

Abstract
Polyglutamine (polygln) diseases are a group of inherited neurodegenerative disorders characterized by protein misfolding and aggregation. Here, we investigate the role in polygln disease of heat shock proteins (Hsps), the major class of molecular chaperones responsible for modulating protein folding in the cell. In transfected COS7 and PC12 neural cells, we show that Hsp40 and Hsp70 chaperones localize to intranuclear aggregates formed by either mutant ataxin-3, the disease protein in spinocerebellar ataxia type 3/Machado-Joseph disease (SCA3/MJD), or an unrelated green fluorescent protein fusion protein containing expanded polygln. We further demonstrate that expression of expanded polygln protein elicits a stress response in cells as manifested by marked induction of Hsp70. Studies of SCA3/MJD disease brain confirm these findings, showing localization of Hsp40 and, less commonly, Hsp70 chaperones to intranuclear ataxin-3 aggregates. In transfected cells, overexpression of either of two Hsp40 chaperones, the DNAJ protein homologs HDJ-1 and HDJ-2, suppresses aggregation of truncated or full-length mutant ataxin-3. Finally, we extend these studies to a PC12 neural model of polygln toxicity in which we demonstrate that overexpression of HDJ-1 suppresses polygln aggregation with a parallel decrease in toxicity. These results suggest that expanded polygln protein induces a stress response and that specific molecular chaperones may aid the handling of misfolded or aggregated polygln protein in neurons. This study has therapeutic implications because it suggests that efforts to increase chaperone activity may prove beneficial in this class of diseases.
Keywords: polyglutamine disease, spinocerebellar ataxia 3, Hsp40, Hsp70, chaperone, cell death
Polyglutamine (polygln) expansion is now recognized to be a major cause of inherited neurodegenerative disease (for review, see Koshy and Zoghbi 1997; Kakizuka, 1998;Paulson, 1999; Perutz, 1999). Eight polygln diseases have been identified to date, including Huntington disease (HD), spinobulbar muscular atrophy (SBMA), dentatorubral pallidoluysian atrophy, and five forms of dominantly inherited spinocerebellar ataxia (SCAs). All eight are caused by expansion of a CAG triplet repeat encoding an abnormally long glutamine repeat in the otherwise unrelated disease proteins. Polygln expansion is thought to cause a conformational change in the polypeptide that promotes misfolding and aggregation of the disease protein (Trottier et al., 1995; Hackam et al., 1998; Kakizuka, 1998; Perutz, 1999). Intranuclear aggregates or inclusions (NI) are a common feature of disease, having been found in six of the eight diseases and in many transgenic animal models (for review, see Paulson, 1999). Aggregates outside the nucleus have also been noted in some polygln diseases, including HD (Gutekunst et al., 1999).
Polygln expansion confers a dominant toxic property on the protein that leads to neuronal dysfunction and degeneration. Longer repeats cause more severe disease and are also more prone to cause aggregation of the disease protein, as demonstrated in numerous studies (Scherzinger et al., 1997; Cooper et al., 1998; Martindale et al., 1998; Moulder et al., 1999). Thus, the toxicity of expanded polygln seems linked to its tendency to assume an abnormal conformation, which promotes aggregation.
However, the precise mechanism of polygln neurotoxicity is still unknown. Although the pathogenic role of aggregation is controversial, misfolding of the disease protein is generally thought to be critical to pathogenesis. This raises the possibility that the cellular machinery that normally recognizes and handles abnormally folded protein may play a role in polygln disease. This machinery includes numerous molecular chaperones that mediate the correct folding, assembly, and degradation of proteins (Hartl, 1996; Hayes and Dice, 1996; Gottesman et al., 1997; Glover and Lindquist, 1998). The heat shock proteins (Hsps) are a family of chaperones inducible by heat and other stressors that serve essential functions under stress and nonstress conditions. Specific Hsp chaperones have been observed to localize to aggregates formed by the disease proteins in spinocerebellar ataxia type 1 (SCA1) and SBMA, ataxin-1 and the androgen receptor (AR) (Cummings et al., 1998; Stenoien et al., 1999). Expression of the Hsp40 chaperone, HDJ-2, suppressed aggregation by both proteins in non-neuronal cells, although whether suppressing aggregation was beneficial to the cell was not determined.
In this study, we investigate the role of Hsp chaperones in polygln disease, focusing on spinocerebellar ataxia type 3 [SCA3/Machado-Joseph disease (MJD)] and its disease protein ataxin-3. We demonstrate that specific Hsp chaperones localize to aggregates formed by mutant ataxin-3 in disease tissue and transfected cells and provide evidence that expanded polygln protein elicits a stress response in cells. We further show that overexpression of Hsp40 chaperones can suppress polygln aggregation and that this suppression correlates with a decrease in neurotoxicity. These results have therapeutic implications, because they indicate that efforts to augment molecular chaperone activity may prove beneficial in this class of degenerative diseases.
MATERIALS AND METHODS
Construction of expression plasmids. The ataxin-3 expression constructs pcDNA3-hemagglutinin (HA)-Q27, pcDNA3-HA-Q78, pAG-nuclear localization signal (NLS)-myc-ataxin-3 (Q27), and pAG-NLS-ataxin-3 (Q78) were described previously (Paulson et al., 1997a; Perez et al., 1998; Chai et al., 1999). Plasmid pcDNA3-myc-ataxin-3 (Q84) was generated by reverse transcription (RT)-PCR amplification of the 3′ segment of ataxin-3 cDNA from patient lymphoblastoid cells and subcloning the sequence-verified product into the appropriate region of pcDNA3-myc-ataxin-3 (Q27) (Paulson et al., 1997a). The green fluorescent protein (GFP)-polyglutamine fusion constructs with Q19, Q35, Q56, or Q80 repeats were generously provided by Dr. Warren Strittmatter (Duke University Medical Center, Durham, NC). Plasmid pcDNA3.1-Hsp70 was constructed by digesting pAG153-human Hsp70 (American Type Culture Collection, Manassas, VA) with BamHI and HindIII and cloning the released Hsp70 cDNA into pcDNA3.1(−) (Invitrogen, Carlsbad, CA). PcDNA3-human Hsp27 expression vector was prepared by digesting pIC-human Hsp27 (generously provided by Dr. Stephen W. Carper, University of Nevada, Las Vegas, NV) withEcoRI and cloning the released fragment into pcDNA3. The pcDNA 3-HDJ-1 expression vector was constructed by digesting pBluescript human HDJ-1 (a generous gift of Dr. K. Ohtsuka, Aichi Cancer Center, Nagoya, Japan) with EcoRI and cloning the released fragment into pcDNA3. Expression vectors for human HDJ-2 and Δ450 (a deleted form of HDJ-2 missing amino acids 9–107 of the protein) were generously provided by Dr. Huda Zoghbi (Baylor College of Medicine, Houston, TX). All plasmid constructs generated by us were verified by restriction enzyme digest analysis and partial sequencing. Western blot analysis with appropriate antisera confirmed correct expression of HDJ-1, HDJ-2, HDJ2 (del9–107), and Hsp70 in transfected cells.
Cell culture and transfection. Cell culture and transfections of HeLa and COS7 were described previously (Chai et al., 1999). For transient transfections in PC12 neural cells, we used the PC6–3 cell line, a PC12 subclone generated by Pittman et al. (1993)that differentiates into a pure population of postmitotic neuronal cells. In the PC6–3 cell line, we developed an improved transfection protocol that we call “sustained transient” because, although transiently transfected, the differentiating PC6–3 neurons show robust expression lasting for 1 week or more. The method takes advantage of the fact that PC6–3 cells, when first exposed to nerve growth factor (NGF), proliferate before extending neurites. Perhaps for this reason, liposome-mediated gene transfer is particularly efficient (∼20% of cells) in the 48 hr after NGF treatment is begun. Briefly, PC12 cells are plated onto collagen-coated dishes and treated with NGF in high serum medium [100 ng/ml NGF added to RPMI medium (Life Technologies, Grand Island, NY) supplemented with 5% fetal bovine serum, 10% horse serum, 100 U/ml penicillin, and 100 μg/ml streptomycin]. Twenty-four to 36 hr after beginning NGF treatment, when cells are undergoing a burst of proliferation and begin to extend neurites, cells are transfected with Lipofectamine-Plus (Life Technologies). Transfected cells are then maintained in a reduced serum medium containing NGF in which PC6–3 cells continue to extend neurites but no longer proliferate (as above but with 3% fetal bovine serum and 6% horse serum). Successfully transfected cells differentiate into neural cells that continue to express the transgene for up to 9 d in reduced serum medium (H. Paulson, unpublished observations).
In cotransfection experiments, constructs encoding Hsp27, HDJ-1, HDJ-2, or Hsp70 were used at a molar ratio of 3:1 with the ataxin-3 or GFP-polygln constructs. For controls, the respective empty expression plasmids were used in a 3:1 ratio with the ataxin-3 or GFP-polyglutamine constructs.
Immunocytochemistry, immunohistochemistry, and immunoblot analysis. Methods for immunofluorescence were described previously (Paulson et al., 1997a; Chai et al., 1999). Hsp40, Hsp70, and Hsp110 were detected with rabbit polyclonal anti-Hsp40 (1:5000; StressGen, Victoria, British Columbia, Canada), anti-Hsp70 (1:2500; StressGen), and anti-Hsp110 (1:250; StressGen), respectively. Hsp27, Hsp60, and Hsp90 were detected using mouse monoclonal anti-Hsp27 (1:250; StressGen), anti-Hsp60 (1:250; StressGen), and anti-Hsp90 antibody (1:250; StressGen), respectively. Endogenous HDJ-2 was detected with the mouse monoclonal anti-HDJ-2/HSDJ (1:2000; Neomarker, Fremont, CA). The FLAG epitope in expressed HDJ-2 was detected using the mouse monoclonal antibody M5 (1:1000; Sigma, St. Louis, MO). Heat shock cognate 70 (Hsc 70) was detected using a rat monoclonal anti-Hsc70 antibody (1:5000; StressGen). The second antibody was either anti-mouse-FITC plus anti-rabbit-tetramethylrhodamine isothiocyanate (TRITC) or anti-rabbit-FITC plus anti-mouse-TRITC except for the Hsc70, which was anti-rat-TRITC (1:2000; Jackson ImmunoResearch, West Grove, PA).
The SCA3/MJD brain tissue used in this study was described previously (Paulson et al., 1997b). Immunohistochemistry was performed as described previously (Paulson et al., 1997a). Concentrations of antisera were 1:200 for anti-Hsp27, anti-Hsp40, anti-HDJ-2, anti-Hsp60, anti-Hsp90, anti-Hsp110, and polyclonal anti-Hsp70 and 1:250 for monoclonal anti-Hsp70 antibody (StressGen). Immunostained samples were lightly counterstained with hematoxylin.
Immunoblot analysis as previously described (Chai et al., 1999) was used to confirm increased Hsp70 levels in cells expressing polyglutamine aggregates. COS7 cells transfected with pcDNA3 control plasmid, pcDNA3-HA-Q27, or pcDNA3-HA-Q78 were harvested 48 hr after transfection, and equal amounts of lysate were electrophoresed on SDS-polyacrylamide gels, transferred to membranes, probed with the monoclonal anti-Hsp70 antibody, and visualized by enzymatic chemiluminescence. Parallel samples were probed with anti-HA antibody to verify expression of HA-Q27 and HA-Q78. In the experiment shown, transfection efficiency was ∼20% as assessed by immunofluorescence of transfected cells.
Quantitation of aggregates and cell viability. In all experiments in which aggregates and cell viability were quantified, cells were scored by the same observer blinded to the treatment condition. For quantitation of all types of polygln aggregates in COS7 cells and for GFP-polygln aggregates in PC12 cells, 200–600 cells were counted per treatment/time point/experiment. For experiments in which HA-Q78 and NLS-ataxin 3 (Q78) were expressed in PC6–3 cells, a lower number of cells were counted because of the lower transfection efficiency (at least 75 cells per treatment/time point/experiment). A transfected cell was scored as “diffuse” if there were no inclusions and only diffuse staining, and as having “inclusions” if there was at least one inclusion (but typically more than one), regardless of whether there was also diffuse staining.
For quantitation of cell viability, dead cells were determined by visual inspection of GFP-positive cells using an Olympus Opticals (Tokyo, Japan) CK40 inverted microscope equipped for fluorescence and phase microscopy. Cells were plated and transfected on chamber slides, and regions of chambers with similar cell density were selected for analysis. The scoring of dead versus live cells was determined by viewing fields under fluorescence and phase microscopy at 400× magnification and scoring all GFP-positive cells as either “dead” (nonadherant and nonrefractile cells that were either shrunken and possessed multiple blebs, or remained large with a single asymmetric bleb) or “live” (all other cells, including any cells that could not be fully visualized because of overlapping cells). By default, poorly visualized cells are labeled as live; hence, this scoring technique may underestimate the percentage of dead cells. The same region of the chamber slide was visualized on consecutive days, with at least 12 consecutive fields counted per sample per time point (at least 200–600 cells per treatment/time point/experiment). All values were expressed as means ± SD. Statistical analysis was performed using the paired t test, with p < 0.05 considered statistically significant.
Trypan blue exclusion experiments confirmed increased toxicity in differentiated PC12 neural cells expressing GFP-Q80. Four days after transfection, identically plated PC12 neural cells expressing GFP, GFP-Q19, or GFP-Q80 were labeled with trypan blue (transfection efficiency ∼25% for each sample). Twenty consecutive fields under high power (400×) were scored for the number of trypan blue-positive cells (i.e., dead cells). The mean ± SD number of cells per field for GFP, GFP-Q19, and GFP-Q80 were 23.2 ± 4.2, 17.3 ± 3.6, and 50.4 ± 11.8, respectively. Similar results were obtained 3 and 5 d after transfection.
RESULTS
Specific Hsp chaperones localize to ataxin-3 aggregates
In SCA3/MJD, the disease protein ataxin-3 forms ubiquitinated NI in selected populations of disease neurons (Paulson et al., 1997a; Schmidt et al., 1998). We and others demonstrated previously that the NI formed in neurons of SCA3 brain can be modeled in cell culture (Ikeda et al., 1996; Paulson et al., 1997a; Perez et al., 1998;Chai et al., 1999; Evert et al., 1999). In transfected cells, truncated ataxin-3 protein consisting of a C-terminal, polygln-containing fragment readily forms aggregates and is cytotoxic in cells and in transgenic mouse and fly models (Ikeda et al., 1996; Paulson et al., 1997a; Warrick et al., 1998). We thus began our analysis of Hsp chaperones in cells expressing this truncated protein.
Preliminary studies in non-neuronal cell lines (COS7 and HEK-293) indicated that specific Hsp chaperones colocalized to aggregates formed by HA-Q78, a truncated ataxin-3 fragment with a Gln repeat of 78 residues (Table 1) (our unpublished observations). To extend these findings to a cellular model that more closely mirrored disease, we expressed a normal or mutant ataxin-3 fragment in differentiating PC12 neural cells and used immunofluorescence to determine whether specific Hsp chaperones colocalized to aggregates. In PC12 cells, a normal ataxin-3 fragment with a Gln repeat of 27 residues, HA-Q27, did not form aggregates (data not shown). In contrast, HA-Q78 readily formed intranuclear aggregates to which four distinct Hsp chaperones localized. The colocalizing chaperones were the heat shock cognate Hsc70 (also known as Hsp73), the inducible form of Hsp70 (also known as Hsp72), and two Hsp40 family members, HDJ-1 and HDJ-2 (Fig.1A). Nearly all aggregates formed by HA-Q78 were intranuclear, but these same four chaperone proteins also localized to the occasionally observed cytoplasmic aggregate (data not shown). Chaperone redistribution into polygln aggregates was only observed for Hsp40 and Hsp70 family members. Several other Hsps did not localize to aggregates or show an appreciable increase in expression: Hsp27, Hsp60, Hsp90, and Hsp110 (Table 1). For all tested chaperones, the results in PC12 neural cells were similar to the results in non-neuronal cells.
Table 1.
Hsp chaperone colocalization to polyglutamine aggregates
| Hsps | Cell culture | SCA3 brain | |||||
|---|---|---|---|---|---|---|---|
| HA-Q78 | NLS-ataxin 3 (Q78) | GFP-Q80 | |||||
| COS 7 | PC12 | COS 7 | PC12 | COS 7 | PC12 | ||
| Hsp27 | − | − | − | − | − | − | − |
| HDJ-1 | + | + | + | + | + | + | + |
| HDJ-2 | ++ | + | ++ | + | ++ | ++ | + |
| Hsp60 | − | − | − | − | − | − | − |
| Hsp70 | ++* | + | ++ | + | ++* | ++* | +/− |
| Hsc70 | ++ | ++ | ++ | ++ | ++ | ++ | NT |
| Hsp90 | − | − | − | − | − | − | − |
| Hsp110 | − | − | − | − | − | − | − |
+ and ++, Moderate and strong immunostaining of NI, respectively; +/−, rare, strongly immunopositive neurons; *, marked induction of Hsp70; NT, not tested.
Fig. 1.

Hsp40 and Hsp70 chaperones localize to intranuclear inclusions formed by mutant ataxin-3. Shown are immunofluorescence images of representative transfected PC12 neural cells containing intranuclear aggregates formed by truncated, HA-tagged ataxin-3 (HA-Q78) (A) or full-length, myc-tagged NLS-ataxin-3 (Q78) (B). Fixed cells were colabeled with anti-HA or anti-myc antisera to label ataxin-3 (green, left panels) and the indicated anti-Hsp antisera to label the chaperones Hsc70, Hsp70, HDJ-1, or HDJ-2 (red, middle panels). All four chaperones localize to polygln aggregates, as indicated by the merged images (right panels). DAPI staining of nuclei (blue) is included in the left andright panels of A andB.
Full-length mutant ataxin-3 also forms aggregates in transfected cells, although much less efficiently and with less toxicity than the truncated ataxin-3 fragment (Ikeda et al., 1996; Paulson et al., 1997a;Evert et al., 1999). We therefore extended our analysis of chaperone involvement to transfected cells expressing the full-length ataxin-3 protein. Similar results were obtained for full-length disease protein in both non-neuronal and neuronal cells (Fig. 1B; Table 1). Ataxin-3 with a Gln repeat of 78 residues, NLS-ataxin-3 (Q78), formed intranuclear aggregates to which the same four chaperones localized: HDJ-1, HDJ-2, Hsc70 and Hsp70. As with truncated ataxin-3, there was no colocalization of Hsp27, Hsp60, Hsp90, or Hsp110 to aggregates formed by full length ataxin-3 (Table 1). We typically expressed a modified ataxin-3 containing an N-terminal NLS to promote intranuclear localization and aggregation of ataxin-3. We have subsequently obtained identical results with full-length ataxin-3 not modified with an NLS and containing a slightly longer Gln repeat of 84 residues. Ataxin-3 (Q84) produced exclusively intranuclear inclusions in ∼20% of transfected PC12 cells, and the same four chaperone proteins localized to these aggregates (data not shown).
Induction of Hsp chaperones by pathogenic polygln protein
In addition to localizing to aggregates, certain chaperone proteins appeared to be upregulated in aggregate-containing cells. For example, the immunofluorescence signals of Hsp70 and Hsc70 were increased compared with the relatively low basal levels in untransfected cells. This was particularly clear for Hsp70, which was essentially undetectable in control PC12 cells but easily detectable in aggregate-containing cells (Fig. 1A and data not shown; see also Fig. 4A). This finding suggested that misfolded and aggregated polygln protein induced a stress response.
Fig. 4.
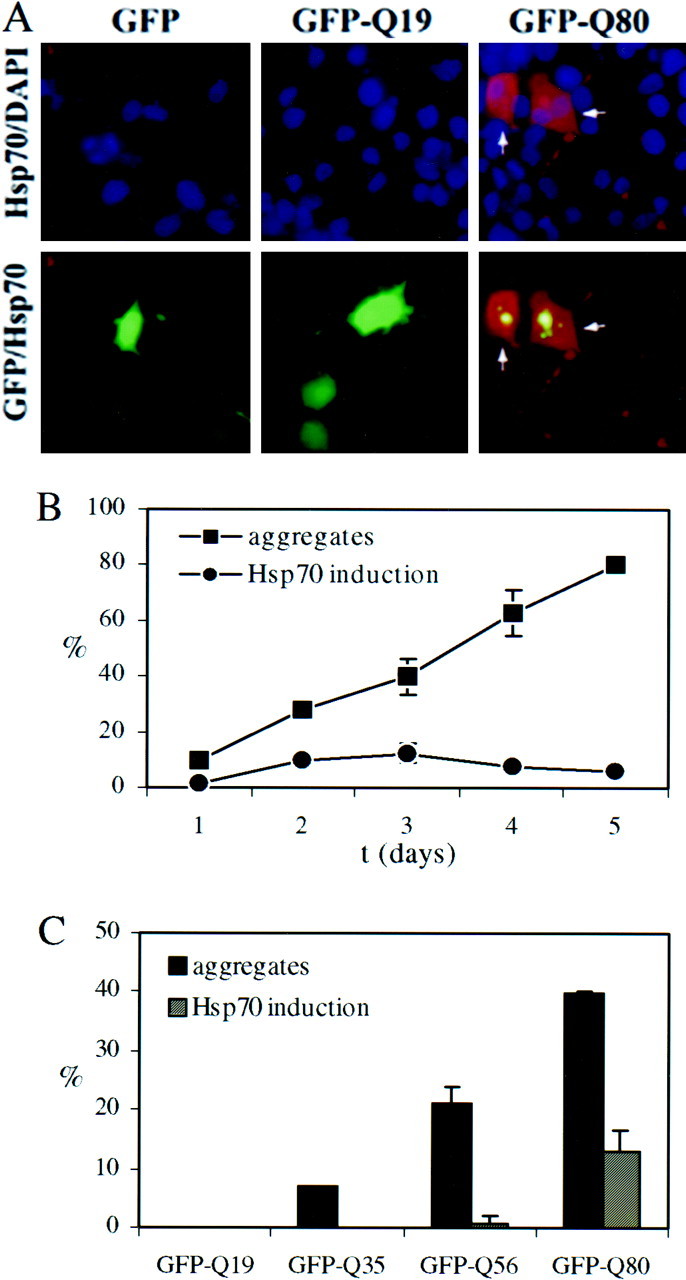
The misfolded polygln stress response in PC12 neurons: Hsp70 induction is correlated with the presence of aggregates.A, Immunofluorescence analysis of Hsp70 induction in transfected PC12 neurons expressing unmodified GFP or GFP-fusion proteins with Gln repeats of 19 or 80 residues (GFP-Q19, GFP-Q80). Hsp70 levels are markedly elevated in neurons containing GFP-Q80 aggregates (arrows) but remain low in cells expressing GFP or GFP-Q19. Depicted are paired images in which the top row shows Hsp70 immunofluorescence (red) merged with nuclear staining (blue), and the bottom row shows Hsp70 immunofluorescence merged with GFP fluorescence (green). B, Time course of GFP-Q80 aggregate formation and Hsp70 induction. The number of transfected PC12 neurons containing aggregates increases over time, reaching ∼80% 5 d after transfection. A subset of transfected cells shows marked Hsp70 induction, peaking 3 d after transfection. Markedly induced Hsp70 only occurred in aggregate-containing cells and never in cells in which the mutant polygln protein remained diffusely distributed.C, Aggregate formation and Hsp70 induction both occur in a glutamine repeat length-dependent manner but with different thresholds. Shown are the percentages of transfected cells with aggregates and the percentages with induced Hsp70, 3 d after transfection. Although GFP-Q35 and Q56 form aggregates, Hsp70 induction was never observed with GFP-Q35 aggregates and only rarely with GFP-Q56 aggregates. Results in B and C represent the means ± SD for two independent experiments.
In studies of COS7 cells transfected with truncated ataxin-3, we confirmed that a polygln-mediated cellular stress response occurred, as detected by immunofluorescence and immunoblot analysis. HA-Q78-expressing cells showed marked induction of Hsp70 protein, with some of the chaperone protein becoming sequestered into aggregates and some remaining diffuse in the cell (Fig.2A). Hsp70 induction was only observed in cells with visible aggregates, never in the small subset of cells in which HA-Q78 remained diffusely distributed. The nonpathogenic control protein, HA-Q27, did not cause induction of Hsp70. Immunoblot analysis confirmed that levels of Hsp70 were increased in cells expressing HA-Q78 aggregates (Fig.2B). Because only ∼ 20% of cells were transfected, the increase in Hsp70 levels shown by immunoblot analysis is likely to be an underestimate of the actual increase. Of the Hsp chaperones tested, only Hsp70 consistently underwent marked induction in aggregate-containing cells. This suggests that the polygln-mediated stress response differs from classic heat shock stress in which many Hsps are upregulated.
Fig. 2.

The polygln-mediated stress response: aggregate-containing cells show marked induction of the inducible chaperone Hsp70. Shown in A are immunofluorescence images of COS7 cells transfected with empty vector, pcDNA3-HAQ27, or pcDNA3-HAQ78 as indicated. Colabeling with anti-HA (green, left panels) and anti-Hsp70 (red, middle panels) antisera demonstrates that cells with aggregates show marked upregulation of Hsp70 with colocalization of induced Hsp70 to these intranuclear aggregates (arrows, middle panel ofbottom row). In contrast, levels of Hsp70 remain low in cells transfected with control vector or cells expressing nonpathogenic HA-Q27. DAPI staining of nuclei (blue) is shown in theleft and right panels. Immunoblot analysis (B) confirmed that Hsp70 levels were increased in cells expressing HA-Q78. The arrowindicates Hsp70.
Chaperone localization in SCA3/MJD disease tissue
The results in transfected cells prompted us to look for immunohistochemical evidence of induction and redistribution of Hsps in SCA3/MJD disease tissue. We focused our analysis on the ventral pons, a region of SCA3/MJD brain with abundant NI (Paulson et al., 1997a). In SCA3 brain, as in the cellular models, Hsp40 and Hsp70 chaperone proteins were found to localize to NI (Fig.3; Table 1). NI were commonly immunoreactive for HDJ-1 and HDJ-2; however, Hsp70-immunoreactive NI were only rarely observed (<5% of NI-containing neurons). The relatively few neurons that did contain NI immunopositive for Hsp70 also invariably showed diffusely increased Hsp70 staining, a finding confirmed with a second Hsp70-specific antibody. As in the cell-based models, NI in SCA3/MJD brain did not immunolabel for Hsp27, Hsp60, Hsp90, or Hsp110 (Table 1). These results indicate that specific Hsps localize to polygln aggregates in SCA3 disease brain and further suggest that, if marked Hsp70 induction does occur in the human disease, it takes place late in the disease process or only in a small subset of cells.
Fig. 3.

Colocalization of Hsp chaperones to NI in SCA3 brain. Shown are representative views of tissue sections from the ventral pons of an SCA3 brain immunohistochemically stained for Hsp70, HDJ-1, and HDJ-2. HDJ-1 and HDJ-2 commonly localize to NI (arrows). Hsp70 induction and colocalization to NI, however, is uncommon, as illustrated in the right panel in which only a single neuron shows increased Hsp70, although several neurons in this field are predicted to contain NI based on ubiquitin staining of neighboring sections (data not shown). The inset shows a higher power view of this NI-containing neuron with markedly increased Hsp70 staining (NI indicated by arrow). Scale bars: left,middle, 5 μm; right, 10 μm.
A PC12 neural model of polygln aggregation, stress response, and toxicity
The preceding findings suggested that polygln-induced changes in Hsp chaperone expression and cellular distribution play a role in disease. To further define the role of Hsp chaperones in disease, we developed a neural model of polygln aggregation and toxicity that would allow us to test the effects of chaperone expression. We chose to use NGF-treated PC12 neural cells (Pittman et al., 1993) transfected with expression constructs encoding GFP fused to Gln repeats of varying length (19, 35, 56, or 80 Gln residues) (Onodera et al., 1997; Moulder et al., 1999). The advantage of using GFP fusion proteins is that it allowed us to identify polygln-expressing neural cells by fluorescence microscopy and to follow this same population of unfixed cells over time for changes in aggregation and cellular morphology.
As anticipated from previous studies, polygln aggregation occurred in PC12 neural cells in a Gln repeat length-dependent manner (Figs.4,5B). With longer Gln repeats, aggregate formation was more efficient, occurring in up to 60–80% of cells expressing GFP-Q80 5 d after transfection. In contrast, GFP fused to a nonpathogenic repeat of 19 residues, GFP-Q19, remained diffusely distributed within the cell. Similar to aggregates of ataxin-3, aggregates formed by GFP-polygln protein showed recruitment of the same four Hsp chaperones, HDJ-1, HDJ-2, Hsc70, and Hsp70 (Table1).
Fig. 5.

Polygln-mediated cell death and relationship to aggregate formation. Polygln toxicity in PC12 neurons occurs in a glutamine repeat length-dependent manner and is correlated with the presence of aggregates. A, Shown are fluorescence images of representative transfected PC12 neurons expressing GFP-Q19 (which remains diffuse in cells) and GFP-Q80 (which readily forms aggregates). Aggregate-containing cells (arrows) become rounded, nonrefractile, and nonadherent and frequently display morphological features of cell death, such as membrane blebbing and condensed nuclei, illustrated here by DAPI staining. B, Time course of inclusion formation by GFP-Q80 versus GFP-Q19 in PC12 neurons.C, Time course of cell death in PC12 cells expressing GFP-Q19 or Q80. The rate of death in GFP-Q80-expressing cells is significantly increased over that in GFP-Q19-expressing cells, which have a low rate of death similar to that in mock-transfected cells analyzed in parallel. D, Cell death is directly correlated with the presence of inclusions. Three days after transfection, cells were identified by fluorescence as diffusely stained or having inclusions. Shown are the percentage of cells in each of these two categories that were then scored as dead. In GFP-Q80-expressing cells, nearly half of all inclusion-containing cells were scored as dead. In contrast, diffusely staining GFP-Q80 cells were scored as dead at a rate similar to the background rate in GFP-Q19 cells. Results in B–D represent the means ± SD of two independent experiments.
This neural model also confirmed the existence of a polygln-induced stress response as measured by Hsp70 immunofluorescence (Fig. 4). In nearly all aggregate-containing cells, Hsp70 was found within aggregates 1 μm or greater in diameter. In most cells, Hsp70 immunoreactivity was confined to the aggregates, consistent with a modest induction. In a subset of cells, however, we observed marked induction with diffuse expression of Hsp70 throughout the cell (Fig.4A). This marked Hsp70 induction initially paralleled the development of aggregates but peaked at 3 d, while aggregation further increased (Fig. 4B). Marked induction occurred exclusively in cells with visible aggregates and never in cells with diffusely distributed polygln protein. The mere presence of aggregates, however, was not sufficient to cause marked induction of Hsp70. For example, induction was never observed with GFP-Q35 and only rarely with GFP-Q56, although both fusion proteins formed aggregates (Fig. 4C). Even with GFP-Q80, maximally only one-third of aggregate-containing cells showed marked Hsp70 induction (Fig.4C). These results suggest that polygln aggregation may be necessary, but not sufficient, to elicit a stress response.
In PC12 neural cells, expression of expanded polygln protein proved to be toxic in a glutamine repeat length-dependent manner (Fig.5A,C). Neural cells expressing GFP-Q80 underwent cell death at a significantly higher rate than did cells expressing the control proteins, GFP-Q19 (Fig. 5C) or GFP (data not shown). When followed over time by phase and fluorescence microscopy, PC12 neural cells expressing GFP-Q80 underwent morphological changes indicative of cell death with mixed features of apoptosis and necrosis. Most commonly, dying cells lost their neurites, became nonadherent and shrunken with numerous blebs, and underwent nuclear condensation as illustrated by 4,6-diamidino-2-phenylindole (DAPI) staining of fixed specimens (Fig. 5A). Although these morphological changes are most consistent with apoptosis, other dying or dead cells remained large and developed a single asymmetric bleb as in necrotic death. Thus, at least in this cellular model, polygln-mediated cell death may represent a mixed type of cell death.
The causal relationship of polygln aggregate formation to cell death remains uncertain. To determine whether the presence of inclusions correlated with cell death in our model, we used morphological criteria to score cells expressing GFP-Q80 as either dying–dead or alive by phase microscopy, and as having inclusions or not (i.e., diffusely stained) by fluorescence microscopy. The population of GFP-Q80-expressing cells with inclusions showed a markedly higher rate of cell death than did diffusely stained cells (Fig. 5D). In contrast, GFP-Q80-expressing cells in which the expanded polygln protein remained diffuse showed a low rate of death similar to control cells expressing the nonpathogenic GFP-Q19. These results indicate that the presence of inclusions is correlated with polygln toxicity in this PC12 neural model.
Hsp40 chaperones suppress ataxin-3 aggregation in neural and non-neural cells
The molecular chaperone HDJ-2 has been shown to suppress aggregation of two other mutant polygln proteins, the androgen receptor and ataxin-1 (Cummings et al., 1998; Stenoien et al., 1999). To determine whether Hsp chaperones could suppress aggregation of mutant ataxin-3, we coexpressed specific chaperones with HA-Q78 or NLS-ataxin-3 (Q78) and assessed the frequency of aggregate formation. These studies were initially performed in COS7 cells and then confirmed in PC12 neural cells. As shown in Figure6, the Hsp40 chaperones HDJ-1 and HDJ-2 suppressed aggregate formation by both truncated and full-length ataxin-3. In contrast to Hsp40 chaperones, neither Hsp70 nor Hsp27 significantly suppressed aggregation (Fig.6E,F and data not shown). Unexpectedly, a partially deleted form of HDJ-2 lacking the N-terminal J domain, HDJ-2 (del9–107), was even more effective than full-length HDJ-2 at suppressing aggregation. In studies by others, this partially deleted HDJ-2 protein did not suppress ataxin-1 aggregation but was modestly effective in suppressing AR aggregation (Cummings et al., 1998; Stenoien et al., 1999). The J domain of Hsp40 is the region of the protein responsible for mediating the binding of Hsp40 to Hsp70. Hsp40 proteins are generally thought to be cochaperones for Hsp70 that bind to and stimulate the ATPase activity of Hsp70, thereby enhancing the chaperone function of Hsp70 (Hartl, 1996). The fact that HDJ-2 is still able to suppress aggregation, even when the J domain is deleted, suggests that this suppression occurs independent of interactions with Hsp70. Moreover, in COS7 cells, coexpression of Hsp70 with HDJ-2 did not enhance suppression above that seen with HDJ-2 alone (data not shown).
Fig. 6.

Overexpression of Hsp40 chaperones reduces aggregation of truncated and full-length ataxin-3. COS7 cells were cotransfected with plasmids encoding HA-Q78 (A,C, E) or NLS-ataxin-3 (Q78) (B, D, F), together with empty control plasmid or plasmids encoding HDJ-1 (A, B), HDJ-2 (C,D), or Hsp70 (E,F). Suppression of aggregate formation occurred with HDJ-1 and HDJ-2 but not Hsp70. A partially deleted form of HDJ-2 lacking the DnaJ domain (del9–107) also suppressed aggregation of mutant ataxin-3. Shown are the mean values of three independent experiments. Statistically significant differences, determined by paired t test, are indicated by asterisks(*p < 0.05; **p < 0.01).
Similarly, in PC12 neural cells, HDJ-1 and HDJ-2 suppressed aggregate formation by both truncated and full-length ataxin-3, whereas Hsp70 and Hsp27 did not. As in COS7 cells, the partially deleted form of HDJ-2 appeared to be more effective than full-length HDJ-2 at suppressing aggregation. In addition, the coexpression of Hsp70 with HDJ-2 did not affect suppression of aggregation (data not shown).
Suppression of polygln aggregation is associated with a decrease in neurotoxicity
The ability to suppress polygln aggregation by overexpressing Hsp40 chaperones allowed us to ask whether, in our PC12 neural model of toxicity, modulating aggregation resulted in a change in toxicity. First, we confirmed that the Hsp40 chaperones suppressed aggregation of GFP-polygln fusion proteins. Both GFP-Q56 and GFP-Q80 readily form aggregates and are associated with an increase in neuronal death (Moulder et al., 1999), so both of these fusion proteins were analyzed for the ability of HDJ-1 and HDJ-2 to suppress aggregation. Of the two Hsp40 proteins, only HDJ-1 proved to be effective at suppressing aggregation of GFP-Q56 and GFP-Q80. Neither HDJ-2 nor HDJ-2 (del9–107) suppressed aggregation (data not shown).
As shown in Figure 7, A andC, coexpression of HDJ-1 with polygln protein resulted in a modest, but statistically significant, decrease in the aggregation of GFP-Q56 and GFP-Q80 in PC12 neural cells. This reduction in aggregation was associated with a parallel, statistically significant decrease in cell death (Fig. 7B,D). As indicated by the results in Table 2, there is inherent variability between experiments with respect to the degree of aggregation and toxicity. However, the suppression of aggregation and toxicity by HDJ-1 proved to be internally consistent in each of three independent experiments for GFP-Q56 and GFP-Q80.
Fig. 7.

Hsp40 chaperone HDJ-1 suppresses aggregation and toxicity of polygln protein in PC12 neural cells. PC12 neurons transfected with plasmids encoding GFP-Q56 (A,B) or GFP-Q80 (C, D), together with HDJ-1 or empty control plasmid, were scored for aggregates and cell death under blinded conditions. HDJ-1 partially suppressed aggregate formation and cell death with both GFP-polygln proteins. Shown are the means of three independent experiments. Statistically significant differences, determined by pairedt test, are indicated by asterisks(*p < 0.05; **p < 0.01).
Table 2.
HDJ-1 suppression of polygln aggregation and toxicity
| Day 2 | Day 3 | Day 4 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| a | b | c | a | b | c | a | b | c | |
| GFP-Q56 | |||||||||
| % aggregates | |||||||||
| Control | 16.9 | 18.5 | 6.1 | 35.8 | 30.3 | 24.5 | 35.8 | 52.2 | 45.7 |
| HDJ-1 | 7.0 | 4.8 | 3.7 | 19.7 | 18.6 | 10.8 | 27.1 | 39.1 | 31.8 |
| NS | p < 0.01 | p < 0.05 | |||||||
| % cell death | |||||||||
| Control | 7.2 | 13.5 | 8.4 | 19.9 | 24.6 | 14.8 | 21.9 | 33.9 | 26.4 |
| HDJ-1 | 6.2 | 7.6 | 3.7 | 14.8 | 15.9 | 10.4 | 19.8 | 29.0 | 22.5 |
| NS | p < 0.05 | p < 0.05 | |||||||
| GFP-Q80 | |||||||||
| % aggregates | |||||||||
| Control | 31.6 | 48.6 | 37.0 | 48.4 | 63.8 | 56.0 | 73.4 | 77.5 | 72.5 |
| HDJ-1 | 24.7 | 32.1 | 24.9 | 38.2 | 54.6 | 45.3 | 52.5 | 66.7 | 64.4 |
| NS (p = 0.05) | p < 0.01 | NS | |||||||
| % cell death | |||||||||
| Control | 10.2 | 21.1 | 9.7 | 22.1 | 32.4 | 23.0 | 38.9 | 41.6 | 36.8 |
| HDJ-1 | 9.0 | 10.1 | 7.2 | 15.9 | 25.9 | 17.2 | 25.8 | 40.3 | 32.0 |
| NS | p < 0.01 | NS | |||||||
GFP-Q56 or GFP-Q80 was cotransfected with empty vector (Control) or HDJ-1 in differentiating PC12 neural cells. Shown are the data from three independent experiments (a, b, c) performed identically; in Figure 7, the mean values for these three experiments are graphically depicted. In each case, the percentage of transfected cells with aggregates and the percentage of transfected cells scored as dead are indicated. Statistical analysis was performed using the pairedt test (p < 0.05 considered statistically significant).
DISCUSSION
Here, we have presented evidence that cellular redistribution of Hsp40 and Hsp70 chaperones and induction of Hsp70 are part of the cellular response to mutant polygln protein and that overexpression of select Hsp chaperones can reduce polygln aggregation and toxicity. Several of our findings support a role for Hsp molecular chaperones in polyglutamine disease. First, Hsp40 and Hsp70 chaperones were shown to localize to NI formed by ataxin-3 in cellular models and in disease tissue. Second, the expression of aggregated polygln protein elicited a stress response in cells, manifested by a marked increase in expression of the inducible chaperone Hsp70. Third, overexpression of Hsp40 chaperones suppressed aggregation by truncated or full-length ataxin-3. Finally, in a PC12 neural model that displays polygln toxicity, overexpression of the Hsp40 chaperone HDJ-1 suppressed aggregation of GFP-polygln fusion protein with a corresponding and parallel decrease in toxicity. Together, these findings support the view that expanded polygln protein is “recognized” by the cell as abnormal polypeptide and that, in response to this apparent cellular stress, there is altered expression and cellular redistribution of specific molecular chaperones. These changes in chaperone activity may enhance the capacity of the cell to trap, refold, disaggregate, or degrade mutant polygln protein.
The polygln stress response
The marked induction of Hsp70 by mutant ataxin-3 and GFP-polygln suggests that misfolded and aggregated polygln protein elicits a stress response in cells. The Hsp70 induction seen here with ataxin-3, and by others with ataxin-1 and AR, indicates that this stress response may be common to polygln disease proteins. Our findings indicate that the polygln stress response does not appear to involve most other recognized heat shock proteins and thus differs in detail from the classic heat shock response. In the latter, induction of stress proteins is mediated by binding of heat shock factor 1 (HSF1) to the heat shock promoter element found in the various Hsp genes (Morimoto, 1998). It will be interesting to determine whether HSF1 or similar mediators of stress responses, such as HSF2 (Mathew et al., 1998), trigger the polygln stress response. Equally important is the identification of other cellular factors that may be induced as part of the polygln stress response. Components of the ubiquitin–proteasome pathway are likely candidates, because various proteasome subunits have already been shown to redistribute to polygln aggregates (Cummings et al., 1998; Chai et al., 1999; Stenoien et al., 1999). Other potential candidates include stress-activated protein kinases and cellular caspases, both of which mediate certain types of neuronal cell death (Xia et al., 1995; Pettmann and Henderson 1998;Sánchez et al., 1999). In HD, expression of mutant huntingtin leads to chronic sublethal activation of caspase-1, which in turn may accelerate disease pathogenesis (Ona et al., 1999). Similar molecular pathways may be activated by polygln protein in transfected primary neurons (Moulder et al., 1999). Therefore, identifying additional components of the stress response may yield clues to the mechanism of polygln toxicity.
Our results suggest that polygln aggregation may be necessary but not sufficient to elicit a stress response, as measured by marked Hsp70 induction (in which the chaperone is present diffusely within the cell and in inclusions). For example, in PC12 neural cells, marked Hsp70 induction was never observed with GFP-Q35 and only rarely with GFP-Q56, although both fusion proteins formed aggregates (Fig. 4C); even with GFP-Q80, only one-third of aggregate-containing cells showed marked Hsp70 induction. One possible explanation is that the stress response mounted by the cell is graded and dependent on aggregate burden. Only when the aggregate burden is high, as it frequently is in GFP-Q80-expressing cells, is there marked induction.
Our finding that Hsp40 and Hsp70 chaperones are recruited into ataxin-3 aggregates extends the observations of others showing chaperone recruitment into aggregates of mutant androgen receptor and ataxin-1 disease proteins (Cummings et al., 1998; Stenoien et al., 1999). The results are not identical for the three proteins; for example, Hsp90 localizes to aggregates of androgen receptor but not of ataxin-3, yet the similarities suggest that chaperone involvement is a general feature of polygln diseases. Moreover, the fact that full-length ataxin-3, truncated ataxin-3, and GFP-polygln fusion proteins all displayed similar chaperone recruitment indicates that it is the expanded polygln domain itself, not the particular protein context, that drives the process. It remains to be determined whether Hsp40 or Hsp70 chaperones directly bind to the expanded polygln domain or to other domains of the disease protein.
The simplest interpretation of the polygln stress response is that it represents an effort by the cell to increase its capacity to refold, disaggregate, or eliminate misfolded polygln protein. This view is supported by the fact that overexpressing Hsp40 chaperones can suppress aggregation of polygln protein, as shown here (Figs. 6, 7) and by others (Cummings et al., 1998; Stenoien et al., 1999). Alternatively, the stress response may signify that the cellular burden of abnormal polygln protein has saturated the normal capacity of the neuron to handle misfolded polypeptides. Polygln diseases have the interesting characteristic of selective neurotoxicity, despite widespread expression of the disease proteins. If neurons are less capable than non-neuronal cells of mounting a sufficient stress response to mutant polygln protein, this could in part explain the neuronal selectivity.
Aggregation and its relationship to toxicity
The relationship of polygln aggregation to cellular toxicity is unclear (Kim and Tanzi, 1998; Sisodia, 1998; Paulson, 1999). Some studies suggest that large aggregates, such as NI, may contribute to pathogenesis; others, however, suggest that polygln aggregates may be irrelevant to pathogenesis or perhaps even serve a protective role (Klement et al., 1998; Saudou et al., 1998; Kim et al., 1999; Li et al., 1999; Moulder et al., 1999). In this study, we have created a PC12 neural cell model that displays aggregation and toxicity upon expression of mutant polygln protein. In this neural model, overexpression of the Hsp40 chaperone, HDJ-1, suppressed aggregate formation. Moreover, this suppression of aggregation was associated with a parallel and corresponding decrease in polygln toxicity. This direct correlation between aggregation and toxicity argues against a protective role for aggregates in this cellular model. However, based on our findings and in view of the results of others, we cannot assert a causal relationship between aggregate formation and toxicity. We suggest that the toxicity of polygln protein is dependent on an abnormal conformation which is also aggregation-prone. Under most cellular conditions, perhaps, this abnormal conformation and resultant toxicity cannot be dissociated from aggregation.
Suppression of polygln aggregation by Hsp40 chaperones has now been observed for three distinct disease proteins, as well as for polygln-GFP fusion proteins. Hsp40 is known to be a cochaperone for Hsp70; it binds to and stimulates the ATPase activity of Hsp70 (Cyr et al., 1994; Hartl, 1996; Bukau and Horwich, 1998). However, Hsp40 can also bind misfolded polypeptide on its own. In our studies, coexpressed Hsp70 did not further enhance suppression by HDJ-2 and, moreover, partially truncated HDJ-2 lacking the J domain still suppressed aggregation. These findings argue that the mechanism by which Hsp40 mediates suppression of polygln aggregation may not depend on interactions with Hsp70. Rather, Hsp40 may directly bind to and trap abnormally folded polygln protein, in the process neutralizing its toxicity. A second possibility is that Hsp40 binding to polyglutamine protein facilitates delivery of the misfolded polypeptide to the cellular machinery for proteolytic degradation, particularly the proteasome (Hayes and Dice, 1996). Lastly, because in our model endogenous Hsp70 is induced to high levels in a subset of transfected cells, we cannot exclude the possibility that induced, endogenous Hsp70 functions together with Hsp40 to mediate suppression. Regardless of the mechanism, Hsp40 chaperones now represent potential therapeutic molecules. If suppression of polygln disease by Hsp40 can be confirmed in animal models of SCA3/MJD (Ikeda et al., 1996; Warrick et al., 1998) and there are no adverse effects of overexpression, then targeted expression of Hsp40 or similar chaperones may become a feasible approach to slow the progression of SCA3/MJD and other fatal polygln diseases.
Footnotes
This work was supported by grants from the Roy J. Carver Charitable Trust and the Howard Hughes Medical Institute (H.L.P.), and the Packard Foundation, the Huntington's Disease Society of America, and the Hereditary Disease Foundation (N.M.B.). We thank K. Ohtsuka, W. Strittmatter, and H. Zoghbi for providing plasmids. We also thank K. Campbell and his laboratory for advice and assistance with graphics.
Correspondence should be addressed to Henry L. Paulson, Department of Neurology, 3160 Med Labs, The University of Iowa College of Medicine, Iowa City, IA 52242. E-mail: henry-paulson@uiowa.edu.
REFERENCES
- 1.Bukau B, Horwich AL. The Hsp 70 and Hsp 60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 2.Chai Y, Koppenhafer S, Shoesmith S, Perez M, Paulson H. Evidence for proteasome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum Mol Genet. 1999;8:673–682. doi: 10.1093/hmg/8.4.673. [DOI] [PubMed] [Google Scholar]
- 3.Cooper J, Schilling G, Peters M, Herring W, Sharp A, Kaminsky Z, Masone J, Khan F, Delanoy M, Borchelt D, Dawson V, Dawson T, Ross C. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum Mol Genet. 1998;7:783–790. doi: 10.1093/hmg/7.5.783. [DOI] [PubMed] [Google Scholar]
- 4.Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of ataxin-1 aggregation and altered subcellular proteosome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- 5.Cyr DM, Langer T, Douglas MG. Dna-J like proteins: molecular chaperones and specific regulators of Hsp 70. Trends Biochem Sci. 1994;19:176–181. doi: 10.1016/0968-0004(94)90281-x. [DOI] [PubMed] [Google Scholar]
- 6.Evert BO, Wüllner U, Schulz JB, Weller M, Groscurth P, Trottier Y, Brice A, Klockgether T. High level expression of expanded full-length ataxin-3 in vitro causes cell death and formation of intranuclear inclusions in neuronal cells. Hum Mol Genet. 1999;8:1169–1176. doi: 10.1093/hmg/8.7.1169. [DOI] [PubMed] [Google Scholar]
- 7.Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 8.Gottesman S, Wickner S, Maurizi MR. Protein quality control: triage by chaperones and proteases. Genes Dev. 1997;11:815–823. doi: 10.1101/gad.11.7.815. [DOI] [PubMed] [Google Scholar]
- 9.Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neurophil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hackam AS, Wellington CL, Hayden MR. The fatal attraction of polyglutamine-containing proteins. Clin Genet. 1998;53:233–242. doi: 10.1111/j.1399-0004.1998.tb02687.x. [DOI] [PubMed] [Google Scholar]
- 11.Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–580. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 12.Hayes SA, Dice JF. Roles of molecular chaperones in protein degradation. J Cell Biol. 1996;132:255–258. doi: 10.1083/jcb.132.3.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ikeda H, Yamaguchi M, Sugai S, Aze Y, Narumiya S, Kakizuka A. Expanded polyglutamine in the Machado-Joseph disease protein induces cell death in vitro and in vivo. Nat Genet. 1996;13:196–202. doi: 10.1038/ng0696-196. [DOI] [PubMed] [Google Scholar]
- 14.Kakizuka A. Protein precipitation: a common etiology in neurodegenerative disorders? Trends Genet. 1998;14:396–402. doi: 10.1016/s0168-9525(98)01559-5. [DOI] [PubMed] [Google Scholar]
- 15.Kim M, Lee H-S, LaForet G, McIntyre C, Martin EJ, Chang P, Kim TW, Williams M, Reddy PH, Tagle D, Boyce FM, Won L, Heller A, Aronin N, DiFiglia M. Mutant huntingtin expression in clonal striatal cells: dissociation of inclusion formation and neuronal survival by caspase inhibition . J Neurosci. 1999;19:964–973. doi: 10.1523/JNEUROSCI.19-03-00964.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim TW, Tanzi RE. Neuronal intranuclear inclusions in polyglutamine diseases: nuclear weapons or nuclear fallout? Neuron. 1998;21:657–659. doi: 10.1016/s0896-6273(00)80581-4. [DOI] [PubMed] [Google Scholar]
- 17.Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 18.Koshy BT, Zoghbi HY. The CAG/polyglutamine tract diseases: gene products and molecular pathogenesis. Brain Pathol. 1997;7:927–942. doi: 10.1111/j.1750-3639.1997.tb00894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li SH, Cheng AL, Li H, Li XJ. Cellular defects and altered gene expression in PC12 cells stably expressing mutant huntingtin. J Neurosci. 1999;19:5159–5172. doi: 10.1523/JNEUROSCI.19-13-05159.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim SU, Bredesen DE, Tufaro F, Hayden MR. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet. 1998;18:150–154. doi: 10.1038/ng0298-150. [DOI] [PubMed] [Google Scholar]
- 21.Mathew A, Mathur SK, Morimoto RI. Heat shock response and protein degradation: regulation of HSF2 by the ubiqutin-proteasome pathway. Mol Cell Biol. 1998;18:5091–5098. doi: 10.1128/mcb.18.9.5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- 23.Moulder KL, Onodera O, Burke JR, Strittmatter WJ, Johnson EM., Jr Generation of neuronal intranuclear inclusions by polyglutamine-GFP: analysis of inclusion clearance and toxicity as a function of polyglutamine length. J Neurosci. 1999;19:705–715. doi: 10.1523/JNEUROSCI.19-02-00705.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ona VO, Li M, Vonsattel JPG, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Friedlander RM. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature. 1999;399:263–267. doi: 10.1038/20446. [DOI] [PubMed] [Google Scholar]
- 25.Onodera O, Burke JR, Miller SE, Hester S, Tsuji S, Roses AD, Strittmatter WJ. Oligomerization of expanded-polyglutamine domain fluorescent fusion proteins in cultured mammalian cells. Biochem Biophys Res Commun. 1997;238:599–605. doi: 10.1006/bbrc.1997.7337. [DOI] [PubMed] [Google Scholar]
- 26.Paulson HL. Protein fate in neurodegenerative proteinopathies: polyglutamine diseases join the (mis)fold. Am J Hum Genet. 1999;64:339–345. doi: 10.1086/302269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, Vig P, Mandel JL, Fischbeck KH, Pittman RN. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997a;19:333–344. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 28.Paulson HL, Das SS, Crino PB, Perez MK, Patel SC, Gotsdiner D, Fischbeck KH, Pittman RN. Machado-Joseph disease gene product is a cytoplasmic protein expressed widely in brain. Ann Neurol. 1997b;41:453–462. doi: 10.1002/ana.410410408. [DOI] [PubMed] [Google Scholar]
- 29.Perez M, Paulson H, Pendse S, Saionz S, Bonini N, Pittman R. Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J Cell Biol. 1998;143:1457–1470. doi: 10.1083/jcb.143.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perutz MF. Glutamine repeats and neurodegenerative diseases: molecular aspects . Trends Biochem Sci. 1999;24:58–63. doi: 10.1016/s0968-0004(98)01350-4. [DOI] [PubMed] [Google Scholar]
- 31.Pettmann B, Henderson CE. Neuronal cell death. Neuron. 1998;20:633–647. doi: 10.1016/s0896-6273(00)81004-1. [DOI] [PubMed] [Google Scholar]
- 32.Pittman RN, Wang S, DiBenedetto AJ, Mills J. A system for characterizing cellular and molecular events in programmed neuronal cell death. J Neurosci. 1993;64:566–575. doi: 10.1523/JNEUROSCI.13-09-03669.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sánchez I, Xu CJ, Juo P, Kakizaka A, Blenis J, Yuan J. Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron. 1999;22:623–633. doi: 10.1016/s0896-6273(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 34.Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 35.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt T, Landwehrmeyer B, Schmitt I, Trottier Y, Auburger G, Laccone F, Klockgether T, Völpel M, Epplen JT, Schöls L, Riess O. An isoform of ataxin-3 accumulates in the nucleus of neuronal cells in affected brain regions of SCA3 patients. Brain Pathol. 1998;8:669–679. doi: 10.1111/j.1750-3639.1998.tb00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sisodia SS. Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? Cell. 1998;95:1–4. doi: 10.1016/s0092-8674(00)81743-2. [DOI] [PubMed] [Google Scholar]
- 38.Stenoien DL, Cummings CJ, Adams HP, Mancini MG, Patel K, DeMartino GN, Marcelli M, Weigel NL, Mancini MA. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum Mol Genet. 1999;8:731–741. doi: 10.1093/hmg/8.5.731. [DOI] [PubMed] [Google Scholar]
- 39.Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L, Agid Y, Brice A, Mandel J-L. Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature. 1995;378:403–406. doi: 10.1038/378403a0. [DOI] [PubMed] [Google Scholar]
- 40.Warrick JM, Paulson HL, Gray-Board G, Bui QT, Fischbeck KH, Pittman RN, Bonini NM. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell. 1998;93:939–949. doi: 10.1016/s0092-8674(00)81200-3. [DOI] [PubMed] [Google Scholar]
- 41.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]