

Abstract
Long-term potentiation (LTP) in the amygdala is a leading candidate mechanism to explain fear conditioning, a prominent model of emotional memory. LTP occurs in the pathway from the auditory thalamus to the lateral amygdala, and during fear conditioning LTP-like changes occur in the synapses of this pathway. Nevertheless, LTP has not been investigated in the thalamoamygdala pathway using in vitro recordings; hence little is known about the underlying mechanisms. We therefore examined thalamoamygdala LTP in vitro using visualized whole-cell patch recording. LTP at these synapses was dependent on postsynaptic calcium entry, similar to synaptic plasticity in other regions of the brain. However, unlike many forms of synaptic plasticity, thalamoamygdala LTP was independent of NMDA receptors, despite their presence at these synapses, and instead was dependent on L-type voltage-gated calcium channels. This was true when LTP was induced by pairing presynaptic activity with either action potentials or constant depolarization in the postsynaptic cell. In addition, the LTP was associative, in that it required concurrent pre- and postsynaptic activity, and it was synapse specific. Thus, although this LTP is different from that described at other synapses in the brain, it is nonetheless well suited to mediate classical fear conditioning.
Keywords: electrophysiology, in vitro, amygdala, LTP, calcium channels, fear, synapse
Classical fear conditioning is a robust form of learning that occurs throughout the animal kingdom. It involves the pairing of an initially innocuous conditioned stimulus (CS), such as a tone, with a noxious unconditioned stimulus (US), such as an electric shock to the skin. After even a single pairing, the tone will, when presented alone, elicit defensive and autonomic responses that are characteristically evoked by stimuli that cause harm or warn of danger.
The neural pathways mediating fear conditioning involve the transmission of sensory information about the CS to the amygdala and the control of conditioned responses by output projections from the amygdala (for review, see LeDoux, 1995). The CS enters the amygdala by way of the lateral nucleus (LA)—principally the dorsal subdivision (LAd)—which then distributes the inputs to other amygdala regions (LeDoux, 1995; Pitkänen et al., 1997). Several lines of evidence suggest that essential aspects of plasticity occur in this early stage of processing within the amygdala (see Quirk et al., 1997).
Long-term potentiation (LTP) is a form of experience-dependent synaptic plasticity that is believed to underlie learning (Milner et al., 1998) and has been studied most extensively in the CA1 region of the hippocampus. However relatively little progress has been made in relating this synaptic plasticity to actual learning processes (Barnes, 1995; Eichenbaum, 1995; Stevens, 1998).
LTP has also been induced in several pathways involving the amygdala, including pathways that transmit sensory inputs to the LA during fear conditioning (Chapman et al., 1990; Clugnet and LeDoux, 1990; Chapman and Bellavance, 1992; Rogan and LeDoux, 1995; Watanabe et al., 1995;Brambilla et al., 1997; Huang and Kandel, 1998; Li et al., 1998). Induction of LTP amplifies the processing of a CS-like acoustic stimulus in the LA (Rogan and LeDoux, 1995), and fear conditioning and LTP induction lead to similar changes in neural responses elicited by a tone in the LA (Rogan et al., 1997). These observations, together with the fact that the LA is required for conditioning (see above) and the similarity between the stimulation protocols used in classical conditioning and those used to induce LTP (Brown et al., 1988), suggest that a link between LTP and learning may be more easily established for LA than hippocampal synapses (see Barnes, 1995; Eichenbaum, 1995;Malenka and Nicoll, 1997; Stevens, 1998).
Studies examining amygdala LTP using in vitro preparations have focused exclusively on the cortical inputs to the LA (Chapman et al., 1990; Chapman and Bellavance, 1992; Gean et al., 1993; Watanabe et al., 1995; Brambilla et al., 1997; Huang and Kandel, 1998; Li et al., 1998). However, the in vivo studies linking amygdala LTP to fear learning have involved the thalamic pathway (Rogan and LeDoux, 1995;Rogan et al., 1997). Therefore, in an effort to uncover the synaptic mechanisms that underlie fear learning via the thalamoamygdala pathway, we examined the properties and mechanisms of LTP at these synapsesin vitro.
MATERIALS AND METHODS
Male Sprague Dawley rats (3–5 weeks old) were deeply anesthetized with halothane, and the brain was rapidly removed and transferred to ice-cold Ringer's solution. The Ringer's solution contained (in mm): 115 NaCl, 3.3 KCl, 1 MgSO4, 2 CaCl2, 25.5 NaHCO3, 1.2 NaH2PO4, 5 lactic acid, and 25 glucose, equilibrated with 95% O2/5% CO2. Picrotoxin (100 μm) was included in all experiments to block fast GABAergic transmission. Coronal slices (400 μm thick) containing the amygdala were cut, and the cortex overlying the amygdala was cut away with a scalpel so that, in the presence of picrotoxin, cortical epileptic burst discharges would not invade the amygdala. The slices were placed in a holding chamber at 32–34°C for 0.5 hr to improve subsequent patch success and quality and then were allowed to return to room temperature for at least another 0.5 hr before recording. Immediately before recording, a slice was transferred to a superfusion recording chamber (Warner Instruments) with a flow rate of 1.5–2.5 ml/min and held immobilized beneath a nylon net stretched over platinum wire. All experiments were done at room temperature. An upright microscope equipped with infrared differential interference contrast optics (Olympus Optical, Tokyo, Japan) was used to perform whole-cell patch recordings under visual guidance. Recording electrodes were filled with (in mm): 130 K-gluconate, 0.6 EGTA, 2 MgCl2, 5 KCl, 10 HEPES, 2 Mg-ATP, 0.3 Na3-GTP, and, in some, 0.25–0.5% biocytin, pH 7.3 (290–300 mOsm). Membrane potentials were adjusted for a 10 mV junction potential. The electrodes typically had resistances of 4–8 MΩ.
Stimuli were delivered through bipolar stainless steel stimulating electrodes (Frederick Haer) that were positioned while using a low-power objective. This allowed easy identification of the LA, triangular in shape in the coronal plane and bounded by the external capsule laterally and the lateral association bundle medially. Thalamic afferents travel rostrally toward the amygdala in the cerebral peduncle and, at the level of the caudal amygdala, can be seen as a bundle running parallel to the medial extent of the optic tract (LeDoux et al., 1990). They then turn laterally, with some ramifying through the striatum en route to the auditory cortex and with others coursing through the ventralmost part of the striatum just above the central nucleus of the amygdala, and terminate in LAd. By placing a stimulating electrode in the ventral striatum, just medial to LAd, we activated fibers that originate, at least in part, in the auditory thalamus (see Fig. 1A). Stimulation intensities were kept low to help avoid nonmonophasic components in the response, and they are indicated for the responses shown in the figures. The stimulus duration was always 150 μsec. Recordings were made using an AxoClamp 2B amplifier (Axon Instruments). Signals were filtered at 3 kHz and digitized at 5 kHz with a National Instruments analog-to-digital board. Data were stored and analyzed using software written with LabVIEW (National Instruments) running on an IBM-compatible personal computer. All cells had membrane potentials more negative than −55 mV and action potentials that exceeded 0 mV. Baseline responses were monitored at 0.05 Hz, and unless otherwise noted, cells were brought to −75 mV with DC current to help avoid action potential generation by synaptic input. Because LTP at synapses between the cells of area CA3 and CA1 of the hippocampus cannot be induced after ∼20 min of recording when using whole-cell recording techniques (Manabe and Nicoll, 1994), in the present experiments we attempted to induce LTP at input synapses to the amygdala within 15 min of entering whole-cell mode. In addition, because changes in response latency may reflect changes in nonmonosynaptic responses, we excluded any experiment in which a latency shift occurred after the LTP induction protocol. In all experiments the series resistance was monitored throughout the experiments, and if it changed by >15%, the data were discarded.
Fig. 1.

Thalamic input to the LA. A,Schematic of stimulating and recording electrode placement. Afferent fibers from the thalamus enter the LA medially. All recordings were made from cells in the dorsal subregion of the LA, which is the main site of termination of fibers from the auditory thalamus (LeDoux et al., 1990). CE, Central nucleus of the amygdala;ec, external capsule; ic, internal capsule; ot, optic tract. B, EPSPs recorded in response to stimuli of increasing intensity (55, 70, 85, and 100 μA). The initial slope is graded with intensity. The action potential contribution to the highest intensity example has been truncated. C, EPSPs in another neuron (120 μA intensity) in control conditions (2 mmCa2+, 1 mm Mg2+;top) and after increasing the Ca2+and Mg2+ concentrations to 12 mm each (bottom). The nonmonosynaptic component is suppressed by the increased divalent concentration. Traces are averages of five responses.
Values are expressed as means ± SEM unless SD is noted, and potentiation is expressed as percent of baseline at the indicated time range after induction. All responses in one cell over the indicated time range were averaged to get one value per cell. These were then averaged, and the indicated n refers to the number of cells. Significance was tested with a two-tailed, two-sample (except where a paired test is indicated) Student's t test. All animals were handled in accordance with National Institutes of Health and New York University guidelines. Drugs were applied by adding them to the superfusing Ringer's solution. Drugs used were MK-801 and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) from Research Biochemicals (Natick, MA); d-2-amino-5-phosphonopentanoic acid (d-APV) from Sigma (St. Louis, MO) and Research Biochemicals; and 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (BAPTA), nifedipine, and picrotoxin from Sigma. Drugs were made up in stock solution and diluted 1000 times into the Ringer's solution. Picrotoxin, MK-801, nifedipine, and CNQX stocks were made up in DMSO.
RESULTS
Recordings were made from LAd neurons, and a stimulating electrode was placed just medial to the LA to activate thalamic afferents as they course through the ventral striatum from near the optic tract to enter the amygdala (Fig. 1A). EPSPs in LA cells generated by this stimulation procedure increased in amplitude as stimulation intensity increased (Fig.1B). A late, presumably polysynaptic, component to the response was also seen sometimes (Fig. 1B; see also, e.g., Fig. 4). Indeed, increasing divalent ion concentrations in the extracellular bathing solution, which has been shown to suppress polysynaptic responses (Jahr and Jessell, 1985; Sah and Nicoll, 1991), was effective at suppressing later components of the response (Fig.1C). In contrast, the early component of the EPSP persisted in the presence of elevated divalent ions. Furthermore, in response to changes in stimulus intensity the initial slope of the EPSP was graded, and its onset latency did not change (Fig. 1B). Together, these results suggest that the initial slope of the EPSP is monosynaptic. Therefore, to eliminate di- or polysynaptic contamination and analyze only monosynaptic input to the LA in the presence of normal levels of divalents, we restricted our analysis to this portion of the EPSPs.
Fig. 4.
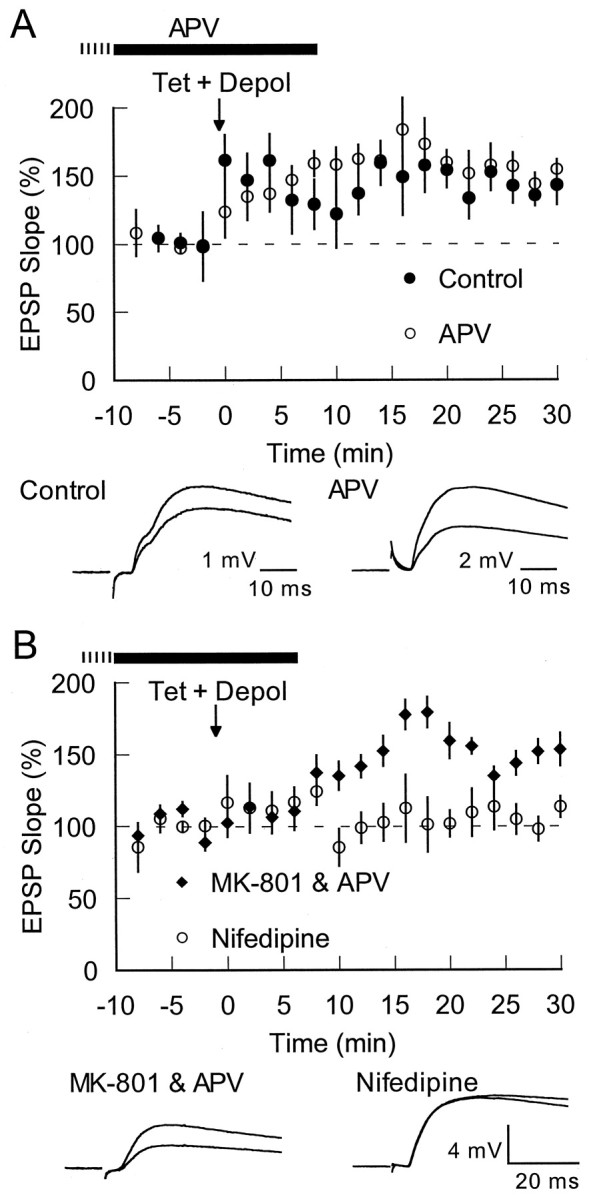
Tetanus with depolarization induces NMDA-independent, VGCC-dependent LTP. A,Top, Tetanization, given while injecting DC current to depolarize the cell near to the action potential discharge threshold (Tet + Depol; arrow), induced LTP in control conditions (filledcircles;n = 6) and in the presence of 50 μmd-APV (opencircles;n = 6; APV applied as indicated by the solidbar, plus variable time before breaking into the cell indicated by thedashedbar). Bottom,Traces from individual experiments before and 25 min after induction are shown (100 and 70 μA intensity for control and APV, respectively). B,Top, Tetanization with DC depolarization (Tet + Depol;arrow) in the presence of both 50 μmd-APV and 40 μm MK-801 still results in LTP (filleddiamonds;n = 6). The L-type calcium channel blocker nifedipine (30 μm) blocked the induction of this LTP at thalamic inputs (opencircles;n = 7). In each case the drug was applied as indicated by the solidbar, plus variable time before breaking into the cell indicated by thedashedbar. Bottom,Traces from individual experiments (averages of 10 responses) for each case (60 and 40 μA stimulation intensity for MK-801 and nifedipine, respectively) before and 25 min after induction are shown.
LTP induction
Although the exact cellular basis of fear conditioning is unclear, presynaptic inputs to LA neurons from CS pathways presumably elicit EPSPs that are somehow paired with postsynaptic action potentials elicited by the US. We therefore used an LTP induction protocol that involved pairing of pre- and postsynaptic activity: trains of presynaptic stimulation were paired with depolarization of a postsynaptic neuron in the LA. Specifically, LTP was induced by pairing trains of 10 stimuli at 30 Hz with 1 nA, 5 msec depolarizations given 5–10 msec after the onset of each EPSP in the train. This pairing was given 15 times at 10 sec intervals (for contrast with a second protocol below we refer to this as “repeated pairing”). This has been shown to be an effective stimulus for enhancing EPSPs between pairs of neocortical neurons (Markram et al., 1997).
An example of a typical cell's response to the 30 Hz stimulation alone is shown in Figure 2A, and the response to the repeated-pairing protocol is shown in Figure2B. Each depolarization given after each EPSP in the train during the repeated-pairing protocol resulted in an action potential (Fig. 2B). Repeated pairing resulted in an increase in EPSP slope to 178 ± 20% of baseline (at 25–30 min;n = 7; see Materials and Methods) (Fig.2C).
Fig. 2.
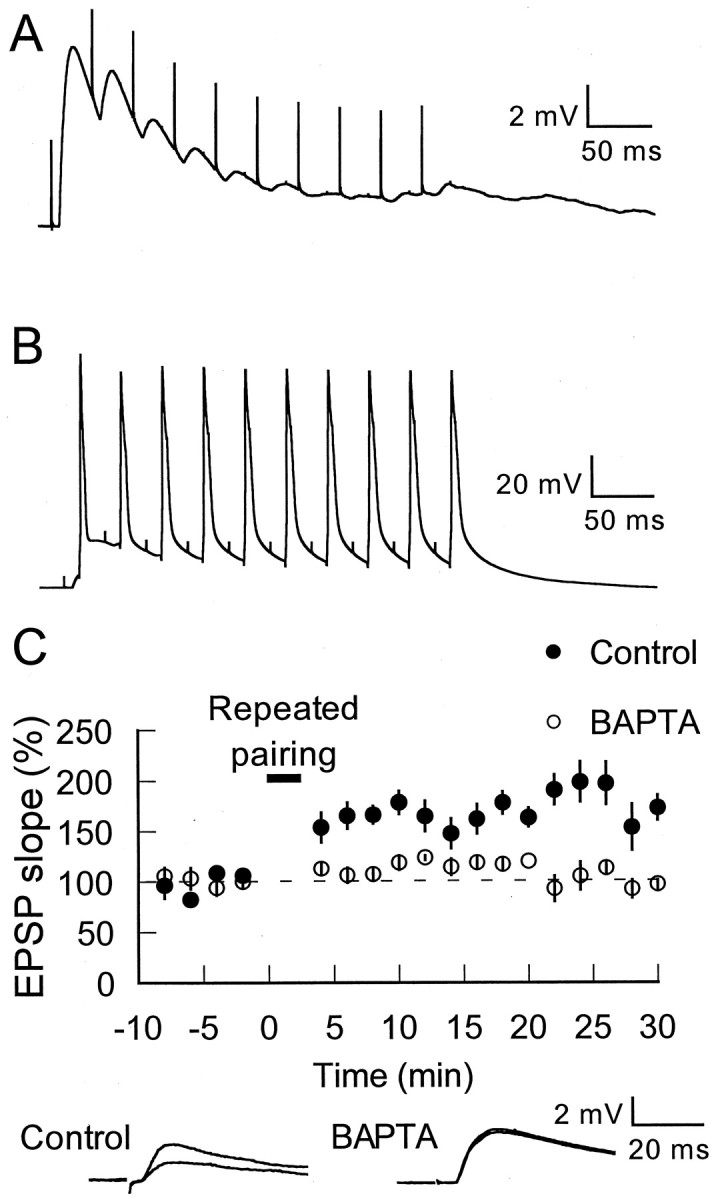
Repeated-pairing LTP of thalamoamygdala inputs.A, Example of one cell's response to 10 stimuli (320 μA) delivered at 30 Hz is shown. B, The response of the same cell when 1 nA, 5 msec depolarizations follow each EPSP in the train (repeated pairing) is shown. Traces are averages of 15 responses. C, Top, At time 0, this repeated pairing (short solidbar) was given 15 times at 10 sec intervals. In control conditions (filledcircles) this induced LTP (n = 7). Including 10 mm BAPTA in the recording pipette (opencircles) prevented this LTP (n = 5). Bottom,Traces before and 25 min after repeated pairing from individual experiments in each case are shown and are averages of 10 responses (150 and 400 μA stimulation intensity for control and BAPTA, respectively).
Calcium-dependent LTP
Because calcium has been implicated in a variety of forms of synaptic plasticity (Lynch et al., 1983; Zucker, 1989; Castillo et al., 1994), we tested its involvement in LTP at thalamic inputs to the amygdala by including the calcium chelator BAPTA in the recording pipette solution. As can be seen in Figure 2C, the presence of BAPTA (10 mm) in the postsynaptic cell blocked the induction of thalamic LTP (106 ± 5% at 25–30 min;n = 5).
In the classic form of LTP seen in the CA1 region of hippocampus and some other brain areas, calcium entry occurs through NMDA receptors (Bliss and Collingridge, 1993; Malenka and Nicoll, 1993). Surprisingly, NMDA receptor blockade with d-APV (50 μm) did not affect thalamoamygdala LTP (control, 178 ± 20%;n = 7; APV, 167 ± 12%; n = 9;p = 0.62; at 25–30 min) (Fig.3A, filled circles), even though one-half this concentration of d-APV effectively blocked the NMDA-mediated responses to 30 Hz stimulation alone (Fig. 3B). To ensure NMDA independence further, we performed another series of experiments in the presence of both the competitive NMDA antagonist d-APV (50 μm) and the noncompetitive antagonist MK-801 (40 μm). Even this increased inhibition of the NMDA receptor did not prevent the induction of LTP (182 ± 45%;n = 6) (Fig. 3A, open diamonds). Thus, thalamoamygdala LTP is independent of NMDA receptors in spite of a contribution of NMDA receptors to baseline synaptic responses (Fig.3B) (see also Weisskopf and LeDoux, 1999). Although calcium entry into the postsynaptic cell is required for LTP induction, the calcium does not seem to enter by way of NMDA receptors.
Fig. 3.
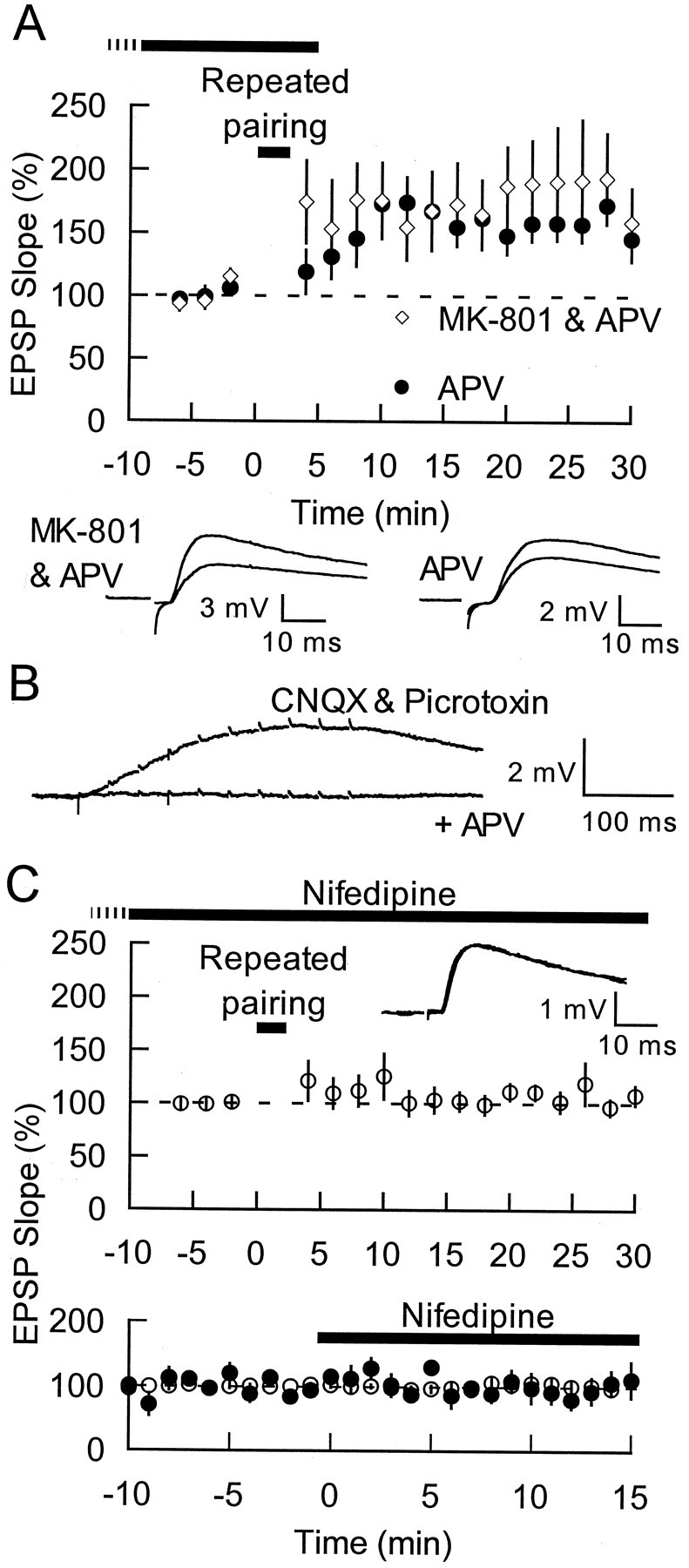
Repeated pairing-induced LTP is NMDA-independent, VGCC-dependent LTP. A,Top, Repeated pairing (shortsolidbar) done at thalamic inputs in the presence of 50 μmd-APV alone (filled circles;n = 9) or with 40 μm MK-801 (open diamonds; n = 6) still results in LTP (drugs applied as indicated by the longsolidbar, plus variable time before breaking into the cell indicated by the dashedbar). Bottom, Traces from individual experiments before and 25 min after induction are shown (80 and 130 μA intensity for APV alone and with MK-801, respectively).B, Example of an LA cell's response to 10 pulses (40 μA intensity) at 30 Hz at −66 mV in the presence of 10 μm CNQX and picrotoxin is shown. The subsequent block of this response after addition of 25 μmd-APV is superimposed. C,Top,The L-type calcium channel blocker nifedipine (30 μm;longsolidbar, plus variable time before breaking into the cell indicated by thedashedbar) blocked the induction of LTP at thalamic inputs (open circles; n= 8). Bottom, Nifedipine (30 μm;longsolidbar) did not affect baseline synaptic transmission (filledcircles; n = 5) or the depolarization induced by a 10 pulse, 30 Hz train (opencircles;n = 6; measured after the fourth pulse). Inset, Traces from an individual experiment before and 25 min after induction are shown (120 μA intensity). Traces are averages of 10 (A, C) and 5 (B) responses.
Significant postsynaptic calcium entry can occur through voltage-gated calcium channels (VGCCs) (Miyakawa et al., 1992; Denk et al., 1996), and the depolarization and action potentials evoked during repeated pairing are likely to be effective in activating these channels. Furthermore, other NMDA-independent forms of plasticity have been shown to depend on L-type VGCCs (Kullmann et al., 1992; Huang and Malenka, 1993; Chen et al., 1998). Therefore, we tested whether blocking L-type VGCCs with nifedipine affected thalamoamygdala LTP. In contrast to APV, nifedipine blocked the induction of thalamoamygdala LTP (106 ± 11% at 25–30 min; n = 8) (Fig. 3C). Nifedipine (30 μm) had no effect on baseline transmission (Fig. 3C). In addition, to determine whether nifedipine has any effects on transmission during the repeated-pairing induction protocol, we tested for effects of nifedipine on a 10 pulse, 30 Hz train. The maximum depolarization caused by this stimulus usually occurred after the fourth pulse in the stimulus, and monitoring the level of this depolarization revealed that nifedipine had no effect (103 ± 9% at 10–15 min; n = 6) (Fig.3C). As an indicator of nifedipine's effects on the overall depolarization induced by the train, we also calculated the integral under the induced envelope of depolarization. This was not affected by nifedipine either (105 ± 17% at 10–15 min; n = 6; data not shown). Thus, nifedipine does not, at this concentration, prevent LTP via an inhibition of presynaptic transmitter release.
Alternate induction protocol
It is possible that the repeated-pairing protocol was so effective at raising calcium levels that the need for calcium entry through NMDA receptors was bypassed. Thus, a different induction protocol might reveal an NMDA-dependent form of LTP. We therefore attempted to induce LTP with a typical tetanic stimulation protocol (4 times 100 Hz for 1 sec, separated by 20 sec). However, this standard LTP protocol did not induce LTP (103 ± 12% at 25–30 min; n = 6; data not shown). Because depolarization played a role in the induction of LTP with the repeated-pairing protocol, we subsequently gave tetanic stimulation while injecting steady depolarizing current to bring the membrane potential near to that at which action potentials were triggered [−63 ± 2 mV (SD); n = 6; compared with −74 ± 5 mV (SD); above]. This protocol effectively induced LTP (143 ± 12% at 25–30 min) (Fig.4A, filled circles).
The NMDA dependence of this tetanically induced LTP was tested by pairing the steady depolarization and the tetanus in the presence of 50 μmd-APV. As with the repeated-pairing protocol described above, d-APV was ineffective at blocking LTP induced by pairing tetanic stimulation with constant depolarization [152 ± 11% at 25–30 min; n = 6; −62 ± 7 mV (SD) membrane potential during tetanus; p = 0.58 compared with control] (Fig. 4A, open circles). As with repeated-pairing LTP, we further tested the NMDA dependence of tetanus LTP by attempting to induce this LTP in the presence ofd-APV and MK-801. As with repeated-pairing LTP, this increased inhibition of the NMDA receptor still did not block tetanus induced LTP [145 ± 13% at 25–30 min; n= 6; −66 ± 3 mV (SD)] (Fig. 4B, filled diamonds). Furthermore, nifedipine blocked this LTP [101 ± 8% at 25–30 min; n = 7; −63 ± 5 mV (SD) membrane potential during tetanus; p = 0.01 compared with control] (Fig. 4B, open circles).
Tetanic stimulation has been shown to induce an NMDA-independent form of LTP at excitatory synapses onto LA inhibitory cells (Mahanty and Sah, 1998). This LTP was shown to be mediated by calcium entry through calcium-permeable AMPA channels at synapses that lacked NMDA receptors. To ensure that the present results were not being contaminated by such LTP, all recorded cells were shown to have an NMDA response by stimulating with trains of 10 stimuli at 30 Hz in the presence of CNQX. The resulting responses were abolished by 25 μmd-APV, indicating that we were not looking at LTP onto inhibitory cells that lack NMDA receptors. This also verified that the APV effectively blocked NMDA-mediated responses (see Fig.3B).
Associative, synapse-specific induction mechanism
The trains of 10 stimuli at 30 Hz, as used in the repeated-pairing protocol, when given without the paired depolarizations, were insufficient to induce LTP (Fig.5A), thus demonstrating the necessity of the postsynaptic neuron to the LTP induction. In addition, the depolarizing pulses given without the concomitant presynaptic stimulation also failed to induce LTP (Fig. 5B), showing the need for presynaptic input. A pre- and postsynaptic contribution to the tetanus-induced LTP is also apparent because it was necessary to depolarize the postsynaptic cell during the tetanus to elicit LTP reliably. These results demonstrate an associative mechanism of LTP induction in the amygdala, but one that does not rely on NMDA receptors.
Fig. 5.

Associative, synapse-specific LTP at thalamic input synapses to the LA. A, The 15 trains of 10 stimuli at 30 Hz (short solidbar) without the paired depolarizations did not induce LTP (open circles;n = 5). B, The 15 trains of 1 nA, 5 msec depolarizations (shortsolidbar) given without paired input stimulation also did not induce LTP (open circles; n = 4).C, LTP induced by repeated pairing (short solidbar) at the thalamic pathway (filledtriangles) did not induce LTP at a second, unpaired input (invertedopentriangles) onto the same cell (n = 6). D, LTP induced by tetanizing while depolarizing (Tet + Depol;arrow) at the thalamic pathway (filledtriangles) did not induce LTP at a second, untetanized input (invertedopentriangles) onto the same cell (n = 6).
In the CA1 region of the hippocampus, NMDA-dependent LTP is restricted to the activated synapses in part because of the need for glutamate release to activate postsynaptic NMDA receptors. Because LTP at thalamic inputs to the amygdala is independent of NMDA receptors, we tested whether synapse specificity still exists. In some of the LTP experiments described above we placed a second stimulating electrode in the external capsule dorsal to the LA to monitor simultaneously a second input onto the recorded cell. This is the pathway most often stimulated in past studies of amygdala LTP (see introductory remarks). These corticoamygdala-elicited EPSPs did not change after LTP induction in the thalamoamygdala pathway by either the repeated-pairing protocol (thalamic, 179 ± 9%; n = 6; cortical, 111 ± 11%; at 25–30 min; p = 0.003, paired ttest) (Fig. 5C) or tetanic stimulation with depolarization (thalamic, 151 ± 12%; n = 6; cortical, 98 ± 5%; at 20–25 min; p = 0.014, paired ttest) (Fig. 5D). This indicates that, in spite of the lack of NMDA receptor involvement in thalamoamygdala LTP, the LTP is nevertheless synapse specific.
DISCUSSION
During fear conditioning, auditory CS signals reach the LA via a direct pathway from the auditory thalamus and indirectly by way of projections from the auditory thalamus to the auditory cortex and from there to the LA (LeDoux et al., 1990; Romanski and LeDoux, 1993). Although the auditory thalamus was not included within the amygdala brain slice, it was possible to stimulate regions in the slice through which thalamic fibers traversed using past findings from tract-tracing studies (LeDoux et al., 1990) to locate the trajectory. Thalamic fibers en route to the LA course medially, entering the LA from the internal capsule (see Fig. 1), and stimulating this site elicits physiological responses that, in comparison with those elicited from external capsule—or cortical—stimulation, resemble thalamic inputs seenin vivo, where the electrodes were actually placed in the auditory thalamus and cortex (Li et al., 1995, 1996; Weisskopf and LeDoux, 1999).
Distinct form of LTP
Given that LTP has been induced in thalamic CS pathways to the LAin vivo (Clugnet and LeDoux, 1990; Rogan and LeDoux, 1995), that the best understood form of LTP in the hippocampus involves NMDA receptors (Bliss and Collingridge, 1993; Malenka and Nicoll, 1993;Huang et al., 1996; Milner et al., 1998), that NMDA receptors are postsynaptic to the thalamic input (Li et al., 1995, 1996; Farb and LeDoux, 1997; Weisskopf and LeDoux, 1999), and that NMDA receptor blockade interferes with fear conditioning (Miserendino et al., 1990;Maren et al., 1996; Gewirtz and Davis, 1997; Lee and Kim, 1998), a natural question to ask is whether LTP in this pathways involves NMDA receptors. Although past studies have found evidence both for (Huang and Kandel, 1998) and against (Chapman and Bellavance, 1992; Watanabe et al., 1995; Li et al., 1998) the involvement of NMDA receptors in LTP of cortical input to the amygdala, LTP of the thalamic pathway has not been studied in vitro previously. Thus no previous information was available about its pharmacological basis. By pairing trains of presynaptic input with trains of postsynaptic depolarizations (repeated pairing) at thalamic input synapses, we found NMDA-independent LTP.
The depolarizations used in the repeated-pairing protocol triggered action potentials, which, in other cells, are known to result in back-propagating action potentials (Stuart et al., 1997). These lead to significant calcium entry into dendrites via VGCCs and even into dendritic spines, particularly if paired with synaptic input (Miyakawa et al., 1992; Denk et al., 1996). A similar sequence of cellular events may be a component of the plasticity-inducing mechanism in LA cells because we found that chelating calcium or blocking L-type VGCCs prevented LTP.
It is interesting that LTP induced by repeated pairing is NMDA independent, in spite of a contribution of NMDA receptors to transmission in this pathway, as seen both in vivo (Li et al., 1995, 1996) and in vitro (present results) (Weisskopf and LeDoux, 1999). One possibility is that the repeated-pairing protocol we used was so effective at raising calcium levels via L-type VGCCs that it obviated a need for calcium entry through NMDA receptors. Indeed, in the CA1 region of the hippocampus, where NMDA-dependent LTP is the predominant form, synapse-specific potentiation that is dependent on L-type VGCCs, but independent of NMDA receptors, can be induced by pairing depolarizations with synaptic stimulation (Kullmann et al., 1992; Chen et al., 1998). The same mechanism has been shown to underlie other forms of NMDA-independent LTP as well (Grover and Tyler, 1990; Aniksztejn and Ben-Ari, 1991; Huang and Malenka, 1993). Therefore, we attempted to induce LTP in the manner in which traditional, NMDA-dependent LTP at the Schaffer collateral–CA1 synapse is induced—with 100 Hz tetanic stimulation.
Tetanic stimulation of thalamoamygdala inputs to the LA, unlike at Schaffer collateral synapses in the hippocampus, did not induce LTP. This may be attributable to the relatively hyperpolarized resting membrane of LA cells compared with hippocampal pyramidal cells. That this was the case is suggested by the fact that tetanic stimulation given while depolarizing LA cells did induce LTP. Intriguingly, however, this LTP was still NMDA independent and L-type VGCC dependent, as with the repeated-pairing induction protocol. This pattern of induction dependence is similar to that seen for a number of forms of LTP in the hippocampus (Grover and Tyler, 1990; Aniksztejn and Ben-Ari, 1991; Kullmann et al., 1992; Huang and Malenka, 1993) and possibly CA3 (Kapur et al., 1998; but see Castillo et al., 1994). However, thalamoamygdala LTP differs from both of these other forms of LTP: mossy fiber–CA3 synapses have only a small NMDA component (Monaghan and Cotman, 1985; Weisskopf and Nicoll, 1995); there is much evidence that LTP at the mossy fiber synapse is entirely presynaptic (Staubli et al., 1990; Zalutsky and Nicoll, 1990; Castillo et al., 1994; Weisskopf and Nicoll, 1995); and in area CA1, L-type VGCC-dependent LTP is induced only under conditions that bypass the primary, NMDA-dependent LTP. In contrast, thalamoamygdala LTP is induced postsynaptically, but the NMDA receptor is unnecessary. L-type VGCC-mediated LTP is clearly the primary form at thalamoamygdala synapses and therefore may be distinct even from VGCC-mediated LTP in CA1 at the molecular level.
Postsynaptic depolarization is a necessary component of thalamoamygdala LTP induced by repeated pairing, but the concurrent presynaptic stimulation is also a necessary component of the induction process, implying that the LTP is associative. The induction conditions for the tetanus-induced LTP are consistent with an associative mechanism as well because LTP was not induced unless the tetanus was done while depolarizing the postsynaptic cell. The LTP is also specific to the stimulated pathway, presumably because of some consequence of the presynaptic activation. In the case of NMDA-dependent hippocampal LTP in the CA1 region, presynaptic activity is required to activate NMDA receptors and achieve synapse specificity. This cannot be the role of presynaptic activity at thalamic input synapses to the LA because the LTP does not depend on NMDA receptors. Synapse specificity without NMDA receptor involvement is also seen with LTP induced via L-type VGCCs in area CA1 of the hippocampus, although the mechanism that provides the synapse specificity has not been determined. At thalamoamygdala synapses presynaptic activity could be necessary for calcium influx through AMPA receptors (Hollmann et al., 1991). However, we found linear I–V curves of thalamic input responses to the LA (Weisskopf and LeDoux, 1999), suggesting that calcium-permeable AMPA receptors do not play a significant role at these synapses (Hollmann et al., 1991). Another possibility, one that we favor, is that presynaptic activity helps achieve synapse specificity by activation of metabotropic glutamate receptors, which have been implicated in LTP and behaviorally mediated plasticity in other brain areas (Bashir et al., 1993; Li et al., 1998).
Functional implications
The present results have a number of implications for the mechanisms of fear conditioning. Although some studies suggest that NMDA blockade in the LA and adjacent areas interferes with the acquisition but not the expression of fear conditioning, others indicate that both acquisition and expression are interfered with (Miserendino et al., 1990; Maren et al., 1996; Gewirtz and Davis, 1997;Lee and Kim, 1998). Given the role of NMDA receptors in synaptic transmission in the LA (see Fig. 3B) (Li et al., 1995, 1996;Danober and Pape, 1998; Weisskopf and LeDoux, 1999), this issue needs to be carefully reconsidered to exclude the possibility that APV disrupts fear learning by interfering with synaptic transmission. On the other hand, if the plasticity underlying fear conditioning is in fact NMDA dependent, then it is possible that fear conditioning involves NMDA-dependent LTP at intrinsic synapses within the LA or other amygdala areas, but not at the thalamic input synapses.
Activation of calcium-dependent kinases has been implicated in fear conditioning (Mayford et al., 1996), and our results clearly implicate calcium in LTP at thalamic input synapses to the LA. Intriguingly, although expression of activated calcium–calmodulin-dependent kinase II (CaMKII) interfered with both amygdala- and hippocampal-dependent behavioral learning, it modified, but did not block, NMDA-dependent LTP in area CA1 of the hippocampus (Mayford et al., 1996). Our results indicate that calcium entry through VGCCs mediates LTP at thalamic input synapses to the LA, which thus may be mechanistically different from the NMDA-dependent form of plasticity found in the hippocampus. On the other hand, it may be a form disrupted by the expression of activated CaMKII, the manipulation that leads to the learning deficits described above. Thus, it would be interesting to determine the dependence of thalamoamygdala LTP on CaMKII.
It has been shown previously that fear conditioning and induction of thalamoamygdala LTP lead to similar changes in field potentials recorded in the LA (Rogan and LeDoux, 1995; Rogan et al., 1997) and that after fear conditioning thalamic input synapses in the LA show enhanced EPSPs (McKernan and Shinnick-Gallagher, 1997). The present findings add to these previous results, showing that the induction requirements for thalamoamygdala LTP are compatible with those proposed for behavioral learning; it is specific to the activated pathway and involves the co-occurrence of activity in the presynaptic CS input and postsynaptic amygdala cell. These observations collectively suggest that LTP occurs at thalamic input synapses in the LA during conditioning. Whether this is a necessary and/or sufficient phenomenon for behavioral learning remains to be determined.
Footnotes
This work was supported by National Institutes of Health Grants R01-MH46516 and K02-MH00956. M.G.W. is supported by National Institutes of Health Grant F32-NS10222.
Correspondence should be addressed to Dr. Joseph E. LeDoux, New York University, Center for Neural Science, 4 Washington Place, Room 809, New York, NY 10003. E-mail: ledoux@cns.nyu.edu.
REFERENCES
- 1.Aniksztejn L, Ben-Ari Y. Novel form of long-term potentiation produced by a K+ channel blocker in the hippocampus. Nature. 1991;349:67–69. doi: 10.1038/349067a0. [DOI] [PubMed] [Google Scholar]
- 2.Barnes CA. Involvement of LTP in memory: are we “searching under the streetlight”? Neuron. 1995;15:751–754. doi: 10.1016/0896-6273(95)90166-3. [DOI] [PubMed] [Google Scholar]
- 3.Bashir ZI, Bortolotto ZA, Davies CH, Berretta N, Irving AJ, Seal AJ, Henley JM, Jane DE, Watkins JC, Collingridge GL. Induction of LTP in the hippocampus needs synaptic activation of glutamate metabotropic receptors. Nature. 1993;363:347–350. doi: 10.1038/363347a0. [DOI] [PubMed] [Google Scholar]
- 4.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 5.Brambilla R, Gnesutta N, Minichiello L, White G, Roylance AJ, Herron CE, Ramsey M, Wolfer DP, Cestari V, Rossi-Arnaud C, Grant SG, Chapman PF, Lipp HP, Sturani E, Klein R. A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nature. 1997;390:281–286. doi: 10.1038/36849. [DOI] [PubMed] [Google Scholar]
- 6.Brown TH, Chapman PF, Kairiss EW, Keenan CL. Long-term synaptic potentiation. Science. 1988;242:724–728. doi: 10.1126/science.2903551. [DOI] [PubMed] [Google Scholar]
- 7.Castillo PE, Weisskopf MG, Nicoll RA. The role of Ca2+ channels in hippocampal mossy fiber synaptic transmission and long-term potentiation. Neuron. 1994;12:261–269. doi: 10.1016/0896-6273(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 8.Chapman PF, Bellavance LL. NMDA receptor-independent LTP in the amygdala. Synapse. 1992;11:310–318. doi: 10.1002/syn.890110406. [DOI] [PubMed] [Google Scholar]
- 9.Chapman PF, Kairiss EW, Keenan CL, Brown TH. Long-term synaptic potentiation in the amygdala. Synapse. 1990;6:271–278. doi: 10.1002/syn.890060306. [DOI] [PubMed] [Google Scholar]
- 10.Chen H-X, Hanse E, Pananceau M, Gustafsson B. Distinct expressions for synaptic potentiation induced by calcium through voltage-gated calcium and N-methyl-d-aspartate receptor channels in the hippocampal CA1 region. Neuroscience. 1998;86:415–422. doi: 10.1016/s0306-4522(98)00042-6. [DOI] [PubMed] [Google Scholar]
- 11.Clugnet MC, LeDoux JE. Synaptic plasticity in fear conditioning circuits: induction of LTP in the lateral nucleus of the amygdala by stimulation of the medial geniculate body. J Neurosci. 1990;10:2818–2824. doi: 10.1523/JNEUROSCI.10-08-02818.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Danober L, Pape H-C. Mechanisms and functional significance of a slow inhibitory potential in neurons of the lateral amygdala. Eur J Neurosci. 1998;10:853–867. doi: 10.1046/j.1460-9568.1998.00092.x. [DOI] [PubMed] [Google Scholar]
- 13.Denk W, Yuste R, Svoboda K, Tank DW. Imaging calcium dynamics in dendritic spines. Curr Opin Neurobiol. 1996;6:372–378. doi: 10.1016/s0959-4388(96)80122-x. [DOI] [PubMed] [Google Scholar]
- 14.Eichenbaum H. The LTP-memory connection. Nature. 1995;378:131–132. doi: 10.1038/378131a0. [DOI] [PubMed] [Google Scholar]
- 15.Farb CR, LeDoux JE. NMDA and AMPA receptors are post-synaptic to auditory thalamic afferents. Synapse. 1997;27:106–121. doi: 10.1002/(SICI)1098-2396(199710)27:2<106::AID-SYN2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 16.Gean P-W, Chang F-C, Huang C-C, Lin J-H, Way L-J. Long-term enhancement of EPSP and NMDA receptor-mediated synaptic transmission in the amygdala. Brain Res Bull. 1993;31:7–11. doi: 10.1016/0361-9230(93)90003-t. [DOI] [PubMed] [Google Scholar]
- 17.Gewirtz JC, Davis M. Second-order fear conditioning prevented by blocking NMDA receptors in amygdala. Nature. 1997;388:471–474. doi: 10.1038/41325. [DOI] [PubMed] [Google Scholar]
- 18.Grover LM, Tyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- 19.Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA-gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 20.Huang YY, Kandel ER. Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron. 1998;21:169–178. doi: 10.1016/s0896-6273(00)80524-3. [DOI] [PubMed] [Google Scholar]
- 21.Huang YY, Malenka RC. Examination of TEA-induced synaptic enhancement in area CA1 of the hippocampus: the role of voltage-dependent Ca2+ channels in the induction of LTP. J Neurosci. 1993;13:568–576. doi: 10.1523/JNEUROSCI.13-02-00568.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang YY, Nguyen PV, Abel T, Kandel ER. Long-lasting forms of synaptic potentiation in the mammalian hippocampus. Learn Mem. 1996;3:74–85. doi: 10.1101/lm.3.2-3.74. [DOI] [PubMed] [Google Scholar]
- 23.Jahr CE, Jessell TM. Synaptic transmission between dorsal root ganglion and dorsal horn neurons in culture: antagonism of monosynaptic excitatory postsynaptic potentials and glutamate excitation by kynurenate. J Neurosci. 1985;5:2281–2289. doi: 10.1523/JNEUROSCI.05-08-02281.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kapur A, Yeckel MF, Gray R, Johnston D. L-type calcium channels are required for one form of hippocampal mossy fiber LTP. J Neurophysiol. 1998;79:2181–2190. doi: 10.1152/jn.1998.79.4.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kullmann DM, Perkel DJ, Manabe T, Nicoll RA. Ca2+ entry via postsynaptic voltage-sensitive Ca2+ channels can transiently potentiate excitatory synaptic transmission in the hippocampus. Neuron. 1992;9:1175–1183. doi: 10.1016/0896-6273(92)90075-o. [DOI] [PubMed] [Google Scholar]
- 26.LeDoux JE. Emotion: clues from the brain. Annu Rev Psychol. 1995;46:209–235. doi: 10.1146/annurev.ps.46.020195.001233. [DOI] [PubMed] [Google Scholar]
- 27.LeDoux JE, Farb CF, Ruggiero DA. Topographic organization of neurons in the acoustic thalamus that project to the amygdala. J Neurosci. 1990;10:1043–1054. doi: 10.1523/JNEUROSCI.10-04-01043.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee H, Kim JJ. Amygdalar NMDA receptors are critical for new fear learning in previously fear-conditioned rats. J Neurosci. 1998;18:8444–8454. doi: 10.1523/JNEUROSCI.18-20-08444.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Weiss SRB, Chuang D-M, Post RM, Rogawski MA. Bidirectional synaptic plasticity in the rat basolateral amygdala: characterization of an activity-dependent switch sensitive to the presynaptic metabotropic glutamate receptor antagonist 2S-alpha-ethylglutamic acid. J Neurosci. 1998;18:1662–1670. doi: 10.1523/JNEUROSCI.18-05-01662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Phillips RG, LeDoux JE. NMDA and non-NMDA receptors contribute to synaptic transmission between the medial geniculate body and the lateral nucleus of the amygdala. Exp Brain Res. 1995;105:87–100. doi: 10.1007/BF00242185. [DOI] [PubMed] [Google Scholar]
- 31.Li XF, Stutzmann GE, LeDoux JL. Convergent but temporally separated inputs to lateral amygdala neurons from the auditory thalamus and auditory cortex use different postsynaptic receptors: in vivo intracellular and extracellular recordings in fear conditioning pathways. Learn Mem. 1996;3:229–242. doi: 10.1101/lm.3.2-3.229. [DOI] [PubMed] [Google Scholar]
- 32.Lynch G, Larson J, Kelso S, Barrionuevo G, Schottler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305:719–721. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- 33.Mahanty NK, Sah P. Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature. 1998;394:683–687. doi: 10.1038/29312. [DOI] [PubMed] [Google Scholar]
- 34.Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- 35.Malenka RC, Nicoll RA. Learning and memory: never fear, LTP is hear. Nature. 1997;390:552. doi: 10.1038/37472. [DOI] [PubMed] [Google Scholar]
- 36.Manabe T, Nicoll RA. Long-term potentiation: evidence against an increase in transmitter release probability in the CA1 region of the hippocampus. Science. 1994;265:1888–1892. doi: 10.1126/science.7916483. [DOI] [PubMed] [Google Scholar]
- 37.Maren S, Aharonov G, Stote DL, Fanselow MS. N-Methyl-d-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of the conditional fear in rats. Behav Neurosci. 1996;110:1365–1374. doi: 10.1037//0735-7044.110.6.1365. [DOI] [PubMed] [Google Scholar]
- 38.Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- 39.Mayford M, Bach ME, Huang Y-Y, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- 40.McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- 41.Milner B, Squire LR, Kandel ER. Cognitive neuroscience and the study of memory. Neuron. 1998;20:445–468. doi: 10.1016/s0896-6273(00)80987-3. [DOI] [PubMed] [Google Scholar]
- 42.Miserendino MJD, Sananes CB, Melia KR, Davis M. Blocking of acquisition but not expression of conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature. 1990;345:716–718. doi: 10.1038/345716a0. [DOI] [PubMed] [Google Scholar]
- 43.Miyakawa H, Ross WN, Jaffe D, Callaway JC, Lasser-Ross N, Lisman JE, Johnston D. Synaptically activated increases in Ca2+ concentration in hippocampal CA1 pyramidal cells are primarily due to voltage-gated Ca2+ channels. Neuron. 1992;9:1163–1173. doi: 10.1016/0896-6273(92)90074-n. [DOI] [PubMed] [Google Scholar]
- 44.Monaghan DT, Cotman CW. Distribution of N-methyl-d-aspartate-sensitive l-(3H)glutamate-binding sites in rat brain. J Neurosci. 1985;5:2909–2919. doi: 10.1523/JNEUROSCI.05-11-02909.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pitkänen A, Savander V, LeDoux JL. Organization of intra-amygdaloid circuitries: an emerging framework for understanding functions of the amygdala. Trends Neurosci. 1997;20:517–523. doi: 10.1016/s0166-2236(97)01125-9. [DOI] [PubMed] [Google Scholar]
- 46.Quirk GJ, Armony JL, Repa JC, Li X-F, LeDoux JE. Emotional memory: a search for sites of plasticity. Cold Spring Harb Symp Quant Biol. 1997;61:247–257. [PubMed] [Google Scholar]
- 47.Rogan M, Staubli U, LeDoux J. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- 48.Rogan MT, LeDoux JE. LTP is accompanied by commensurate enhancement of auditory-evoked responses in a fear conditioning circuit. Neuron. 1995;15:127–136. doi: 10.1016/0896-6273(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 49.Romanski LM, LeDoux JE. Information cascade from primary auditory cortex to the amygdala: corticocortical and corticoamygdaloid projections of temporal cortex in the rat. Cereb Cortex. 1993;3:515–532. doi: 10.1093/cercor/3.6.515. [DOI] [PubMed] [Google Scholar]
- 50.Sah P, Nicoll RA. Mechanisms underlying potentiation of synaptic transmission in rat anterior cingulate cortex in vitro. J Physiol (Lond) 1991;433:615–630. doi: 10.1113/jphysiol.1991.sp018446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Staubli U, Larson J, Lynch G. Mossy fiber potentiation and long-term potentiation involve different expression mechanisms. Synapse. 1990;5:333–335. doi: 10.1002/syn.890050410. [DOI] [PubMed] [Google Scholar]
- 52.Stevens CF. A million dollar question: does LTP = memory? Neuron. 1998;20:1–2. doi: 10.1016/s0896-6273(00)80426-2. [DOI] [PubMed] [Google Scholar]
- 53.Stuart G, Spruston N, Sakmann B, Hausser M. Action potential initiation and backpropagation in neurons of the mammalian CNS. Trends Neurosci. 1997;20:125–131. doi: 10.1016/s0166-2236(96)10075-8. [DOI] [PubMed] [Google Scholar]
- 54.Watanabe Y, Saito H, Abe K. Roles of GABAA, NMDA and muscarinic receptors in induction of long-term potentiation in the medial and lateral amygdala in vitro. Neurosci Res. 1995;21:317–322. doi: 10.1016/0168-0102(94)00867-f. [DOI] [PubMed] [Google Scholar]
- 55.Weisskopf MG, LeDoux JE. Distinct populations of NMDA receptors at subcortical and cortical inputs to principal cells of the lateral amygdala. J Neurophysiol. 1999;81:930–934. doi: 10.1152/jn.1999.81.2.930. [DOI] [PubMed] [Google Scholar]
- 56.Weisskopf MG, Nicoll RA. Presynaptic changes during mossy fibre LTP revealed by NMDA receptor-mediated synaptic responses. Nature. 1995;376:256–259. doi: 10.1038/376256a0. [DOI] [PubMed] [Google Scholar]
- 57.Zalutsky RA, Nicoll RA. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science. 1990;248:1619–1624. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]
- 58.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]