

Abstract
Neuropilins have recently been characterized as receptors for secreted semaphorins. Here, we report the generation of a dominant negative form of neuropilin-1 by the deletion of one of its extracellular domains. Expression of this variant in cultured primary sympathetic neurons blocks the paralysis of growth cone motility normally induced by SEMA-3A (collapsin-1, semaphorin III, semaphorin D) and SEMA-3C (collapsin-3, semaphorin E) but not that induced by SEMA-3F (semaphorin IV). A truncated form of neuropilin-1 that is missing its cytoplasmic domain fails to act as a dominant negative receptor component. These results suggest that neuropilin-1 is a necessary component of receptor complexes for some, but not all, secreted semaphorin family members. Overexpression of dominant negative neuropilins should provide a powerful new method of blocking the functions of secreted semaphorins.
Keywords: semaphorin, collapsin, neuropilin-1, sympathetic neuron, growth cone guidance, growth cone collapse, dominant negative receptor
The development of a functional nervous system requires that axons navigate through a complex environment, sometimes over long distances, to locate their correct targets. At the growing tips of axons, motile growth cones detect and respond to a multitude of attractive and repellent guidance cues in their environment. Chick SEMA-3A (chick collapsin-1, human semaphorin-III, mouse semaphorin-D) is a member of the semaphorin family of signaling proteins and is thought to act as a repellent guidance cue for a variety of specific axons. Recombinant SEMA-3A inhibits the motility of growth cones from explanted dorsal root ganglia (DRGs), sympathetic ganglia, several but not all cranial sensory ganglia, olfactory sensory epithelial neurons, and spinal motor neurons (Luo et al., 1993; Kobayashi et al., 1997; Shepherd et al., 1997). In collagen-stabilized cultures, it has been shown to repel sensory axons projecting from explanted DRGs (Messersmith et al., 1995) and motor axons from most motor nuclei in the brainstem (Varela-Echavarria et al., 1997). Knocking out the homologous protein (semaphorin-D) in embryonic mice results in defasciculation and aberrant pathfinding of peripheral axon projections (Behar et al., 1996; Taniguchi et al., 1997).
The semaphorin family now includes more than 20 members. Several of these are secreted proteins structurally related to SEMA-3A. SEMA-3C (chick collapsin-3, mouse semaphorin-E) and SEMA-3F (human sema-IV) have overall domain structures identical to SEMA-3A and amino acid sequences that are ∼50% identical to SEMA-3A and to each other (Adams et al., 1997; Chen et al., 1997; Koppel et al., 1997). All three of these semaphorin family members induce the collapse of sympathetic growth cones, but only SEMA-3A induces the collapse of DRG growth cones (Chen et al., 1997; Koppel et al., 1997; Giger et al., 1998b) All three are expressed in overlapping patterns in the developing embryo (Adams et al., 1996; Shepherd et al., 1996; Giger et al., 1998a,b) and are likely to act as repellents that help guide peripheral axons, particularly sympathetic axons, along their appropriate trajectories.
Significant progress has been made in identifying receptors for these axonal guidance molecules. A SEMA-3A binding protein, neuropilin-1, has been identified by expression cloning (He and Tessier-Lavigne, 1997; Kolodkin et al., 1997). Neuropilin-1 is expressed in SEMA-3A-sensitive neurons as they extend their axons during development (Takagi et al., 1995). Antibodies directed against neuropilin-1 inhibit SEMA-3A-induced collapse of growth cones from DRGs (He and Tessier-Lavigne, 1997; Kolodkin et al., 1997), and DRGs from neuropilin-1 knock-out mice are unresponsive to SEMA-3A when tested in the growth cone collapse assay (Kitsukawa et al., 1997). Neuropilin-1 knock-out mice have a phenotype that is similar to the SEMA-3A knock-out mouse until they die between 10.5 and 13.5 d postcoitus (dpc). These results demonstrate that neuropilin-1 is required in neurons for SEMA-3A responsiveness.
Although neuropilin-1 is necessary for SEMA-3A function, several lines of evidence suggest that, by itself, it is unlikely to comprise the complete SEMA-3A receptor (Feiner et al., 1997). First, it has an extremely short cytoplasmic tail lacking any known signaling motifs. Second, a wide variety of secreted semaphorin family members bind to neuropilin-1 with approximately equal affinities, although, as described above, they do not all have the same biological specificities. Third, alkaline phosphatase (AP)-tagged versions of these semaphorins bind in overlapping but distinct patterns on sectioned embryonic tissues, suggesting that binding specificities are determined by more than the distribution of neuropilin-1. These considerations suggest that neuropilin-1 could be a common component for receptors responsive to secreted semaphorins, although additional receptor components help determine binding specificities and biological responsiveness. As addressed later in Discussion, some or all of the required specificity could be determined by the presence or absence of the neuropilin-1-related protein neuropilin-2 (Chen et al., 1998; Giger et al., 1998b; Takahashi et al., 1998).
The objective of this study was to engineer a dominant negative form of neuropilin-1 that could be used to explore its functional role in semaphorin receptors. If neuropilin-1 is a common component of receptor complexes that are specific for different secreted semaphorins, then it should follow that a dominant negative neuropilin-1 would block responsiveness to multiple secreted semaphorins. We have generated neuropilin-1 constructs missing specific structural domains, expressed them in cultured primary sympathetic cells responsive to several secreted semaphorins, and tested whether the constructs interfere with semaphorin activities. We have determined that deleting a portion of the extracellular domain of neuropilin-1 generates a dominant negative construct that blocks the activities of both SEMA-3A and SEMA-3C but not SEMA-3F. This finding is consistent with the hypothesis that neuropilin-1 is a component of receptors for some but not all secreted semaphorins. In future studies, the overexpression of this construct could be used to simultaneously block the activities of SEMA-3A and SEMA-3C during embryogenesis in vivo.
MATERIALS AND METHODS
Generation of neuropilin-1 deletion constructs. PCR was used to generate constructs of neuropilin-1 with specific domains deleted. PCR products were cloned into the modified mammalian expression vector pAG-NT as described previously, containing an N-terminal tag consisting of a signal sequence (from the first 25 amino acids of chick SEMA-3A), two myc epitope tags, and a 6xHis tag (Koppel et al., 1997). The oligonucleotide primers for the neuropilin deletion constructs were made containing the appropriate restriction enzyme sites so that the amplified products could be cloned directly into the BamHI and Not1 restriction sites of pAG-NT. Standard PCR amplification between oligonucleotide primers, all of which placed a BglII restriction site 5′ and a Not1 site 3′, was used to make the following constructs: full-length neuropilin-1 between cgaagcgataaatgcggcgac (F1) and tcatgcttccgagtaagaattctg (R1); a1/a2 domain deletion neuropilin-1 between atggaaccactaggtatggag (F2) and R1; and cytoplasmic domain deletion neuropilin-1 between F1 and gcaggcacagtacaggcaaac (R2).
Constructs that required the deletion of internal domains were made using a two-step PCR protocol described by Koppel et al. (1997). Briefly, the sequence on either side of the deleted region is amplified in the first step. The 5′ end of the internal reverse primer is complimentary to the internal forward primer. The second step involves annealing the two primers at the complimentary sequence and amplifying the final product using the outermost primers. The following constructs were made: b1/b2 domain deletion neuropilin-1 [step 1, between F1 and ttcggaaacagtagggacgacagcgcactgga-aatcttctgatac (R3) and between gctgtccctactgtttccgaa (F3) and R1] and (step 2, between F1 and R1); C-domain deletion neuropilin-1 [step1, between F1 and tgcactcatggctatgatggtcgtgggagcttcaagttcaca (R4) and between aacatcatagccatgagtgca (F4) and R1] and (step 2, between F1 and R1).
Protein expression in cultured cells. Human embryonic kidney (HEK) 293T or Cos-7 cells were grown to ∼70% confluency in a 10 cm dish in DMEM (Life Technologies, Gaithersburg, MD) with 1% penicillin–streptomycin (P–S) (Life Technologies) and 10% fetal bovine serum (FBS) (Hyclone Laboratories, Logan, UT) and transfected using calcium phosphate in the presence of 25 μm of chloroquine (Sigma, St. Louis, MO). Approximately 50 μg of DNA was added per 10 cm dish. Cells were incubated in the transfection mix for at least 4 hr and then changed into fresh media. HEK293T cells expressing AP-SEMA-3A or AP-SEMA-3C were allowed to grow for 2 d. Conditioned media were then collected, spun down to remove cell debris, and stored in frozen aliquots before their use in the growth cone collapse assay. Cos-7 cells were transfected with the neuropilin-1 deletion constructs, grown overnight, and assayed for AP-SEMA-3A binding the next day.
Culture of sympathetic neurons. Sympathetic chain ganglia were dissected from E7–E8 chick embryos and placed in ice cold HBSS (Life Technologies). The ganglia were carefully cleaned of connective tissue and placed in DMEM containing 1% P–S and 10% FBS and preincubated at 37°C with 5% CO2. The ganglia were spun down, resuspended in 0.05% trypsin, and incubated at 37°C for 15 min. The ganglia were again spun down and then dissociated by trituration in 100 μl of fresh medium. The dissociated cells were plated on 10 mm round coverslips coated with laminin (Life Technologies) at an approximate density of 104 cells per coverslip and cultured in 500 μl of media. Cells were incubated at 37°C with 5% CO2 for at least 1 hr to allow them to adhere to the coverslip before transfection (see below).
Protein expression in sympathetic cells. Cultured sympathetic cells were transfected using calcium phosphate. Approximately 1 μg of plasmid DNA was added to 500 μl of medium with 25 μm chloroquin in the well of a 48-well cluster plate. The cells were incubated for no longer than 5 hr at 37°C in 5% CO2. To stop the transfection, the media was removed and replaced with F-12 (Life Technologies) supplemented with glutamine, glucose, bovine pituitary extract, nerve growth factor, insulin, transferrin, selenium, 1% P–S, and 10% FBS (Baird and Raper, 1995). Cells were grown overnight in supplemented F-12. The next day, the dissociated sympathetic cells were washed with warm HBSS and then treated in 0.25% trypsin for 1–2 min. After the cells had dislodged from the coverslip, supplemented F-12 was added, and the cells were either replated as dissociated cells on fresh laminin coated coverslips or suspended in drop culture for reaggregation. Dissociated cells were grown for 5–6 hr and then assayed for collapse and/or protein expression. Sympathetic cells in drop cultures were allowed to reaggregate for 4–5 hr and then plated onto fresh laminin coated coverslips. Reaggregates were grown overnight (18 hr) in supplemented F-12 at 37°C with 5% CO2 and assayed the next day for growth cone collapse and/or protein expression.
Collapse assay. The collapse assay was performed as described by Luo et al. (1993) with slight alterations. In brief, an amount of recombinant secreted semaphorin, 10 times greater than that required to induce 50% of DRG or sympathetic growth cones to collapse [10 collapsing units (c.u.)], or as a control an equal volume of media, was added to each well in a volume not exceeding 250 μl. The cells were incubated at 37°C in 5% CO2 for 35 min and then fixed in 4% paraformaldehyde in PBS containing 10% sucrose. Cells were then stained for the myc epitope tag (see below) to identify transfected cells. Neurites from transfected cells that had a length of greater than two times the width of the cell body were analyzed. The tips of neurites without lamellipodia or filopodia were scored as being collapsed.
Immunohistochemistry. Cells were fixed as described above and incubated with PBS containing a mix of polyvinyl–pyrolidone (Sigma) with molecular weights of 10,000, 40,000, and 360,000 and 3% BSA. Cells were incubated with a mouse anti-myc ascites (9E10 from American Type Culture Collection, Manassas, VA) diluted 1:500 in blocker for 3 hr at room temperature or overnight at 4°C. Cells were then washed with PBS and incubated with a Cy3-conjugated donkey anti-mouse IgG/IgM secondary antibody (Jackson ImmunoResearch, West Grove, PA) for 2 hr at room temperature or overnight at 4°C.
AP fusion protein binding assay. AP-SEMA-3A, AP-Sema, and AP-Ig-basic were tested for their ability to bind to neuropilin-1 deletion constructs expressed in Cos-7. In brief, cells expressing truncated neuropilins were washed gently with PBS and incubated with AP-SEMA-3A, AP-Sema, or AP-Ig-basic diluted in PBS with 10% FBS. The AP fusion proteins were produced by HEK293T cells transiently transfected with the appropriate expression vector. The concentration of AP-SEMA-3A was determined by measuring the amount of conditioned media required to cause 50% collapse in the growth cone collapse assay. The concentrations of AP-Sema and AP-Ig-basic were estimated by comparing their AP activities with that of AP-collapsin. Cells were incubated with probe for 1 hr. After three 10 min washes with PBS, the cells were fixed in 4% paraformaldehyde in PBS with 10% sucrose. Inactivation of endogenous alkaline phosphatases was accomplished by heating the samples to 65°C for 3 hr. Binding of the AP-tagged ligands was visualized by reacting with nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate (Sigma).
Membrane preparations and Western blots. Neuropilin-1 deletion constructs were expressed in HEK293T cells as described above and grown overnight (18 hr). Cells were harvested in lysis buffer (Hallak et al., 1994) containing 20 mm HEPES, 2 mm MgCl2, 1 mm EDTA, leupeptin (2ug/ul), and PMSF (0.1 mm). Cells were incubated on ice for 5 min and then lysed by passing them through a 20 gauge needle. The lysed cells were spun down at 1000 × g for 5 min to pellet unbroken cells and nuclei. The supernatant was transferred to an ultracentrifuge tube and spun at 100,000 × g for 60 min to pellet the membranes. The pellet was resuspended in 100 μl of lysis buffer. Ten microliters of this sample were extracted with SDS-sample buffer and analyzed by Western blot using an anti-myc antibody.
RESULTS
Generation of neuropilin-1 deletion constructs
Neuropilin-1 is a cell surface protein recently identified as a receptor or receptor component for SEMA-3A. It has a large extracellular domain containing five distinct sub-domains, a single transmembrane domain, and a short cytoplasmic domain (Fig.1A). The five extracellular domains, a1, a2, b1, b2, and C, have been defined by their homology to other proteins (Takagi et al., 1991). Domains a1 and a2 are related to each other and to the noncatalytic region of the complement components C1r and C1s. The b1 and b2 domains are related to each other and to the C1 and C2 domains of coagulation factors VIII and V. A portion of the C domain shares homology to meprin, A5, μ (MAM) domains found in a variety of proteins that are thought to mediate homophilic protein–protein interactions (Beckmann and Bork, 1993; Zondag et al., 1995).
Fig. 1.
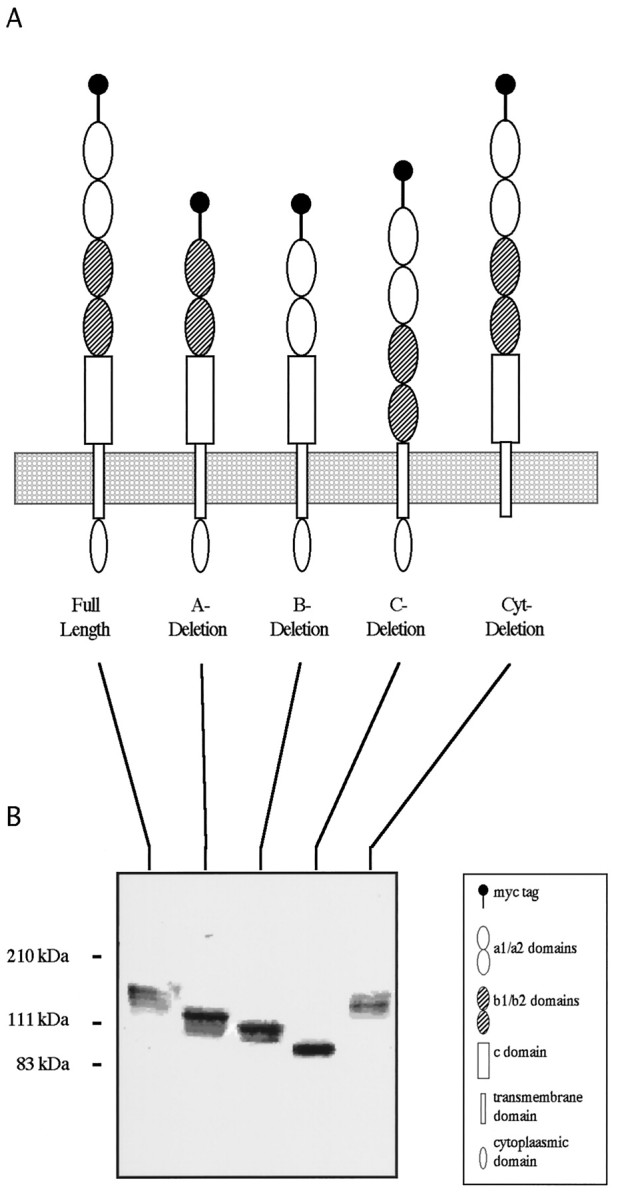
Neuropilin-1 deletion constructs and their expression. A, The domain structure of full-length neuropilin-1 is shown on the left, and the deletion constructs used in this study are arrayed to the right. The domains are described in the boxed key, and the boundaries between domains are defined in Results.B, HEK293-T cells were transfected with the neuropilin-1 constructs in A. Blotted membrane preparations were probed for the myc epitope. All of the constructs generate membrane-associated recombinant proteins that are close to their expected sizes.
We used PCR to delete specific portions of chick neuropilin-1 sequences corresponding to these domains. The boundaries of the domains were defined approximately as described by Takagi et al. (1991). Specifically, for this study, we defined the boundaries as follows: in the A-deletion construct, the a1 and a2 domains from Arg21 to Glu254 are missing; in the B-deletion construct, the b1 and b2 domains from Gly255 to Thr587 are missing; in the C-deletion construct, the C domain from Ala588 to Ile851 is missing; and in the Cyt-deletion construct, the cytoplasmic domain from Trp875 to the C terminus is deleted. Membranes from HEK293T cells transfected with these deletion constructs were purified and extracted, and the myc-tagged recombinant deletion products were analyzed by Western blots of reducing gels. All the constructs copurified with the cell membrane and, with the exception of the C-deletion product, are close to their predicted size (Fig.1B). C-deletion neuropilin-1 is reduced in weight to a larger degree than predicted by the loss of the deleted amino acids alone. This is most likely explained by the loss of three predicted glycosylation sites within the C domain. In addition, a band with the approximate molecular weight of a dimer is present in all of the constructs except C-deletion neuropilin-1. This band is more prominent in nonreducing SDS-PAGE gels (data not shown). This finding is consistent with the suggestion of Giger et al. (1998b) andNakamura et al. (1998) that the C domain mediates dimer formation, which occurs in the absence of ligand.
Neuropilin-1 contains more than one binding site for SEMA-3A
In an effort to determine which domains within neuropilin-1 are responsible for binding SEMA-3A, we expressed neuropilin-1 deletion constructs in Cos-7 cells and probed them with alkaline phosphatase-tagged versions of full-length SEMA-3A (AP-SEMA-3A), the semaphorin domain of SEMA-3A (AP-Sema), or the Ig-basic tail of SEMA-3A (AP-Ig-basic). Previous studies have shown AP-Sema has an ∼30-fold reduced activity compared with full-length SEMA-3A and that AP-Ig-basic has no detectable biological activity (Koppel and Raper, 1998). AP-SEMA-3A and AP-Ig-basic bind to cells transfected with full-length, A-deletion, C-deletion, and Cyt-deletion neuropilin-1 equally well (Fig. 2A). Reaction conditions that detect strong binding of AP-SEMA-3A and AP-Ig-basic to these neuropilin-1 constructs detect no binding to the B-deletion construct. The b1 and b2 domains therefore contribute very strongly to the binding of SEMA-3A to neuropilin-1, and the Ig-basic regions of SEMA-3A appear to mediate this binding.
Fig. 2.

Mapping SEMA-3A binding sites to neuropilin-1 domains. A, HEK293T cells were transiently transfected with A-, B-, or C- deletion neuropilin-1 and probed with ∼1.5 nm AP-SEMA-3A (top) or an anti-myc antibody (bottom). After 1 hr development in NBT/BCIP, AP-SEMA-3A was visualized bound to cells expressing A-deletion and C-deletion neuropilin-1 but not to those expressing B-deletion neuropilin-1. Staining live cells with anti-myc demonstrates that all constructs are expressed on the cell surface. B, The same neuropilin-1 deletion constructs were probed with ∼3 nm AP-Sema (top two rows) or 1.5 nm AP-Ig-basic (bottom). After 2 d development, AP-Sema was visualized bound to A- and B-deletion neuropilin-1 but not to C-deletion neuropilin-1. After 1 hr development, AP-Ig-basic was visualized bound to A- and C-deletion neuropilin-1 but not B-deletion neuropilin-1. Scale bars: A, 62.5 μm;B, 100 μm.
Commensurate with its much lower biological potency, AP-Sema binds full-length neuropilin-1 more weakly than does AP-SEMA-3A (Fig.2B). Surprisingly, it binds to B-deletion neuropilin-1, indicating that it binds outside the b1 and b2 domains recognized by the Ig-basic portion of full-length SEMA-3A. It is also possible to detect weak binding of AP-Sema to A-deletion neuropilin-1. No binding of AP-Sema to C-deletion neuropilin-1 was detected in our experiments. Experiments using the Sema domain fused to a fragment that crystalizes (Fc) as a probe produced an identical binding pattern (data not shown). These results argue that the C domain is the primary locus of semaphorin domain binding on neuropilin-1.
Overexpression of neuropilin-1 without the C domain in sympathetic neurons blocks their responsiveness to SEMA-3A
Each of the neuropilin-1 deletion constructs was transfected into cultured primary sympathetic cells in an effort to identify a dominant negative neuropilin-1 variant that blocks SEMA-3A function. Dissociated sympathetic neurons from E7 to E8 chicks were grown on laminin-coated coverslips. Eighteen hours after transfection, the cells were treated with trypsin and replated to ensure that all neurites were newly formed and would therefore incorporate proteins generated from the transfected plasmids. Neurites were allowed to extend for an additional 5–6 hr before adding control medium or medium containing ∼300 pmrecombinant AP-SEMA-3A. This is ∼10 c.u. of SEMA-3A. A collapsing unit is defined as the amount of SEMA-3A required to induce 50% of growth cones to collapse in our standard explant assay. Neurons that stained positive for the myc epitope tag incorporated into every neuropilin-1 construct were assayed for their ability to respond to SEMA-3A.
The engineered recombinant proteins can be visualized with anti-myc antibodies and are seen to be expressed on the growth cone, as well as on the neurites and cell bodies of all transfected cells (Fig.3). Expression levels are generally high and uniform between cells as judged by the intensity of myc staining. The anti-myc antibody did not label untransfected cells.
Fig. 3.
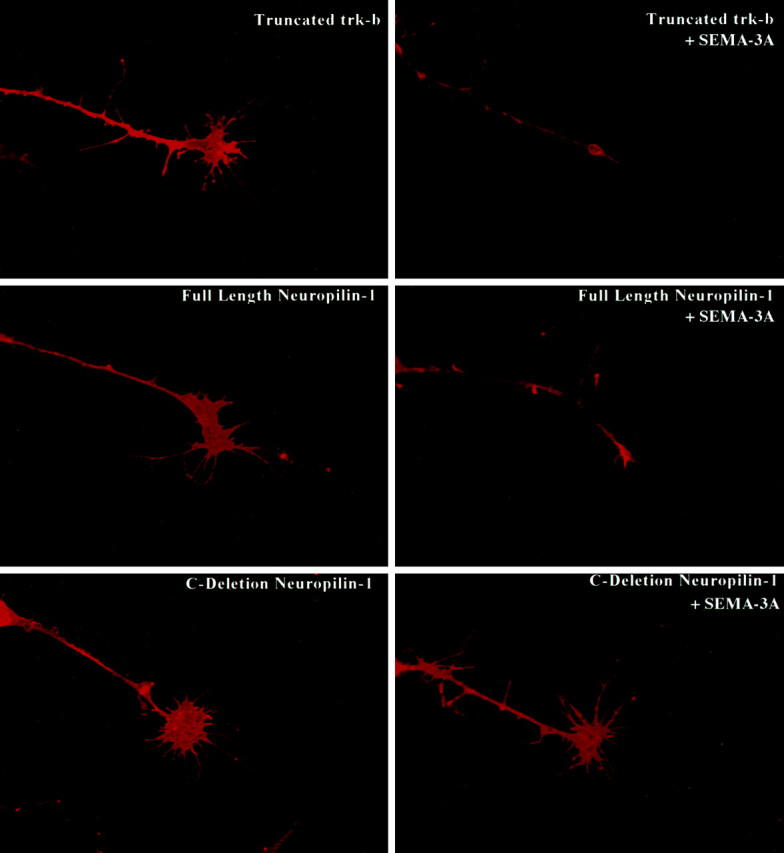
Expression of neuropilin-1 deletion constructs in cultured sympathetic growth cones. Myc-tagged recombinant proteins were visualized using an anti-myc ascites and a Cy3-conjugated secondary antibody. Recombinant protein is expressed throughout the cell, including the lamellipodia and filopodia of the growth cone. The addition of SEMA-3A induces the collapse of growth cones expressing either a control truncated Trk-B protein or full-length neuropilin-1. Sympathetic neurons expressing C-deletion neuropilin-1 are resistant to collapse when exposed to SEMA-3A. Scale bar, 20 μm.
As expected, sympathetic neurons transfected with a control plasmid producing an inactive, myc-tagged Trk-B missing its kinase domain respond normally to SEMA-3A. SEMA-3A induces a loss of motile growth cones (Fig. 4A). A similar dramatic collapsing effect is induced by SEMA-3A in sympathetic cells expressing full-length neuropilin-1. SEMA-3A induces normal collapse of sympathetic neurons expressing A-deletion neuropilin-1 as well. In contrast, sympathetic neurons expressing B-deletion neuropilin-1 are partially resistant to SEMA-3A-induced collapse. More dramatically, sympathetic neurons expressing C-deletion neuropilin-1 do not collapse to any significant degree when exposed to SEMA-3A.
Fig. 4.
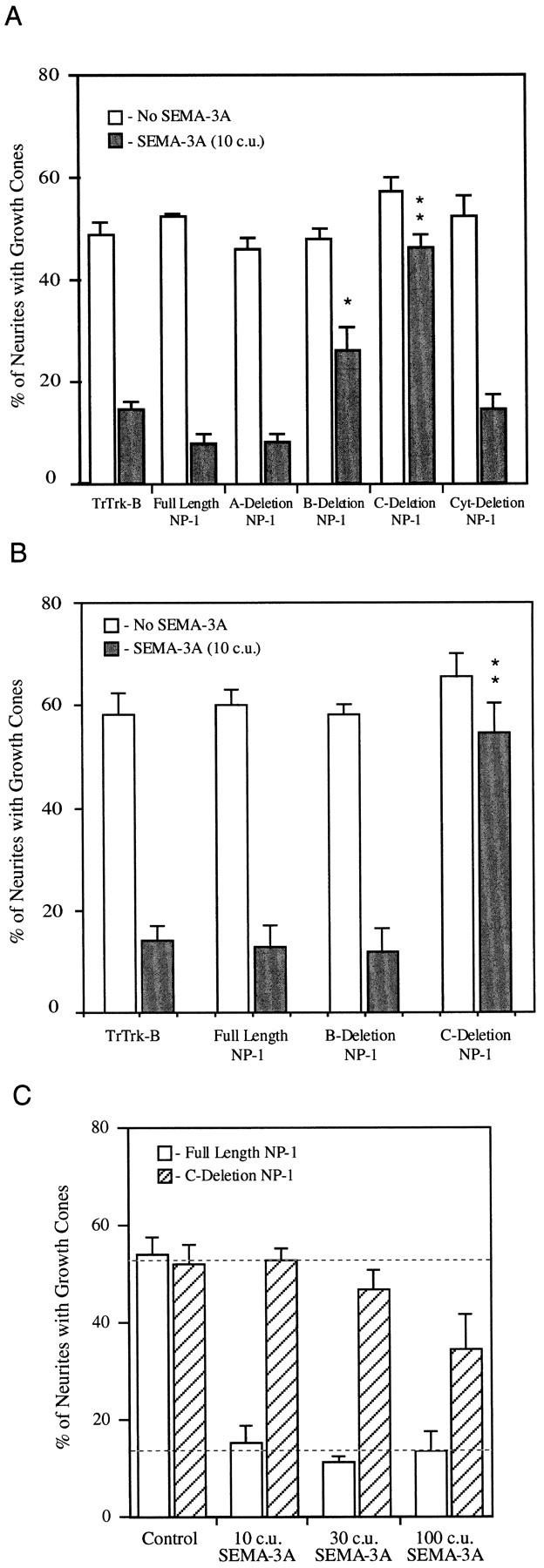
C-deletion neuropilin-1 is a dominant negative receptor component for SEMA-3A. Sympathetic neurons were transfected with the indicated constructs and then exposed to either control media or media containing AP-SEMA-3A. After 35 min, the cultures were fixed, and the percentage of myc-labeled neurites with growth cones were counted. A, The addition of 10 c.u. of AP-SEMA-3A induces collapse in growth cones expressing truncated Trk-B, full length neuropilin-1, and cytoplasmic-deletion neuropilin-1. Growth cones expressing C-deletion neuropilin-1 do not respond to SEMA-3A. Expression of B-deletion neuropilin-1 caused a partial block of SEMA-3A-induced collapse. B, Sympathetic neurons were transfected with test constructs and then reaggregated. Growth cones expressing truncated Trk-B, full-length neuropilin-1, and B-deletion neuropilin-1 all collapse in response to AP-SEMA-3A. Growth cones expressing C-deletion neuropilin-1 do not respond to SEMA-3A. The SEM of three to eight experiments is shown for each condition. *p ≤ 0.01; **p ≤ 0.001 by Student’s two-tailed t test. C, Sympathetic neurons were transfected with either full-length or C-deletion neuropilin-1 and then reaggregated. Ten, 30, or 100 c.u. of SEMA-3A induced growth cone collapse in neuropilin-1-transfected neurons. Neurons transfected with C-deletion neuropilin-1 did not respond to 10 or 30 c.u. of AP-SEMA-3A and were partially responsive at 100 c.u. The concentration of SEMA-3A required to induce 50% collapse of sympathetic neurons was shifted to ∼100-fold higher concentrations by C-deletion neuropilin-1.
The percentage of neurites with recognizable growth cones in dissociated sympathetic cultures is only ∼50%. One possible explanation for this relative paucity of growth cones is that they may collapse on contact with other neuronal processes and with non-neuronal cells in these cultures (Ivins and Pittman, 1989). To decrease these contacts, sympathetic cells were reaggregated after transfection and plated as large clumps of cells. Neurites extend from these reaggregates in a manner similar to that observed from explanted sympathetic ganglia, whereas most non-neuronal cells remain associated with the reaggregates.
The effects of SEMA-3A on sympathetic neurons transfected with full-length neuropilin-1, B-deletion, and C-deletion neuropilin-1, as well as truncated Trk-B, were reexamined in this assay. As expected, neither the Trk-B control nor the full-length neuropilin-1 constructs affected SEMA-3A responsiveness (Fig. 4B). The B-deletion neuropilin-1 construct that partially blocks SEMA-3A activity in the dissociated assay has no detectable blocking effect in the reaggregate assay. This result suggests that any dominant negative effect induced by the overexpression of this construct is weak and of doubtful utility. The C-deletion neuropilin-1 construct strongly suppresses SEMA-3A activity in the reaggregate assay. The ability of C-deletion neuropilin-1 to suppress SEMA-3A activity in sympathetics persists, even when very high concentrations of SEMA-3A are used. Fifty percent responsiveness is shifted to 100-fold higher concentrations of SEMA-3A when C-deletion neuropilin-1 is expressed in sympathetic neurons with our expression vector (Fig. 4C). Thus, C-deletion neuropilin-1 acts as a powerful dominant negative SEMA-3A receptor.
C-deletion neuropilin-1 blocks collapse of sympathetic growth cones induced by SEMA-3C
SEMA-3C, like SEMA-3A, induces a dose-dependent collapse of cultured sympathetic growth cones. Sympathetic neurons transfected with the C-deletion or appropriate control constructs were tested for their ability to respond to SEMA-3C. Sympathetic neurons transfected with truncated Trk-B or full-length neuropilin-1 collapse normally when exposed to 10 c.u. of SEMA-3C (Fig.5A). Sympathetic neurons expressing C-deletion neuropilin-1 do not respond to SEMA-3C. These results indicate that the C-deletion neuropilin-1 construct acts as a dominant negative receptor component for both SEMA-3A and SEMA-3C and suggests that neuropilin-1 may participate in signaling mediated by each of these two ligands.
Fig. 5.
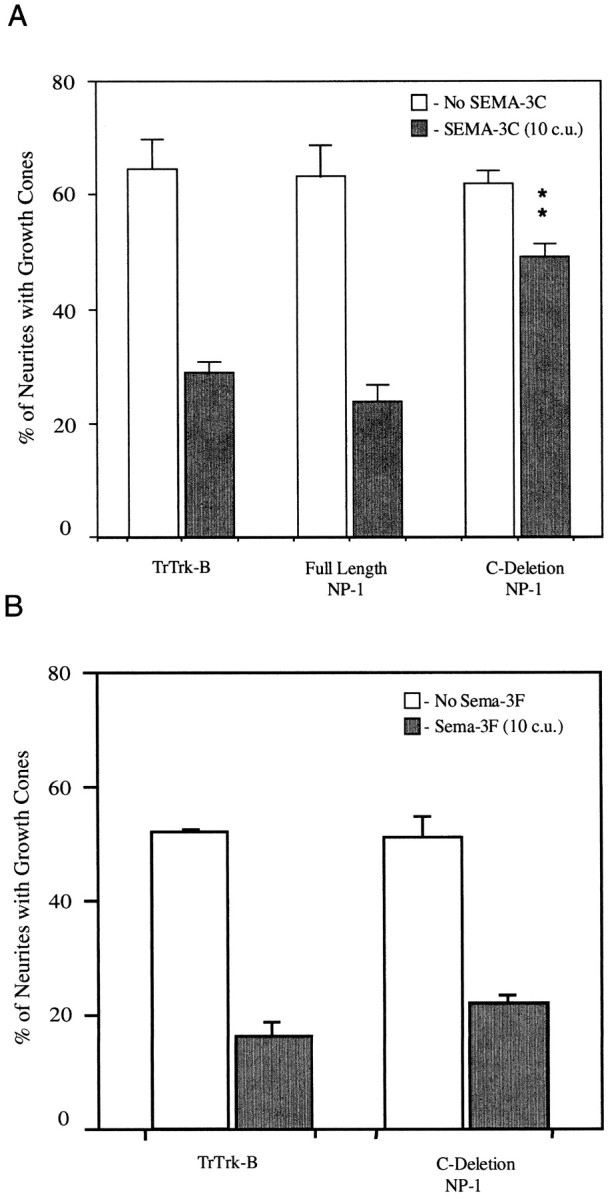
C-deletion neuropilin-1 is a dominant negative receptor component for SEMA-3C but not for SEMA-3F. Sympathetic reaggregates were transfected with truncated Trk-B, full-length neuropilin-1, or C-deletion neuropilin-1 and treated with 10 c.u. of AP-SEMA-3C or 10 c.u. of AP-SEMA-3F. A, Growth cones expressing truncated Trk-B or full-length neuropilin-1 collapse when exposed to AP-SEMA-3C. Neurons expressing C-deletion neuropilin-1 do not respond to AP-SEMA-3C. B, Growth cones expressing truncated Trk-B or C-deletion neuropilin-1 collapse normally in response to AP-SEMA-3F. The SEM of four experiments is shown. **p ≤ 0.001 by Student’s two-tailedt test.
C-deletion neuropilin-1 does not block collapse of sympathetic growth cones induced by SEMA-3F
We have shown that C-deletion neuropilin-1 acts as a dominant negative receptor for at least two secreted semaphorin family members. SEMA-3F is another secreted semaphorin family member that induces the collapse of sympathetic growth cones (Chen et al., 1997; Giger et al., 1998b). SEMA-3F-induced collapse is thought to be mediated not by neuropilin-1 but by neuropilin-2 (Chen et al., 1998; Giger et al., 1998b; Takahashi et al., 1998). C-deletion neuropilin-1 was therefore tested for its ability to prevent SEMA-3F-induced collapse of sympathetic growth cones. Sympathetic neurons transfected with truncated Trk-B or C-deletion neuropilin-1 collapse normally when exposed to 10 c.u. of SEMA-3F (Fig. 5B). The C-deletion neuropilin-1 construct therefore does not act as a dominant negative receptor component for SEMA-3F, consistent with the proposal that neuropilin-2 mediates SEMA-3F signaling without any involvement of neuropilin-1.
C-deletion neuropilin-1 does not block collapse of sympathetic growth cones induced by the semaphorin domain of SEMA-3A
Neuropilin-1 appears to have at least two binding sites for SEMA-3A, one located in the b1 and b2 domains that bind the Ig-basic tail of SEMA-3A and at least one outside the b1 and b2 domains required for the binding of the semaphorin domain (see above). The semaphorin domain of SEMA-3A forms a biologically active dimer when made as a fusion protein with an Fc fragment (Koppel and Raper, 1998). This semaphorin domain dimer is ∼30-fold less potent than full-length SEMA-3A, presumably because it is missing the Ig-basic portion of SEMA-3A that binds so strongly to the B domain of neuropilin-1. Sympathetic neurons transfected with truncated Trk-B or C-deletion neuropilin-1 collapse normally when exposed to 5 c.u. of the Sema-Fc construct (Fig. 6). The C-deletion neuropilin-1 construct therefore only acts as an effective dominant negative receptor component when the SEMA-3A ligand contains the Ig-basic domains.
Fig. 6.

C-deletion neuropilin-1 is not a dominant negative receptor component for collapse induced by the semaphorin domain of SEMA-3A. Sympathetic reaggregates were transfected with truncated Trk-B or C-deletion neuropilin-1 and treated with 5 c.u. of the semaphorin domain from Sema-Fc. This truncated form of SEMA-3A that is missing the Ig-basic domains induces growth cone collapse, even in growth cones expressing C-deletion neuropilin-1. The SEM of four experiments is shown for each condition.
DISCUSSION
Neuropilin-1 has been identified as a candidate receptor for SEMA-3A (He and Tessier-Lavigne, 1997; Kolodkin et al., 1997). We have generated a variety of neuropilin-1 deletion constructs and tested them for both their ability to bind full-length and truncated forms of SEMA-3A and their ability to interfere with SEMA-3A function in sympathetic neurons.
Table 1 presents a compendium of the results of our binding studies, along with the results of similar experiments reported by others (Giger et al., 1998b; Nakamura et al., 1998). Although the neuropilin-1 and SEMA-3A constructs used by the various groups are not always strictly comparable, several interesting conclusions can be drawn. (1) Neither the A nor the C domains of neuropilin-1 are required for full-length SEMA-3A binding. (2) The same appears to be true for both the Ig and the basic portions of SEMA-3A. (3) In contrast, neither the A nor the B domains of neuropilin-1 appear to be required for the binding of the semaphorin portion of SEMA-3A. These results suggest that the Ig-basic portion of SEMA-3A binds to the B domain, whereas the semaphorin portion of SEMA-3A binds to the C domain. Further tentative conclusions can be drawn about the necessity of smaller stretches of amino acids. The amino acids 254–274 near the junction of the A and B domains of neuropilin-1 appear to be essential for the binding of the Ig-basic portion of SEMA-3A. It would also appear that the semaphorin portion of SEMA-3A binds to sequences within the C domain that are exclusive of the MAM domain. These findings reveal a complex multi-site interaction between SEMA-3A and neuropilin-1.
Table 1.
Summary of the domain mapping of SEMA-3A binding to neuropilin-1

A compendium of the results reported for the binding of SEMA-3A to neuropilin-1 as described: 1this paper;2Giger et al., 1998b; and 3Nakamura et al., 1998. Full-length and partial AP-tagged SEMA-3A constructs (top row) were tested for binding to various neuropilin-1 constructs (first column). Schematics of neuropilin-1 deletion constructs from different laboratories are shown with the amino acid numbers used to define domain deletion boundaries. Neuropilin-1 deletion constructs with break points differing by 12 amino acids or less are grouped together. Binding results are expressed as follows: +++, strong binding; +, weak binding; −, no detectable binding; and ( ), not tested. §, Results that differed between two laboratories in experiments using similar reagents. Giger et al. (1998b) observed no binding, whereas we detected binding after developing the reactions for very long times.
The mechanism by which neuropilin-1 mediates semaphorin function is not yet certain. Most simply, neuropilin-1 could act as a simple type I receptor in which the binding of ligand to the extracellular portion of neuropilin-1 causes the direct activation of an intracellular signaling pathway via the cytoplasmic tail. Alternatively, neuropilin-1 could be an essential component of one or more receptor complexes, each of which is activated by specific semaphorin ligands (Feiner et al., 1997). The primary objective of this work was to generate a dominant negative neuropilin-1 that could be used to help distinguish between these possibilities. Our results are consistent with the hypothesis that neuropilin-1 binds specific semaphorins and then presents them to additional receptor components that initiate signal transduction.
If neuropilin-1 were a simple type I receptor (Fig.7A), then it is reasonably straightforward to predict the kinds of truncations that are likely to generate dominant negative variants. Previous experiments with type I receptors have shown that the deletion of their cytoplasmic domains generally makes a dominant negative receptor. When overexpressed in cells that would normally respond to a ligand, the truncated receptor interferes with normal receptor function by either sequestering ligand on inactive receptors (Ross et al., 1997; Moriggl et al., 1998) or forming inactive multimers with wild-type receptors (Ueno et al., 1993; Perrot-Applanat et al., 1997). Alternatively, overexpression of receptor variants in which the cytoplasmic domains are intact and extracellular domains are missing can sometimes generate dominant negative receptors that sequester downstream signaling components in an inactive complex (Dosil et al., 1998; Maruyama et al., 1998).
Fig. 7.

Models for how neuropilin-1 could be involved in Semaphorin signaling. A, Neuropilin-1 is unlikely to be acting as a type I receptor. Semaphorin signaling is more likely to involve a second component. Additional factor(s) may be present as preformed complexes with neuropilin-1 on the cell surface (B) or may be recruited into the neuropilin–semaphorin complex after ligand binding (C). Together, our data are most consistent with the third model.
We therefore constructed truncated versions of neuropilin-1 missing either the cytoplasmic or all extracellular domains. Neuropilin-1 missing the cytoplasmic domain reaches the cell surface and binds AP-SEMA-3A, suggesting that the presence or absence of the cytoplasmic domain does not affect its ability to bind ligand. Expression of neuropilin-1 missing the cytoplasmic domain in cultured sympathetic neurons does not alter their responsiveness to SEMA-3A. It therefore does not interfere with the functional activity of endogenous neuropilin-1 and does not act as a dominant negative receptor. Similarly, a variant of neuropilin-1 that includes only its transmembrane and cytoplasmic portions reaches the surface of sympathetic neurons but does not alter their responsiveness to SEMA-3A (M. J. Renzi, unpublished data; data not shown).
These results argue strongly that neuropilin-1 is unlikely to act as a type I receptor. Further evidence supporting this conclusion is the recent observation that SEMA-3A responsiveness can be conferred on otherwise unresponsive retinal ganglion cell axons by the expression of either full-length neuropilin-1 or a variant of neuropilin-1 in which the cytoplasmic domain is missing (Nakamura et al., 1998; Takahashi et al., 1998).
These findings suggest that neuropilin-1 interacts with an additional receptor component that in turn initiates signal transduction. This conclusion is further strengthened by our surprising finding that a version of neuropilin-1 missing its extracellular C domain blocks the response of sympathetic neurons to SEMA-3A. There are several mechanisms by which this variant could act as a dominant negative receptor component. The deleted portion of the molecule contains within it a single MAM-like domain. MAM domains have been shown to be involved in protein–protein interactions. Several studies have implicated MAM domains in the formation of homodimeric complexes of receptor proteins (Zondag et al., 1995; Marchand et al., 1996), and recent evidence indicates that the MAM domains in neuropilins cause them to associate with one another in a ligand-independent manner (Chen et al., 1998;Giger et al., 1998b; Takahashi et al., 1998). One possible explanation for our finding is that C-deletion neuropilin-1 binds SEMA-3A into incompletely organized receptor complexes that are not functional. The association of neuropilin-1 with itself, with neuropilin-2, or with an as yet unknown receptor component could be prevented by the absence of the C domain.
An alternative model could explain how C-deletion neuropilin-1 acts as a dominant negative receptor component for SEMA-3A. Previous studies demonstrate that the semaphorin domain contains a short stretch of sequence that determines the biological specificity of each secreted semaphorin, and it is possible that this sequence triggers signaling activity when presented by neuropilin-1 to a nearby transducing molecule (Koppel et al., 1997). The Ig-basic domains of SEMA-3A bind to the B domain of neuropilin-1, whereas the semaphorin domain of SEMA-3A binds outside the B domain (Chen et al., 1998; Giger et al., 1998b; Nakamura et al., 1998; and this paper). Our data indicate that semaphorin domain binding is greatly reduced in the absence of the C domain. This suggests that the C domain contributes to a semaphorin domain binding site. A comparison of our binding data with those of Giger et al. (1998b) suggests that this semaphorin domain binding site must be outside the MAM domain. It is possible that, in the absence of this site, the semaphorin domain of SEMA-3A is incapable of activating the signal transducing component, because either it is mispositioned when bound to neuropilin-1 or it fails to assume a required conformational configuration. C-deletion neuropilin-1 would then block SEMA-3A activity by binding the ligand but failing to present it properly to a transducing receptor component.
Both of these models predict that neuropilin-1 must interact with additional receptor components to form a functional receptor. However, are these components preassembled with neuropilin-1 (Fig.7B) or are they recruited after neuropilin-1 binds its ligand (Fig. 7C)? Our data are consistent with the latter model. This conclusion is inferred from the observation that, although C-deletion neuropilin-1 acts as a dominant negative receptor component for full-length SEMA-3A, it does not interfere with the action of the Sema-Fc fusion protein.
The failure of C-deletion neuropilin-1 to block the activity of this truncated ligand implies that Sema-Fc can either (1) act directly on the presumptive transducing receptor component or (2) use native full-length neuropilin-1 on the cell surface to access the transducer. The first of these possibilities is unlikely because the presence of neuropilin-1 has been shown to be absolutely required for full-length SEMA-3A to induce collapse (Kitsukawa et al., 1997; Chen et al., 1998;Giger et al., 1998b). This result suggests that the presumptive transducing receptor component cannot be activated without the cooperation of neuropilin-1. Sema-Fc should therefore be unable to activate a transducing receptor component directly. Our data show that the C-deletion neuropilin-1 we engineered does not bind and therefore cannot sequester Sema-Fc. We therefore hypothesize that Sema-Fc instead interacts, as it normally would, with native neuropilin-1 that is still present on sympathetic neurons overexpressing C-deletion neuropilin-1. The native neuropilin-1 then presents Sema-Fc to the presumptive transducing receptor component. This could only happen if native neuropilin-1 still has access to the transducing receptor component. This then implies that overexpressed C-deletion neuropilin-1 is not preassembled with, and therefore does not prevent access of native neuropilin-1 to, the transducer. A model consistent with these observations is that neuropilin-1 does not preassemble with the transducing component but recruits the transducing unit after SEMA-3A is bound.
We previously proposed that neuropilin-1 acts as a common component of receptor complexes that are specific for different secreted semaphorins (Feiner et al., 1997). If so, then C-deletion neuropilin-1 should act as a dominant negative receptor component for more than one secreted semaphorin. Sympathetic neurons are known to be responsive to at least three secreted semaphorins: SEMA-3A, SEMA-3C, and SEMA-3F. C-deletion neuropilin-1 effectively blocks the collapse of sympathetic growth cones induced by SEMA-3A and SEMA-3C but does not block collapse induced by SEMA-3F. These results demonstrate that C-deletion neuropilin-1 is specific in its dominant negative effects and is unlikely to act through nonspecific artifactual mechanisms. These results are also consistent with our original hypothesis that neuropilin-1 acts as a receptor component for multiple secreted semaphorins but demonstrates that is not involved in the activities of all.
The information currently available suggests that the specificity of the response of a neuron to particular secreted semaphorins is partially or even wholly determined by the combination of neuropilins present on its cell surface. Neuropilins form ligand-independent dimers. Homodimers have been shown to form when either neuropilin-1 or neuropilin-2 are expressed alone, and heterodimers have been shown to form when neuropilin-1 and neuropilin-2 are expressed together (Kitsukawa et al., 1997; Chen et al., 1998; Giger et al., 1998b; Takahashi et al., 1998). Several lines of experimental evidence are consistent with a model in which the expression of neuropilin-1 homodimers confer responsiveness to SEMA-3A, neuropilin-2 homodimers confer responsiveness to SEMA-3F, and neuropilin-1/2 heterodimers confer responsiveness to SEMA-3C. One line of evidence is that responsiveness can be altered in predictable ways by the overexpression of neuropilins in primary neurons. For example, only neuropilin-1 is normally expressed in DRG neurons, and they are sensitive to SEMA-3A but not to SEMA-3C or SEMA-3F. Transfection of neuropilin-2 into DRGs makes them, like sympathetic neurons in which both neuropilin-1 and neuropilin-2 are normally expressed, sensitive to all three of these semaphorins (Giger et al., 1998b; Takahashi et al., 1998). Another line of evidence is that responsiveness can be altered in predictable ways by incubating sympathetic neurons with neuropilin-1- and neuropilin-2-specific antibodies. Antibodies against neuropilin-1 block responsiveness to SEMA-3A and reduce responsiveness to SEMA-3C, whereas antibodies against neuropilin-2 block responsiveness to SEMA-3F (Chen et al., 1998; Giger et al., 1998b). Finally, overexpression of C-deletion neuropilin-1 in sympathetic neurons specifically blocks responsiveness to SEMA-3A and SEMA-3C, indicating that sensitivity to each is neuropilin-1-dependent. C-deletion neuropilin-1 does not affect responsiveness to SEMA-3F, however, consistent with the expectation that SEMA-3F sensitivity is mediated solely by neuropilin-2 homodimers.
What is the nature of the missing receptor component(s) that, along with neuropilin-1, forms a functional semaphorin receptor? It is interesting to note that neuropilin-1 also binds the chemoattractant vascular endothelial growth factor (VEGF) (Soker et al., 1998). VEGF can activate the transmembrane tyrosine kinase KDR directly, but when neuropilin-1 is present, VEGF activation of KDR is potentiated. VEGF has no chemoattractant effect on cells expressing neuropilin-1 alone. Thus, just as we propose to be the case for growth cone collapse, neuropilin-1 only acts in endothelial chemotaxis through an additional transducing receptor component.
A striking difference between the role of neuropilin-1 in growth cone collapse and endothelial chemotaxis, however, is that neuropilin-1 is essential for inducing growth cone collapse but not for kinase insert domain-containing receptor (KDR) activation in chemotaxis. For this reason, neuropilin-1 may interact with a signal transducing receptor component in growth cone collapse, just as the interleukin-6 receptor (IL-6R) interacts with its signal-transducing component gp130 (Taga et al., 1989). The IL-6 receptor by itself is unable to transduce a signal. IL-6 binding to the IL-6 receptor causes the recruitment of a third component, gp130, that is responsible for signal transduction. As with neuropilin-1, a truncated form of the IL-6 receptor that is missing its cytoplasmic domain fails to act as a dominant negative receptor component. This truncated IL-6 receptor is functionally intact and retains the ability to interact with gp130. Interestingly, gp130 transduces signals mediated by several different cytokines that first bind to additional, specific receptors (Davis et al., 1993). By analogy, the missing semaphorin receptor element could be a common signal-transducing protein that is recruited and activated by neuropilins once they bind their ligand.
A dominant negative form of neuropilin-1 may be of considerable practical use in studying the role semaphorins play in growth cone guidance. If semaphorins have overlapping functions in vivo, as seems likely from their overlapping patterns of expression (Adams et al., 1996; Shepherd et al., 1996; Giger et al., 1998b), their ability to share at least one receptor component (Feiner et al., 1997), and their similar biological activities (Koppel et al., 1997), then the analysis of animals in which any one of them is knocked out may be relatively uninformative. One way to avoid this difficulty would be to examine axon trajectories in animals missing the shared receptor component neuropilin-1. However, neuropilin-1 knock-out embryos die before the formation of many of the projections likely to be affected. Overexpression of a dominant negative neuropilin-1 in older embryos will provide a very useful alternative to a knock-out strategy.
Footnotes
This work was supported by National Institutes of Health Grant RO1-NS-26527 to J.A.R. and Training Grant T32HD07516 to M.J.R. and L.F.
Correspondence should be addressed to Dr. Jonathan A. Raper, University of Pennsylvania, School of Medicine, 215 Stemmler Hall, 36th and Hamilton Walk, Philadelphia, PA 19104.
REFERENCES
- 1.Adams RH, Betz H, Puschel AW. A novel class of murine semaphorins with homology to thrombospondin is differentially expressed during early embryogenesis. Mech Dev. 1996;57:33–45. doi: 10.1016/0925-4773(96)00525-4. [DOI] [PubMed] [Google Scholar]
- 2.Adams RH, Lohrum M, Klostermann A, Betz H, Puschel AW. The chemorepulsive activity of secreted semaphorins is regulated by furin-dependent proteolytic processing. EMBO J. 1997;16:6077–6086. doi: 10.1093/emboj/16.20.6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baird JL, Raper JA. A serine proteinase involved in contact mediated repulsion of retinal growth cones by DRG neurites. J Neuroscience. 1995;15:6605–6618. doi: 10.1523/JNEUROSCI.15-10-06605.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beckmann G, Bork P. An adhesive domain detected in functionally diverse receptors. Trends Biochem Sci. 1993;18:40–41. doi: 10.1016/0968-0004(93)90049-s. [DOI] [PubMed] [Google Scholar]
- 5.Behar O, Golden JA, Mashimo H, Schoen FJ, Fishman MC. Semaphorin III is needed for normal patterning and growth of nerves, bones and heart. Nature. 1996;383:525–528. doi: 10.1038/383525a0. [DOI] [PubMed] [Google Scholar]
- 6.Chen H, Chedotal A, He Z, Goodman CS, Tessier-Lavigne M. Neuropilin-2, a novel member of the neuropilin family, is a high affinity receptor for the semaphorins Sema E and Sema IV but not Sema III. Neuron. 1997;19:547–559. doi: 10.1016/s0896-6273(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 7.Chen H, He Z, Bagri A, Tessier-Lavigne M. Semaphorin–neuropilin interactions underlying sympathetic axon responses to class III semaphorins. Neuron. 1998;21:1283–1290. doi: 10.1016/s0896-6273(00)80648-0. [DOI] [PubMed] [Google Scholar]
- 8.Davis S, Aldrich TH, Stahl N, Pan L, Taga T, Kishimoto T, Ip NY, Yancopoulos GD. LIFR β and gp130 as heterodimerizing signal transducers of the tripartite CNTF receptor. Science. 1993;260:1805–1808. doi: 10.1126/science.8390097. [DOI] [PubMed] [Google Scholar]
- 9.Dosil M, Giot L, Davis C, Konopka JB. Dominant-negative mutations in the G-protein-coupled α-factor receptor map to the extracellular ends of the transmembrane segments. Mol Cell Biol. 1998;18:5981–5991. doi: 10.1128/mcb.18.10.5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feiner L, Koppel AM, Kobayashi H, Raper JA. Secreted chick semaphorins bind recombinant neuropilin with similar affinities but bind different subsets of neurons in situ. Neuron. 1997;19:539–545. doi: 10.1016/s0896-6273(00)80370-0. [DOI] [PubMed] [Google Scholar]
- 11.Giger RJ, Pasterkamp RJ, Heijnen S, Holtmaat AJ, Verhaagen J. Anatomical distribution of the chemorepellent semaphorin III/collapsin-1 in the adult rat and human brain: predominant expression in structures of the olfactory–hippocampal pathway and motor system. J Neurosci Res. 1998a;52:27–42. doi: 10.1002/(SICI)1097-4547(19980401)52:1<27::AID-JNR4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 12.Giger RJ, Urquhart ER, Gillespie SK, Levengood DV, Ginty DD, Kolodkin AL. Neuropilin-2 is a receptor for semaphorin IV: insight into the structural basis of receptor function and specificity. Neuron. 1998b;21:1079–1092. doi: 10.1016/s0896-6273(00)80625-x. [DOI] [PubMed] [Google Scholar]
- 13.Hallak H, Brass LF, Manning DR. Failure to myristolate the a subunit of GZ is correlated with an inhibition of palmitoylation and membrane attachment, but has no affect on phosphorylation by protein kinase C. J Biol Chem. 1994;269:4571–4576. [PubMed] [Google Scholar]
- 14.He Z, Tessier-Lavigne M. Neuropilin is a receptor for the axonal chemorepellent semaphorin III. Cell. 1997;90:739–751. doi: 10.1016/s0092-8674(00)80534-6. [DOI] [PubMed] [Google Scholar]
- 15.Ivins JK, Pittman RN. Growth cone-growth cone interactions in cultures of rat sympathetic neurons. Dev Biol. 1989;135:147–157. doi: 10.1016/0012-1606(89)90165-6. [DOI] [PubMed] [Google Scholar]
- 16.Kitsukawa T, Shimizu M, Sanbo M, Hirata T, Taniguchi M, Bekku Y, Yagi T, Fujisawa H. Neuropilin-semaphorin III/D-mediated chemorepulsive signals play a crucial role in peripheral nerve projection in mice. Neuron. 1997;19:995–1005. doi: 10.1016/s0896-6273(00)80392-x. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi H, Koppel AM, Luo Y, Raper JA. A role for collapsin-1 in olfactory and cranial sensory axon guidance. J Neurosci. 1997;17:8339–8352. doi: 10.1523/JNEUROSCI.17-21-08339.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolodkin AL, Levengood DV, Rowe EG, Tai YT, Giger RJ, Ginty DD. Neuropilin is a semaphorin III receptor. Cell. 1997;90:753–762. doi: 10.1016/s0092-8674(00)80535-8. [DOI] [PubMed] [Google Scholar]
- 19.Koppel AM, Raper JA. Collapsin-1 covalently dimerizes, and dimerization is necessary for collapsing activity. J Biol Chem. 1998;273:15708–15713. doi: 10.1074/jbc.273.25.15708. [DOI] [PubMed] [Google Scholar]
- 20.Koppel AM, Feiner L, Kobayashi H, Raper JA. A 70 amino acid region within the semaphorin domain activates specific cellular response of semaphorin family members. Neuron. 1997;19:531–537. doi: 10.1016/s0896-6273(00)80369-4. [DOI] [PubMed] [Google Scholar]
- 21.Luo Y, Raible D, Raper JA. Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell. 1993;75:217–227. doi: 10.1016/0092-8674(93)80064-l. [DOI] [PubMed] [Google Scholar]
- 22.Marchand P, Volkmann M, Bond JS. Cysteine mutations in the MAM domain result in monomeric meprin and alter stability and activity of the proteinase. J Biol Chem. 1996;271:24236–24241. doi: 10.1074/jbc.271.39.24236. [DOI] [PubMed] [Google Scholar]
- 23.Maruyama K, Endoh H, Sasaki-Iwaoka H, Kanou H, Shimaya E, Hashimoto S, Kato S, Kawashima H. A novel isoform of rat estrogen receptor β with 18 amino acid insertion in the ligand binding domain as a putative dominant negative regular of estrogen action. Biochem Biophys Res Commun. 1998;246:142–147. doi: 10.1006/bbrc.1998.8590. [DOI] [PubMed] [Google Scholar]
- 24.Messersmith EK, Leonardo ED, Shatz CJ, Tessier-Lavigne M, Goodman CS, Kolodkin AL. Semaphorin III can function as a selective chemorepellent to pattern sensory projections in the spinal cord. Neuron. 1995;14:949–959. doi: 10.1016/0896-6273(95)90333-x. [DOI] [PubMed] [Google Scholar]
- 25.Moriggl R, Erhardt I, Kammer W, Berchtold S, Schnarr B, Lischke A, Groner B, Friedrich K. Activation of STAT6 is not dependent on phosphotyrosine-mediated docking to the interleukin-4 receptor and can be blocked by dominant-negative mutants of both receptor subunits. Eur J Biochem. 1998;251:25–35. doi: 10.1046/j.1432-1327.1998.2510025.x. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura F, Tanaka M, Takahashi T, Kalb RG, Strittmatter SM. Neuropilin-1 extracellular domains mediate semaphorin D/III-induced growth cone collapse. Neuron. 1998;21:1093–1100. doi: 10.1016/s0896-6273(00)80626-1. [DOI] [PubMed] [Google Scholar]
- 27.Perrot-Applanat M, Gualillo O, Pezet A, Vincent V, Edery M, Kelly PA. Dominant negative and cooperative effects of mutant forms of prolactin receptor. Mol Endocrinol. 1997;11:1020–1032. doi: 10.1210/mend.11.8.9954. [DOI] [PubMed] [Google Scholar]
- 28.Ross RJ, Esposito N, Shen XY, Von Laue S, Chew SL, Dobson PR, Postel-Vinay MC, Finidori J. A short isoform of the human growth hormone receptor functions as a dominant negative inhibitor of the full-length receptor and generates large amounts of binding protein. Mol Endocrinol. 1997;11:265–273. doi: 10.1210/mend.11.3.9901. [DOI] [PubMed] [Google Scholar]
- 29.Shepherd I, Luo Y, Raper JA, Chang S. The distribution of collapsin-1 mRNA in the developing chick nervous system. Dev Biol. 1996;173:185–199. doi: 10.1006/dbio.1996.0016. [DOI] [PubMed] [Google Scholar]
- 30.Shepherd IT, Luo Y, Lefcort F, Reichardt LF, Raper JA. A sensory axon repellent secreted from ventral spinal cord explants is neutralized by antibodies raised against collapsin-1. Development. 1997;124:1377–1385. doi: 10.1242/dev.124.7.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- 32.Taga T, Hibi M, Hirata Y, Yamasaki K, Yasukawa K, Matsuda T, Hirano T, Kishimoto T. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell. 1989;58:573–581. doi: 10.1016/0092-8674(89)90438-8. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi T, Nakamura F, Jin Z, Kalb RG, Strittmatter SM. Semaphorins A and E act as antagonists of neuropilin-1 and agonists of neuropilin-2 receptors. Nat Neurosci. 1998;1:487–493. doi: 10.1038/2203. [DOI] [PubMed] [Google Scholar]
- 34.Takagi S, Hirata T, Agata K, Mochii M, Eguchi G, Fujisawa H. The A5 antigen, a candidate for the neuronal recognition molecule, has homologies to complement components and coagulation factors. Neuron. 1991;7:295–307. doi: 10.1016/0896-6273(91)90268-5. [DOI] [PubMed] [Google Scholar]
- 35.Takagi S, Kasuya Y, Shimizu M, Matsuura T, Tsuboi M, Kawakami A, Fujisawa H. Expression of a cell adhesion molecule, neuropilin, in the developing chick nervous system. Dev Biol. 1995;170:207–222. doi: 10.1006/dbio.1995.1208. [DOI] [PubMed] [Google Scholar]
- 36.Taniguchi M, Yuasa S, Fujisawa H, Naruse I, Saga S, Mishina M, Yagi T. Disruption of semaphorin III/D gene causes severe abnormality in peripheral nerve projection. Neuron. 1997;19:519–530. doi: 10.1016/s0896-6273(00)80368-2. [DOI] [PubMed] [Google Scholar]
- 37.Ueno H, Escobedo JA, Williams LT. Dominant-negative mutations of platelet-derived growth factor (PDGF) receptors. Inhibition of receptor function by ligand-dependent formation of heterodimers between PDGF α- and β-receptors. J Biol Chem. 1993;268:22814–22819. [PubMed] [Google Scholar]
- 38.Varela-Echavarria A, Tucker A, Puschel AW, Guthrie S. Motor axon subpopulations respond differentially to the chemorepellents netrin-1 and semaphorin D. Neuron. 1997;18:193–207. doi: 10.1016/s0896-6273(00)80261-5. [DOI] [PubMed] [Google Scholar]
- 39.Zondag GC, Koningstein GM, Jiang YP, Sap J, Moolenaar WH, Gebbink MF. Homophilic interactions mediated by receptor tyrosine phosphatases μ and κ. A critical role for the novel extracellular MAM domain. J Biol Chem. 1995;270:14247–14250. doi: 10.1074/jbc.270.24.14247. [DOI] [PubMed] [Google Scholar]