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kraszewski K, Mundigl O, Daniell L, Verderio C, Matteoli M, De Camilli P. Synaptic vesicle dynamics in living cultured hippocampal neurons visualized with CY3-conjugated antibodies directed against the luminal domain of synaptotagmin. J Neurosci. 1995;15:4328–4342. doi: 10.1523/JNEUROSCI.15-06-04328.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin RC, Scheller RH. Structural organization of the synaptic exocytosis core complex. Neuron. 1997;19:1087–1094. doi: 10.1016/s0896-6273(00)80399-2. [DOI] [PubMed] [Google Scholar]
- 21.Lippincott-Schwartz J, Yuan LC, Tipper C, Amherdt M, Orci L, Klausner R. Brefeldin A's effects on endosomes, lysosomes, and the TGN suggest a general mechanism for regulating organelle structure and membrane traffic. Cell. 1991;67:601–616. doi: 10.1016/0092-8674(91)90534-6. [DOI] [PubMed] [Google Scholar]
- 22.Mandelkow E, Mandelkow E-M. Microtubules and microtubule-associated proteins. Curr Opin Cell Biol. 1995;7:72–81. doi: 10.1016/0955-0674(95)80047-6. [DOI] [PubMed] [Google Scholar]
- 23.Matter KS, Mellman I. Mechanisms of cell polarity: sorting and transport in epithelian cells. Curr Opin Cell Biol. 1994;6:545–554. doi: 10.1016/0955-0674(94)90075-2. [DOI] [PubMed] [Google Scholar]
- 24.Mayor S, Presley JF, Maxfield FR. Sorting of membrane components from endosomes and subsequent recycling to the cell surface. J Cell Biol. 1993;121:1257–1269. doi: 10.1083/jcb.121.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mellman I. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- 26.Mundigl O, Matteoli M, Daniell L, Thomas-Reetz A, Metcalf A, Jahn R, De Camilli P. Synaptic vesicle proteins and early endosomes in cultured hippocampal neurons: differential effects of brefeldin A in axon and dendrites. J Cell Biol. 1993;122:1207–1221. doi: 10.1083/jcb.122.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parton RG, Simons K, Dotti CG. Axonal and dendritic endocytic pathways in cultured neurons. J Cell Biol. 1992;119:123–137. doi: 10.1083/jcb.119.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prekeris R, Klumperman J, Chen YA, Scheller RH. Syntaxin 13 mediates cycling of plasma membrane proteins via tubulovesicular recycling endosomes. J Cell Biol. 1998;143:957–971. doi: 10.1083/jcb.143.4.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reaves B, Banting G. Perturbation of the morphology of the trans-Golgi network following brefeldin A treatment: redistribution of a TGN-specific integral membrane protein TGN38. J Cell Biol. 1992;116:85–94. doi: 10.1083/jcb.116.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson MS, Watts C, Zerial C. Membrane dynamics in endocytosis. Cell. 1996;84:13–21. doi: 10.1016/s0092-8674(00)80988-5. [DOI] [PubMed] [Google Scholar]
- 31.Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly–disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 32.Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4A resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 33.Vale RD, Fletterick RJ. The design plan of kinesin motors. Annu Rev Cell Dev Biol. 1997;13:745–777. doi: 10.1146/annurev.cellbio.13.1.745. [DOI] [PubMed] [Google Scholar]
- 34.Vale RD, Reese TS, Sheetz MP. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 1985;42:39–50. doi: 10.1016/s0092-8674(85)80099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vale RD, Funatsu T, Pierce DW, Romberg L, Harada Y, Yanagida TY. Direct observation of a single kinesin molecule moving along microtubules. Nature. 1996;380:451–453. doi: 10.1038/380451a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verde F, Dogterom M, Stelzer E, Karsenti E, Leibler S. Control of microtubule dynamics and length by cyclin A- and cyclin B-dependent kinases in Xenopus egg extracts. J Cell Biol. 1992;118:1097–1108. doi: 10.1083/jcb.118.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang H, Frelin L, Pevsner J. Human syntaxin 7: a pep12p/Vps6p homologue implicated in vesicle trafficking to lysosomes. Gene. 1997;199:39–48. doi: 10.1016/s0378-1119(97)00343-0. [DOI] [PubMed] [Google Scholar]
- 38.Wong SH, Xu Y, Zhang T, Hong W. Syntaxin 7, a novel syntaxin member associated with the early endosomal compartment. J Biol Chem. 1998;273:375–380. doi: 10.1074/jbc.273.1.375. [DOI] [PubMed] [Google Scholar]
- 39.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamashiro D, Tycko B, Fluss S, Maxfield FR. Segregation of transferrin to a mildly acidic (pH 6.5) para-Golgi compartment in the recycling pathway. Cell. 1984;37:789–800. doi: 10.1016/0092-8674(84)90414-8. [DOI] [PubMed] [Google Scholar]
- 41.Zakharenko S, Popov S. Dynamics of axonal microtubules regulate the topology of new membrane insertion into the growing neurites. J Cell Biol. 1998;143:1077–1086. doi: 10.1083/jcb.143.4.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]