

Abstract
Target-derived neurotrophins initiate signals that begin at nerve terminals and cross long distances to reach the cell bodies and regulate gene expression. Neurotrophin receptors, Trks, themselves serve as retrograde signal carriers. However, it is not yet known whether the retrograde propagation of Trk activation reflects movement of Trk receptors from neurites to cell bodies or reflects serial activation of stationary Trk molecules. Here, we show that neurotrophins selectively applied to distal neurites of sensory neurons rapidly induce phosphorylation of the transcription factor cAMP response element-binding protein (CREB) and also cause a slower increase in Fos protein expression. Both nuclear responses require activation of neurotrophin receptors (Trks) at distal nerve endings and retrograde propagation of Trk activation to the nerve cell bodies. Using photobleach and recovery techniques to follow biologically active, green fluorescent protein (GFP)-tagged BDNF receptors (TrkB-GFP) in live cells during retrograde signaling, we show that TrkB-GFP moves rapidly from neurites to the cell bodies. This rapid movement requires ligand binding, Trk kinase activity, and intact axonal microtubules. When they reach the cell bodies, the activated TrkB receptors are in a complex with ligand. Thus, the retrograde propagation of activated TrkB from neurites to cell bodies, although rapid, reflects microtubule-dependent transport of phosphorylated Trk–ligand complexes. Moreover, the relocation of activated Trk receptors from nerve endings to cell bodies is required for nuclear signaling responses. Together, these data support a model of retrograde signaling whereby rapid vesicular transport of ligand–receptor complex from the neurites to the cell bodies mediates the nuclear responses.
Keywords: retrograde signaling, retrograde transport, c-fos, neurotrophin, Trk, DRG
Neurons and the targets they innervate develop coordinately. Target tissues release neurotrophins and other trophic factors critical for the survival and differentiation of presynaptic cells (Levi-Montalcini, 1987; Barde, 1989; Oppenheim, 1991). Neurotrophins initiate these cellular responses by binding and activating specific receptor tyrosine kinases, Trks, embedded within the plasma membrane (Kaplan and Stephens, 1994; Barbacid, 1995;Bothwell, 1995; Kaplan and Miller, 1997). NGF preferentially binds TrkA (Cordon-Cardo et al., 1991), BDNF and neurotrophin (NT) 4/5 bind TrkB (Klein et al., 1991), and NT3 binds TrkC (Lamballe et al., 1991). Binding of ligand to receptor causes receptor dimerization and autophosphorylation (Jing et al., 1992; Clary et al., 1994). In neurons with long axons, target-derived neurotrophins bind to Trk receptors at axonal terminals that can be located up to a meter away from the cell bodies. How then do trophic factors inform the cell bodies of events occurring at the distal axon terminals?
Classical experiments established that neurotrophins are taken up at nerve terminals and transported to the cell bodies (Schwab, 1977;Johnson et al., 1987). Functional consequences of NGF uptake were demonstrated recently. Riccio et al. (1997) showed that soluble NGF applied selectively to the neurites of sympathetic neurons could initiate a nuclear response in distant cell bodies, but NGF linked to beads could not do so. These studies indicate that ligand endocytosis is required to initiate remote signaling in neurons with long axons. It is possible that the process of endocytosis itself initiates a signaling cascade (Vieria et al., 1996; Saragovi et al., 1998) critical for a retrograde response. Alternatively, the physical transport of an endocytosed ligand–receptor complex from the neurites to the cell bodies may indeed be required for nuclear responses.
Recent studies show that stimulation of neurites also initiates an activation-state change in Trk receptors that propagates rapidly through axons in a retrograde direction (Ehlers et al., 1995;Bhattacharyya et al., 1997; Riccio et al., 1997; Senger and Campenot, 1997). Surprisingly, the velocity of this retrograde wave of Trk activation was faster than expected based on published rates of neurotrophin transport. The rate of ligand transport, determinedin vivo and in vitro using I125-NGF, has been calculated as 0.2–2 μm/sec, or 2–20 mm/hr (Hendry et al., 1974; Claude et al., 1982;Korsching and Thoenen, 1983; Palmatier et al., 1984; Richardson and Riopelle, 1984; Yip and Johnson, 1986; Ure and Campenot, 1997). The rapidity of the Trk response could reflect the use of transport systems quicker than those hitherto appreciated or could reflect a novel mechanism of signal propagation involving sequential activation of stationary Trk molecules.
To define the mechanisms whereby retrograde signals elicit nuclear responses, we used dorsal root ganglion (DRG) neurons grown in compartmented cultures. The compartmented culture system (Campenot, 1977) allows selective stimulation of either cell bodies or distal neurites. Here, we show that selective application of neurotrophin to neurites acts at a distance to induce two nuclear events: rapid phosphorylation of the transcription factor c-AMP response element binding protein (CREB) and subsequent expression of the immediate early gene product c-fos. We show that both of these nuclear events require activation of Trk receptors in the cell bodies, as well as in the neurites. Furthermore, using photobleach and recovery to follow green fluorescent protein (GFP)-tagged receptors in living cells, we demonstrate that activated Trk receptors use a microtubule-dependent process to move from the neurites to the cell bodies. These data support a model of retrograde signaling in which signaling from nerve terminal to nuclei is achieved by rapid vesicular transport of a ligand–receptor complex.
MATERIALS AND METHODS
Materials and antibodies. BDNF and NGF were a gift from Dr. Andrew Welcher (Amgen, Thousand Oaks, CA). High molecular weight marker proteins were purchased from Life Technologies (Gaithersburg, MD). Acrylamide was purchased from National Diagnostics (Atlanta, GA). SuperSignal chemiluminescent substrate (ECL) was purchased from Pierce (Rockford, IL) and enhanced chemifluorescence (ECF) reagents were purchased from Amersham Pharmacia Biotech (Arlington Heights, IL). K252a was purchased from Calbiochem (San Diego, CA). Colchicine and all other reagents were purchased from Sigma (St. Louis, MO).
In previous studies, we characterized an affinity-purified polyclonal antibody [(pY490 or phosphorylated Trk (pTRK)] that recognizes the phosphorylated Tyr 490 within the Shc recognition motif of Trks A, B, and C (Segal et al., 1996). The pCREB antibody was a generous gift from Dr. Michael Greenberg (Harvard Medical School, Boston, MA). The pCREB antibody recognizes CREB when the transcription factor is phosphorylated on serine 133 residue (Ginty et al., 1993, 1994). A polyclonal anti-c-fos antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and from Upstate Biotechnology (Lake Placid, NY). An anti-platelet-derived growth factor receptor antibody (PDGF-R) was purchased from Santa Cruz Biotechnology. Anti-GFP was purchased from Clontech Laboratories (Palo Alto, CA). Compartmented cultures, pin rake, and grease loader were purchased from Tyler Research (Edmonton, Alberta, Canada).
Cell culture. HeLa cells were maintained in DMEM containing 5% fetal calf serum, 5% glutamine, and 1% penicillin and streptomycin at 37°C, 5% CO2. For receptor activation experiments, HeLa cells were incubated for 1 hr in DMEM and then exposed to NGF at a final concentration of 100 ng/ml for 5 min.
Compartmented cultures (Campenot cultures) were set up as described byCampenot (1977, 1992, 1997). Briefly, the compartmented cultures consist of a Teflon divider that is fixed with grease to a collagen-coated 35 mm tissue culture dish. DRG neurons were dissected from embryonic day 14 rat, dissociated, and plated at ∼5.5 × 104 cells per compartmented culture. Dissociated cells were plated into the center compartment and, over the course of 4 d, the neurons extended their neurites through the grease to the side compartments. In this way, the cell bodies are separated from the neurites by a 0.5 mm Teflon divider. Cells were plated in DMEM, 5% horse serum, 1% penicillin–streptomycin, and 1 × 10−5m5-fluoro-5′-deoxyuridine containing 100 ng/ml BDNF and 100 ng/ml NGF. On day 2 (D2), the neurotrophin concentration in the media over the cell bodies was reduced to 10 ng/ml BDNF and 10 ng/ml NGF in the center compartment while maintaining a 100 ng/ml neurotrophin concentration in the adjacent side compartments containing the neurites. On D5 in vitro, the DRG neurons were progressively deprived of neurotrophins by reducing the neurotrophin concentration to 2.5 ng/ml at the neurites (side compartments) and 0 ng/ml at the cell bodies (center compartment). These starved DRG neurons were stimulated and used on D8 or D9.
A critical feature of this system is that the media bathing the neurites do not communicate with the media bathing the cell bodies. Here, we used three methods to ensure that the remote activation of Trk at the cell bodies did not result from leakage of neurotrophin-rich media between compartments. First, we added media to the side compartments for 2 d before plating the cells and used only the compartmented cultures that showed no leakage. Second, we added I125-NGF to the side compartments of cultures in which the neurites of the DRGs are located. We then assayed the media in the center compartment (in which the cell bodies are located) for radioactivity. Over the course of 60 min, no detectable radioactive ligand was recovered in the media bathing the cell bodies (data not shown). Finally, we applied neurotrophin to the neurites in the side compartment and then collected the media overlying the cell bodies from the center compartment. We tested this media for biological activity, by adding it directly to DRG cell bodies. The media was unable to induce increases in pTrk, Fos, or pCREB, indicating that leakage could not account for our results (negative data not shown).
Cell stimulation and drug treatment. Cells were selectively stimulated with either DMEM containing 100 ng/ml neurotrophin (NGF and BDNF) or a vehicle control [0.1 ng/ml bovine serum albumin (BSA)]. Inhibitors (K252a, colchicine) were applied for either 30 or 60 min before stimulation as indicated. To control for effects of inhibitors on downstream signal transducers such as c-fos, inhibitors were applied to both vehicle control- and neurotrophin-stimulated cultures. After stimulation, DRG neurons were rinsed once in ice-cold 1× Tris-buffered saline (TBS), pH 7.4, with 1 mm vanadate before harvesting for biochemical or immunofluorescence analysis.
Immunoblot analysis. DRG neurons were lysed in 20 mm Tris, pH 7.4, 137 mmNaCl, 1% SDS, 10% glycerol, 5% β-mercaptoethanol containing 1 mm phenylmethylsulfonyl fluoride, 1 mm leupeptin, 10 μg/ml aprotinin, and 1 mm sodium orthovanadate for 20 min on ice. The protein lysates from the neurites and cell bodies were removed separately from the compartmented cultures, and a final concentration of 0.1% bromophenol blue was added. The protein lysates were size-fractionated through a 10% SDS-acrylamide gel and transferred to Immobilon-P membrane (Millipore, Bedford, MA). The blots were incubated with primary antibodies, processed with secondary antibodies, and visualized using either the ECL kit (Pierce) as per the manufacturer’s instructions or the ECF kit (Amersham) in conjunction with a Molecular Dynamics (Sunnyvale, CA) Storm system (Storm 840).
Coimmunoprecipitation. Neurites of DRG neurons grown in compartmented cultures were stimulated with BDNF for 20 or 40 min. Neurons were lysed in nondenaturing buffer containing 20 mm Tris, pH 8.0, 137 mmNaCl, 1% Nonidet P-40 (NP-40), 10% glycerol containing 1 mm phenylmethylsulfonylfluoride, 1 mm leupeptin, 10 μg/ml aprotinin, and 1 mm sodium orthovanadate. Lysates were immunoprecipitated using either anti-BDNF or anti-PDGF-R for 2 hr at 4°C. Then, 25 μl of a 50% solution of protein A Sepharose beads in the nondenaturing lysis buffer was added to each sample, and samples were rotated at 4°C for 1 hr and washed once in lysis buffer and twice in 10 mm Tris, pH 7.5, containing 1 mm phenylmethylsulfonylfluoride, 1 mm leupeptin, 10 μg/ml aprotinin, and 1 mm sodium orthovanadate. After the final wash, samples were resuspended in 125 mm Tris HCl, pH 6.8, 4% SDS, 20% glycerol, 5% β-mercaptoethanol, and 0.004% bromophenol blue, boiled 5 min, separated on a 7.5% polyacrylamide gel, transferred to Immobilon-P membrane, and immunoblotted with anti-pTrk.
Immunofluorescence. DRG neurons immunostained with the pTrk antibody were fixed with 4% paraformaldehyde and 1 mm vanadate in TBS. For immunostaining with the pCREB antibody, cells were fixed using 10% paraformaldehyde and 1 mm vanadate in TBS, washed with TBS, and permeabilized using 100% methanol at −20°C for 10 min. After fixation, cells were washed with TBS, blocked, and permeabilized (5% normal goat serum, 0.5% NP-40, and 10 mm glycine in TBS) for 1 hr at room temperature. Cells were rinsed in TBS and incubated with primary antibodies, anti-pCREB (1:1000) and anti-pTrk (anti-pY490; 1:100), overnight at 4°C. Subsequently, cells were washed in TBS and incubated with a Cy3-conjugated goat anti-rabbit antibody (1:1000 in 5% normal goat serum in TBS; Jackson ImmunoResearch, West Grove, PA) for 1 hr at room temperature. Cells were washed and mounted in Immu-Mount (Shandon Inc., Pittsburgh, PA) and 4% 1,4-diazabicyclo-[2.2.2]octane (Sigma).
Adenovirus constructs. To construct a TrkB enhanced GFP fusion protein, a fragment of the mouse TrkB cDNA was excised with StuI and AvrII, which cut in the 5′ UTR and in the last two codons of the coding sequence, respectively, filled in with Klenow, and cloned into the SmaI site in the polylinker of pEGFP-N1 (Clontech). The resulting construct, pmTrkB-GFP, encodes full-length TrkB fused in frame to EGFP via a linker derived from the polylinker of pEGFP-N1 with the amino acid sequence DPPVAT.
To construct adenovirus expressing TrkB-GFP, the TrkB-GFP cDNA was excised from an EcoRI site in the 5′ polylinker to aDraI site in the 3′ UTR and cloned into the EcoRI and SwaI polylinker sites downstream of the human cytomegalovirus promoter in the high recombination efficiency adenovirus shuttle vector pMZL5 (M. Z. Lin, unpublished data). This construct, pmTrkB-EGFP/5, was cotransfected with pBHG10 (Microbix Biosystems, Toronto, Ontario, Canada) into a 24-well plate of HEK293 cells grown in DMEM with 5% calf serum, and virus from an isolated plaque was amplified once on a 10 cm plate of HEK293 cells (Bett et al., 1994). Details of the construction and complete sequences of the plasmids are available on request.
HeLa cells and DRG neurons grown in culture were infected with 50 transducing units per cell for 2 hr. Then, media containing 10% fetal calf serum in DMEM was added to HeLA cells, and media containing 5% horse serum, 1% penicillin–streptomycin, 50 ng/ml BDNF, and 50 ng/ml NGF in DMEM was added to DRG neurons for 48 hr. Two days later, HeLa cells were deprived of serum for 1 hr and then stimulated with 100 ng/ml BDNF or vehicle control for 5 min. Protein extracts were prepared as described above, immunoprecipitated with anti-GFP, separated on SDS-PAGE, and transferred to Immobilon. The blots of the immunoprecipitated proteins were visualized with anti-pTrk (anti-pY490). For visualizing TrkB-GFP expression in DRG neurons, 2 d after infection, DRG neurons were fixed in 4% paraformaldehyde for 20 min, and the fluorescence of the TrkB-GFP was visualized.
The cell bodies of DRG neurons grown in compartmented cultures were infected at 100 transducing units per cell for 2 hr, followed by addition of 5% horse serum and 1% penicillin–streptomycin in DMEM for 48 hr. After 48 hr, ∼5% of the DRG neurons were fluorescent. DRG neurons were washed once in ice-cold 1× TBS with 1 mmvanadate, fixed in 4% paraformaldehyde, and immunostained with pTrk as described above.
TrkB-GFP fluorescence measurements and receptor photobleaching. Cell bodies of DRG neurons were bleached by exposure to high-intensity fluorescent light (infrared laser) for 1–6 min until the fluorescence of the cell bodies had decreased to 10–40 intensity units, ∼90% decrease in fluorescence. To ensure that photobleaching did not cause significant toxicity, after photobleaching, the cells were tested for the ability to exclude trypan blue dye. The photobleach–recovery techniques used here did not cause any detectable cell toxicity, and similar results were obtained with and without photobleaching.
The fluorescence emitted from cell bodies for both bleached and unbleached DRG neurons was sampled every 2 min for 16 min. A baseline light intensity of cell bodies was measured for 6 min, after which neurites were stimulated with either 100 ng/ml neurotrophin (BDNF and NGF) or vehicle control (0.1 ng/ml BSA in DMEM). Cell body fluorescence was measured over the course of 10 min after stimulation. Neurotrophin-induced change in fluorescence was calculated as follows. First, background fluorescence was subtracted from the measured fluorescence at each time point. Second, percent change in fluorescence was calculated as the difference between the corrected fluorescence at the indicated time after stimulation and the corrected baseline fluorescence divided by the corrected baseline fluorescence. Finally, the neurotrophin-induced change in cell body fluorescence equals the change in cell body fluorescence in neurotrophin-stimulated cultures minus the change in cell body fluorescence in control-stimulated cultures. Comparison between the fluorescence emitted by DRG neurons from neurotrophin-stimulated and from that of the unstimulated (vehicle control) cultures was done using a two-sample t test assuming unequal variance.
To detect the recovery of TrkB-GFP within the cell over time, images taken as described above were compiled, and the baseline image was subtracted from all subsequent images (Inoue, 1986). The baseline is defined as the first image after photobleaching. The images were colorized using a blue, red, and green scale that varied according to pixel intensity. All manipulations were performed using the two-dimensional analysis module Oz confocal laser scanning imaging system (Noran Instruments, Middleton, WI).
RESULTS
Application of neurotrophins to neurites of DRG neurons rapidly induces remote Trk activation and CREB phosphorylation
In a previous study, we showed that activated TrkB functions as a retrograde signal carrier in sciatic nerve axons, and the retrograde signal is propagated more rapidly than expected based on known rates of neurotrophin transport (Bhattacharyya et al., 1997). To develop an analogous in vitro system, we use embryonic DRG neurons grown in compartmented cultures (Kimpinski et al., 1997). Because DRG neurons respond to multiple neurotrophins, we can assess retrograde signaling by both BDNF and NGF in this system. The DRG neurites grown in the compartmented cultures were primarily axonal in that they express acetylated tubulin, an axonal marker (data not shown) (Chitnis and Kuwada, 1990). Previous studies have used compartmented cultures of sympathetic neurons to study retrograde signaling in vitro(Riccio et al., 1997; Senger and Campenot, 1997; Toma et al., 1997).
To determine whether the response of DRG neurons to neurotrophin stimulation was similar to that described for sympathetic neurons to NGF stimulation, we first examined Trk phosphorylation in embryonic DRG neurons grown in compartmented cultures. We applied neurotrophin or a vehicle control either directly to the DRG cell bodies or to the neurites and visualized the activated Trk by immunofluorescence with anti-pTrk (pY490). This antibody selectively recognizes Trk receptors phosphorylated at the critical Shc binding site, a major and persistent site of autophosphorylation (Segal et al., 1996). Phosphorylation at this site initiates the Ras–mitogen-activated protein kinase (MAPK) signaling pathway and the subsequent phosphorylation of transcription factors, such as CREB (Obermeier et al., 1993; Stephens et al., 1994). After selective neurotrophin stimulation at the neurites, Trk is activated at both the neurites and the cell bodies within 20 min (Fig.1A). Similar results were obtained by immunoblotting (Fig. 1B). Quantitation of immunoblot data indicates that remote activation of Trk within the cell bodies occurs as early as 5 min after neurite stimulation and is maximal at 20 min (Fig.1B,C).
Fig. 1.
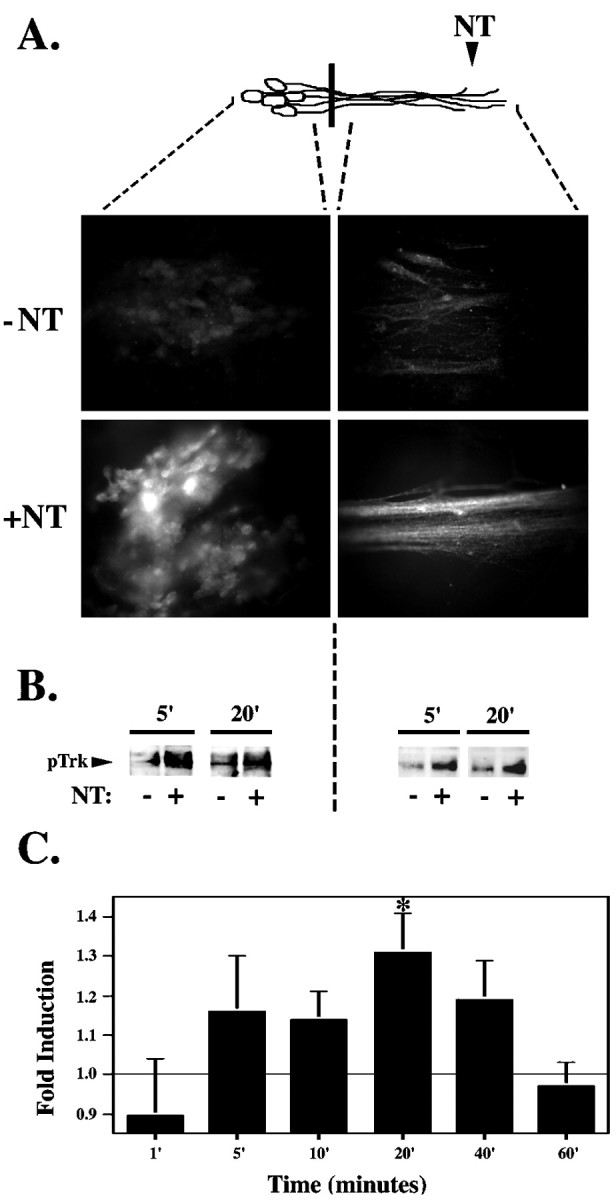
Trk is rapidly activated in response to neurotrophin stimulation. A, Selective neurotrophin stimulation of DRG neurites results in pTrk immunostaining in neurites and cell bodies of DRG neurons. After a 20 min neurotrophin (+NT) or vehicle control (−NT) (0.1 ng/ml BSA) stimulation at the neurites, DRG neurons were fixed in 4% paraformaldehyde and immunostained using the anti-pTrk antibody in conjunction with a Cy3-conjugated secondary antibody. B, Trk phosphorylation is detected in the cell body as early as 5 min after selective neurotrophin (+NT) or vehicle control (−NT) (0.1 ng/ml BSA) stimulation at the neurites. DRG neuron lysates from the neurites and from the cell bodies were pooled from nine compartmented cultures, separated on 10% SDS-PAGE gel, transferred to Immobilon-P membrane, and immunoblotted with anti-pTrk. C, Time course of Trk phosphorylation at the cell body in response to neurite stimulation. Intensity of the pTrk band was quantitated with the Molecular Dynamics Storm 860 imaging system using Image Quant (version 2.0; Molecular Dynamics) and normalized for protein loading. The time course of pTrk induction (calculated as the ratio between pTrk in neurotrophin- and control-stimulated cultures) was calculated from several experiments (n = 3–10 for each time point; *p < 0.05). Diagram (top) is a schematic representation of neurons; black bar indicates presence of the Teflon divider. Area of DRG neurons analyzed is indicated by dashed lines.
Phosphorylation of the transcription factor CREB at Ser 133 has been shown to be a critical nuclear response to neurotrophins (Ginty et al., 1994; Bonni et al., 1995; Finkbeiner et al., 1997). Increased CREB phosphorylation can be detected by immunoblot analysis 5 min after neurotrophin stimulation of the DRG cell bodies (data not shown). CREB also becomes phosphorylated at the cell bodies as early as 5 min after selective neurotrophin stimulation of the neurites and is maximal at 20 min after stimulation (Fig. 2B,C). Remote activation of nuclear CREB in response to neurotrophin stimulation of the neurites was also demonstrated by immunostaining using the phosphorylated Ser 133 CREB (pCREB) antibody (Fig. 2A). Of note, pCREB is only detectable in nuclei and was never seen in the neurites by either immunostaining or immunoblot analysis.
Fig. 2.
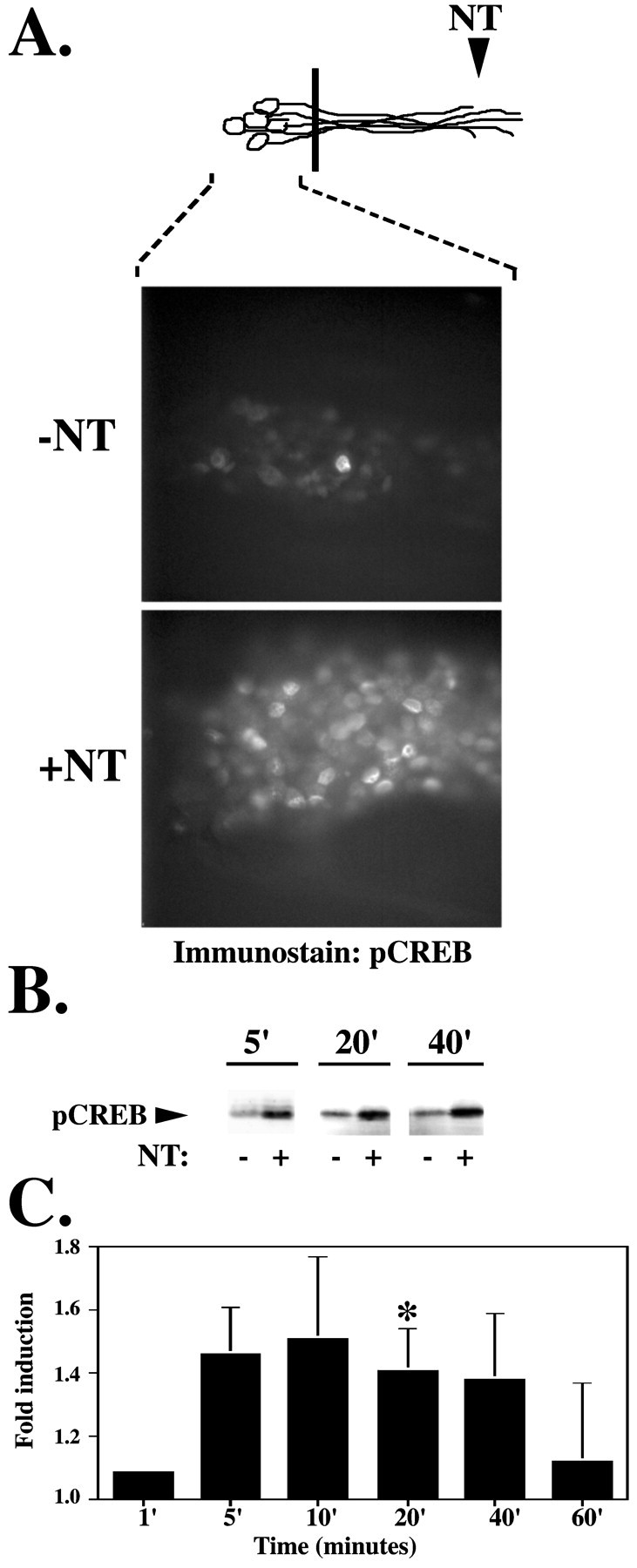
Neurite stimulation evokes a rapid nuclear response. A, Selective neurotrophin stimulation of DRG neurites results in pCREB immunostaining of DRG neuron cell bodies. After a 20 min neurotrophin (+NT) or vehicle control (−NT) (0.1 ng/ml BSA) stimulation at the neurites, DRG neuron cell bodies were fixed in 10% paraformaldehyde and immunostained using the anti-pCREB antibody in conjunction with a Cy3-conjugated secondary antibody. B, CREB phosphorylation is detected in the cell body within 5 min after selective neurotrophin (+NT) or vehicle control (−NT) (0.1 ng/ml BSA) stimulation at the neurites. Cell body lysates from nine compartmented cultures were pooled, separated on an SDS-PAGE gel, transferred to Immobilon membrane, and immunoblotted with anti-pCREB. C, Time course of CREB phosphorylation at the cell body in response to neurotrophin stimulation at the neurite. The intensity of the pCREB band was quantitated with the Molecular Dynamics Storm 860 imaging system using Image Quant (version 2.0; Molecular Dynamics), and normalized for protein loading. The time course of pCREB induction in response to neurotrophin stimulation at the neurites (calculated as the ratio between pCREB in neurotrophin and control-stimulated cultures) was calculated from several experiments (n = 4–10 for each time point; *p < 0.05). Diagram (top) is a schematic representation of neurons; black bar indicates presence of the Teflon divider. Area of DRG neurons analyzed is indicated by dashed lines.
If activated Trk is the signal carrier leading to this nuclear response, then an increase in pTrk at the cell bodies should precede or coincide with CREB phosphorylation. As shown, both Trk and CREB phosphorylation at the cell bodies were first detectable at 5 min after neurite stimulation and were maximal at 20 min after stimulation. This is somewhat earlier than the time course reported for pCREB immunoreactivity in sympathetic neurons after selective stimulation of the neurites (Riccio et al., 1997). This difference in kinetics is likely to reflect the sensitivity of pCREB immunoblotting compared with the sensitivity of immunostaining. By immunoblotting, we detect CREB phosphorylation in the cell bodies in response to neurotrophin stimulation of the neurites as early as 5 min, but by immunostaining we do not detect significant CREB phosphorylation until 20 min after neurite stimulation. Together, these studies indicate that BDNF and NGF applied to the neurites can induce rapid and coincident phosphorylation of both Trk and CREB in the distant cell bodies.
Induction of immediate early genes
As shown, neurotrophins, acting at nerve terminals, can initiate CREB phosphorylation at Ser 133 (Fig. 2). Phosphorylation at this site has previously been shown to activate the transcription factor CREB, leading to de novo transcription of immediate early genes in response to neurotrophins (Greenberg et al., 1985; Ginty et al., 1994;Bonni et al., 1995; Finkbeiner et al., 1997). Although phosphorylation of CREB has been shown to be necessary for induction of immediate early genes, it is not always sufficient to initiate new transcription (Bonni et al., 1995). To determine whether neurite stimulation and the subsequent CREB phosphorylation can initiate transcription, we chose to examine induction of the immediate early gene c-fos. Thec-fos immediate early gene family encode transcription factors that are rapidly and transiently induced in response to a wide array of extracellular stimuli, including the neurotrophins (Greenberg et al., 1985). To determine whether the retrograde signal can induce immediate early genes, neurotrophins were applied to DRG neurons grown in compartmented cultures. Protein lysates from the cell bodies of the stimulated or control cultures were analyzed for the presence of c-fos by immunoblot. As shown, neurotrophins applied selectively to the neurites or applied globally to both the neurites and cell bodies each induce c-fos protein (Fig. 3). Expression of c-fos protein in the cell bodies is detected 5 and 7 hr after neurotrophin stimulation at the neurites but is not detected at 2.5 hr (Fig. 3A). In contrast, neurotrophins applied globally to both the cell bodies and the neurites induces Fos proteins at 2.5 hr (Fig. 3B). Thus, neurotrophins, whether applied to neurites alone or to the whole cells, upregulate the c-fos immediate early gene. However, the timing of the response varies greatly depending on the location of stimulation.
Fig. 3.

Neurotrophins applied to the neurites induce immediate early genes. Neurotrophins (+NT) or a vehicle control (−NT) (0.1 ng/ml BSA) were applied to the distal neurites of DRG neurons grown in compartmented tissue cultures for 2.5, 5, and 7 hr (A) or were applied globally to both the DRG cell bodies and the neurites for 2.5 hr (B). Cell body protein lysates from five compartmented cultures were separated on a 10% polyacrylamide gel, transferred to membrane, and blotted with anti-c-fos. Similar results were obtained by applying BDNF or NGF alone to the distal neurites (data not shown). Diagram (top) is a schematic representation of neurons; black bar indicates presence of the Teflon divider. Area of DRG neurons analyzed is indicated bydashed lines.
Nuclear responses to neurite stimulation require that Trk receptor activation be propagated from neurites to cell bodies
The data demonstrate that, in response to neurotrophin stimulation of the neurites, Trk is rapidly activated at both the neurites and the cell bodies, and pCREB and c-fos are induced in the nuclei. To determine whether activation of Trk is required in both locations to elicit these nuclear responses, we selectively pretreated either the neurites or the cell bodies with a Trk kinase inhibitor, K252a. At 20 min after neurotrophin stimulation of neurites, we analyzed cell body lysates for pCREB by immunoblot analysis. Treatment of either the neurites or cell bodies with K252a abolishes the remote induction of CREB phosphorylation (Fig.4A). To verify that K252a only acts at the site of application, we monitored pTrk in these cultures. Trk phosphorylation in the neurites was not inhibited by K252a applied to the cell bodies (Fig. 4B). To determine whether activation of Trk is also required in both locations to elicit Fos induction, we again selectively pretreated either the neurites or the cell bodies with K252a. At 7 hr after neurotrophin stimulation of neurites, we analyzed cell body lysates for c-fos by immunoblot analysis. Treatment of either the neurites or cell bodies with K252a abolishes the remote induction of Fos protein in response to neurotrophin (Fig. 4C). Because there is variability in background c-fos expression, we show results of both K252a in control-stimulated cultures and K252a in neurotrophin-stimulated cultures. We again verified that Trk phosphorylation in the neurites was not inhibited by K252a applied to the cell bodies (data not shown). These data indicate that Trk activity is required at both the neurites and the cell bodies to elicit Fos induction, as well as pCREB activation.
Fig. 4.

Trk activity is required at both the neurites and the cell bodies to elicit a nuclear response.A, K252a applied to either the neurites or the cell bodies abolishes neurotrophin-induced CREB phosphorylation at the cell body. Neurotrophins (+NT) or a 0.1 ng/ml BSA control (−NT) was applied to the neurites for 20 min after selective pretreatment of the neurites (1) or the cell bodies (2) with K252a for 30 min. Cell body protein lysates from eight compartmented cultures were pooled and separated on SDS-PAGE and immunoblotted using a pCREB antibody. B, K252a applied to neurites blocks Trk phosphorylation in the neurites, whereas K252a applied to cell bodies does not interfere with Trk phosphorylation at the neurites. Lysates from neurites of eight compartmented cultures were pooled and separated on 10% SDS-PAGE and immunoblotted using a pTrk antibody. C, K252a applied to neurites, cell bodies, or both before distal neurotrophin stimulation at the neurites abolishes c-fos induction. Neurotrophins (+NT) or a 0.1 ng/ml BSA control (−NT) were applied to the neurites for 7 hr after selective pretreatment of the neurites (1), the cell bodies (2), or both (global; 1 & 2) with K252a for 30 min. Cell body protein lysates from eight compartmented cultures were pooled, separated on SDS-PAGE, and immunoblotted using a c-fos antibody. D, K252a applied globally before global neurotrophin stimulation at the cell body and neurite inhibits neurotrophin-induced c-fos induction. Neurotrophins (+NT) or a 0.1 ng/ml BSA control (−NT) were applied globally to the cell bodies and neurites for 7 hr after pretreatment of the both the cell bodies and the neurites (global; 1 & 2) with K252a for 30 min. Cell body protein lysates from eight compartmented cultures were pooled, separated on SDS-PAGE, and immunoblotted using a c-fos antibody. Diagram (top) is a schematic representation of neurons; black bar indicates presence of the Teflon divider. Area of DRG neurons analyzed is indicated by dashed lines.
TrkB is retrogradely transported from the neurites to the cell body
How then is the activated Trk signal conveyed to the cell body? Previous studies have suggested that the signal might be conveyed by retrograde vesicular transport (Ehlers et al., 1995; Grimes et al., 1996, 1997). However, the remote responses to neurotrophin stimulation of neurites occur more rapidly than predicted by this model. Alternatively, rapid retrograde signaling could reflect the sequential activation of Trk receptors distributed along the axons. To determine whether Trk receptors are physically transported from neurites to the cell bodies to elicit a nuclear response, we developed a method for following Trk receptors in live cells in real time. To do this, we constructed an adenovirus that encodes the full-length TrkB receptor fused at the C terminus to enhanced GFP. The adenovirus was first tested in HeLa cells, which express no endogenous neurotrophin receptors. HeLa cells, infected with the adenovirus, were stimulated with BDNF or vehicle control. Protein lysates were immunoprecipitated with an antibody to GFP and blotted with anti-pTrk. As shown, the fusion protein displays ligand-dependent phosphorylation (Fig.5A). This indicates that the tagged receptor is inserted into the plasma membrane, can bind ligand, and becomes autophosphorylated. The adenovirus was then used to infect cultures of DRG neurons. Two days after infection, TrkB-GFP expression was visible throughout the DRG neurons, in both neurites and cell bodies (Fig. 5B). Thus, the tagged receptor localizes appropriately in neurons, as previously demonstrated for a GFP-tagged TrkA (Nakata et al., 1998).
Fig. 5.

pTrk localized to the neurites is transported to the cell body. A, Uninfected HeLa cells (lanes 1, 2) or HeLa cells expressing TrkB-GFP (lanes 3, 4) were treated with PBS control (−) or 100 ng/ml BDNF (+) for 5 min. Cell lysates were immunoprecipitated with anti-GFP, followed by immunoblotting with anti-pTrk. B, Three days after infection of DRG neurons, expression of TrkB-GFP is visible in the cell bodies and the neurites as fluorescence. C–E, DRG cell bodies of neurons expressing TrkB-GFP grown in compartmented cultures were bleached until the fluorescence was reduced by 90% (C) or left unbleached (D, E). Fluorescence emitted by the cell bodies was measured after addition of 100 ng/ml BDNF and 100 ng/ml NGF or vehicle control to the neurites. Cell body fluorescence at 10 min after neurite stimulation was calculated as a percent of cell body fluorescence at time 0. Neurotrophin-induced change in cell body fluorescence equals mean change in cell body fluorescence in neurotrophin-stimulated cultures minus mean change in cell body fluorescence in control-stimulated cultures. The experiment was repeated using DRG neurons expressing GFP alone (C,D). The kinase inhibitor K252a was applied to neurites 30 min before neurotrophin stimulation of DRG neurons expressing TrkB-GFP (D). To test whether the TrkB-GFP is transported from the neurites to the cell bodies, we repeated the experiment in the presence of colchicine or a vehicle control (DMSO). Colchicine (125 μm) or DMSO was applied to neurites 60 min before neurotrophin stimulation (E). The percent change in fluorescence over the course of 10 min was compared between the neurotrophin- and control-stimulated cells using a two tailed t test assuming unequal variance (n >15 for each point; *p < 0.05).
DRG neurons were grown in compartmented cultures and infected with the TrkB-GFP adenovirus vector. Two days later, we photobleached the cell bodies of individual DRG neurons expressing TrkB-GFP. We then monitored the recovery of cell body fluorescence in response to 10 min of neurotrophin or control stimulation of the neurites (Fig.5C). A significant increase in fluorescence was detected in the cell bodies of neurotrophin-stimulated DRG neurons compared with cultures stimulated with a vehicle control. To ensure that photobleaching did not cause significant toxicity, after photobleaching, the cells were tested for the ability to exclude trypan blue dye. The photobleach–recovery techniques used here did not cause any detectable cell toxicity. Furthermore, similar results were obtained when TrkB-GFP was followed without previous photobleaching (Fig. 5D,E). These results indicate that a rapid accumulation of TrkB in cell bodies is initiated by neurotrophin stimulation of the neurites.
To verify that these results reflect specific rapid transport of Trk receptors, we used an adenovirus encoding enhanced GFP only. When DRG neurons were infected with the adenovirus expressing GFP, no difference in fluorescence was detected between the neurotrophin- and the vehicle control-stimulated cells (Fig. 5C,D). These results demonstrate that TrkB is rapidly and specifically transported to the cell bodies in response to neurotrophin stimulation of the neurites.
The TrkB-GFP provides a unique system to analyze the steps required for rapid retrograde transport of receptor. We therefore performed a series of experiments to determine whether ligand stimulation, receptor kinase activity, and microtubule-dependent transport were required. As shown, the rapid increase in fluorescence was only seen in response to neurotrophin stimulation of the neurites. Control stimulation did not induce an increase in fluorescence after photobleaching (Fig.5C) or in unbleached cells (Fig. 5D), indicating that neurotrophins, acting at the neurites, induce rapid and specific movement of Trk receptors to the cell bodies.
Ligand binding to receptor stimulates Trk kinase catalytic activity (Barbacid, 1995; Bothwell, 1995). To determine whether the increase in TrkB-GFP requires receptor kinase activity, we first pretreated the neurites with K252a, an inhibitor of the Trk kinase. As shown, K252a prevented the neurotrophin-induced increase in fluorescence (Fig.5D), indicating that receptor kinase activity is required for retrograde transport.
The data presented indicate that the rapid retrograde propagation of activated Trks reflects ligand-induced translocation of receptors from neurites to the cell bodies. Does this translocation require microtubule-dependent transport of endocytosed receptor? To address this question, we used colchicine, a pharmacological agent that causes depolymerization of microtubule formation. This drug has been used extensively to disrupt retrograde vesicular transport (Paulson and McClure, 1975). To determine whether the increase in TrkB-GFP within the cell bodies is caused by microtubule-dependent transport of TrkB-GFP from the neurites, we pretreated neurites with colchicine and then applied neurotrophin or vehicle control to the neurites. Pretreatment with colchicine prevented the neurotrophin-induced increase in fluorescence (Fig. 5E). Together, these data indicate that the increase in fluorescence at the cell body reflects microtubule-dependent movement of activated TrkB-GFP initiated by neurotrophin stimulation at the neurites.
It was surprising to see such rapid movement of the TrkB-GFP receptors. To monitor the speed of the signal, we performed a time course experiment with photobleach recovery (Fig.6A). A comparison of the time course seen in stimulated cells (n = 29) and unstimulated controls (n = 23) indicates that the two lines diverge at 4 min. Within 8 min of stimulation, there is a significant increase in cell body fluorescence in neurotrophin-stimulated cells that is not seen in unstimulated cells. Thus, the movement of Trk receptors occurs rapidly enough to account for retrograde propagation of Trk phosphorylation initiated by neurotrophin stimulation of neurites (Fig. 1).
Fig. 6.

TrkB-GFP accumulates in the cell body after neurite stimulation. DRG cell bodies were photobleached, and their fluorescent recovery was monitored. A, A time course experiment with photobleach recovery. Neurotrophin stimulation of the neurites induced a significant increase in fluorescence within 8 min. The percent change in fluorescence was compared between neurotrophin (black)- and control (red)-stimulated cells at each time point using a two-tailed t test assuming unequal variance (n > 23 for each time point; *p < 0.05). B, Colorized image of a single DRG neuron after photobleaching. The first image in the series (top left, time 0) represents baseline after cell body bleaching. Neurites are stimulated with neurotrophin (100 ng/ml) after acquisition of baseline image. Using confocal microscopy, images are captured sequentially every 2 min. TrkB-GFP fluorescence increases within the interior of the cell beginning at the axon hillock. Background has been subtracted from images, and the change in fluorescence over time was colorized according to fluorescence intensity. The color scale (left) indicates the relative fluorescence intensity. Scale bar, 2 μm.
An advantage of using GFP to follow the Trk receptors in live cells is that we can monitor the intracellular location of Trk receptors during photobleach recovery. By following fluorescent receptors within an individual neuron over time, we can see that the fluorescence increases within the interior of the cell (Fig. 6B). The increase is first seen near the axon hillock and then accumulates unevenly within the cell body, excluding the nucleus. These experiments using a tagged receptor suggest that neurotrophin stimulation of the neurites induces a very rapid movement of Trk receptors from the axons to the cell body by intracellular vesicular transport.
Active receptor, pTrk, in the cell bodies is associated with ligand after neurite stimulation
If the activated Trk receptor reaches the cell bodies by retrograde vesicular transport, then we would expect activated Trk to remain in a ligand–receptor complex. However, Ure and Campenot (1997), using sympathetic ganglia grown in compartmented cultures, showed that I125-NGF applied to the neurites of sympathetic neurons only accumulates in the cell bodies after 60 min. We repeated these experiment in DRG neurons grown in the compartmentalized cultures and found no detectable accumulation of I125-NGF in the cell bodies before 60 min (data not shown). We therefore chose another approach to examine transport of a ligand–receptor complex. To determine whether activated receptors at the cell bodies were traveling alone or in a ligand–receptor complex, we used an antibody to BDNF to immunoprecipitate lysates from DRG cell bodies 20 (Fig.7) and 40 (data not shown) min after stimulation at the neurites with BDNF alone. We then analyzed immunoprecipitates on SDS-PAGE and immunoblotted with an antibody to pTrk. As shown, the antibody to BDNF coprecipitates pTrk. The pTrk band was not precipitated from unstimulated cells or from stimulated cells immunoprecipitated with an unrelated antibody (Fig. 7A). Similarly, activated receptors in rat sciatic nerve axon traveling in a retrograde direction can be coprecipitated with their ligands (A. Bhattacharyya, unpublished observations). Although we cannot exclude the possibility that the receptor–ligand complex forms after cell lysis, it is more likely that this complex forms in vivogiven the relatively low concentration of both ligand and receptors in the lysates. These data suggest that activated Trks reach the cell bodies as part of ligand–receptor complexes and further support a model in which rapid retrograde vesicular transport conveys a signal from the targets to distant cell bodies.
Fig. 7.

pTrk in the cell bodies is associated with ligand after neurite stimulation. Neurites of DRG neurons were stimulated with 100 ng/ml BDNF (+) or a 0.1 ng/ml BSA control (−) for 20 min. After neurite stimulation, cell body lysates were collected from compartmented cultures (A), and whole-cell lysates were collected from mass cultures (B) in nondenaturing lysis buffer as described in Materials and Methods. These lysates were immunoprecipitated using either anti-BDNF (B) or anti-PDGFR (P), separated on a 7.5% SDS-PAGE, and immunoblotted with anti-pTrk. Diagram (top) is a schematic representation of neurons;black bar indicates presence of the Teflon divider. Area of DRG neurons analyzed is indicated by dashed lines.
Activation of Trk is unidirectional
Axonal transport systems are directional and require an intact microtubule scaffold (Wang et al., 1995; Hirokawa, 1998). If the propagation of Trk phosphorylation reflects axonal transport, then Trk activation should propagate only in one direction. To test this prediction, we selectively stimulated DRG neurons at the cell bodies. After neurotrophin stimulation of the cell bodies, Trk is locally activated in the cell bodies (Fig. 8). However, Trk activation does not propagate to the neurites in response to neurotrophin stimulation of the DRG cell bodies (Fig. 8). Even in the presence of neurotrophin stimulation at the cell bodies for up to 7 hr, we were unable to detect pTrk at the neurites in response to stimulation of cell bodies (data not shown). Thus, activation of Trk propagates only in the retrograde direction and does not propagate in the anterograde direction. In contrast, activation of Trk in the neurites spreads rapidly in the retrograde direction and reaches the cell bodies (Fig. 1). The unidirectional propagation of pTrk is consistent with motor-driven transport.
Fig. 8.

Activation of Trk is unidirectional. To determine the direction of Trk phosphorylation, neurotrophins were applied to the cell bodies for 20 min. A, After neurotrophin stimulation of cell bodies, Trk phosphorylation is detected only at the cell body. DRG neurons were fixed in 4% paraformaldehyde, immunostained using anti-pTrk, and visualized using a Cy3-conjugated secondary antibody. B, After neurotrophin stimulation of cell bodies, protein lysates from the cell bodies and the neurites from eight compartmented cultures were pooled, separated on SDS-PAGE, and immunoblotted using anti-pTrk. Compare with Figure 1. Diagram (top) is a schematic representation of neurons;black bar indicates presence of the Teflon divider. Area of DRG neurons analyzed is indicated by dashed lines.
Colchicine blocks retrograde neurotrophin-induced Trk signaling
We have shown that the rapid transport of Trk receptors from the neurites to the cell bodies is blocked by the microtubule depolymerizing agent colchicine (Fig. 5E). To determine whether the activated Trk required for pCREB induction travels to the cell bodies by microtubule-dependent transport, DRG neurons were treated with 125 μm colchicine for 60 min before neurotrophin stimulation. After colchicine treatment, neurotrophin was added to neurites for 20 min and then lysates of DRG cell bodies or neurites were immunoblotted with pTrk. Colchicine treatment did not interfere with local Trk phosphorylation in response to neurotrophin stimulation. However, this treatment did inhibit remote Trk phosphorylation at the cell bodies in response to neurite stimulation (Fig. 9A). Control experiments showed that colchicine did not interfere with cell body Trk phosphorylation in response to stimulation of the cell bodies (Fig.9B). The fact that the propagation of activated Trk is disrupted by colchicine is consistent with a model of signal propagation by retrograde transport of activated Trk–ligand complexes.
Fig. 9.

Colchicine blocks the neurotrophin-induced nuclear response. The cell bodies and neurites of DRG neurons were pretreated with 125 μm colchicine or DMSO control for 60 min. Neurons were selectively stimulated at the neurites (A) or at the cell bodies (B) with neurotrophin (+NT) or 0.1 ng/ml BSA control (−NT) after global colchicine treatment. DRG cell body lysates were separated on 10%SDS-PAGE and immunoblotted using anti-pTrk and anti-pCREB. Diagram (top) is a schematic representation of neurons; black bar indicates presence of the Teflon divider. Area of DRG neurons analyzed is indicated by dashed lines.
To determine whether the transport of activated Trk receptors is required for nuclear responses, we analyzed CREB phosphorylation. Colchicine blocked CREB phosphorylation in response to neurite stimulation, consistent with a model of retrograde transport (Fig.9A). Surprisingly however, whereas colchicine blocked Trk phosphorylation of cell bodies only in response to neurite stimulation, colchicine blocked CREB phosphorylation in response to stimulation of the cell bodies, as well as the neurites (Fig. 9B). These studies suggest that there are two microtubule-dependent processes in retrograde signaling. The first microtubule-dependent step is retrograde transport of activated Trk receptors through the axons. The second microtubule-dependent step is an intermediate event between receptor activation and CREB phosphorylation that is required regardless of the site of stimulation. The colchicine effect on CREB phosphorylation complicates the interpretation of the effect of colchicine in this system. However, when taken together, the data presented here demonstrating that colchicine and K252a inhibit pTrk propagation, Trk-GFP movement, and nuclear responses, suggest that retrograde microtubule-dependent transport of activated Trk-ligand complexes is required to initiate nuclear responses to target-derived neurotrophins.
DISCUSSION
We used compartmented cultures of DRG neurons to study the nature and mechanism of retrograde signaling in neurons with long axons. Neurotrophins selectively applied at the neurites rapidly induce phosphorylation of CREB, and this is followed by c-fos induction. These responses require retrograde propagation of Trk receptor activation. Using GFP-tagged TrkB receptors and activation-state-specific antibodies to Trk receptors, we show that this retrograde propagation requires microtubule-dependent transport of receptors from the neurites to the cell bodies. These data demonstrate that a neurotrophin-induced remote signal is propagated to the nucleus by rapid retrograde vesicular transport of an activated receptor–ligand complex.
Activated Trk is the retrograde signal carrier leading to pCREB induction
Selective neurite stimulation with the neurotrophins NGF and BDNF, applied together or individually, results in the phosphorylation of Trk in both DRG neuron cell bodies and neurites. Trk is phosphorylated on a Tyr 490, a docking site for Shc, and initiates subsequent signaling cascades, including the Ras–MAP kinase pathway (Obermeier et al., 1993; Stephens et al., 1994; Segal et al., 1995; Segal and Greenberg, 1996).
Both NGF and BDNF induce phosphorylation of CREB at Ser 133 and thereby promote new gene expression (Ginty et al., 1994; Bonni et al., 1995;Finkbeiner et al., 1997). As shown here in DRG neurons, neurotrophins induce nuclear CREB phosphorylation and c-fos expression when neurotrophins are selectively presented at neurite endings, as well as when neurotrophins are applied to cell bodies. We have found that the time course with which neurotrophins applied to neurites initiate Trk phosphorylation in cell bodies (Fig. 1), TrkB-GFP movement from neurites to cell bodies (Fig. 6A), and CREB phosphorylation (Fig. 2) closely coincide. Furthermore, blocking Trk receptor activity using K252a at either neurites or cell bodies abolishes CREB phosphorylation and Fos induction. These data indicate that Trk phosphorylation is required at both the neurites and the cell bodies for remote signaling to occur. Similarly, Riccio et al. (1997)showed that NGF presented to neurites of sympathetic neurons elicits Trk activation at the cell bodies, and this activation is required for CREB phosphorylation. Thus, although the timing and functions of the Ras–MAP kinase pathway differ when comparing the neurotrophin-induced signaling used by sympathetic and sensory neurons (Borasio et al., 1993; Klinz and Heumann, 1995), the role of Trk as a signal carrier appears to be a conserved mechanism in retrograde signaling.
The rapidity of remote responses shown here is consistent with our previous study investigating the neurotrophin-induced activation of Trk along sciatic nerve axons in vivo (Bhattacharyya et al., 1997) and with studies on sympathetic neurons grown in compartmented cultures (Senger and Campenot, 1997). Although the rate of retrograde signaling in DRG neurons grown in compartmented cultures is extremely rapid (3–16 μm/sec), it overlaps the range for vesicular retrograde transport kinetics (0.5–7 μm/sec) (Breuer et al., 1992; Wang et al., 1995).
Retrograde transport of a ligand–receptor complex
Does the neurotrophin-induced signal generated at neurite endings travel to the cell body by vesicular retrograde transport? In studies on sympathetic neurons, Riccio et al. (1997) demonstrated that NGF linked to beads could induce CREB phosphorylation if the beads were applied to the cell bodies but not if applied to neurites. NGF linked to beads binds and activates receptors but cannot be endocytosed. Thus, ligand internalization is required for retrograde signaling. However, these data do not directly address whether the signal is conveyed to the cell bodies by retrograde vesicular transport.
Several lines of evidence presented here indicate the signal reaches the cell bodies by retrograde transport of activated Trk receptors. First, increases in pTrk only travel in a retrograde direction. Using the pTrk antibody for both immunostaining and immunoblotting, we show activated Trk does not move in the anterograde direction, even in response to neurotrophin stimulation of the cell bodies. The directional nature of signal propagation is consistent with an energy-dependent transport system, such as dynein-based motility (Wang et al., 1995; Hirokawa, 1998).
Experiments with the microtubule depolymerizing agent colchicine provide a second line of evidence that the signal is propagated by retrograde transport. Colchicine inhibits movement of GFP-tagged Trk receptors and also inhibits the translocation of activated Trk receptors from neurites to cell bodies. Colchicine also blocked the nuclear response to neurotrophin stimulation of neurites, consistent with the model that retrograde transport of activated Trk receptors mediates the nuclear response. We noted that CREB phosphorylation in response to cell body stimulation was also blocked by colchicine. These data suggest that there is another microtubule-sensitive step downstream of the receptor. For example, there could be microtubule-associated kinases located downstream of Trk or nuclear translocation of MAPKs and ribosomal S6 kinase (Rsks) could require microtubules (Gundersen and Cook, 1999).
A third line of evidence supporting retrograde transport is the demonstration that pTrk arrives at the cell bodies in a complex with ligand. By immunoprecipitating cell body lysates with anti-BDNF followed by immunoblotting with anti-pTrk, we detected a ligand–receptor complex in the cell bodies. This is consistent with a model in which neurotrophin binding induces Trk internalization, and vesicles containing both ligand and activated receptor are then transported to the cell bodies. A surprising finding is that accumulation of the ligand–receptor complex occurs earlier than predicted based on the transport rate of I125-NGF (Ure and Campenot, 1997). This result most likely reflects improved sensitivity by the methods used here.
The final line of evidence implicating transport of receptors in retrograde signaling, is the demonstration that individual Trk receptors are physically translocated from neurites to cell bodies during retrograde signaling. Using GFP to tag Trk receptors in DRG neurons, we show TrkB is transported from neurites to cell bodies as early as 8 min after neurotrophin stimulation. Control studies indicate that the increase in cell body fluorescence reflects specific transport of Trk receptors in response to ligand stimulation. Similar results were not seen with GFP alone. The rapid transport required ligand stimulation. Furthermore, pretreatment of neurites with colchicine or K252a abolished neurotrophin-stimulated transport of Trk receptors, indicating that receptor kinase activity and intact microtubule scaffolding are required for retrograde signaling. Thus, TrkB-GFP is transported to the cell body by microtubule-dependent retrograde transport in response to ligand stimulation at a time consistent with the initial appearance of pTrk and consistent with the earliest nuclear response. Together, these four lines of evidence indicate that the rapid nuclear responses to target-derived neurotrophin use an unusually fast method of microtubule-dependent transport of ligand–receptor complexes.
The colorized image showing the location of newly transported TrkB-GFP molecules suggests that receptors arrive at the cell bodies in an intracellular compartment, such as an endosome. The images do not suggest that receptors reinsert into the plasma membrane at early time points. Therefore, activated Trk receptors may relay their signal from discrete vesicles inside the cell. This could allow differential pathways to be induced in response to neurite stimulation compared with cell body stimulation. Further experiments will be needed to address this possibility.
Induction of new gene transcription
Many of the critical effects that neurotrophins exert on neurons require changes in gene expression (Segal and Greenberg, 1996;Finkbeiner et al., 1997). Increased expression of c-fos requires CREB activation and is one of the earliest neurotrophin-induced transcriptional responses (Greenberg et al., 1985; Ginty et al., 1994;Bonni et al., 1995). Application of K252a to neurites or to cell bodies blocked both Fos induction and CREB phosphorylation. Thus, pTrk is a necessary signal carrier for both nuclear responses. Because activation of CREB is first detected as early after neurite stimulation as after cell body stimulation, it is surprising that a 2.5 hr time lag exists in the Fos response to neurite stimulation. The delay in c-fos may represent a threshold effect. In sympathetic neurons grown in compartmented cultures, NGF increases expression of two delayed response genes, tyrosine hydroxylase (TH) and the neurotrophin receptor p75 (Toma et al., 1997). The magnitude of the TH and p75 responses are significantly lower after NGF stimulation of neurites than after NGF stimulation of cell bodies (Toma et al., 1997). Thus, at early time points, detectable increases in p75 and TH are only seen in response to cell body stimulation. A similar scenario is possible for c-fos induction. However, because the c-fos response elicited by selective neurotrophin stimulation at the neurites is similar in magnitude to the response elicited by applying neurotrophin directly to cell bodies, the delay seems unlikely to represent a threshold effect.
Alternatively, the delay may signify that CREB phosphorylation is not sufficient to induce Fos expression in response to neurite stimulation, and an additional slower component of retrograde signaling is necessary for induction of immediate early genes. This suggests the intriguing possibility that the location of stimulation could alter the nature of the biological response. In studies on PC12 cells, activation of CREB works in cooperation with other transcription factors, such as serum response factor (SRF) and ets-like transcription factor (Elk-1) to activate transcription of c-fos (Bonni et al., 1995). However, in primary cultures of cortical neurons, BDNF-mediated CREB phosphorylation is sufficient to induce c-fos transcription (Finkbeiner et al., 1997). The DRG neurons selectively stimulated at the neurites may resemble PC12 cells and require activation of other transcription factors, as well as CREB. Further experiments will be required to define additional slower retrograde signaling mechanisms that might be responsible for the delay in Fos induction and to determine whether other genes are also differentially regulated depending on the location of the stimulus. Nonetheless, the data presented here demonstrate that physical transport of activated receptors is a required step for nuclear responses to target-derived neurotrophins.
Footnotes
This work was funded by National Institutes of Health Grants NS35148 (R.A.S., F.L.W., D.B.M., A.B.) and NS35701 (S.L.P.) and the Robert Ebert Scholar award from the Klingenstein Foundation (R.A.S.). Additional support was supplied from Mental Retardation Research Center Grant HD 18655. F.L.W. was supported by a Lefler Fellowship, H.M.H. was supported by a predoctoral fellowship from the National Science Foundation, and M.Z.L. was supported by a National Defense and Engineering Graduate Fellowship. We thank A. Myerov, J. Fiala, and S. Finkbeiner for help with the confocal microscope, T. Bradlee for technical assistance, and R. Campenot for help with the compartmented cultures. We also thank C. D. Stiles, M. E. Greenberg, T. Roberts, and members of R. A. Segal’s laboratory for helpful discussion.
Correspondence should be addressed to Rosalind Segal, Department of Pediatric Oncology, Dana-Farber Cancer Institute, 44 Binney Street, Boston, MA 02115.
REFERENCES
- 1.Barbacid M. Neurotrophic factors and their receptors. Curr Opin Cell Biol. 1995;7:148–155. doi: 10.1016/0955-0674(95)80022-0. [DOI] [PubMed] [Google Scholar]
- 2.Barde YA. Trophic factors and neuronal survival. Neuron. 1989;2:1525–1534. doi: 10.1016/0896-6273(89)90040-8. [DOI] [PubMed] [Google Scholar]
- 3.Bett AJ, Haddara W, Prevec L, Graham FL. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc Natl Acad Sci USA. 1994;91:8802–8806. doi: 10.1073/pnas.91.19.8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattacharyya A, Watson FL, Bradlee TA, Pomeroy SL, Stiles CD, Segal RA. Trk receptors function as rapid retrograde signal carriers in the adult nervous system. J Neurosci. 1997;17:7007–7016. doi: 10.1523/JNEUROSCI.17-18-07007.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonni A, Ginty DD, Dudek H, Greenberg ME. Serine 133-phosphorylated CREB induces transcription via a cooperative mechanism that may confer specificity to neurotrophin signals. Mol Cell Neurosci. 1995;6:168–183. doi: 10.1006/mcne.1995.1015. [DOI] [PubMed] [Google Scholar]
- 6.Borasio GD, Markus A, Wittinghofer A, Barde YA, Heumann R. Involvement of ras p21 in neurotrophin-induced response of sensory, but not sympathetic neurons. J Cell Biol. 1993;121:665–672. doi: 10.1083/jcb.121.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci. 1995;18:223–253. doi: 10.1146/annurev.ne.18.030195.001255. [DOI] [PubMed] [Google Scholar]
- 8.Breuer A, Bond M, Atkinson M. Fast axonal transport is modulated by altering trans-axolemmal calcium flux. Cell Calcium. 1992;13:249–262. doi: 10.1016/0143-4160(92)90013-i. [DOI] [PubMed] [Google Scholar]
- 9.Campenot RB. Local control of neurite development by nerve growth factor. Proc Natl Acad Sci USA. 1977;74:4516–4519. doi: 10.1073/pnas.74.10.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campenot RB. Compartmented culture analysis of nerve growth. In: Stevenson B, Paul D, Gallin W, editors. Cell–cell interactions: a practical approach. IRL; Oxford: 1992. pp. 275–298. [Google Scholar]
- 11.Campenot RB. Construction and use of compartmented cultures. In: Federoff S, Richardson A, editors. Protocols for neural cell culture. Humana; Totowa, NJ: 1997. pp. 107–116. [Google Scholar]
- 12.Chitnis AB, Kuwada JY. Axogenesis in the brain of zebrafish embryos. J Neurosci. 1990;10:1892–1905. doi: 10.1523/JNEUROSCI.10-06-01892.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clary D, Weskamp G, Austin L, Reichardt L. TrkA cross-linking mimics neuronal responses to nerve growth factor. Mol Biol Cell. 1994;5:549–563. doi: 10.1091/mbc.5.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Claude P, Hawrot E, Dunis DA, Campenot RB. Binding, internalization, and retrograde transport of 125I-nerve growth factor in cultured rat sympathetic neurons. J Neurosci. 1982;2:431–442. doi: 10.1523/JNEUROSCI.02-04-00431.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cordon-Cardo C, Tapley P, Jing S, Nanduri V, O’Rourke E, Lamballe F, Kovary K, Klein R, Jones KR, Reichardt LF, Barbacid M. The trk tyrosine protein kinase mediates the mitogenic properties of nerve growth factor and NT3. Cell. 1991;66:173–183. doi: 10.1016/0092-8674(91)90149-s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ehlers M, Kaplan D, Price D, Koliatsos V. NGF-stimulated retrograde transport of trk A in the mammalian nervous system. J Cell Biol. 1995;130:149–156. doi: 10.1083/jcb.130.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1037. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- 18.Ginty DD, Kornhauser JM, Thompson MA, Bading H, Mayo KE, Takahashi JS, Greenberg ME. Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and a circadian clock. Science. 1993;260:238–241. doi: 10.1126/science.8097062. [DOI] [PubMed] [Google Scholar]
- 19.Ginty DD, Bonni A, Greenberg ME. Nerve growth factor activates a ras-dependent protein kinase that stimulates c-fos transcription via phosphorylation of CREB. Cell. 1994;77:713–725. doi: 10.1016/0092-8674(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 20.Greenberg ME, Greene LA, Ziff EB. Nerve growth factor and epidermal growth factor induce rapid transient changes in proto-oncogene transcription in PC12 cells. J Biol Chem. 1985;260:14101–14110. [PubMed] [Google Scholar]
- 21.Grimes M, Zhou J, Beattie E, Yuen E, Hall D, Valletta J, Topp K, LaVail J, Bunnett N, Mobley W. Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signaling endosomes. J Neurosci. 1996;16:7950–7964. doi: 10.1523/JNEUROSCI.16-24-07950.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grimes ML, Beattie E, Mobley WC. A signaling organelle containing the nerve growth factor-activated receptor tyrosine kinase, TrkA. Proc Natl Acad Sci USA. 1997;94:9909–9914. doi: 10.1073/pnas.94.18.9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gundersen GG, Cook TA. Microtubules and signal transduction. Curr Opin Cell Biol. 1999;11:81–94. doi: 10.1016/s0955-0674(99)80010-6. [DOI] [PubMed] [Google Scholar]
- 24.Hendry IA, Stockel K, Thoenen H. The retrograde axonal transport of nerve growth factor. Brain Res. 1974;68:103–121. doi: 10.1016/0006-8993(74)90536-8. [DOI] [PubMed] [Google Scholar]
- 25.Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- 26.Inoue S. Video microscopy. Plenum; New York: 1986. [Google Scholar]
- 27.Jing S, Tapley P, Barbacid M. Nerve growth factor mediates signal transduction through trk homodimer receptors. Neuron. 1992;9:1067–1079. doi: 10.1016/0896-6273(92)90066-m. [DOI] [PubMed] [Google Scholar]
- 28.Johnson E, Taniuchi M, Clark H, Springer J, Koh S, Tayrien M, Loy R. Demonstration of the retrograde transport of nerve growth factor receptor in the peripheral and central nervous system. J Neurosci. 1987;7:923–929. doi: 10.1523/JNEUROSCI.07-03-00923.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaplan D, Miller F. Signal transduction by the neurotrophin receptors. Curr Biol. 1997;9:213–221. doi: 10.1016/s0955-0674(97)80065-8. [DOI] [PubMed] [Google Scholar]
- 30.Kaplan DR, Stephens RM. Neurotrophin signal transduction by the Trk receptor. J Neurobiol. 1994;25:1404–1417. doi: 10.1002/neu.480251108. [DOI] [PubMed] [Google Scholar]
- 31.Kimpinski K, Campenot RB, Mearow K. Effects of the neurotrophins nerve growth factor, neurotrophin-3, and brain-derived neurotrophic factor (BDNF) on neurite outgrowth from adult sensory neurons in compartmented cultures. J Neurobiol. 1997;33:395–410. doi: 10.1002/(sici)1097-4695(199710)33:4<395::aid-neu5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 32.Klein R, Nanduri V, Jing SA, Lamballe F, Tapley P, Bryant S, Cordon CC, Jones KR, Reichardt LF, Barbacid M. The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell. 1991;66:395–403. doi: 10.1016/0092-8674(91)90628-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klinz FJ, Heumann R. Time-resolved signaling pathways of nerve growth factor diverge downstream of the p140trk receptor activation between chick sympathetic and dorsal root ganglion sensory neurons. J Neurochem. 1995;65:1046–1053. doi: 10.1046/j.1471-4159.1995.65031046.x. [DOI] [PubMed] [Google Scholar]
- 34.Korsching S, Thoenen H. Quantitative demonstration of the retrograde axonal transport of endogenous nerve growth factor. Neurosci Lett. 1983;39:1–4. doi: 10.1016/0304-3940(83)90155-6. [DOI] [PubMed] [Google Scholar]
- 35.Lamballe F, Klein R, Barbacid M. TrkC, a new member of the trk family of tyrosine protein kinases, is a receptor for neurotrophin 3. Cell. 1991;66:967–979. doi: 10.1016/0092-8674(91)90442-2. [DOI] [PubMed] [Google Scholar]
- 36.Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- 37.Nakata T, Terada S, Hirokawa N. Visualization of the dynamics of synaptic vesicle and plasma membrane proteins in living axons. J Cell Biol. 1998;140:659–674. doi: 10.1083/jcb.140.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Obermeier A, Lammers R, Wiesmuller K, Jung G, Schlessinger J, Ullrich A. Identification of trk binding sites for SHC and phosphatidylinositol 3′-kinase and formation of a multimeric signaling complex. J Biol Chem. 1993;268:22963–22966. [PubMed] [Google Scholar]
- 39.Oppenheim R. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–480. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 40.Palmatier MA, Hartman BK, Jonhson EM., Jr Demonstration of retrogradely transported endogenous growth factor in axons of sympathetic neurons. J Neurosci. 1984;4:751–756. doi: 10.1523/JNEUROSCI.04-03-00751.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paulson J, McClure W. Microtubules and axoplasmic transport. Inhibition of transport by podophyllotoxin: an interaction with microtubule protein. J Cell Biol. 1975;67:461–467. doi: 10.1083/jcb.67.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riccio A, Pierchala BA, Ciarallo CL, Ginty DD. An NGF-TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science. 1997;277:1097–1100. doi: 10.1126/science.277.5329.1097. [DOI] [PubMed] [Google Scholar]
- 43.Richardson P, Riopelle R. Uptake of nerve growth factor along peripheral and spinal axons of primary sensory neurons. J Neurosci. 1984;4:1683–1689. doi: 10.1523/JNEUROSCI.04-07-01683.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saragovi H, Zheng W, Maliartchouk S, DiGugliemo G, Mawal Y, Kamen A, Woo S, Cuello A, Debeir T, Neet K. A TrkA-selective, fast internalizing nerve growth factor-antibody complex induces trophic but not neuritogenic signals. J Biol Chem. 1998;273:34933–34940. doi: 10.1074/jbc.273.52.34933. [DOI] [PubMed] [Google Scholar]
- 45.Schwab ME. Ultrastructural localization of a nerve growth factor-horseradish peroxidase (NGF-HRP) coupling product after retrograde axonal transport in adrenergic neurons. Brain Res. 1977;130:190–196. doi: 10.1016/0006-8993(77)90857-5. [DOI] [PubMed] [Google Scholar]
- 46.Segal R, Greenberg M. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 47.Segal R, Pomeroy S, Stiles C. Axonal growth and fasciculation linked to differential expression of BDNF and NT3 receptors in developing cerebellar granule cells. J Neurosci. 1995;15:4970–4981. doi: 10.1523/JNEUROSCI.15-07-04970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Segal R, Bhattacharyya A, Rua L, Alberta J, Stephens R, Kaplan D, Stiles C. Differential utilization of Trk autophosphorylation sites. J Biol Chem. 1996;271:20175–20181. doi: 10.1074/jbc.271.33.20175. [DOI] [PubMed] [Google Scholar]
- 49.Senger DL, Campenot RB. Rapid retrograde tyrosine phosphorylation of Trka and other proteins in rat sympathetic neurons in compartmented cultures. J Cell Biol. 1997;138:411–421. doi: 10.1083/jcb.138.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stephens R, Loeb D, Copelan T, Pawson T, Greene L, Kaplan D. Trk receptors use redundant signal transduction pathways involving SHC and PLC gamma 1 to mediate NGF responses. Neuron. 1994;12:691–705. doi: 10.1016/0896-6273(94)90223-2. [DOI] [PubMed] [Google Scholar]
- 51.Toma JG, Rogers D, Senger DL, Campenot RB, Miller FD. Spatial regulation of neuronal gene expression in response to nerve growth factor. Dev Biol. 1997;184:1–9. doi: 10.1006/dbio.1997.8515. [DOI] [PubMed] [Google Scholar]
- 52.Ure DR, Campenot RB. Retrograde transport and steady-state distribution of I-125-nerve growth factor in rat sympathetic neurons in compartmented cultures. J Neurosci. 1997;17:1282–1290. doi: 10.1523/JNEUROSCI.17-04-01282.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vieria A, Lamaze C, Schmid S. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science. 1996;274:2086–2089. doi: 10.1126/science.274.5295.2086. [DOI] [PubMed] [Google Scholar]
- 54.Wang C, Asai DJ, Robinson KR. Retrograde but not anterograde bead movement in intact axons requires dynein. J Neurobiol. 1995;27:216–226. doi: 10.1002/neu.480270208. [DOI] [PubMed] [Google Scholar]
- 55.Yip HK, Johnson EMJ. Comparative dynamics of retrograde transport of nerve growth factor and horseradish peroxidase in rat lumbar dorsal root ganglia. J Neurocytol. 1986;15:789–798. doi: 10.1007/BF01625195. [DOI] [PubMed] [Google Scholar]