

Abstract
Cognitive decline is apparent in the early stages of Huntington's disease and progressively worsens throughout the course of the disease. Expression of the human Huntington's disease mutation in mice (R6/2 line) causes a progressive neurological phenotype with motor symptoms resembling those seen in Huntington's disease. Here we describe the cognitive performance of R6/2 mice using four different tests (Morris water maze, visual cliff avoidance, two-choice swim tank, and T-maze). Behavioral testing was performed on R6/2 transgenic mice and their wild-type littermates between 3 and 14.5 weeks of age, using separate groups of mice for each test. R6/2 mice did not show an overt motor phenotype until ∼8 weeks of age. However, between 3.5 and 8 weeks of age, R6/2 mice displayed progressive deterioration in specific aspects of learning in the Morris water maze, visual cliff, two-choice swim tank, and T-maze tasks. The age of onset and progression of the deficits in the individual tasks differed depending on the particular task demands. Thus, as seen in humans with Huntington's disease, R6/2 mice develop progressive learning impairments on cognitive tasks sensitive to frontostriatal and hippocampal function. We suggest that R6/2 mice provide not only a model for studying cognitive and motor changes in trinucleotide repeat disorders, but also a framework within which the functional efficacy of therapeutic strategies aimed at treating such diseases can be tested.
Keywords: transgenic mice, Huntington's disease, cognition, behavior, Morris water maze, T-maze
Expansions of unstable CAG trinucleotide repeats within the coding regions of target genes cause at least eight different genetic neurodegenerative diseases, the most prevalent being Huntington's disease (HD) (for review, see Paulson, 1999). HD is characterized by motor, cognitive, and psychological disturbances (Vonsattel and DiFiglia, 1998). Although HD is traditionally considered to be a “motor” disorder, cognitive decline is an early and pivotal feature (Mohr et al., 1991; Foroud et al., 1995; Lange et al., 1995; Lawrence et al., 1996, 1998).
The primary neuropathology in HD is a selective neuronal loss in the striatum and cortex followed by more widespread atrophy and neuronal loss in other brain regions in the later stages of the disease (Vonsattel and DiFiglia, 1998). Several animal models for HD have been described, including lesions of the striatum induced by excitotoxins (e.g., quinolinic acid) and metabolic poisons (e.g., 3-nitropropionic acid). These models show striatal pathology similar to that seen in HD (Coyle and Schwarcz, 1976; McGreer and McGreer, 1976; Beal et al., 1986, 1993; Bossi et al., 1993; Brouillet et al., 1993, 1995), and also replicate some of the motor and cognitive symptoms of the disease (Borlongan et al., 1995; Brouillet et al., 1995; Furtado and Mazurek, 1996; Palfi et al., 1996; Emerich et al., 1997; Kodsi and Swerdlow, 1997; Shear et al., 1998a,b). However, a major disadvantage of these neurotoxic models is that they lack the genetic pathogenesis and the progressive nature of HD.
The recent development of transgenic mouse models of HD (Mangiarini et al., 1996; Reddy et al., 1998; Hodgson et al., 1999; Schilling et al., 1999; Shelbourne et al., 1999) provide new ways of examining the mechanisms underlying the progression of HD as well as the genetic basis of the disease. The R6/2 transgenic mouse, which expresses the first exon of the human HD gene carrying 141–157 CAG repeat expansions, develops a number of the key features of HD, including the progressive motoric deterioration (Mangiarini et al., 1996; Dunnett et al., 1998; Carter et al., 1999) and the appearance of neuronal intranuclear inclusions (NIIs) (Davies et al., 1997).
Cognitive function in R6/2 mice has not been well studied. We reported recently that 8-week-old R6/2 transgenic mice are impaired in a spatial navigation cognitive task using the Morris water maze (Murphy et al., 1998). However, although R6/2 mice showed a clear deficit in performance of this task, the design of the Morris water maze task was such that the relative contribution of motor, sensory, and cognitive factors could not easily be resolved. Hence, the slower learning of R6/2 mice may have reflected, in part, a motor and/or visual deficit, rather than a purely cognitive impairment.
In the present study, a series of tests were chosen in which the cognitive tasks could be manipulated separately from the sensory, motor, and motivational conditions within each test. We have used four cognitive tests (the Morris water maze, visual cliff avoidance, two-choice swim tank, and T-maze) to examine spatial, visual, reversal, and alternation discrimination learning and memory.
MATERIALS AND METHODS
Animals (R6/2 transgenic mice). The R6/2 line of transgenic mice was generated as previously described (Mangiarini et al., 1996). A colony of R6/2 transgenic mice was established in the Department of Pharmacology, University of Cambridge, and the line was maintained by back-crossing onto CBA × C57BL/6 F1 animals. All mice used in the study were taken from the 14–19th generations of back-crossing.
Mice were housed together in numerical birth order in groups of mixed genotype, and data were recorded for analysis by mouse number. All mice were tested during the light phase of a 12 hr light/dark cycle. Until the appearance of the hindlimb grooming behavior, transgenic mice could not be distinguished by observation from littermates in their home cage. Therefore, until the grooming behavior appeared (between 8 and 9 weeks of age; see Mangiarini et al., 1996), the experimenters were blind to the genotype of the mice. Although data collected after the onset of an overt phenotype was not collected blind, it should be noted that the home cage observation for overt phenotype was performed and recorded separately from the behavioral tests. Because the abnormal grooming in its early stage is difficult to distinguish from normal grooming behaviors, this meant that until the abnormal grooming movements occurred regularly, the experimenters did not routinely know the genotype of a particular mouse (see below). Once the grooming behavior and other phenotypic changes became pronounced (usually between 10 and 14 weeks), the experiments could no longer be conducted blindly.
R6/2 mice suffer from diabetes (Hurlbert et al., 1999). In our colony, diabetes is common in R6/2 mice >12–14 weeks of age. However, we have never seen elevated blood glucose levels in mice <8 weeks of age (our unpublished observations). Therefore, during the critical testing period of our study (3–8 weeks of age), any deleterious neurological effects reported to be associated with diabetes are unlikely to contribute to the behavioral phenotype that we describe in the present study.
Genotyping. Genotyping was confirmed by PCR based on a modification of the method of Mangiarini et al. (1996). Tail tips were removed from each mouse at 3 weeks of age, and DNA extraction and PCR were subsequently performed as described (Mangiarini et al., 1996), with the exception that the following primers were used in the place of those previously published: 31329 HD (5′ to 3′: ATG AAG GCC TTC GAG TCC CTC AAG TCC TTC) and 33934 HD (5′ to 3′: GGC GGC TGA GGA AGC TGA GGA). In all cases, mice that exhibited a progressive impairment in the behavioral tests (see below) were genotyped to be transgenic (and vice versa).
Behavioral testing. Behavioral testing for spatial, visual, reversal, and alternation discrimination learning and memory was assessed in R6/2 transgenic mice using the Morris water maze, visual cliff avoidance, two-choice swim tank, and T-maze tests. Testing started at 3 weeks of age, at which time transgenic mice displayed no overt detectable motor deficits.
Separate groups of mice were tested for each of the spatial, visual, reversal, and alternation discrimination learning and memory tasks. During the acquisition phase of the Morris water maze and two-choice swim tank tests, five transgenic mice consistently failed to show motivation for swimming and made no attempt to escape from the water tank. These mice (designated “floaters” on the two tests) consistently exhibited latencies of >60 or >120 sec, respectively. Data from these mice were excluded from the analysis. During habituation in the T-maze, two transgenic mice did not eat the banana-flavored pellets each day, hence data from these mice were also excluded from the analysis.
Morris water maze. Spatial and nonspatial learning was assessed in a Morris water maze modified for use in mice (Stewart and Morris, 1994). A circular water tank, made from black polypropylene (diameter, 100 cm; height, 40 cm) was filled to a depth of 25 cm with water (23°C) and rendered opaque by the addition of a small amount of nontoxic white paint powder. Four positions around the edge of the tank were arbitrarily designated north (N), south (S), east (E), and west (W); this provided four alternative start positions and also defined the division of the tank into four quadrants: NE, SE, SW, and NW. A square clear Perspex escape platform (10 × 10 × 2 cm) was submerged 0.5 cm below the water surface and placed at the midpoint of one of the four quadrants. A video camera was fixed 1.6 m above the center of the swim tank, and all swimming trials were recorded. Mice were tested daily, from 3 weeks of age, over 19 d.
On the first 3 d, mice were trained on a visible platform task. The platform was made visible by the attachment of a high-contrast striped flag. Mice were trained for 4 trials per day (with an intertrial interval of ∼10 min). The start position (N, S, E, or W) and the location of the platform (NE, SE, SW, or NW) were pseudorandomized across trials. Mice were allowed up to 60 sec to locate the escape platform, and their escape latency and pathlengths were recorded. Mice that failed to locate the platform within the time limit were ascribed an escape latency of 60 sec and were placed on the platform by hand. All mice were then allowed to stay on the platform for 15 sec, before being removed and returned to the home cage during the intertrial interval.
From day 4, mice were trained for four trials per day to swim to the submerged platform, which was now “hidden” by the removal of the flag. The platform remained in the midpoint of the SW quadrant. Training continued for 11 d. On day 14 the mice received a single probe trial, during which the escape platform was removed from the tank, and the swimming path of each mouse was recorded over 60 sec while it searched for the missing platform. The mice then received a further four trials with the hidden escape platform returned to the SW quadrant.
From day 15, reversal training commenced. The escape platform was moved to the midpoint of the NE quadrant, and the mice were trained to swim to this new position for four trials per day, over days 15–19. All trials were videotaped and subsequently analyzed manually using purpose-designed image analysis software (HVS, Hampton, UK).
Visual cliff avoidance. To assess visual acuity, several groups of age-matched mice were tested for their ability to avoid a visual cliff. Visual cliff avoidance was tested in an open-topped box (60 × 60 cm square × 30 cm high; Dunnett et al., 1998). The four walls of the box were made from white plywood, and the base was made from clear Perspex. The box was positioned on the edge of a laboratory bench so that half of the base was placed on the bench (“bench side”), and the other half over the edge of the bench, suspended 1 m above the floor (“open side”). Anglepoise lamps (60 W) were positioned 60 cm above and 60 cm below the base of the box. The lamps were positioned in such a way to highlight the edge of the bench (the “visual cliff”) and to illuminate both the bench and open side of the box. Mice were placed in a central 7 × 5 cm “start area”, in the middle of the base at the edge of the cliff, and their activity was recorded for 5 min using a video camera fixed 1 m above the center of the box. Three separate groups of mice (aged 3, 4, and 6 weeks of age at the start of testing) were used, with each group of mice thereafter being retested on a weekly basis until 10 weeks of age. The videos were subsequently analyzed for the percentage of time each mouse spent in the start area, bench side and open side of the box, and in which direction the first step outside of the start area was taken.
Two-choice swim tank. Acquisition of a simple left–right visual discrimination task was performed in a modified version of the swim tank test as described previously (Carter et al., 1999). Briefly, the two-choice swim tank is a water-filled corridor adapted from a glass aquarium, 100-cm-long and 6-cm-wide, and filled to a depth of 20 cm with water maintained at a temperature of 23°C. The swim tank was completely surrounded by 1-m-high gray boards, thus totally obscuring surrounding spatial cues in the experimental room. Two vertical black lines on the side of the swim tank marked a horizontal distance 40 cm from either end of the tank and provided a 20 cm start area in the middle. A visible escape platform made from black Perspex (6-cm-square, 20.5-cm-high, with the top surface 0.5 cm above the water level) was placed in a pseudorandom order at either the left or right end of the swim tank for each trial. At the beginning of each trial, mice were placed in the start area facing one side wall so that no directional bias for swimming was given.
In acquisition training, a 60 W Anglepoise light was positioned over the escape platform; in reversal learning, the light was positioned over the end of the swim tank opposite to the platform. The main light source in the experimental room was dimmed to provide a greater contrast between the lit and unlit ends of the swim tank. Thus, mice were trained to swim toward or away from the light stimulus to reach the escape platform in the acquisition and reversal training, respectively.
During acquisition, mice were given 10–20 trials per day (with an intertrial interval of 5–30 min) for 7 d. On each trial, a mouse was considered to have made a correct choice if, and only if, it swam directly to the platform. An incorrect choice was recorded if (1) the mouse swam out of the start area in the opposite direction, (2) the mouse swam out of the start area in the correct direction but returned across the start area, or (3) the mouse failed to reach the platform within 120 sec. Analysis was based on the percentage of correct choices of the first 10 trials performed each day. Additional training trials were given each day until the mice made 10 correct choices to a maximum of 20 trials in total.
A first study was performed in which 3-week-old mice were trained for 7 d in this manner, within which time they reached criterion level (≥90% correct choices of their first 10 trials). These mice were then retested twice weekly to assess retention of learning and ability to discriminate a light stimulus over time. On their final day of testing, at 9 weeks of age, the light stimulus over the escape platform was removed to investigate the importance of the light cue in this task.
In the second experiment, four additional groups of mice were subjected to 7 d of acquisition training. These mice were aged 3, 5, 7, and 10 weeks at the start of testing. After acquisition training, each group was allowed to rest for 3 d before undergoing a single day of reversal training. During reversal training all mice were given a total of 30 trials, with an intertrial interval of 5–30 min. Data were analyzed in blocks of five trials, and the mean number of correct choices within each trial block was used for analysis. The performance criterion was set at achieving four correct trials in a five trial block within the 30 trials. On each trial, mice were considered to have made a correct choice when they swam directly toward the escape platform and an incorrect choice when they swam in the opposite direction to the escape platform.
T-maze. Alternation, spatial, and nonspatial learning was assessed in a T-maze adapted from a radial eight-arm maze for mice (Molinari et al., 1996). A T-maze was formed by using the central enclosure and three of the arms; two at 180° to each other, and one at 90° to these (the stem of the T). The maze was constructed from clear Perspex with access to the remaining five arms prevented by clear Perspex guillotine doors that remained lowered throughout the study. The stem and arms of the T were each 45-cm-long and 8-cm-wide and surrounded by a 20-cm-high clear Perspex enclosure. Clear Perspex guillotine doors provided entry to each arm, and these doors could be raised and lowered remotely. A 10-cm-long area at the end of the stem of the T provided the start area. The maze was elevated 50 cm above the floor. A number of distal cues were placed around the maze. Reinforcement was provided by a 20 mg banana-flavored pellet (Bio-serve) placed in a white cup at the end of each arm. Mice were maintained on a 16 hr schedule of food deprivation throughout the period of testing in the T-maze; water was available ad libitum. Body weights were monitored daily and maintained at 85% free-feeding weight.
Before training, all mice were given 3 d of 6 min/d access to the baited maze with the three doors open to habituate them to the environment. The number of arms entered and the number of pellets consumed during this phase did not differ between R6/2 and control mice, providing evidence against any form of nonspecific motor and motivational impairment. Mice were then given 3 d of 10 “forced” alternation trials before reinforced alternation training began.
At the start of each alternation training session, both arms were baited, and all guillotine doors were closed. The mouse was then placed in the start area and left for 10 sec before all three doors were raised simultaneously. After the mouse's entry into an arm (defined as placing all four feet in the arm), all doors were reclosed, confining the mouse to its chosen arm. The mouse was allowed to receive its food reward and then returned to the start area. The first trial of any session each day was a “free choice” trial in which a food pellet was placed in both left and right food cups, and the result was taken as the mouse's initial choice, after which the mouse was required to alternate 10 times to receive its food reward. On each subsequent trial, a mouse was considered to have made a correct choice if it entered the opposite arm to that last visited. An error was recorded if the mouse entered the same arm to that last visited. The process was repeated with as little intertrial delay as possible, until the mouse had completed the required number of trials (criterion being 10 correct choices). Analysis was based on the number of correct trials of the first 10 trials and the total number of errors made. The latency between leaving the start area and entering the chosen arm was also recorded. Mice were given one training session a day, and additional trials were given until the mice made 10 correct choices. Training continued until mice reached a performance level of eight or nine correct choices of the first 10 trials.
Black–white discrimination. Acquisition of the black–white discrimination task was also performed in the T-maze. One of the arms was entirely covered with black cardboard (black), and the other was covered with thick translucent tracing paper with a 60 W Anglepoise lamp positioned directly above (white). Although these coverings totally obscured surrounding spatial cues in the room, they provided a dark arm and a contrasting light arm. One half of each group of mice received reinforcement in the dark arm; the other half received reinforcement in the light arm. The position of the arms in the apparatus remained on the same side from trial to trial with the constraint that for half of each group the reinforced arm was on the left, and for other half on the right. This allowed mice to solve the discrimination from spatial, visual, or positional information. Each mouse was placed in the start area and remained there for 10 sec until all doors were raised simultaneously. After the mouse's entry into one of the arms, all doors were reclosed, confining the mouse to its chosen arm. The mouse was allowed to receive its food reward and then returned to the start area. On each trial, a mouse was considered to have made a correct choice if it entered into the same reinforced arm to that last visited. An error was recorded if the mouse entered into the opposite arm to that last visited; hence, the mice had to learn which arm was correct. Mice received up to 20 trials each day until the criterion of five consecutive correct trials had been achieved. The choice of arm and latency between leaving the start area and entering the chosen arm was recorded on each trial. After acquisition, the dark and light arm positions were reversed, whereby the respective reinforced arm was positioned on the opposite side of the maze to the previous session. Training continued again to a criterion of five consecutive correct trials. This reversal procedure was repeated five times. Analysis was based on the number of trials to criterion of five consecutive correct choices and the number of errors made.
After black–white discrimination, mice were trained on the same procedure without the black–white cues. Distal cues were reintroduced around the maze as for alternation training. On the sixth and eleventh reversal the maze remained in the same position, but a fourth arm, 180° to the stem of the T, was introduced as an alternative start arm. Mice were placed in each start arm on alternate trials. On each trial, a mouse was considered to have made a correct choice if it entered into the same arm to that last visited. An error was recorded if the mouse entered into the opposite arm to that last visited. Hence, on each trial the mice now had to make a different turn to make a correct choice, relying entirely on spatial information and removing any possible contribution to solving the task from positional information. In this way, the proportion with which each group of mice used spatial versus positional cues to solve the task was assessed.
Statistical analysis. Behavioral data were subjected to ANOVA (Genstat software package, version 3.2) with two between-subject factors (genotype and gender) and with repeated measures on one within-subject factor (age/day/block of trials) as appropriate to the particular test. In cases of a significant interaction (genotype × age/day/block of trials) Sidak's test was used for multiple independent post hoc pairwise comparisons between transgenic and wild-type mice at each relevant age, day, or block of trials (Rohlf and Sokal, 1995). Within each group, changes in performance with training were evaluated using Dunnett's test. A critical value for significance of p < 0.05 was used throughout the study.
In no case was there any significant genotype × gender interactions. Consequently, although the data from males and females were separated in all analyses, data has been pooled for clarity of presentation of the results.
RESULTS
Morris water maze
Nonspatial learning
The visible platform version of the Morris water maze test was used to test nonspatial learning and to assess whether spatial learning deficits detected in the R6/2 line are a result of deficient escape motivation or impairment of visual and/or motor performance. As illustrated in Figure 1, both groups showed marked improvements in escape latencies over the 3 d of training (day, F(2,82) = 54.21;p < 0.001). However, the two groups did not differ either in overall level of performance or change across days [genotype, F(1,41) = 0.00; genotype × day, F(2,82) = 2.24, both not significant (NS)]. An identical pattern of results was seen in the pathlength measure (Fig. 1B; day,F(2,82) = 69.74, p < 0.001; genotype, F(1,41) = 0.21; genotype × day, F(2,82) = 3.48, both NS). The swim speed of R6/2 mice did not differ from that of controls on any day (Fig. 1C; genotype,F(1,41) = 0.95; genotype × day,F(2,82) = 0.66, both NS).
Fig. 1.
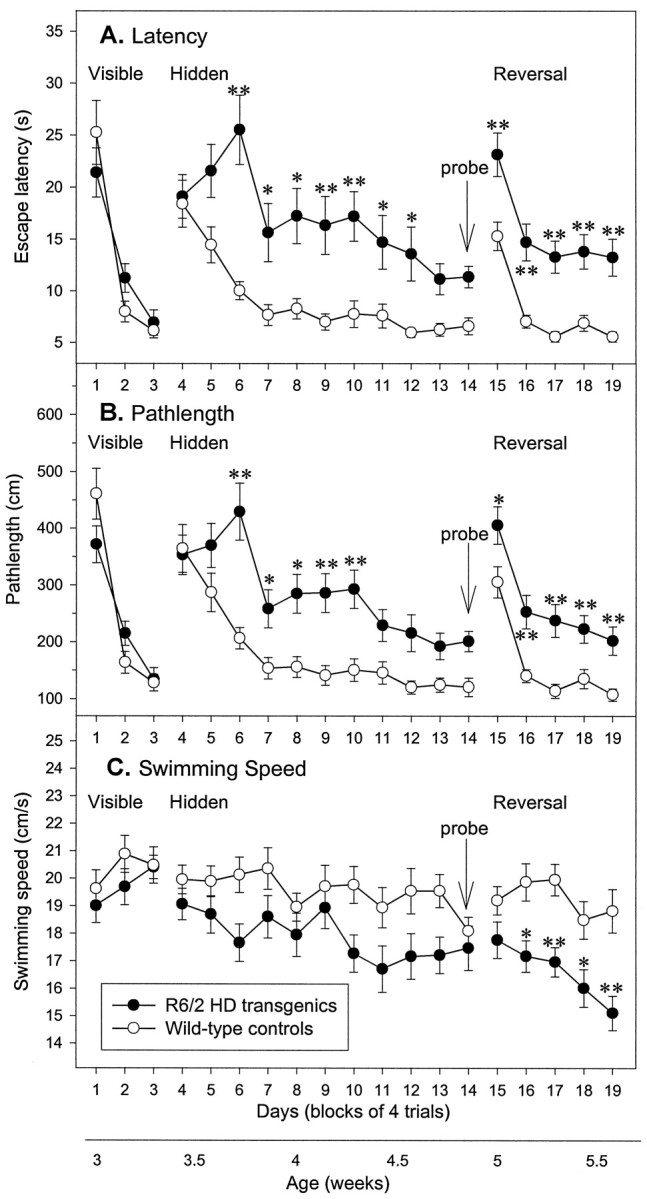
Impairment of Morris water maze learning in R6/2 mice. Escape latency (A), pathlength (B), and swimming speed (C) of control (n = 20) and R6/2 (n= 23) mice during acquisition of visible (days 1–3), hidden (days 4–14), and reversal (days 15–19) learning. R6/2 mice were unimpaired in swimming to a visible platform compared with controls (A–C). The latency (A) and pathlength (B) to escape to the hidden platform was impaired in R6/2 mice relative to control mice. Note that control and R6/2 mice did not differ in initial escape latency (A), pathlength (B), or swimming speed (C), indicating that R6/2 mice did not show nonspecific sensory impairment. R6/2 mice were impaired relative to controls in ability to learn reversal place learning of a hidden platform. There was no significant difference in swimming speed between R6/2 and control mice during the visible and hidden trials, but during reversal trials swimming speeds were significantly different between the two groups. Symbols indicate means ± SEM by mice of each group on each measure. Asterisksindicate significant differences between control and R6/2 mice (*p < 0.05; **p < 0.01).
Spatial learning: acquisition
The results of the spatial learning task in the Morris water maze are also shown in Figure 1. In learning to find the fixed hidden platform, control and R6/2 mice exhibited comparable latency and pathlength measures on the first 2 d of spatial learning. However, the R6/2 mice failed to improve on subsequent days as rapidly as the control mice and, from the third day of spatial training (day 6 onward), R6/2 mice showed significantly longer latency and pathlength measures than controls (genotype × day: latency,F(10,410) = 2.97, p < 0.001; pathlength, F(10,410) = 2.85,p < 0.01). The swim speed of R6/2 mice did not differ from that of controls on any day (Fig. 1C; genotype × day: F(10,410) = 1.46, NS).
On the probe trial, both control and R6/2 mice exhibited evidence for spatial learning, spending above chance time swimming in the platform quadrant (Fig. 2A), and the two groups did not differ significantly (genotype,F(1,41) = 0.05; genotype × quadrant, F(3,123) = 0.71, both NS). However on a more stringent measure of spatial navigation, measured by counting the “crossings” (the number of times the mice cross the correct location of the training platform), control mice made significantly more crossings than R6/2 mice (Fig. 2B; genotype, F(1,41) = 22.39,p < 0.001; genotype × platform,F(3,123) = 4.50, p < 0.01). Furthermore, R6/2 mice spent significantly less time swimming in the exact location of platform compared with controls (control, 10.4 ± 0.8 sec; R6/2 transgenic, 6.0 ± 1.0 sec;p < 0.01). An alternative measure of navigation accuracy is the proportion of time spent in the inner, middle, and outer zones of the maze. R6/2 mice spent significantly more time in the outer annuli and significantly less time in the middle annuli than control mice (Fig. 2C; genotype × zone,F(2,82) = 25.54, p < 0.001), indicating that transgenic and control mice used different platform-searching strategies. During the probe trial, R6/2 mice swam more slowly over the full 60 sec test than their wild-type littermates (control, 22.8 ± 0.5 cm/sec; R6/2 transgenic, 19.0 ± 0.6 cm/sec; t = 5.05; df = 41; p < 0.001).
Fig. 2.
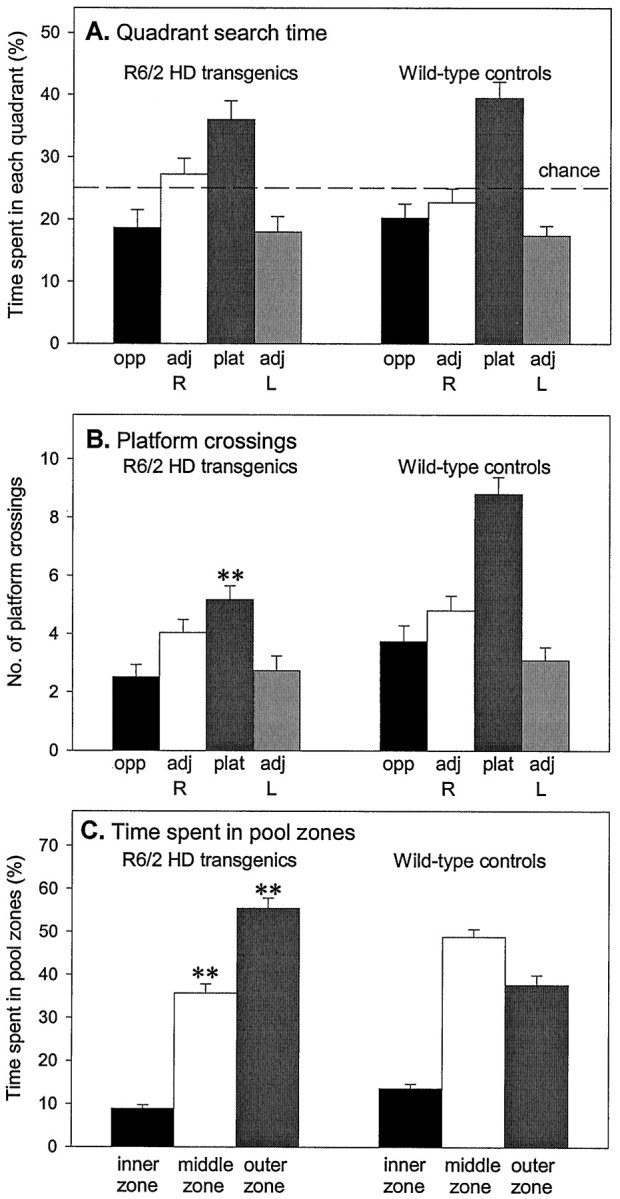
Impairment of probe trial performance in R6/2 mice. During the probe trials, R6/2 and control groups spent significantly >25% of their time in the platform quadrant, indicating that all mice had learned the platform location (A). The implications of A are refuted by the observation that R6/2 mice crossed what had been the exact location of the platform significantly less frequently than did controls (B) and furthermore that R6/2 mice did not show a preference for platform crossings over equivalent sites in the other quadrants. R6/2 mice spent significantly more time in the peripheral outer pool zone and significantly less time in the middle pool zone relative to control mice (C).Vertical bars indicate the SEM. Asterisksindicate significant preferences between measures (**p < 0.01).
Spatial learning: reversal
On the first day of reversal training (day 15), when the escape platform was moved from the center of the SW quadrant to the center of the NE quadrant, escape latency and pathlength increased transiently in both control and R6/2 transgenic groups, as the mice initially continued to search for the platform in the previous location. Thereafter, both control and R6/2 mice rapidly learned to swim to the “new” NE location. However, as before, R6/2 mice showed significantly longer latencies and pathlengths compared with the wild-type mice (Fig. 2A,B; genotype,F(1,41) = 35.07 and 32.34, bothp values < 0.001, for latency and pathlength, respectively).
Whereas control mice maintained stable swimming speeds throughout the study, R6/2 mice swam significantly more slowly from day 16, and their swim speeds continued to decline (Fig. 1C; genotype × day, F(4,164) = 3.40,p < 0.01). This early deficiency in motor performance is consistent with our previous findings (Carter et al., 1999).
Visual cliff avoidance
To assess whether the impaired performance of 3.5- to 5.5-week-old R6/2 transgenic mice in the Morris water maze could be attributed to impaired vision using more complex stimuli than simple light brightness discrimination, we studied mouse behavior in the visual cliff avoidance test. In this test, the amount of time spent in the open side of the box is used as a measure of impairment in visual acuity, although motivational changes in fear or an inability to learn to avoid the cliff edge may also lead to failure in this test. Here, control and R6/2 mice were tested in the visual cliff between 3 and 10 weeks of age. Our previous data show that only the performances of R6/2 mice at the final time point were likely to be affected by motor deficits. Between 3 and 7 weeks of age, there were no significant differences between control and R6/2 mice in the proportion of time spent on the bench versus open side of the visual cliff box (Fig.3A). Additionally, the amount of time spent on the bench side of the box increased significantly in both groups of mice from week 4 (effect of age,F(7,284) = 20.32, p < 0.01). However, whereas the wild-type mice continued to spend the majority of their time on the bench side during all subsequent trials, reflecting habituation, the R6/2 mice regularly ventured into the center of the visual cliff box and frequently crossed the cliff edge without hesitation from 8 weeks of age (Fig. 3; genotype,F(1,62) = 24.74; genotype × age,F(7,284) = 17.45, both pvalues < 0.01).
Fig. 3.

Deficient visual cliff avoidance in R6/2 mice. Separate groups of mice were tested between 3 and 10 weeks of age. Visual cliff avoidance of mice was measured using the percentage of time spent on the bench side of the visual cliff open field, as well as the direction of the first step outside of the start area (0 equates to open side step and 1 equates to bench side step). Control (n = 34) and R6/2 (n = 30) mice spent significantly more time on the bench side of the visual cliff box from 3–7 weeks of age (A). From 8 weeks of age, R6/2 mice spent significantly less time on the bench side relative to controls. A similar profile was seen for control and R6/2 mice for the direction of first step measure (B).Symbols indicate means ± SEM at each age.Asterisks indicate significant differences between control and R6/2 mice (**p < 0.01).
A similar pattern of behavior was shown when the direction of the first step outside the start area was analyzed. There were no significant differences between control and R6/2 mice from 3 and 7 weeks of age (Fig. 3B), with mice in both groups stepping onto the bench side, in preference to the open side, of the visual cliff box. However, after 7 weeks of age, whereas control mice continued to step onto the bench side first, the direction of first step taken by R6/2 mice reverted to chance (genotype, F(1,62)= 11.25; genotype × day,F(7,284) = 4.43, both pvalues < 0.01).
Two-choice swim tank
In the first experiment, 3-week-old control and R6/2 mice learned to discriminate bright and dimmed light and respond accordingly within 7 d of training. The mean percentage of correct choices made of the first 10 trials for R6/2 mice did not differ significantly from controls on any day (Fig.4A; genotype,F(1,29) = 0.99; genotype × day,F(16,464) = 0.64, both NS). Subsequent retesting revealed that control and R6/2 mice retained their learning, performing at criterion until 8.5 weeks of age. On the final retest at 9 weeks of age, the light stimulus over the visible escape platform was removed. Performance declined to chance levels for both groups (Fig.4A), suggesting that all mice were using the light stimulus as their visual cue, and that R6/2 mice are able to detect a distant bright light until at least 8.5 weeks of age. It is noteworthy that R6/2 mice swam significantly more slowly than controls from 5 weeks of age, as shown by their increased latency to swim the 40 cm distance (Fig. 4B; genotype,F(1,29) = 28.94, p < 0.001; genotype × day, F(16,464)= 11.02, p < 0.01), although this did not affect choice accuracy.
Fig. 4.

Normal visual discrimination learning and memory independent of swimming dysfunction in R6/2 mice. Control (n = 17) and R6/2 (n = 14) mice were tested between 3 and 9 weeks of age. Control and R6/2 mice displayed a comparable degree of acquisition of a two-choice swim tank visual discrimination task at 3–4 weeks of age, and both groups maintained performance criterion until 8.5 weeks of age (A). On removal of the light stimulus at 9 weeks of age, performance declined to chance levels for both groups. R6/2 mice displayed swimming impairments from 5 weeks of age, which progressively worsened, as compared with controls (B). Motoric dysfunction failed to impair performance of R6/2 mice in the two-choice swim tank task.Vertical bars indicate means ± SEM of each measure across all trials at each age. Asterisks indicate significant differences between control and R6/2 mice (*p < 0.05; **p < 0.01).
In the second experiment, the two-choice swim tank task was used to examine visual and reversal discrimination learning in more detail. Separate groups of mice were used, aged 3, 5, 7, and 10 weeks old at the start of testing. R6/2 mice were not impaired in task acquisition at 3–6 weeks of age (Fig.5A,B; genotype × day, 3–4 weeks: F(6,168) = 1.54; 5–6 weeks: F(6,174) = 2.07, both NS). R6/2 mice exhibited significant deficits at 7–8 weeks of age, although they could still learn the task (Fig. 5C; genotype × day, 7–8 weeks: F(6,174) = 4.82,p < 0.001). By 10–11 weeks of age, the ability of R6/2 mice to acquire this task was severely impaired compared with their age-matched littermate controls (Fig. 5D; genotype × day, F(6,168) = 7.13,p < 0.001).
Fig. 5.

Selective deficits in visual and reversal discrimination learning in R6/2 mice. Separate groups of control (n = 10–18) and R6/2 (n = 12–21) mice were tested between 3 and 10 weeks of age. Control and R6/2 mice displayed a comparable degree of acquisition of a two-choice swim tank visual discrimination task at 3–4 (A) and 5–6 (B) weeks of age, however R6/2 mice displayed significantly slower learning than controls at 7–8 weeks of age (C), and by 10–11 weeks of age (D) acquisition was severely impaired in R6/2 mice. In contrast, acquisition of reversal discrimination was impaired in R6/2 mice from 6.5 weeks of age (E–H).Symbols indicate means ± SEM by mice of each group at each age on both measures. Asterisks indicate significant differences between control and R6/2 mice (*p < 0.05; **p < 0.01).
When the task was reversed, both control and R6/2 mice initially exhibited a performance deficit at all ages tested, as they continued to swim toward the previously reinforced visual cue, the light stimulus. However, for control mice, this performance deficit was transient, and at all ages tested they learned to swim away from the light stimulus within 15–20 trials and performed to criterion within 30 trials (Fig. 5E–H). In contrast, whereas 4.5-week-old R6/2 mice learned to dissociate the original visual cue from reinforcement and learned to swim away from the light stimulus as rapidly as age-matched controls (Fig. 5E; genotype,F(1,28) = 0.24; genotype × trial bin, F(5,140) = 1.01, both NS), older R6/2 mice showed performance deficits. Although 5- to 6-week-old R6/2 mice could learn the simple visual discrimination during acquisition as well as their littermate controls, they were impaired when they were subsequently forced to reverse the discrimination (Fig. 5F; genotype, F(1,29) = 26.69; genotype × trial bin, F(5,145) = 16.83, both p values < 0.01). These R6/2 mice failed to learn the reversal task after 30 trials, persistently swimming in the direction of the light. This deficit in reversal discrimination learning was also seen in 8.5- and 11.5-week-old R6/2 mice (Fig.5G,H; 8.5 week: genotype,F(1,29) = 19.28; genotype × trial bin, F(5,145) = 9.72, bothp values < 0.01; 11.5 week: genotype,F(1,28) = 36.19; genotype × trial bin, F(5,140) = 7.06, bothp values < 0.01).
T-maze
The T-maze test was used to test alternation, spatial, and nonspatial learning in R6/2 mice. R6/2 mice made significantly fewer alternations in the T-maze and more errors relative to controls from day 1 onwards (Fig. 6A; genotype, F(1,26) = 55.54 and 59.10, both p values < 0.001; genotype × block of days,F(5,130) = 55.54 and 59.10, bothp values < 0.001, for the number of alternations and errors, respectively). Initial and final levels of performance significantly differed between R6/2 mice and controls, indicating that the observed distinction may be attributable to nonspecific sensory, motor, or motivational deficits of the R6/2 mice. Neither group differed in their mean latency to enter an arm on any day (data not shown). This indicated that R6/2 mice did not have impairments in locomotor activity and that motivation was normal.
Fig. 6.
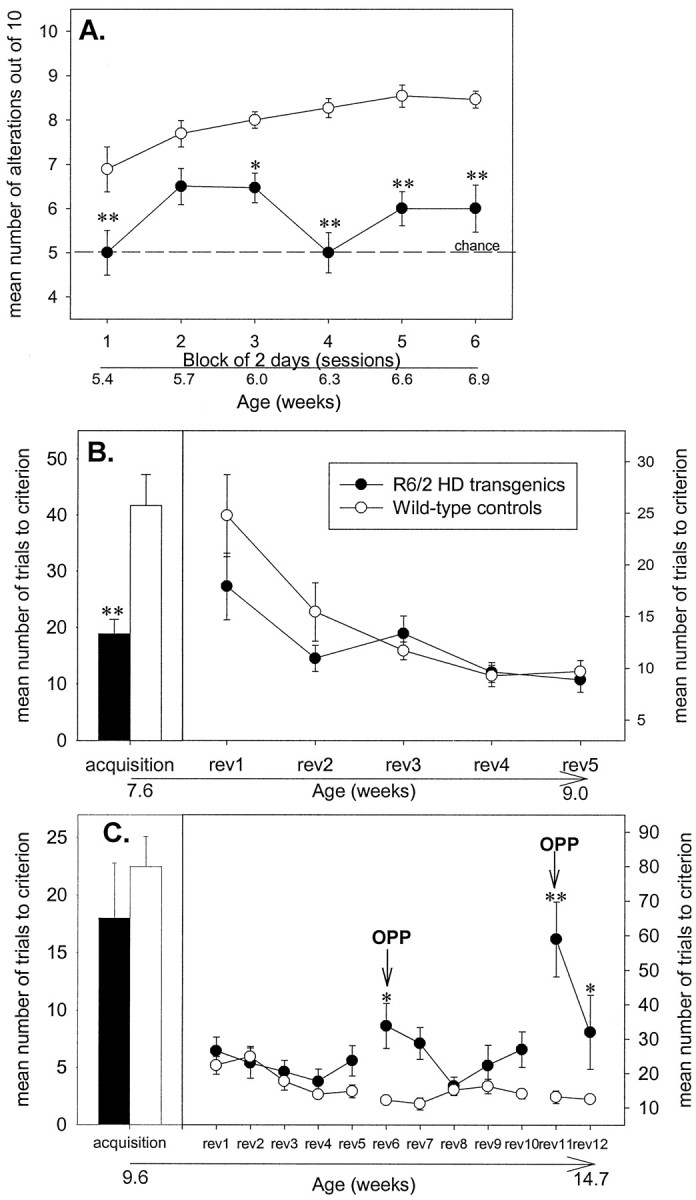
Alternation learning impairments in R6/2 mice. R6/2 mice (n = 15) were impaired in T-maze alternation (A) but unimpaired in a simple T-maze black–white visual discrimination test (B), compared with controls (n = 13). Whereas control mice adopted spatial strategies, R6/2 mice used nonspatial strategies (C). Symbols indicate means ± SEM by mice of each group on each measure. Asterisksindicate significant differences between control and R6/2 mice (*p < 0.05; **p < 0.01).
In the second phase of the T-maze study, whereby mice were rewarded for discriminating a black or white arm, there was no evidence of any nonspecific deficit in R6/2 mice compared with controls (Fig.6B). In fact, R6/2 mice learned the black–white discrimination more rapidly than controls, requiring significantly fewer trials to reach criterion and producing fewer errors (t = 4.14 and 3.51; both df = 25 andp < 0.01 for the number of trials to criterion and errors, respectively). On the subsequent reversals of this task there was no significant difference on the number of trials to criterion and errors made between each group (Fig. 6B; genotype,F(1,25) = 3.59 and 0.54, both NS, for the number of trials to criterion and errors, respectively).
On removal of the black/white cues, the number of trials to criterion and errors made by R6/2 mice did not differ from controls on acquisition (Fig. 6C; t = 0.76 and 0.44, both df = 24, and NS, for the number of trials to criterion and errors, respectively) and any reversal, except the “opposite” reversals six and eleven (in which all mice were placed in the opposite start arm on alternate trials) (Fig. 6C; number of trials to criterion: genotype, F(1,24) = 28.80; genotype × reversal, F(11,264) = 4.11, both p values < 0.001; number of errors: genotype, F(1,24) = 24.71; genotype × reversal, F(11,264) = 3.10, both p values < 0.001). Hence, whereas control mice spatially learned when placed in the opposite start arm to make a turn in the opposite direction to enter the same arm as on the previous trial, R6/2 mice persistently turned in the same direction irrespective of their start position. Furthermore, the deficit displayed by R6/2 mice on this task was significantly worse on reversal 11 compared with that on reversal 6 (p < 0.01).
DISCUSSION
This study provides the first evidence for progressive learning and memory deficits in R6/2 mice. We describe deficits in a number of tasks assessing spatial, visual, reversal, and alternation discrimination learning and memory. These deficits cannot be attributed to a loss of motor or visual function because impairments in spatial, reversal, and alternation learning were observed as early as 3.5–6.5 weeks of age, when R6/2 mice were not impaired in the motor and visual components of these tasks. Furthermore, different patterns of deficit were seen in tests with similar sensory and motor demands, with the extent of the deficit depending on the cognitive demands of the task (Fig. 7).
Fig. 7.

Schematic representation of the earliest age of onset of impairment in spatial, visual, reversal, and alternation learning in R6/2 mice. R6/2 mice showed spatial learning impairments in the Morris water maze from 3.5 weeks (A), alternation learning impairments in the T-maze from 5 weeks (B), and reversal learning impairments in the two-choice swim tank from 6.5 weeks (C). Visual discriminative learning impairments were first observed from 7–8 weeks (D, E), and deterioration in retention of a previously learned visual task was not seen until 8.5 weeks (F). Filled bars indicate onset of impairment.
Cognitive deficits in R6/2 mice
Behavioral testing of R6/2 mice started at 3 weeks of age, several weeks before subtle (5–6 weeks), or overt (8–9 weeks) motor symptoms appeared (Carter et al., 1999). At the earliest age tested (3.5 weeks), it was apparent that R6/2 mice have a spatial learning deficit in the Morris water maze test. This is consistent with our previous study, which found that older R6/2 mice (7–8 weeks) exhibit severe impairments in spatial learning of the hidden platform task (Murphy et al., 1998). The slower learning of older R6/2 mice may reflect a motor deficit, rather than a purely cognitive impairment. However, the impaired performance of young R6/2 mice reported here cannot be secondary to motor deficits or lack of motivation to escape from the water, because control and R6/2 mice initially showed identical pathlengths and swim speeds. Moreover, although R6/2 mice swim more slowly with increasing age, impaired water maze learning was associated with increased pathlengths, indicating that older R6/2 mice swam further than controls without apparent difficulty. It was also notable during the probe trial that R6/2 mice exhibited a different platform search strategy from controls, swimming preferentially in the periphery of the water tank. “Thigmotaxis” (seen commonly in normal animals in the early phase of learning Morris water maze tasks) is exacerbated in rodents with striatal lesions (Devan et al., 1999). This suggests there is a striatal component to the dysfunction in the R6/2 mice.
The possibility that visual impairments in R6/2 mice contribute to impairments in water maze performance is an important confound. Thus, we tested R6/2 mice on two visual discrimination tasks: visual cliff avoidance and two-choice swim tank. The visual cliff task showed that R6/2 mice avoid a visual cliff until at least 7 weeks of age but display a progressive reduction in avoidance thereafter. The increased time spent over the edge of the cliff is unlikely to reflect increased exploratory activity, because control and R6/2 mice have comparable levels of locomotor activity in an open field until 10 weeks of age (Dunnett et al., 1998). The performance of older (>8 weeks) R6/2 mice in the visual cliff may be confounded by visual dysfunction, although it is possible that an impaired ability to learn to avoid the cliff edge is responsible for their deteriorating performance. Reduced anxiety in R6/2 mice (File et al., 1998) may also contribute to these deficits.
The performance of R6/2 mice on the two-choice swim tank task showed that mice trained at 3–4 weeks can perceive a visual light cue until at least 8.5 weeks of age. Subsequently removing the light cue causes the performance of control and R6/2 mice to decline to chance levels, demonstrating that all mice use the light as their definitive visual cue. Data from both cliff and swim tank tests support the idea that young R6/2 mice have sufficiently normal vision to perform the tasks and support the suggestion that poor performance of R6/2 mice in the Morris water maze and swim tank is attributable to cognitive rather than nonspecific sensory or motor deficits.
Impairments in cognitive performance in the two-choice swim tank were particularly revealing. The ability of young R6/2 mice to learn the task (acquisition) was not affected; however, by 7–8 weeks, learning was mildly impaired, and by 10 weeks learning was severely compromised. Impairments on the reversal trials were also seen, but were much more severe, with maximum impairment seen by 6.5 weeks. The reversal learning deficits in the swim tank at least, cannot be attributed to a loss of motor or visual function, because pretrained R6/2 mice continue to perform accurately in this task at that same age. It is more likely that R6/2 mice have an age-dependent deficit in cognitive flexibility, because they appear to be incapable of making this strategy switch.
Between 5 and 6.5 weeks R6/2 mice are deficient in their ability to alternate in a T-maze. This may reflect either a disturbance in the expression of innate motivational program or a tendency of R6/2 mice to perseverate. When the same mice were trained subsequently in a specific visual discrimination paradigm, R6/2 mice reached criterion more rapidly than controls, although this may simply reflect the fact that they were starting from a lower baseline of alternation performance. Certainly, once trained at 7–9 weeks, R6/2 mice and controls showed comparable ability to learn the reversal, again indicating that the acquisition deficit is not attributable to a visual deficit. Spontaneous alternation represents an innate species-specific pattern of motivated behavior that may be related to optimal foraging strategies in the wild (Dember and Fowler, 1958). However, to alternate, animals must have a functionally intact short-term memory. Significantly, spontaneous alternation is disrupted by striatal, prefrontal, and hippocampal lesions (Divac et al., 1975; Johnson et al., 1977).
To determine whether a spatial and/or a nonspatial strategy is adopted by mice in this task, they were placed in the opposite start arm on alternate trials. R6/2 mice appear to be deficient in spatial learning, because they continued to adopt nonspatial intramaze (and/or positional) cues, eventually solving the task to a very low level of accuracy. In contrast, control mice adopt normal spatial learning and reach criterion within 10 trials.
Similarities between cognitive deficits in R6/2 mice and HD patients
The cognitive impairment in HD is believed to be a consequence of the profound frontostriatal pathology associated with the disease (Vonsattel and DiFiglia, 1998). Striatal dysfunction is implicated in the specific cognitive deficits seen in HD patients (Lange et al., 1995; Lawrence et al., 1996, 1998). The nature of cognitive decline seen in patients with HD includes a decline in visuospatial skills, cognitive flexibility, and recall memory. Notably, Lawrence et al. (1996, 1998) reported impairments involving changing a previously learned response, with HD patients showing deficits in a visual discrimination/set-shifting task by exhibiting perseverative responding to the original stimulus. This response may be analogous to the impaired performance of R6/2 mice in the reversal phase of the swim tank and their inability to alternate in the T-maze. An inability to switch from one set of learned responses to another therefore appears to be a fundamental feature of patients with HD as well as R6/2 mice. HD patients (Swerdlow et al., 1995) and R6/2 mice (Carter et al., 1999) also exhibit deficient inhibitory control in a number of neuropsychological paradigms, including prepulse inhibition. However the neural basis for these deficits remains unresolved.
Possible mechanisms underlying cognitive dysfunction
The behavioral tests used here were designed to test frontostriatal function in R6/2 mice. Although R6/2 mice show no striatal neuronal loss (Mangiarini et al., 1996; Davies et al., 1997), inclusions (Davies et al., 1997) and neurotransmitter abnormalities (Cha et al., 1998) are found in the frontostriatal region of R6/2 mice, suggesting that the cognitive changes reported here are attributable to neuronal dysfunction (rather than cell death). The cognitive deficits in HD are associated with frontostriatal pathology, however striatal dysfunction and cognitive deficits in the absence of cell death are seen in early stage HD (Myers et al., 1988). Interestingly, animal models of HD with frontostriatal lesions display cognitive deficits, attributed to the selective striatal damage, which are similar to those reported here (Divac et al., 1967; Furtado and Mazurek, 1996; Palfi et al., 1996; Emerich et al., 1997; Shear et al., 1998a,b). The present data are consistent with a frontostriatal basis for the deficit.
NII formation in R6/2 mice is not restricted to the striatum, and an alternative explanation for the observed learning deficits is the existence of hippocampal impairments in R6/2 mice. Lesions of the hippocampus can cause maze-learning deficits in rodents (Morris et al., 1982; Logue et al., 1997), and R6/2 mice show NII pathology in this area (Davies et al., 1997). In addition, we have shown that R6/2 mice exhibit a reduced long-term potentiation and a lasting long-term depression in the CA1 area of the hippocampus (Murphy et al., 1998), electrophysiological phenomena that have been implicated in learning and memory (Tsien et al., 1996; Stevens and Sullivan, 1998). Similar changes in plasticity have been seen in other transgenic models of HD, although these groups did not report on the cognitive phenotype (Hodgson et al., 1999; Usdin et al., 1999). Thus, it seems possible that changes in synaptic plasticity, or other aspects of hippocampal dysfunction, contribute to the deficits in learning and memory that we describe here.
Conclusion
Our study describes specific deficits in several forms of learning in R6/2 mice. We provide evidence for a specific and progressive pattern of discriminative learning impairments in R6/2 mice that, as in humans, have no clear “age of onset”, because the age at which an impairment becomes apparent is dependent on the sensitivity of the test and the particular demands of each task. These results indicate that transgenic mice, such as the R6/2 strain, may not only be useful for studying the relationship between cognitive and motor components of neurological dysfunction, but also for providing models of specific diseases for assessing potential therapeutic agents.
Footnotes
This work was supported by grants from the Hereditary Disease Foundation, Medical Research Council, Parke-Davis Neuroscience Research Centre (UK), and the Wellcome Trust (UK). L.A.L. is supported by Parke-Davis Neuroscience Research Centre. R.J.C. is supported by the Medical Research Council. We thank Mrs. Chris Riches and Mr. Trevor Humby for valuable technical assistance.
L.A.L. and R.J.C. contributed equally to this work.
Correspondence should be addressed to Dr. Lisa Lione, Parke-Davis Neuroscience Research Centre, Cambridge University Forvie Site, Robinson Way, Cambridge, CB2 2QB, UK. E-mail: Lisa.Lione@wl.com.
REFERENCES
- 1.Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature. 1986;321:168–171. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- 2.Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterisation of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borlongan CV, Koutouzis TK, Freeman TB, Cahill DW, Sanberg PR. Behavioral pathology induced by repeated systemic injections of 3-nitropropionic acid mimics the motoric symptoms of Huntington's disease. Brain Res. 1995;697:254–257. doi: 10.1016/0006-8993(95)00901-2. [DOI] [PubMed] [Google Scholar]
- 4.Bossi SR, Simpson JR, Isacson O. Age dependence of striatal neuronal death caused by mitochondrial dysfunction. NeuroReport. 1993;4:73–76. doi: 10.1097/00001756-199301000-00019. [DOI] [PubMed] [Google Scholar]
- 5.Brouillet E, Jenkins BG, Hyman BT, Ferrante RJ, Kowall NW, Srivastava R, Roy DS, Rosen BR, Beal MF. Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J Neurochem. 1993;60:356–359. doi: 10.1111/j.1471-4159.1993.tb05859.x. [DOI] [PubMed] [Google Scholar]
- 6.Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal MF. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci USA. 1995;92:7105–7109. doi: 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterisation of progressive motor deficits in mice transgenic for the Human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cha JJ, Kosinski CH, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington's disease gene. Proc Natl Acad Sci USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coyle JT, Schwarcz R. Lesion of striatal neurones with kainic acid provides a model of Huntington's chorea. Nature. 1976;263:244–246. doi: 10.1038/263244a0. [DOI] [PubMed] [Google Scholar]
- 10.Davies SW, Turmaine M, Cozens B, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 11.Dember WN, Fowler H. Spontaneous alternation behaviour. Psychol Bull. 1958;55:412–427. doi: 10.1037/h0045446. [DOI] [PubMed] [Google Scholar]
- 12.Devan BD, McDonald RJ, White NM. Effects of medial and lateral caudate-putamen lesions on place- and cue-guided behaviour in the water maze: relation to thigmotaxis. Behav Brain Res. 1999;100:5–14. doi: 10.1016/s0166-4328(98)00107-7. [DOI] [PubMed] [Google Scholar]
- 13.Divac I, Rosvold HE, Schwarcbart M. Behavioural effects of selective ablation of the caudate nucleus. J Comp Physiol Psychol. 1967;63:184–190. doi: 10.1037/h0024348. [DOI] [PubMed] [Google Scholar]
- 14.Divac I, Wikmark RGE, Gade A. Spontaneous alternation in rats with lesions in the frontal lobe: an extension of the frontal lobe syndrome. Physiol Psychol. 1975;3:39–42. [Google Scholar]
- 15.Dunnett SB, Carter RJ, Watts C, Torres EM, Mahal A, Mangiarini L, Bates GP, Morton AJ. Striatal transplantation in a transgenic mouse model of Huntington's disease. Exp Neurol. 1998;154:31–40. doi: 10.1006/exnr.1998.6926. [DOI] [PubMed] [Google Scholar]
- 16.Emerich DF, Cain CK, Greco C, Saydoff JA, Hu ZY, Liu H, Lindner MD. Cellular delivery of human CNTF prevents motor and cognitive dysfunction in a rodent model of Huntington's disease. Cell Transplant. 1997;6:249–266. doi: 10.1177/096368979700600308. [DOI] [PubMed] [Google Scholar]
- 17.File SE, Mahal A, Mangiarini L, Bates GP. Striking changes in anxiety in Huntington's disease transgenic mice. Brain Res. 1998;805:234–240. doi: 10.1016/s0006-8993(98)00736-7. [DOI] [PubMed] [Google Scholar]
- 18.Foroud T, Siemers E, Kleindorfer D, Bill DJ, Hode ME, Norton JA. Cognitive scores in carriers of Huntington's disease gene compared to noncarriers. Ann Neurol. 1995;37:657–664. doi: 10.1002/ana.410370516. [DOI] [PubMed] [Google Scholar]
- 19.Furtado JC, Mazurek MF. Behavioural characterisation of quinolinate-induced lesions of the medial striatum: relevance for Huntington's disease. Exp Neurol. 1996;138:158–168. doi: 10.1006/exnr.1996.0054. [DOI] [PubMed] [Google Scholar]
- 20.Hodgson JG, Agopyan N, Gutekunst C, Leavitt BR, LePaine F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li X, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington's disease with full-length mutant Huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 21.Hurlbert MS, Zhou W, Wasmeier C, Kaddis FG, Hutton JC, Freed CR. Mice transgenic for an expanded CAG repeat in the Huntington's disease gene develop diabetes. Diabetes. 1999;48:649–651. doi: 10.2337/diabetes.48.3.649. [DOI] [PubMed] [Google Scholar]
- 22.Johnson CT, Olton DS, Gage FH, Jenko PG. Damage to hippocampus and hippocampal connections: effects of DRL and spontaneous alternation. J Comp Physiol Psychol. 1977;91:508–522. doi: 10.1037/h0077346. [DOI] [PubMed] [Google Scholar]
- 23.Kodsi MH, Swerdlow NR. Mitochondrial toxin 3-nitropropionic acid produces startle reflex abnormalities and striatal damage in rats that model some features of Huntington's disease. Neurosci Lett. 1997;231:103–107. doi: 10.1016/s0304-3940(97)00482-5. [DOI] [PubMed] [Google Scholar]
- 24.Lange KW, Sahakian BJ, Quinn NP, Marsden CD, Robbins TW. Comparison of executive and visuospatial memory function in Huntington's disease and dementia of Alzheimer type matched for degree of dementia. J Neurol Neurosurg Psychiatry. 1995;58:598–606. doi: 10.1136/jnnp.58.5.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lawrence AD, Sahakian BJ, Hodges JR, Rosser AE, Lange KW, Robbins TW. Executive and mnemonic functions in early Huntington's disease. Brain. 1996;119:1633–1645. doi: 10.1093/brain/119.5.1633. [DOI] [PubMed] [Google Scholar]
- 26.Lawrence AD, Hodges JR, Rosser AE, Kershaw A, ffrench-Constant C, Rubinsztein DC, Robbins TW, Sahakian BJ. Evidence for specific cognitive deficits in preclinical Huntington's disease. Brain. 1998;121:1329–1341. doi: 10.1093/brain/121.7.1329. [DOI] [PubMed] [Google Scholar]
- 27.Logue SF, Paylor R, Wehner JM. Hippocampal lesions cause learning deficits in inbred mice in the Morris water maze and conditioned-fear task. Behav Neurosci. 1997;111:104–113. doi: 10.1037//0735-7044.111.1.104. [DOI] [PubMed] [Google Scholar]
- 28.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 29.McGreer EG, McGreer PL. Duplication of biochemical changes of Huntington's chorea by intrastriatal injections of glutamic and kainic acids. Nature. 1976;263:517–519. doi: 10.1038/263517a0. [DOI] [PubMed] [Google Scholar]
- 30.Mohr E, Brouwers P, Claus JJ, Mann UM, Fedio P, Chase TN. Visuospatial cognition in Huntington's disease. Mov Disord. 1991;6:127–132. doi: 10.1002/mds.870060207. [DOI] [PubMed] [Google Scholar]
- 31.Molinari S, Battini R, Ferrari S, Pozzi L, Killcross AS, Robbins TW, Jouvenceau A, Billard J-M, Dutar P, Lamour Y, Baker WA, Cox H, Emson PC. Deficits in memory and hippocampal long-term potentiation in mice with reduced calbindin D28K expression. Proc Natl Acad Sci USA. 1996;93:8028–8033. doi: 10.1073/pnas.93.15.8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris RG, Garrud P, Rawlins JN, O'Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- 33.Murphy KPSJ, Lione LA, Carter RJ, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Altered hippocampal synaptic plasticity at CA1 synapses in vitro and impaired spatial learning in a transgenic mouse model of Huntington's disease. J Physiol (Lond) 1998;513:12P. [Google Scholar]
- 34.Myers RH, Vonsattel J, Stevens TJ, Cupples LA, Richardson EP, Martin JB, Bird ED. Clinical and neuropathologic assessment of severity in Huntington's disease. Neurology. 1988;38:341–347. doi: 10.1212/wnl.38.3.341. [DOI] [PubMed] [Google Scholar]
- 35.Palfi S, Ferrante RJ, Brouillet E, Beal MF, Dolan R, Guyot M, Peschanski M, Hantraye P. Chronic 3-nitropropionic acid treatment in baboons replicates the cognitive and motor deficits of Huntington's disease. J Neurosci. 1996;16:3019–3025. doi: 10.1523/JNEUROSCI.16-09-03019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paulson HL. Protein fate in neurodegenerative proteinopathies: Polyglutamine diseases join the (mis)fold. Am J Hum Genet. 1999;64:339–345. doi: 10.1086/302269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reddy PH, Williams M, Charles V, Garrett L, Pike-Buchanan L, Whetsell WO, Miller G, Tagle DA. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat Genet. 1998;20:198–202. doi: 10.1038/2510. [DOI] [PubMed] [Google Scholar]
- 38.Rohlf FJ, Sokal RR. Statistical tables. Freeman; New York: 1995. [Google Scholar]
- 39.Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR. Intranuclear inclusions and neuritic aggregates in a transgenic mice expressing an N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 40.Shear DA, Dong J, Haik-Creguer KL, Bazzett TJ, Albin RL, Dunbar GL. Chronic administration of quinolinic acid in the rat striatum causes spatial learning deficits in a radial arm water maze task. Exp Neurol. 1998a;150:305–311. doi: 10.1006/exnr.1998.6767. [DOI] [PubMed] [Google Scholar]
- 41.Shear DA, Haik-Creguer KL, Brandel SM, Dupont J, Dunbar GL. Systemic administration of 3-nitropropionic acid in progressively increasing concentrations produces spatial learning impairment and motor abnormalities relevant to Huntington's disease. Soc Neurosci Abstr. 1998b;24:970. [Google Scholar]
- 42.Shelbourne PF, Killeen N, Hevner RF, Johnston HM, Tecott L, Lewandoski M, Ennis M, Ramirez L, Li Z, Iannicola C, Littman DR, Myers RH. A Huntington's disease CAG expansion at the murine Hdh locus is unstable and associated with behavioural abnormalities in mice. Hum Mol Genet. 1999;8:763–774. doi: 10.1093/hmg/8.5.763. [DOI] [PubMed] [Google Scholar]
- 43.Stevens CF, Sullivan J. Synaptic plasticity. Curr Biol. 1998;8:R151–R153. doi: 10.1016/s0960-9822(98)70097-1. [DOI] [PubMed] [Google Scholar]
- 44.Stewart CA, Morris RG. Behavioural neuroscience (Sahgal A, ed), pp 107–121. IRL; Oxford: 1994. [Google Scholar]
- 45.Swerdlow NR, Paulson J, Braff DL, Butters N, Geyer MA, Swenson MR. Impaired prepulse inhibition of acoustic and tactile startle response in patients with Huntington's disease. J Neurol Neurosurg Psychiatry. 1995;58:192–200. doi: 10.1136/jnnp.58.2.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- 47.Usdin MT, Shelbourne PF, Myers RH, Madison DV. Impaired synaptic plasticity in mice carrying the Huntington's disease mutation. Hum Mol Genet. 1999;8:839–846. doi: 10.1093/hmg/8.5.839. [DOI] [PubMed] [Google Scholar]
- 48.Vonsattel J, DiFiglia M. Huntington's disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]