

Abstract
We investigated the role of caffeine-sensitive intracellular stores in regulating intracellular calcium ([Ca2+]i) and glutamatergic synaptic transmission from rod photoreceptors. Caffeine transiently elevated and then markedly depressed [Ca2+]i to below prestimulus levels in rod inner segments and synaptic terminals. Concomitant with the depression was a reduction of glutamate release and a hyperpolarization of horizontal cells, neurons postsynaptic to rods. Caffeine did not affect the rods’ membrane potentials indicating that caffeine likely acted via some mechanism(s) other than a voltage-dependent deactivation of the calcium channels. Most of caffeine’s depressive action on [Ca2+]i, on glutamate release, and on ICa in rods can be attributed to calcium release from stores: (1) caffeine’s actions on [Ca2+]i andICa were reduced by intracellular BAPTA and barium substitution for calcium, (2) other nonxanthine store-releasing compounds, such as thymol and chlorocresol, also depressed [Ca2+]i, and (3) the magnitude of [Ca2+]i depression depended on basal [Ca2+]i before caffeine. We propose that caffeine-released calcium reducesICa in rods by an as yet unidentified intracellular signaling mechanism. To account for the depression of [Ca2+]i below rest levels and the increased fall rate of [Ca2+]i with higher basal calcium, we also propose that caffeine-evoked calcium release from stores activates a calcium transporter that, via sequestration into stores or extrusion, lowers [Ca2+]i and suppresses glutamate release. The effects of store-released calcium reported here operate at physiological calcium concentrations, supporting a role in regulating synaptic signaling in vivo.
Keywords: photoreceptor, rod, calcium, caffeine, intracellular calcium stores, glutamate release
It is now generally recognized that intracellular calcium concentration ([Ca2+]i) is an important regulator of neurotransmitter release. Extensive studies have delineated a role for calcium influx in gating transmitter release (Katz and Miledi, 1969; Matthews, 1997; Neher, 1998). Much less study has been directed at a possible role of intracellular calcium stores in exocytosis, particularly in glutamatergic neurons. In chromaffin cells, Ca2+ released from caffeine-sensitive intracellular storage compartments can both enhance and reduce exocytosis of catecholamines. The stimulation of exocytosis by caffeine may occur in the absence of extracellular calcium (Cheek et al., 1990). However, caffeine treatment can also reduce the size of depolarization-evoked release of catecholamines from these same cells (Lara et al., 1997). On the basis of the finding that caffeine reduced the depolarization-evoked rises of [Ca2+]i in bullfrog neurons, Friel and Tsien (1992) articulated the hypothesis that caffeine-induced depletion of stores stimulated uptake back into these compartments and that this sequestration acted as a sink for incoming calcium. Cseresnyes et al. (1997) concluded that in frog sympathetic ganglion neurons there is a release-activated calcium transport (RACT) that significantly increases sequestration into stores when calcium is released from caffeine-sensitive compartments. When activated, RACT transports calcium at rates 1.6 and 4 times faster than the conventional sarcoplasmic–endoplasmic calcium ATPases (SERCA) and the plasma membrane extrusion pumps, respectively. This indicates that RACT can play a significant role in controlling [Ca2+]i, and possibly exocytosis as well. In this present study we used the rod–horizontal cell synapse of the amphibian retina to examine the possibility that caffeine-sensitive Ca2+stores contribute to the regulation of glutamate exocytosis.
A hallmark feature of synaptic transmission at the photoreceptor and bipolar cell synapses of the retina is the continuous release of glutamate. It is generally accepted that the release from rods is directly controlled by an influx of Ca2+, predominantly through L-type calcium channels located in inner segments and synaptic terminals of these cells (Copenhagen and Jahr, 1989; Rieke and Schwartz, 1996; Schmitz and Witkovsky, 1997; Witkovsky et al., 1997). In general, caffeine-sensitive stores are localized to smooth endoplasmic reticulum (ER) (Golovina and Blaustein, 1997; Meldolesi and Pozzan, 1998), an intracellular compartment widely distributed throughout the cell body, dendritic trees, and synaptic terminals of neurons (Walton et al., 1991; Krijnse-Locker et al., 1995). Although there are no reports of caffeine-sensitive stores in photoreceptors, cisternae of smooth ER have been noted in synaptic nerve terminals of photoreceptors (Mercurio and Holtzman, 1982; Ungar et al., 1984).
We studied the effects of caffeine on both presynaptic and postsynaptic cells at the photoreceptor synapse. We investigated the effects of caffeine on [Ca2+]i in rods using fura-2, on L-type calcium currents in rods using whole-cell recording methods, on rod and horizontal cell membrane potentials using microelectrode techniques, and on the release of endogenous glutamate using a newly developed preparation consisting of a sheet of photoreceptors separated from the rest of the retina (Cahill and Besharse, 1992; Schmitz and Witkovsky, 1996). Our data strongly suggest that calcium release from caffeine-sensitive calcium stores can modulate synaptic transmission from these glutamatergic neurons.
Parts of this study have been published previously in abstract form (Krizaj et al., 1997).
MATERIALS AND METHODS
The experiments were performed on rod photoreceptors from two amphibian species: the clawed frog (Xenopus laevis) and the tiger salamander (Ambystoma tigrinum). All imaging experiments were performed on salamander photoreceptors because of their large size and experimental tractability; we used theXenopus retina because it can be used as a “reduced preparation” to study glutamate release (see below) (Schmitz and Witkovsky, 1996). Intracellular recording was performed on bothXenopus and Ambystoma horizontal cells (HC). We found no significant species differences in relation to the light-evoked responses or membrane potential of HCs when exposed to caffeine.
Preparation of isolated cells. Larval stage tiger salamanders were decapitated and pithed. Retinas were dissected at room temperature (20–22°C) in room light, incubated on a shaker in 0 Ca2+ and papain (7 U/ml; Worthington, Freehold, NJ) saline for 25 min, and triturated with a BSA-coated Pasteur pipette. The outer segments of many rods were shorn during the isolation procedure, resulting in the absence of the dark current and hyperpolarization of their membrane potentials. Dissociated cells were kept at 4°C in 80% L-15 medium supplemented with 10 mmHEPES, 20 mm glucose, 1 mm pyruvic acid, 1 mg/ml bovine serum albumin, and 1 μl/ml Liquid Media Supplement containing transferrin and selenium (Sigma, St. Louis, MO). Cells were plated onto acid-cleaned glass coverslips coated with IgG and/or IgM (Jackson ImmunoResearch, West Grove, PA) and the Sal-1 antibody [a kind gift from Dr. Peter MacLeish (MacLeish et al., 1983)]. The control saline solution contained (in mm): 97 NaCl, 2 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 20 glucose, 1 pyruvic acid, 0.3 ascorbic acid, and 1 glutathione at 240 mOsm. pH was adjusted to 7.6 with NaOH. In the high (20 mm) potassium saline the concentration of NaCl was correspondingly reduced. The volume around cells was superfused through either a Y-tube mechanism in which solution exchange was completed within 10 sec or a multibarrel quartz perfusion system that allowed solution exchange within 100 msec.
[Ca2+] measurement and image acquisition. These measurements are described in more detail elsewhere (Krizaj and Copenhagen, 1998). Briefly, photoreceptors were loaded for 10 min with 3–5 μm fura-2 AM (Molecular Probes, Eugene, OR) supplemented with 0.005% Pluronic F-127 in L-15 and then washed for 20 min in L-15. The fluorescence signals were acquired at 0.3–1 Hz by a cooled 12-bit digital CCD camera (PXL; Photometrics, Tucson, AZ) controlled by commercial software (Metafluor; Universal Imaging Corporation, West Chester, PA). Ratios between the 340 and 380 nm excitation wavelengths were calculated after subtraction of the background fluorescence. Free Ca2+ levels were calibrated in vivo with 10 μm ionomycin using the standard relationship developed by Grzynkiewicz et al. (1985). TheKd values for Ca2+ (224 nm) and Ba2+ (780 nm) binding to fura-2 were taken from the literature (Grzynkiewicz et al., 1985; Schilling et al., 1989; Neher, 1995). The actual calcium concentrations should be considered estimates, because a standardized value for the Kd of fura-2 was used throughout these experiments. Most measurements were taken in a region encompassing most of the inner segment of isolated rods. Loss of synaptic terminals during dissociation and low signal/noise ratios made it difficult to test routinely every protocol on the terminals. The waveforms of the transient elevations and depressions of calcium were not discernibly different from those in the inner segments. The lower signal/noise ratio prevented us from quantitatively assessing whether the responses were faster in the terminals, as we had reported previously for potassium-evoked rises of calcium (Krizaj and Copenhagen, 1998).
Patch-clamp recording. Recording electrodes were pulled in four steps on a horizontal pipette puller (P97; Sutter Instruments, Novato, CA) from 1.7 mm borosilicate capillary glass (TW 150F; World Precision Instruments, Sarasota, FL). Electrodes were filled with (in mm): 75 Cs MeSO4, 5 EGTA, 30 HEPES, 0.5 CaCl2, 2 Mg-ATP, 0.5 Na3GTP, and 20 TEA-Cl, with pH adjusted to 7.5 with CsOH. The electrode resistance was 10–20 mΩ. The extracellular solution contained (in mm): 55 NaCl, 2.5 KCl, 10 CaCl2, 1 MgCl2, 8 glucose, 10 HEPES, and 30 TEA-Cl, with pH buffered to 7.6 with NaOH. In some experiments, 10 mm CaCl2 was replaced by 10 mm BaCl2. Seal resistance ranged from 1 to 20 GΩ. Series resistances were typically 4–12 mΩ. The membrane current was measured with an Axopatch 2-D amplifier (Axon Instruments, Foster City, CA). Capacitance currents were canceled electronically. Current records were low-pass filtered at 2 kHz (−3 dB) and digitized at 5× the filter cutoff frequency. Data were acquired via an analog-to-digital interface (Indec Systems, Sunnyvale, CA). Junction potentials were 1–2 mV, as measured using a low-resistance 3 m KCl reference electrode in the bath; membrane potentials were not corrected for junction potentials. Leak subtraction was performed on whole-cell currents as follows. Ca2+ currents were blocked by adding 100 μm Cd2+, and the remaining leak currents were subtracted from the equivalent experimental records from the same cell. The leak currents usually were obtained immediately after each set of experimental records. The perforated-patch recordings were performed as described above, except that the electrode was filled with 2 ng·ml−1 gramicidin.
Rods were held at −70 mV. In most experiments, Ca2+ currents were induced by depolarizing the cells with voltage ramps (−70 to +50 mV) scanned at a rate of 1.02 V/sec. Ramps were given at 20 sec intervals throughout the experiments, and the drugs under study were applied onto cells after Ca2+ currents stabilized, which typically occurred within a few minutes after break-in. Solutions were exchanged via a gravity-driven multibarrel microperfusion system positioned within 1 mm of the tested cell.
The percentage inhibition of Ca2+ currents was expressed as [1 −ICa(test)/ICa(control)] × 100. Cadmium-subtracted currents were used, whenever possible, to calculate the peak amplitude. Student’s ttests were performed for paired or unpaired groups, whereas one-way ANOVA was done for three or more groups.
Intracellular recording. Eyecups were prepared as described previously (Krizaj et al., 1994). Microelectrodes were backfilled with 4 m potassium acetate and had an average resistance of 150–200 mΩ. After isolation in room light, the eyecups were dark adapted in the superfusion chamber for >1 hr before recording. The eyecups were superfused continuously at 1.5 ml/min and stimulated with diffuse 200 msec steps of light emitted by a green light-emitting diode (λmax = 567 nm). Light intensity was controlled by a neutral density wedge. Data were stored on digital tape for off-line analysis with SPIKE software (Modular Instruments, Taunton, MA). Isolated cells were obtained as described above and impaled with borosilicate microelectrodes attached to a motorized manipulator (MP-285; Sutter Instruments). Under these conditions photoreceptors are hyperpolarized to −55 to −65 mV (Bader et al., 1979; Barnes and Hille, 1989) (D. Krizaj and D. R. Copenhagen, unpublished observations) that is close to the light-evoked plateau potentials measured in intact rods in vivo (Fain, 1976; Witkovsky et al., 1997). Because we were interested in the modulation of glutamate release, whose magnitude is higher at more depolarized membrane potentials, we usually depolarized the test rod by elevating extracellular [K+]. In isolated rods, 20 mm KCl raised Vm from approximately −60 mV to approximately −40 mV, a value that is close to dark potentials recorded in intact rods.
Glutamate release. The preparation of the reduced retina was performed as described previously (Schmitz and Witkovsky, 1996). Briefly, the cornea, the inner ring of the iris, and the lens were removed, and the eyecup was flushed successively with 0.5% Triton X-100 in distilled water, distilled water alone, and culture medium. After ∼1 hr of incubation in culture medium, the retina split apart in the inner nuclear layer, permitting the inner retinal layers to be removed with fine forceps and thereby preserving a laminar sheet of photoreceptor cells. The preservation of physiological function in this preparation is documented in Schmitz and Witkovsky (1996) andWitkovsky et al. (1997).
All experiments were done in room light (40 μW/cm2) in the plane of the retina. The reduced retinas were maintained in aerated chambers (95% O2/5% CO2) and superfused at 1 ml/hr with culture medium containing (in mm): 82 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 35 NaHCO3, and 1 NaH2PO4, pH 7.55. The basic salt medium was supplemented with 5 mm glucose, 100 μm ascorbic acid, and a mixture of 14 amino acids including 5 μm glutamine. Dihydrokainate (2 mm) was added to all media to reduce glutamate reuptake. Two-position valves permitted a rapid switch from control to test solution. Test solutions flowed for at least 30 min before samples, each representing 5 min of perfusion, were collected.
The glutamate content was analyzed by the method of Fosse et al. (1986)that uses glutamate dehydrogenase coupled via FMN reductase to bacterial luciferase. The resulting light production was measured by a luminometer (Monolight 2010; Analytical Luminescence Lab, San Diego, CA). l-glutamate standards were prepared in control and test solutions. For statistical analysis, ANOVA with the subsequent Tukey test was used. All data were normalized to the glutamate released during superfusion with control saline containing 2 mmKCl.
Chemicals. Nifedipine (Sigma), ryanodine (9,21-didehydro-ryanodine:ryanodine ratio of 60:40; Research Biochemicals, Natick, MA), and 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (BAPTA) AM (Molecular Probes) were dissolved in DMSO. BAPTA AM was made as a 1 mm stock and was diluted to 5–20 μm before the experiment. Dilutions were made fresh with final concentrations of DMSO < 0.001%. The free-acid form of BAPTA used in whole-cell experiments was dissolved directly into the internal pipette solution. 4-Chloro-m-cresol [4-chloro-3-methylphenol (4-CmC)] was obtained from Aldrich (Milwaukee, WI); thymol, glutamate dehydrogenase, gramicidin, and luciferase were from Sigma; and FMN reductase was obtained from Boehringer Mannheim (Indianapolis, IN).
RESULTS
Caffeine is a xanthine that releases [Ca2+]i from intracellular stores by increasing the affinity of the ryanodine receptor for cytoplasmic Ca2+ (Pozzan et al., 1994; Hernandez-Cruz et al., 1995). The caffeine-evoked release of Ca2+ from intracellular stores is typically observed as a transient increase in [Ca2+]i. We observed caffeine-evoked transient [Ca2+]i increases in inner segments and synaptic terminals but not in outer segments of rods. Figure 1Aillustrates, in the inner segment of a rod, transient increases of [Ca2+]i produced by brief puffs of caffeine (50 mm in the pipette). In control saline containing 2 mm KCl, [Ca2+]i was elevated transiently by ∼50 nm. Caffeine-evoked increases of [Ca2+]i ranged from 20 to 80 nm under these same conditions (n = 10 cells). Responses to repeated puffs of caffeine in calcium-free saline gradually diminished over time but began to increase after a return to control saline (Fig. 1A). These findings are not only indicative of a caffeine-sensitive store in the rods but demonstrate that the stores can be depleted if calcium influx from the extracellular milieu is diminished. The major influx pathway for calcium influx into rods is via L-type calcium channels (Corey et al., 1984; Krizaj and Copenhagen, 1998). In agreement with a requirement for influx through these channels to fill the caffeine-sensitive stores, we found that repeated caffeine puffs in nifedipine, an L-type channel antagonist, caused the transient [Ca2+]i elevations to run down (data not shown) similar to the rundown in 0 Ca2+ conditions shown in Figure1A.
Fig. 1.

Caffeine evokes a transient increase of [Ca2+]i in rods. A,Responses of [Ca2+]i to brief puffs of caffeine (50 mm in the pipette; indicated by thestars). Isolated cells were perfused continuously first with control saline containing 2 mm KCl and 2.0 mm Ca2+, then with calcium-free (0 Ca2+ + EGTA) saline (indicated by thehorizontal bar), and then with control. Caffeine-induced transients became progressively smaller in calcium-free saline but grew progressively larger after return to control, consistent with a rundown and then replenishment of caffeine-sensitive stores in the absence and reestablishment, respectively, of calcium influx from outside the rod. B,Amplitude of the caffeine-induced transient peaks of [Ca2+]i as a function of prestimulus calcium concentration. Peak magnitude was correlated positively with basal concentration, suggesting that the caffeine-sensitive stores were more completely filled at higher [Ca2+]i (n = 70 measurements in 62 different cells; data summed with [K+]o ranging from 2 to 45 mm).
The magnitude of caffeine-evoked [Ca2+]i transients was positively correlated to the basal calcium concentration in the rods. Figure 1B plots the peak amplitude of the caffeine transient versus prestimulus [Ca2+]i. This finding agrees with previous studies indicating that the releasable pool of calcium from stores is enhanced in proportion to the amount of calcium in the cytosol (Sitsapesan and Williams, 1990; Hua et al., 1993; Garaschuk et al., 1997).
Caffeine also evokes a prolonged [Ca2+]i depression in rods
At membrane potentials equivalent to the resting potential of rods in darkness (approximately −40 mV), caffeine evoked a prolonged depression in [Ca2+]i after the initial peak (Fig. 2A). Basal [Ca2+]i in isolated cells in 2 mm KCl was 49 ± 3 nm (mean ± SE; n = 70). We found that 20 mm KCl raised the membrane potential from approximately −55 mV to approximately −40 mV and elevated the resting [Ca2+]i to 325 ± 16 nm (n = 54). At 20 mm KCl caffeine elicited a stereotyped two-phase response in [Ca2+]i. The first phase was a transient increase in [Ca2+]i peaking at ∼5–30 sec, whereas the second phase was a prolonged [Ca2+]iundershoot. Figure 2A shows an example from an inner segment of a rod in which caffeine was applied in the presence of 20 mm KCl. Here [Ca2+]i rose transiently from ∼480 to ∼1450 nm and then rapidly dropped to a level around 250 nm, well below the precaffeine level. Figure 2B shows an example of a caffeine-evoked increase and subsequent depression in a synaptic terminal of a rod. These results demonstrate that for rods maintained near their dark resting potential in these cells, caffeine evoked a transient rise, followed by a prolonged depression of [Ca2+]i in inner segments and synaptic terminals. Caffeine did not evoke a transient peak or depression in rod outer segments.
Fig. 2.

Caffeine also evokes prolonged depression in the inner segments and synaptic terminals of rods when basal [Ca2+]i is higher. A, Response in a rod inner segment to a puff of caffeine (50 mm in the pipette) in 20 mm KCl saline. [Ca2+]i started at ∼480 nm, peaked at ∼1450 nm, and dropped to ∼250 nm. [Ca2+]i recovered to the prestimulus level ∼5 min after the puff. B, Response to caffeine measured in the synaptic terminal of a rod. Calibration with ionomycin was not completed in this cell; therefore only the relative ratio of 340/380 nm fluorescence, known to be directly proportional to [Ca2+]i, is shown.
Higher basal levels of [Ca2+]i not only increased the size of the transient peak but increased the speed of the falling phase after the peak and the magnitude of the depression. Figure 3A–C shows a caffeine response recorded from the same rod at [Ca2+]i of ∼50, ∼350, and ∼530 nm, respectively. The magnitudes of the transient peaks and the size of the depression were larger at higher basal [Ca2+]i. Notably, the fall of calcium after the peak was significantly faster for higher basal [Ca2+]i. Single-exponential fits to the falling phase revealed time constants of 97 sec in 2 mm KCl and 42 and 40 sec, respectively, in 30 and 90 mm KCl. The depression below the prestimulus levels and the faster decline at raised [Ca2+]i suggest the activation of a second process, subsequent to the transient peak, that actively lowers calcium in the cytosol.
Fig. 3.
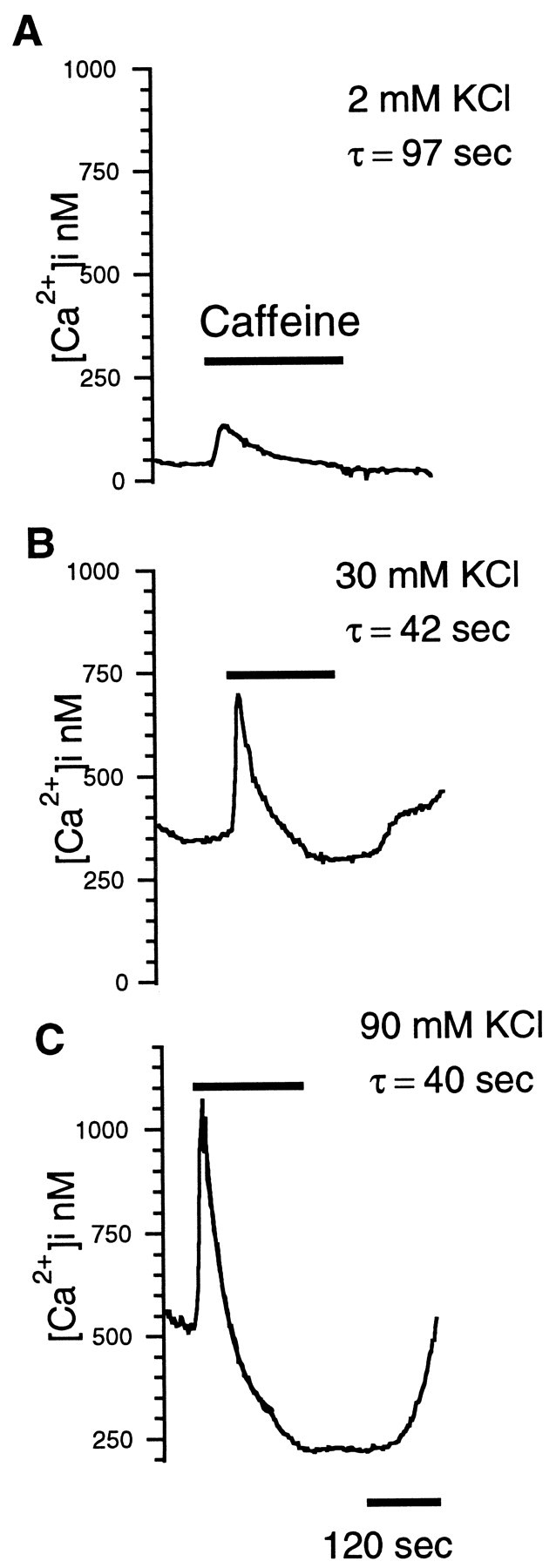
Caffeine-induced depression is higher for increased basal calcium. A–C, [Ca2+]i responses of a rod inner segment to bath-applied caffeine in 2 mm (A), 30 mm (B), and 90 mm(C) KCl. The magnitudes of the transient peak and depression were positively correlated to basal [Ca2+]i before caffeine. The decay from the initial peaks was fitted with single-exponential functions having time constants of 97, 42, and 40 sec.
In some types of cell, ryanodine can block caffeine-induced release of Ca2+ from intracellular stores (McPherson et al., 1991). However, there is also precedent for caffeine releasing calcium from ryanodine-insensitive stores (Schmid et al., 1990; McNulty and Taylor, 1993; Orkand and Thomas, 1995). We tested the effects of ryanodine on the caffeine-induced rise and depression of [Ca2+]i and found that, by itself, ryanodine slightly raised baseline [Ca2+]i in 7 of 21 rods. It also irreversibly eliminated caffeine-mediated elevations of [Ca2+]i in the great majority of cells (n = 19/21). Ryanodine, however, only reduced and did not completely eliminate the caffeine-induced suppression of [Ca2+]i. In the control experiments caffeine suppressed [Ca2+]i by 75 ± 5% of prestimulus levels. After exposure to 20–100 μm ryanodine, [Ca2+]i was reduced to 51 ± 4% of baseline (n = 12 rods). In Figure 4, the left trace shows the response to caffeine in control saline. Themiddle and right traces show the caffeine response 12 and 30 min after ryanodine application, respectively. This differential effect of ryanodine on the two phases suggests that the transient elevation may be from a predominantly ryanodine-sensitive store but the depressive action might involve caffeine effects on both ryanodine-sensitive and -insensitive stores. Accordingly, the depression may result from the activation of a pump or transporter that is triggered by the release of calcium from both types of stores (Schmid et al., 1990; Friel and Tsien, 1992; McNulty and Taylor, 1993; Orkand and Thomas, 1995).
Fig. 4.

Ryanodine completely blocks caffeine-induced transient peaks in [Ca2+]i but only partially blocks the caffeine-induced depression of [Ca2+]i. Left, Caffeine (10 mm) elicited the two-phased [Ca2+]i response in a rod constantly depolarized with 20 mm KCl. Middle,Right, Caffeine was reapplied at t = 12 and 30 min after exposure to 50 μm ryanodine. After ryanodine, the first phase of the caffeine-mediated response was irreversibly blocked, and the [Ca2+]iundershoot was reduced to 56 and 44% of control att = 12 and 30 min, respectively.
In the following experiments we studied the effects of caffeine on synaptic transmission from rods to second-order cells.
Caffeine hyperpolarizes horizontal cells but not rods
In dark-adapted amphibian retinas, the membrane potentials of HCs in darkness reflect the activation of non-NMDA glutamate receptors by the tonic release of glutamate from photoreceptors (Krizaj et al., 1994). We tested the effects of caffeine on synaptic transmission by recording from HCs and rods in dark-adapted eyecup preparations of both Xenopus and tiger salamander retinas. The waveform and the chromatic sensitivity of HC light-evoked responses indicated that the HCs were driven solely by rods. The membrane potentials of rods in darkness were monitored during exposure to 10 mm caffeine. The initial dark membrane potentials varied from −39 to −45 mV, and they were essentially unchanged throughout the exposure to caffeine-containing saline (Fig.5A, open circles). Figure 5B shows that the light-evoked responses of rods were altered little by exposure to 10 mm caffeine (n = 6). In HCs, caffeine elicited a brief period of depolarization followed by a more prolonged period of hyperpolarization. The resting potentials of HCs were −43 ± 4 mV. Figure 5A (filled circles) shows that the HC membrane began to depolarize within 2 min after the switch to caffeine-containing saline. After reaching a mean peak depolarization of 4 mV, the HCs hyperpolarized by 26 ± 2.5 mV over the next 4–5 min. The filled circles (Fig. 5A) show the mean and SE of seven HCs. These data indicate that caffeine has little effect on the rod membrane potential in darkness but strongly modulates the membrane potential of HCs. Given that the depolarization and subsequent hyperpolarization of HCs have a time course similar to the caffeine-induced rise and depression of [Ca2+]i measured in the rods, we hypothesize that caffeine-evoked changes of calcium in the presynaptic neurons modulates the release of neurotransmitter from these cells. Because the membrane potentials of rods are not affected, the data tend to rule out a caffeine-dependent, voltage-induced suppression of transmitter release in rods.
Fig. 5.

Caffeine hyperpolarizes horizontal cells but not rods. Intracellular recordings from rods and HCs in the eyecup preparation are shown. A, Caffeine (10 mm) was added at t = 0 min. No effect on the membrane potential of rods was discerned (open circles), whereas horizontal cells (filled circles) had a two-phased response—transient depolarization followed by a peak hyperpolarization of 26 ± 2.5 mV (n = 7). The effects of caffeine were reversible. In five of six cells recorded for >20 min after caffeine washout, the membrane potential returned to within 3 mV of prestimulus values. B, Light responses of a rod to 200 msec, 567 nm flash (top, −3.5 log;bottom, −1.0 log quanta) before and during caffeine exposure are shown. In caffeine, slightly slower rise times were observed for dim but not for stronger flashes. No significant effect of caffeine was observed on either the transient hyperpolarization of the rod or the rod “tail” for brighter flashes. Plotted membrane potentials were normalized to the resting potential before caffeine.
Caffeine inhibits glutamate release
We tested directly whether caffeine could modulate endogenous glutamate release from photoreceptors. These experiments were performed using a photoreceptor sheet preparation (Schmitz and Witkovsky, 1996). We approximated the same conditions used for isolated rods (Fig. 2) by suppressing the dark current with light and then adding 20 mm[K+]o to increase glutamate release. Figure 6 shows that 35 min after a switch from normal to 20 mm KCl saline, glutamate release increased 3.12 ± 0.35 times (n= 4). After addition of 10 mm caffeine to the 20 mm K+-containing saline, glutamate release declined to 47% of baseline levels over a 15–20 min period. The decline was statistically significant (p < 0.001). After return to control saline without caffeine, recovery was complete after ∼40–45 min. These data indicate that the effect of caffeine on synaptic transmission between rods and horizontal cells is consistent with a presynaptic locus of caffeine action, in which caffeine directly or indirectly suppresses exocytosis from the rods.
Fig. 6.

Glutamate release is reduced during exposure to 10 mm caffeine. The time course of glutamate release from the retina prepared as a photoreceptor sheet is shown. These reduced preparations were depolarized with 20 mm KCl (gray bars) att = 0 min after stabilization in 2 mmKCl (white bars). In these experiments, glutamate release stabilized after 35 min in 20 mm KCl, which is when the first measurements were made. Glutamate release was markedly suppressed by caffeine added at t = 45 min (to 47% of control; black bars). Each barrepresents the glutamate content of 5 min samples of the perfusate. The effect of caffeine was reversible; during the washout (att = 65 min) a consistent rebound increase in glutamate release was observed (n = 15).
The concentration dependence of caffeine is shown in Figure7A. Superfusion with 1 mm caffeine had no effect on glutamate release. On the other hand, exposure to either 5 or 10 mmcaffeine resulted in very similar reductions in glutamate release. The concentration dependence of caffeine on glutamate release matches well that on [Ca2+]i. Because ryanodine reduced the caffeine-mediated depression of [Ca2+]i, it might be expected to reduce the action of caffeine on release of glutamate from rods. We found that ryanodine alone at 20 μm had no effect on glutamate release, either in normal saline or in the 20 mm KCl saline (Fig.7B). In the presence of ryanodine, moreover, 10 mm caffeine still produced a 59% inhibition of glutamate release (4.17 ± 0.38 vs 1.72 ± 0.17;n = 8) (Fig. 7B). Although ryanodine blocked approximately one-third of caffeine-evoked [Ca2+]idepression, these experiments did not reveal an effect of ryanodine on caffeine-suppressed glutamate release.
Fig. 7.

Dose dependence and ryanodine sensitivity of the caffeine-evoked reduction in glutamate release. A, The effect of caffeine (filled bars) versus control (open bars) on release was observed at 5 mm(2.14 ± 0.14 vs 4.37 ± 0.35 control) and 10 mm(1.96 ± 0.23 vs 4.67 ± 0.41 control) caffeine but not with 1 mm caffeine (2.86 ± 0.22 vs 2.64 ± 0.19 in control). B, Ryanodine (20 μm) had no effect on glutamate release from reduced retinas under normal conditions or when depolarized by KCl (20 mm). Data are normalized to the average of the control samples (open bars). In the presence of ryanodine, 10 mm caffeine still suppressed glutamate release by 42.5%.
Caffeine inhibits calcium current in isolated rods
The data presented so far suggest that caffeine affects exocytosis by modulating Ca2+ release from stores, resulting in alterations of [Ca2+]i. An alternative, but not mutually exclusive, hypothesis is that caffeine modulates Ca2+ influx through the voltage-dependent Ca2+ channels in rods. The following experiments tested that hypothesis. Figure8 illustrates an I–V curve for an L-type calcium current recorded from a rod in whole-cell mode. This current, ICa, peaked at ∼0 mV (Fig. 8A) and showed little inactivation during 120 msec voltage steps when the pipette solution contained 10 mm EGTA (see Fig. 8A, inset).ICa was completely blocked by Cd2+ (100 μm;n = 26; data not shown). Caffeine (5 and 10 mm) reversibly suppressed peakICa by 35.4 ± 3.1 and 50.0 ± 2.7%, respectively (n = 29; Fig.8A,B). Caffeine had no detectable effect on the voltage range over whichICa was activated. Often during caffeine washout, a rebound increase in Ca2+ currents was observed (Fig.8B, arrow), with the Ca2+ current returning to its precaffeine level after a 4–6 min wash (Fig.8A,B).
Fig. 8.

Caffeine reduces ICa in rods. Calcium current was recorded with whole-cell patch pipettes. A, Calcium currents were evoked with voltage-clamp ramps from −70 to +50 mV and with voltage-clamp steps (inset, −70 to +50 mV in 10 mV increments). Caffeine (CAF; 10 mm) reduced the peak amplitude (at −10 mV) but did not affect the activation voltage. Note the rebound increase in ICa after washout of caffeine.B, Time course of caffeine action onICa is shown. The cell was held at −70 mV and stepped for 120 msec to 0 mV. Test pulses were applied every 20 sec. Sequential application of 10 mm caffeine reversibly reduced ICa. After the first washout,ICa rebounded to levels above control.
The predominant action of caffeine on ICain rods requires changes in [Ca2+]i
The suppression of ICa by caffeine could result from either a direct caffeine action on the Ca2+ channel or an indirect one mediated by release of Ca2+ from intracellular stores (Pacaud et al., 1987; Kramer et al., 1994; Adachi-Akahane et al., 1996) or both. The indirect action of caffeine should be reduced or eliminated by blockers of Ca2+ release from stores. Figure 9A shows that in the presence of ryanodine the inhibitory action of 10 mm caffeine on peakICa was reduced by approximately one-half. In ryanodine the mean reduction of peak current was 25.7 ± 5.8% (n = 8) versus 50.0 ± 2.7% in control. These data indicate that more than one-half of the caffeine-induced suppression of ICa results from release from ryanodine-sensitive stores.
Fig. 9.
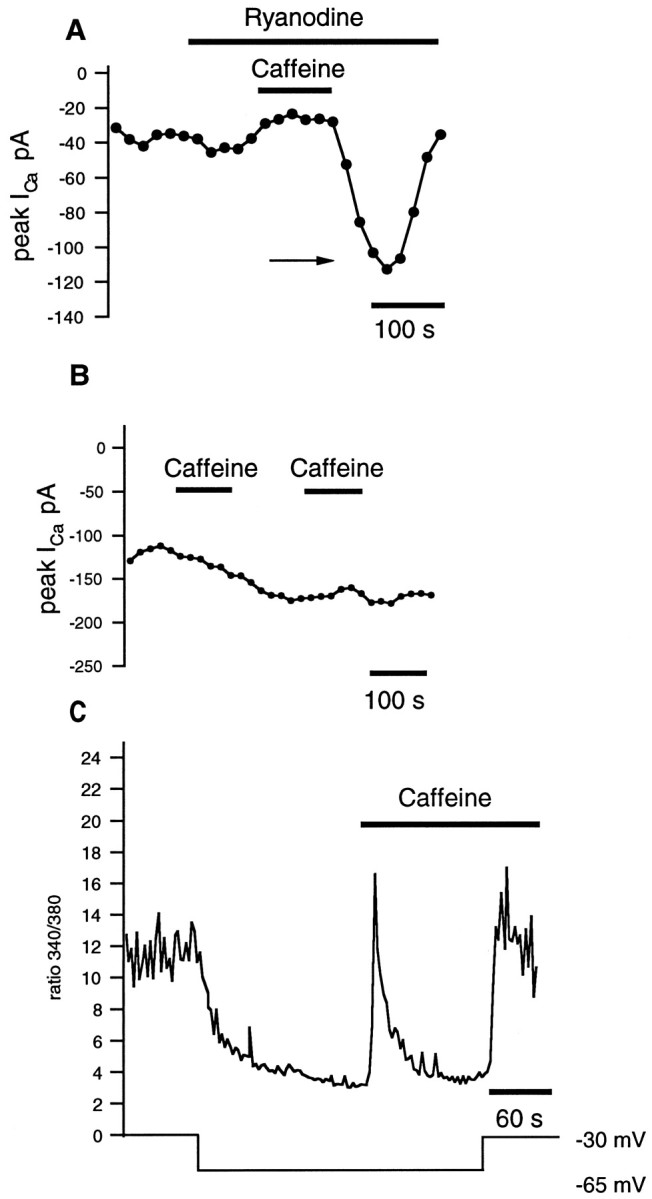
Ryanodine and BAPTA reduce the effectiveness of caffeine on ICa. The rods were held at −70 mV and periodically (0.02 Hz) depolarized with a voltage ramp to +50 mV. The peak of ICa is plotted inA and B. A, Ryanodine (20 μm) suppressed the caffeine-mediated decrease inICa. B, This rod was dialyzed with 10 mm BAPTA in the patch pipette. Repetitive applications of caffeine did not reduce peakICa under these conditions.C, [Ca2+]i was monitored in a rod held under voltage clamp with a perforated patch electrode. The switch in potential from −30 to −65 mV reduced [Ca2+]i. Caffeine evoked a transient increase in [Ca2+]i. Switching the membrane potential back to −30 mV increased [Ca2+]i, indicating that the voltage-dependent channels were not closed by caffeine.
The inhibitory effect of caffeine onICa was much more pronounced in the presence of intrapipette EGTA compared with BAPTA, a Ca2+ chelator with a higher affinity for Ca2+ and faster kinetics of binding (Tsien, 1980). In the rod shown in Figure 9B, intracellular BAPTA blocked virtually all the caffeine-induced suppression ofICa. In summary, we found that when rods were loaded with 10–20 mm BAPTA, the average peak amplitude of ICa was significantly larger than that in controls (186.6 ± 74.5 pA;n = 5). This larger current is consistent with the idea that ICa is being tonically inhibited by cytosolic calcium. In these BAPTA-loaded cells, the inhibitory effect of caffeine on Ca2+ current was substantially reduced. Caffeine reducedICa by 20.3 ± 7.7%. The difference between control and caffeine’s action was not significant statistically. These experiments demonstrate that a major proportion of caffeine’s suppression of ICa can be attributed to effects of caffeine on intracellular calcium.
Further indication that Ca2+ released by caffeine acts to inhibit ICa was provided by experiments in which [Ca2+]o was replaced by 10 mm[Ba2+]o. Although Ba2+ currents are well supported by neuronal L-type calcium channels, including those of photoreceptors (Corey et al., 1984; Tsien et al., 1988), several features of cellular Ba2+ regulation differ from the regulation of Ca2+. Ba2+, for example, is not recognized by SERCAs and is thus not sequestered into intracellular stores (Schilling et al., 1989; Kwan and Putney, 1990). Moreover, in contrast to Ca2+, cytoplasmic Ba2+ ions do not inactivate calcium channels (Haack and Rosenberg, 1994) and may inhibit release of Ca2+ through ryanodine receptor-gated channels, thus allowing one to differentiate between direct caffeine effects on the conductance or open probability of the L-type calcium channel and its indirect effects on the channel via Ca2+ released from intracellular stores. In rod photoreceptors, peak ICaelicited by a depolarizing voltage ramp was 98.7 ± 13.1 pA. After a switch of the superfusate from [Ca2+]o to [Ba2+]o, peakICa increased to 300.8 ± 39.8 pA. Caffeine inhibited the Ba2+ current by 27.6 ± 6.6% (n = 8; data not shown) compared with 50 ± 2.7% in the Ca2+-containing saline. This result suggests that the upper bound for caffeine-mediated inhibition ofICa by direct block of the channels is 55%. In these experiments, if there were residual calcium in the stores after the change to Ba2+, this calcium could have been released by caffeine to inhibit the channels via the indirect pathway (Przywara et al., 1993), resulting in an overestimate of caffeine’s direct effect on the channels.
As an alternative method to test whether calcium influx viaICa was suppressed in caffeine, we measured [Ca2+]iwith fura-2 while recording under voltage clamp with perforated-patch recording pipettes. We found that a depolarization-induced influx of Ca2+ was evident in the presence of caffeine. Figure 9C illustrates that [Ca2+]i decreased when the membrane potential was lowered from −30 to −65 mV, a reflection of deactivation of ICa. After the caffeine-evoked transient increase in [Ca2+]i, the membrane potential was switched back to −30 mV. [Ca2+]i rose to a slightly higher level than the precaffeine level at −30 mV. These data illustrate that depolarization-induced calcium influx is not suppressed by caffeine, indicating that caffeine did not significantly block the L-type channels directly. These data, taken as a whole, indicate that a large proportion of caffeine’s effect is a result of caffeine acting inside the rods, presumably by releasing calcium from stores. Experiments discussed below add support to the idea that it is calcium release from stores that suppresses [Ca2+]i .
Chlorocresol, a nonxanthine store-releasing compound, and spontaneous calcium spikes produce a depression in [Ca2+]i
Xanthines, such as caffeine, inhibit phosphodiesterases (PDEs) in addition to releasing calcium from stores (Nehlig et al., 1992). To test whether the caffeine-induced changes in [Ca2+]i were separable from any inhibition of PDEs, we used 4-CmC, a nonxanthine compound that releases calcium from stores (Zorzato et al., 1993; Cseresnyes et al., 1997). Figure10 shows the action of 4-CmC on [Ca2+]i in a rod. 4-CmC elevated [Ca2+]itransiently and evoked a prolonged decrease, similar to the undershoot elicited by caffeine (n = 13). 4-CmC was shown to release [Ca2+]ifrom the stores at a much slower rate than caffeine (Cseresnyes et al., 1997); hence the transient rise in calcium might be expected to be smaller than that in caffeine. Similar depressions in [Ca2+]i were observed in thymol, another nonxanthine compound known to release Ca2+ from stores (n = 4; data not shown).
Fig. 10.

[Ca2+]i is depressed by nonxanthine store-releasing compounds and after spontaneous calcium spikes. A, 4-Chloro-m-cresol (500 μm) evoked a transient peak and a subsequent depression in [Ca2+]i.B, Ratios (340/380 nm) from two simultaneously recorded rods in 20 mmKCl are plotted. In this particular recording session, we were not able to perform absolute calibrations of [Ca2+]i on these particular cells and so have used the 340/380 ratio as the indicator of calcium concentration. Caffeine (10 mm) was applied at 14.9 and 35.4 min. At t = 23.7 min, rod 2 (bottom solid line) exhibited a spontaneous regenerative increase in [Ca2+]i(thick open arrow) followed by an undershoot (thin filled arrow). No such phenomenon was exhibited by rod 1 (top dotted line).
Spikes of [Ca2+]iobserved in neuronal and non-neuronal cells primarily result from the spontaneous release of calcium from ryanodine-sensitive stores (Berridge, 1998). We observed that spontaneous spikes in rod [Ca2+]i often were followed by a depression of [Ca2+]i below the resting level. Figure 10B illustrates caffeine-evoked increases and depression of [Ca2+]i recorded from two rods simultaneously. In between caffeine applications, a spontaneous increase in [Ca2+]i, termed a calcium spike, occurred in one of the rods but not in the other. The increase (Fig. 10B, thick open arrow) was followed by an undershoot (thin filled arrow). The existence of the depressive phases after a spike suggests that even a noncaffeine-mediated release can cause a depression of [Ca2+]i, supporting the hypothesis that it is the transient release of calcium that triggers the prolonged decrease in intracellular calcium concentration.
As a further test of a possible role for caffeine-evoked calcium release in the depression of [Ca2+]i, we used fura-2 to monitor intracellular Ba2+(Schilling et al., 1989). As mentioned above, because barium is sequestered poorly into stores (Kwan and Putney, 1990; Adachi-Akahane et al., 1996), the effects of caffeine on [Ba2+]i provide a measure of how caffeine affects influx through the calcium channels. Figure 11 shows that replacement of extracellular Ca2+ by Ba2+ increased fura-2 ratios. This increase reflects the higher permeability ofICa to barium. However, Ba2+ substitution for Ca2+ resulted in a block of the caffeine-evoked transient increase and a significant decrease of [Ba2+ + Ca2+]i depression (16 ± 4% relative to the baseline in 2 mm[Ba2+]o compared with control 82 ± 3% relative to the baseline in 2 mm[Ca2+]i;n = 6). This blockage of the initial transient increase is consistent with a diminished release of Ca2+ from the stores. The small degree of caffeine-induced depression in barium indicates that direct effects of caffeine on ICa were <19% (=16/82) of the depression mediated by calcium released from the stores.
Fig. 11.

Extracellular barium reduces the effects of caffeine on [Ca2+]i. Caffeine was applied before, during, and after barium substitution for extracellular calcium.Left, Caffeine elicited a typical response consisting of a transient increase in [Ca2+]ifollowed by a depression. Middle, Caffeine evoked a much smaller change in [Ca2+]i (measured here as 340/380 ratios) during Ba2+ substitution.Right, The washout response to caffeine is shown and demonstrates a partial recovery of both the transient elevation and the more prolonged undershoot of [Ca2+]iin Ca2+-containing saline.
DISCUSSION
We report here that caffeine transiently raises and then depresses [Ca2+]i in rod photoreceptors. Exposure to caffeine also reduces the L-type calcium current. These presynaptic events are paralleled by a transient depolarization and subsequent hyperpolarization of the membrane potential of a second-order retinal neuron, the horizontal cell. Release of endogenous glutamate from rods is also depressed by caffeine. The poor temporal resolution of our glutamate measurements did not allow us to observe a transient increase in release. Our data provide novel evidence that intracellular calcium stores play a role in mediating glutamate release. The main question raised by our findings is the mechanism(s) by which caffeine modulates [Ca2+]i and the rate of transmitter release. Figure 12schematizes the probable mechanisms. These are discussed in detail below.
Fig. 12.

Schematic diagram of the proposed action of caffeine on ICa and intracellular stores and the action of store-released calcium on glutamate release,ICa, and the release-activated calcium transport. Caffeine was shown to suppressICa moderately via a direct action and more significantly via an indirect one in which intracellular calcium led to a decrease in ICa. Caffeine also caused a transient peak in calcium that is attributable to a caffeine-induced release from stores. The depression of [Ca2+]i can be explained by a release-activated uptake into stores and the suppression ofICa.
Caffeine-evoked transient [Ca2+]iincreases reflect discharge of calcium from ryanodine-sensitive stores
The caffeine-mediated transient increase in [Ca2+]i observed in rods has been reported in a multitude of other cell types and reflects immediate calcium release from the stores (Friel and Tsien, 1992; Orkand and Thomas, 1995; Garaschuk et al., 1997). The observation that the magnitude of the transient depended on the basal level of [Ca2+]i (Fig.1B) is consistent with greater amounts of calcium being pumped into the caffeine-sensitive stores by the constitutively active SERCA pumps. Supporting evidence of store release comes from our finding that the transient increase in [Ca2+]i is substantially reduced when Ca2+ is replaced with Ba2+ (Fig. 11), a divalent cation that is poorly pumped into stores (Kwan and Putney, 1990). In addition, ryanodine blocked the transient increases of [Ca2+]i, suggesting that caffeine acts on an intracellular store expressing a conventional class of ryanodine receptors (Pozzan et al., 1994;Berridge, 1998). It is noteworthy that ryanodine receptors have been reported recently in photoreceptor synaptic terminals (Gabriel et al., 1998). Caffeine-sensitive stores in rods may coexist with the other major class of calcium stores, gated by the inositol trisphosphate receptor (Peng et al., 1991) (Krizaj and Copenhagen, unpublished observations). Ca2+ release from these stores is inhibited by caffeine (Ehrlich et al., 1994) and thus is unlikely to contribute to caffeine-mediated changes in rod [Ca2+]i.
Caffeine-evoked calcium release contributes significantly to the reduction of ICa and the depression of [Ca2+]i
We show that caffeine-induced calcium release suppressesICa and depresses [Ca2+]i. We propose that a portion of the depression of [Ca2+]i can be attributed to the reduction of calcium entry viaICa; however there must be additional mechanisms by which [Ca2+]i is depressed. First we will discuss the suppression ofICa by caffeine. Intracellular BAPTA, ryanodine, and the substitution of calcium by barium significantly reduced the suppressive action of caffeine onICa (Fig. 9). These experiments can be taken as strong evidence that an intracellular calcium-dependent mechanism, triggered by caffeine, leads to suppression ofICa. A smaller component of caffeine’s action on ICa could be attributed to a direct effect in the channels (Hughes et al., 1990). Mechanistically, ICa could be inactivated by a localized and restricted rise in caffeine-released calcium near the channel (Adachi-Akahane et al., 1996; Sham, 1997) or by a release-triggered activation of a second messenger cascade. In consideration of the first mechanism, L-type voltage-gated calcium channels are inhibited by direct binding of Ca2+ to the α1 subunit of the channel (de Leon et al., 1995). Calcium release from both caffeine-sensitive and IP3-gated stores inactivate Ca2+ channels in other cell types (Kramer et al., 1994; Adachi-Akahane et al., 1996; Lara et al., 1997). Consistent with the required proximity of the release site to the calcium channel is our finding that BAPTA, but not EGTA, reduced the caffeine-evoked suppression of ICa. Similar close proximities were demonstrated in other excitable cell types (Henkart et al., 1976; Walton et al., 1991; Sham, 1997; Tse et al., 1997) including hair cells (Sridhar et al., 1996) and invertebrate photoreceptors (Minke and Selinger, 1996). Alternatively, caffeine-released calcium might activate a second messenger, such as calcineurin (Schumann et al., 1997), or a calcium dependent kinase/phosphatase cascade that regulates the channel directly. If store-released calcium activated a second messenger cascade, the source of calcium need not be as close to the channel.
To depress [Ca2+]i, caffeine must either reduce ongoing influx, via an action onICa, or stimulate extrusion from the rods or uptake into the stores. An examination of the time course and pharmacology of caffeine-mediated depression indicates that simple suppression of ongoing influx is an insufficient explanation for caffeine’s action. As evidence we found that (1) during 250 msec puffs of caffeine, the caffeine-induced depression lasted much longer (>120 sec) than what would be predicted from the washout (∼10 sec) and from the time necessary for equilibration by diffusion of caffeine between the extracellular space and the cytoplasm [∼1 sec (Lipscombe et al., 1988; O’Neill et al., 1990; Friel and Tsien, 1992)]; (2) the fall of [Ca2+]i from the transient peak was much more rapid for larger excursions from the baseline (Fig. 3); (3) similar depressions in [Ca2+]i were observed after spontaneous regenerative spikes in rod [Ca2+]i (Fig. 10) in the absence of caffeine; (4) depressions in [Ca2+]i were observed with 4-chloro-m-cresol and thymol, two nonxanthine compounds known to release Ca2+ from caffeine-sensitive stores (Zorzato et al., 1993; Cseresnyes et al., 1997); and (5) the depression was substantially reduced by substitution of Ca2+ by Ba2+.
The caffeine-evoked depression of [Ca2+]i is also unlikely to result from an inhibitory action of caffeine on a phosphodiesterase that reduces intracellular cyclic nucleotides. We found that exposure to caffeine did not markedly affect either the membrane potential or the light responses of dark-adapted rods. This strongly suggests that the cGMP-dependent phosphodiesterases in rod outer segments were not inhibited significantly by 10 mmcaffeine. Furthermore, we found that 100 μm IBMX, a dose that should block PDEs, did not affect release of Ca2+ from caffeine-sensitive channels (Capovilla et al., 1983; Cervetto and McNaughton, 1986; Cseresnyes et al., 1997) and had no effect on the steady-state [Ca2+]i in rods (n = 6). It should be noted that our findings contrast to a previous report by Capovilla et al. (1983) in which 0.3 mm caffeine increased the amplitude of rod light responses in toad. Finally, the experiments with 4-CmC (0.5 mm) and thymol (0.1 mm), two nonxanthine compounds not known to interfere with phosphodiesterases, mimicked caffeine by generating a depression of [Ca2+]i.
Voltage-clamp and microelectrode recordings eliminate the possibility that caffeine depressed [Ca2+]i by activating Ca2+-dependent conductances that hyperpolarized the rods to decrease the activation of the L-type calcium channels and hence calcium influx (Akaike et al., 1983; Sah and McLachlan, 1991; Marrion and Adams, 1992; Sridhar et al., 1996). Salamander rods possess calcium-activated chloride and potassium conductances (Bader et al., 1982). Although these conductances may have increased after caffeine-mediated release of Ca2+ from the stores, they could not have contributed significantly to the reduction inICa that persisted under voltage clamp (Fig. 8). Moreover, intracellular recording from rods showed no effect of caffeine on rod membrane potential, whereas both glutamate release and horizontal cell membrane potential were markedly depressed under those conditions (Figs. 5, 6). This indicates that a change in the rod membrane potential is not likely to be responsible for generating the depression.
Yet another explanation might be proposed that caffeine suppresses the IP3 receptor on the inner segment ER (Peng et al., 1991; Ehrlich et al., 1994) and thus inhibits a possible depolarization-activated tonic IP3-gated release of Ca2+ (Gan and Iuvone, 1997). However, an effect similar to that of caffeine was obtained with nonxanthine compounds, such as 4-CmC, that are not thought to inhibit IP3 receptors.
A release-activated stimulation of calcium uptake or extrusion likely contributes to caffeine-evoked depression of [Ca2+]i
It has been postulated that caffeine-induced calcium release from stores stimulates calcium uptake into ER or into mitochondria (Friel and Tsien, 1992; Orkand and Thomas, 1995; Cseresnyes et al., 1997; Lara et al., 1997; Golovina and Blaustein, 1998). Our finding that 1 mm caffeine slightly potentiated glutamate release whereas 10 mm caffeine inhibited it is similar to a previous report on the effect of caffeine on [Ca2+]i in sympathetic neurons (Friel and Tsien, 1992). Friel and Tsien were the first to propose that “depending on its Ca2+ content, the caffeine-sensitive store can either attenuate or potentiate responses to depolarization.” Recently, Cseresnyes et al. (1997) reported that in sympathetic neurons caffeine greatly potentiates calcium removal from the cytosol. After being triggered by caffeine-induced release, this mechanism continues removing calcium from the cytosol, resulting in a substantial lowering of [Ca2+]i. Our findings in rods that caffeine evokes a depression of [Ca2+]i to values well below prestimulus levels and that the falling phase of the caffeine-evoked Ca2+ signal is faster (Fig. 3) are consistent with release-stimulated sequestration or extrusion.
Why did ryanodine not completely eliminate the decreases in [Ca2+]i and glutamate release?
In isolated cells, ryanodine eliminated caffeine-induced increases in [Ca2+]i, consistent with its action as a blocker of the ryanodine receptor. Ryanodine also reduced the caffeine-mediated decrease in [Ca2+]i andICa, suggesting that at least part of the undershoot in [Ca2+]i and glutamate release is mediated by Ca2+release from ryanodine-gated channels. The finding that ryanodine did not completely block the [Ca2+]i undershoot suggests that at least part of the Ca2+release, activated by caffeine, may be from another, ryanodine-insensitive, class of calcium stores, similar to those described in several other cell types (Marrion and Adams, 1992; McNulty and Taylor, 1993; Orkand and Thomas, 1995; Pessah et al., 1997). This release may be missed by the global fura-2 measurements yet significant enough to evoke a depression in [Ca2+]i (e.g., Fig. 4). Surprisingly, ryanodine had no effect on the caffeine-mediated reduction of glutamate release. Possibly this result was a consequence of poor tissue permeability of ryanodine, as has been hypothesized for tissue slices (Llano et al., 1995). Consistent with this interpretation of poor tissue penetration was the observation that even the caffeine effect on horizontal cell potential was somewhat delayed.
The role of calcium stores in the regulation of transmitter release from rods
Our results demonstrate that intracellular stores in rods are capable of storing significant amounts of calcium. This finding complements the cytochemical and EM studies that localized the highest concentrations of calcium in amphibian and teleost rods to the inner segment ER (Mercurio and Holtzman, 1982; Ungar et al., 1984; Somlyo and Walz, 1985). Our finding that the pool size of caffeine-releasable intracellular calcium depends on the basal levels of [Ca2+]i suggests that the participation of calcium stores in synaptic signaling will be more significant in darkness, when [Ca2+]i is high (Ratto et al., 1988; Gray-Keller and Detwiler, 1994), than in the light, when inner segment [Ca2+]i drops to levels as low as 20–50 nm (Krizaj and Copenhagen, 1998).In vivo, calcium is released from ryanodine-sensitive stores by cytosolic calcium, by a process termed calcium-induced calcium release. The concentration range over which calcium activates the ryanodine receptor, an approximately sigmoidal activation by Ca2+ at concentrations between 10 nm and 10 μm(Bezprozvanny et al., 1991; Hernandez-Cruz et al., 1995), indicates that calcium stores in rod inner segments and synaptic terminals could be activated in both dark and light.
In darkness, steady influx of Ca2+ through the L-type Ca2+ channels (Rieke and Schwartz, 1996) would trigger Ca2+ release from the stores. The high density of ryanodine receptors, the long open times and high single-channel conductances of ryanodine receptor-gated channels [100 to ∼400 pS (Bezprozvanny et al., 1991; Hernandez-Cruz et al., 1995)], and the close proximity of Ca2+ stores to the plasma membrane (Sridhar et al., 1996; Sham, 1997) would create a local negative feedback loop. Ca2+ released from the stores would inactivate the L-type channels and thus reduce calcium influx. The resulting depression of [Ca2+]i is accompanied by facilitation of Ca2+ uptake into the stores (Orkand and Thomas, 1995; Cseresnyes et al., 1997; Lara et al., 1997). Thus, the compounded action of Ca2+-induced inactivation of Ca2+ channels and enhancement of calcium uptake would result in a reduced [Ca2+]i and a reduction of transmitter release. Conversely, in the light, when Ca2+ channels are closed and the Ca2+ influx is low, the stores would be relatively quiescent, and each Ca2+ ion would be proportionally more efficient in triggering release of vesicles filled with glutamate. The stores could therefore act as a negative feedback regulator, increasing the dynamic range of the synapse via its control of [Ca2+]i and transmitter release.
Footnotes
This work was supported by the National Institutes of Health (D.R.C. and P.W.), Research to Prevent Blindness (D.R.C. and P.W.), the Hoffritz Foundation (P.W.), an Association for Research in Vision and Ophthalmology / CIBA Vision Corporation award (D.K.), and the Deutsche Forschungsgemeinschaft (Y.S.). Additional support was provided by That Man May See, Inc. We thank Drs. Fred Rieke and Richard Lewis for valuable comments on previous versions of this manuscript.
Correspondence should be addressed to Dr. David Krizaj, Department of Ophthalmology, University of California at San Francisco School of Medicine, Box 0730, Room K-140, 10 Kirkham Street, San Francisco, CA 94143-0730.
Dr. Schmitz’s present address: Department of Neurology, Columbia University, New York, NY 10032.
REFERENCES
- 1.Adachi-Akahane S, Cleeman L, Morad M. Cross-signaling between L-type Ca2+ channels and ryanodine receptors in rat ventricular myocytes. J Gen Physiol. 1996;108:435–454. doi: 10.1085/jgp.108.5.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akaike N, Brown AM, Dahl G, Higashi H, Isenberg G, Tsuda Y, Yatani A. Voltage-dependent activation of potassium current in Helix neurones by endogenous cellular calcium. J Physiol (Lond) 1983;334:309–324. doi: 10.1113/jphysiol.1983.sp014496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bader CR, MacLeish PR, Schwartz EA. A voltage-clamp study of the light response in solitary rods of the tiger salamander. J Physiol (Lond) 1979;296:1–26. doi: 10.1113/jphysiol.1979.sp012988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bader CR, Bertrand D, Schwartz EA. Voltage-activated and calcium-activated currents studied in solitary rod inner segments from the salamander retina. J Physiol (Lond) 1982;331:253–284. doi: 10.1113/jphysiol.1982.sp014372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnes S, Hille B. Ionic channels of the inner segment of tiger salamander cone photoreceptors. J Gen Physiol. 1989;94:719–743. doi: 10.1085/jgp.94.4.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 7. Bezprozvanny I, Watras J, Ehrich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351 1991. 751 754 J Physiol (Paris) 76:459–470. [DOI] [PubMed] [Google Scholar]
- 8.Cahill GM, Besharse JC. Light-sensitive melatonin synthesis by Xenopus photoreceptors after destruction of the inner retina. Vis Neurosci. 1992;8:487–490. doi: 10.1017/s0952523800005009. [DOI] [PubMed] [Google Scholar]
- 9.Capovilla M, Cervetto L, Torre V. The effect of phosphodiesterase inhibitors on the electrical activity of toad rods. J Physiol (Lond) 1983;343:277–294. doi: 10.1113/jphysiol.1983.sp014892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cervetto L, McNaughton PA. The effects of phosphodiesterase inhibitors and lanthanum ions on the light-sensitive current of toad retinal rods. J Physiol (Lond) 1986;370:91–109. doi: 10.1113/jphysiol.1986.sp015924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheek TR, O’Sullivan AJ, Moreton RB, Berridge MJ, Burgoyne RD. The caffeine-sensitive Ca2+ store in bovine adrenal chromaffin cells; an examination of its role in triggering secretion and Ca2+ homeostasis. FEBS Lett. 1990;266:91–95. doi: 10.1016/0014-5793(90)81514-o. [DOI] [PubMed] [Google Scholar]
- 12.Copenhagen DRC, Jahr CE. Release of endogenous excitatory amino acids from turtle photoreceptors. Nature. 1989;341:536–539. doi: 10.1038/341536a0. [DOI] [PubMed] [Google Scholar]
- 13.Corey DP, Dubinsky JM, Schwartz EA. The calcium current in inner segments of rods from the salamander (Ambystoma tigrinum) retina. J Physiol (Lond) 1984;354:557–575. doi: 10.1113/jphysiol.1984.sp015393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cseresnyes Z, Bustamante AI, Klein MG, Schneider MF. Release-activated Ca2+ transport in neurons of frog sympathetic ganglia. Neuron. 1997;19:403–419. doi: 10.1016/s0896-6273(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 15.de Leon M, Wang Y, Jones L, Perez-Reyes E, Wei X, Soong TW, Snutch T, Yue D. Essential Ca2+-binding motif for Ca2+-sensitive inactivation of L-type Ca2+ channels. Science. 1995;270:1502–1506. doi: 10.1126/science.270.5241.1502. [DOI] [PubMed] [Google Scholar]
- 16.Ehrlich BE, Kaftan E, Bezprozvannaya S, Bezprozvanny I. The pharmacology of intracellular Ca2+-release channels. Trends Pharmacol Sci. 1994;15:145–149. doi: 10.1016/0165-6147(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 17.Fain GL. Sensitivity of toad rods: dependence on wave-length and background illumination. J Physiol (Lond) 1976;261:71–101. doi: 10.1113/jphysiol.1976.sp011549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fosse VM, Kolstad J, Fonnum F. A bioluminescence method for measurement of L-glutamate: applications to the study of changes in release of L-glutamate from lateral geniculate nucleus and superior colliculus after visual ablation in rats. J Neurochem. 1986;47:340–349. doi: 10.1111/j.1471-4159.1986.tb04507.x. [DOI] [PubMed] [Google Scholar]
- 19.Friel DD, Tsien RW. A caffeine- and ryanodine-sensitive Ca2+ store in bullfrog sympathetic neurones modulates effects of Ca2+ entry on [Ca2+]i. J Physiol (Lond) 1992;450:217–246. doi: 10.1113/jphysiol.1992.sp019125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gabriel R, Akopian A, Witkovsky P. Co-localization of ryanodine- and inositol triphosphate receptors in retinal ganglion cells. IOVS Suppl. 1998;39:4564. [Google Scholar]
- 21.Gan J, Iuvone PM. Depolarization and activation of dihydropyridine-sensitive Ca2+ channels stimulate inositol phosphate accumulation in photoreceptor-enriched chick retinal cell cultures. J Neurochem. 1997;68:2300–2307. doi: 10.1046/j.1471-4159.1997.68062300.x. [DOI] [PubMed] [Google Scholar]
- 22.Garaschuk O, Yaari Y, Konnerth A. Release and sequestration of calcium by ryanodine-sensitive stores in rat hippocampal neurones. J Physiol (Lond) 1997;502:13–30. doi: 10.1111/j.1469-7793.1997.013bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golovina VA, Blaustein MP. Spatially and functionally distinct Ca2+ stores in sarcoplasmic and endoplasmic reticulum. Science. 1998;275:1643–1648. doi: 10.1126/science.275.5306.1643. [DOI] [PubMed] [Google Scholar]
- 24.Gray-Keller MP, Detwiler PB. The calcium feedback signal in the phototransduction cascade of vertebrate rods. Neuron. 1994;13:849–861. doi: 10.1016/0896-6273(94)90251-8. [DOI] [PubMed] [Google Scholar]
- 25.Grzynkiewicz G, Poenie M, Tsien RY. A new generation of Ca indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 26.Haack JA, Rosenberg RL. Calcium-dependent inactivation of L-type calcium channels in planar lipid bilayers. Biophys J. 1994;66:1051–1060. doi: 10.1016/S0006-3495(94)80886-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henkart ML, Landis DMD, Reese TS. Similarity of junctions between plasma membrane and endoplasmic reticulum in muscle and neurons. J Cell Biol. 1976;70:338–347. doi: 10.1083/jcb.70.2.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hernandez-Cruz A, Diaz-Muñoz M, Gomez-Chavarin M, Cañedo-Merino R, Protti DA, Escobar AL, Sierralta J, Suarez-Isla BA. Properties of the ryanodine-sensitive release channels that underlie caffeine-induced Ca2+ mobilization from intracellular stores in mammalian sympathetic neurons. Eur J Neurosci. 1995;7:1684–1699. doi: 10.1111/j.1460-9568.1995.tb00690.x. [DOI] [PubMed] [Google Scholar]
- 29.Hua SY, Nohmi M, Kuba K. Characteristics of Ca2+ release induced by Ca2+ influx in cultured bullfrog sympathetic neurones. J Physiol (Lond) 1993;464:245–272. doi: 10.1113/jphysiol.1993.sp019633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hughes A, Hering S, Bolton TB. The action of caffeine on inward barium current through voltage-dependent calcium channels in single rabbit ear artery cells. Pflügers Arch. 1990;416:462–466. doi: 10.1007/BF00370755. [DOI] [PubMed] [Google Scholar]
- 31.Katz B, Miledi R. Tetrodotoxin-resistant electrical activity in presynaptic terminals. J Physiol (Lond) 1969;203:459–487. doi: 10.1113/jphysiol.1969.sp008875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kramer RH, Mokkapatti R, Levitan ES. Effects of caffeine on intracellular calcium, calcium current and calcium-dependent potassium current in anterior pituitary GH3 cells. Pflügers Arch. 1994;426:12–20. doi: 10.1007/BF00374665. [DOI] [PubMed] [Google Scholar]
- 33.Krijnse-Locker J, Parton RG, Fuller SD, Griffiths G, Dotti CG. The organization of the endoplasmic reticulum and the intermediate compartment in cultured rat hippocampal neurones. Mol Biol Cell. 1995;6:1315–1332. doi: 10.1091/mbc.6.10.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krizaj D, Copenhagen DR. Compartmentalization of calcium extrusion in the outer and inner segments of vertebrate photoreceptors. Neuron. 1998;21:249–256. doi: 10.1016/s0896-6273(00)80531-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krizaj D, Akopian A, Witkovsky P. The effects of l-glutamate, AMPA, quisqualate, and kainate on retinal horizontal cells depend on adaptational state: implications for rod–cone interactions. J Neurosci. 1994;14:5661–5671. doi: 10.1523/JNEUROSCI.14-09-05661.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krizaj D, Bao JX, Copenhagen DR. Intracellular calcium stores in vertebrate rods and cones. Soc Neurosci Abstr. 1997;23:2360. [Google Scholar]
- 37.Kwan CY, Putney JW. Uptake and intracellular sequestration of divalent cations in resting and methacholine-stimulated mouse lacrimal acinar cells. Dissociation by Sr2+ and Ba2+ of agonist-stimulated divalent cation entry from the refilling of the agonist-sensitive intracellular pool. J Biol Chem. 1990;265:678–684. [PubMed] [Google Scholar]
- 38.Lara B, Lopez MG, Villaroya M, Gandia L, Cleeman L, Morad M, Garcia AG. A caffeine-sensitive Ca store modulates K+-evoked secretion in chromaffin cells. Am J Physiol. 1997;272:C1211–C1221. doi: 10.1152/ajpcell.1997.272.4.C1211. [DOI] [PubMed] [Google Scholar]
- 39.Lipscombe D, Madison DV, Poenie M, Reuter H, Tsien RY, Tsien RW. Spatial distribution of calcium channels and cytosolic calcium transients in growth cones and cell bodies of sympathetic neurons. Proc Natl Acad Sci USA. 1988;85:2398–2402. doi: 10.1073/pnas.85.7.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Llano I, DiPolo R, Marty A. Calcium-induced calcium release in cerebellar Purkinje cells. Neuron. 1995;12:663–673. doi: 10.1016/0896-6273(94)90221-6. [DOI] [PubMed] [Google Scholar]
- 41.MacLeish P, Barnstable CJ, Townes-Anderson E. Use of a monoclonal antibody as a substrate for mature neurons in vitro. Proc Natl Acad Sci USA. 1983;80:7014–7018. doi: 10.1073/pnas.80.22.7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marrion NV, Adams PR. Release of intracellular calcium and modulation of membrane currents by caffeine in bullfrog sympathetic neurones. J Physiol (Lond) 1992;445:515–535. doi: 10.1113/jphysiol.1992.sp018937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matthews G. Neurotransmitter release. Annu Rev Neurosci. 1997;19:219–233. doi: 10.1146/annurev.ne.19.030196.001251. [DOI] [PubMed] [Google Scholar]
- 44.McNulty TJ, Taylor CW. Caffeine-stimulated Ca2+ release from the intracellular stores of hepatocytes is not mediated by ryanodine receptors. Biochem J. 1993;291:799–801. doi: 10.1042/bj2910799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McPherson PS, Kim YK, Valdivia H, Knudson CM, Takekura H, Franzini-Armstrong C, Coronado R, Campbell KP. The brain ryanodine receptor: a caffeine-sensitive calcium release channel. Neuron. 1991;7:17–25. doi: 10.1016/0896-6273(91)90070-g. [DOI] [PubMed] [Google Scholar]
- 46.Meldolesi J, Pozzan T. The endoplasmic reticulum: a view from the lumen. Trends Biochem Sci. 1998;23:10–14. doi: 10.1016/s0968-0004(97)01143-2. [DOI] [PubMed] [Google Scholar]
- 47.Mercurio AM, Holtzman E. Smooth endoplasmic reticulum and other agranular reticulum in frog retinal photoreceptors. J Neurocytol. 1982;11:263–293. doi: 10.1007/BF01258247. [DOI] [PubMed] [Google Scholar]
- 48.Minke B, Selinger Z. The roles of trp and calcium in regulating photoreceptor function in Drosophila. Curr Opin Neurobiol. 1996;6:459–466. doi: 10.1016/s0959-4388(96)80050-x. [DOI] [PubMed] [Google Scholar]
- 49.Neher E. The use of fura-2 for estimating Ca buffers and Ca fluxes. Neuropharmacology. 1995;34:1423–1442. doi: 10.1016/0028-3908(95)00144-u. [DOI] [PubMed] [Google Scholar]
- 50.Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 51.Nehlig A, Daval JL, Debry G. Caffeine and the central nervous system: mechanisms of action, biochemical, metabolic and psychostimulant effects. Brain Res Rev. 1992;17:139–170. doi: 10.1016/0165-0173(92)90012-b. [DOI] [PubMed] [Google Scholar]
- 52.O’Neill SC, Donoso P, Eisner DA. The role of [Ca2+]i and [Ca2+] sensitization in the caffeine contracture of rat myocytes: measurement of [Ca2+]i and [caffeine]i. J Physiol (Lond) 1990;425:55–70. doi: 10.1113/jphysiol.1990.sp018092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Orkand RK, Thomas RC. Effects of low doses of caffeine on [Ca2+]i in voltage-clamped snail (Helix aspersa) neurones. J Physiol (Lond) 1995;489:19–28. doi: 10.1113/jphysiol.1995.sp021026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pacaud P, Loirand G, Mironneau C, Mironneau J. Opposing effects of noradrenaline on the two classes of voltage-dependent calcium channels of single vascular smooth muscle cells in short term primary culture. Pflügers Arch. 1987;410:557–559. doi: 10.1007/BF00586539. [DOI] [PubMed] [Google Scholar]
- 55.Peng YW, Sharp AH, Snyder SH, Yau KW. Localization of the inositol 1,4,5-triphosphate receptor in synaptic terminals in the vertebrate retina. Neuron. 1991;6:525–531. doi: 10.1016/0896-6273(91)90055-5. [DOI] [PubMed] [Google Scholar]
- 56.Pessah IN, Molinski TF, Meloy TD, Wong P, Buck ED, Allen PD, Mohr FC, Mack MM. Bastadins relate ryanodine-sensitive and -insensitive Ca2+ efflux pathways in skeletal SR and BC3H1 cells. Am J Physiol. 1997;272:C601–C614. doi: 10.1152/ajpcell.1997.272.2.C601. [DOI] [PubMed] [Google Scholar]
- 57.Pozzan T, Rizzuto R, Volpe P, Meldolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiol Rev. 1994;74:595–636. doi: 10.1152/physrev.1994.74.3.595. [DOI] [PubMed] [Google Scholar]
- 58.Przywara DA, Chowdhury PS, Bhave SV, Wakade TD, Wakade AR. Barium-induced exocytosis is due to internal calcium release and block of calcium efflux. Proc Natl Acad Sci USA. 1993;90:557–561. doi: 10.1073/pnas.90.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ratto GM, Payne R, Owen WG, Tsien RY. The concentration of cytosolic free calcium in vertebrate rod outer segments measured with fura-2. J Neurosci. 1988;8:3240–3246. doi: 10.1523/JNEUROSCI.08-09-03240.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rieke F, Schwartz EA. Asynchronous transmitter release: control of exocytosis and endocytosis at the salamander rod synapse. J Physiol (Lond) 1996;493:1–8. doi: 10.1113/jphysiol.1996.sp021360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sah P, McLachlan EM. Ca2+-activated K+ currents underlying the afterhyperpolarization in guinea pig vagal neurons: a role for Ca2+-activated Ca2+ release. Neuron. 1991;7:257–264. doi: 10.1016/0896-6273(91)90264-z. [DOI] [PubMed] [Google Scholar]
- 62.Schilling WP, Rajan L, Strobl-Jager E. Characterization of the bradykinin-stimulated calcium influx pathway of cultured vascular endothelial cells. J Biol Chem. 1989;264:12838–12848. [PubMed] [Google Scholar]
- 63.Schmid A, Dehlinger-Kremer M, Schulz I, Gogelein H. Voltage-dependent Ins3-insensitive calcium channels in membranes of pancreatic endoplasmic reticulum vesicles. Nature. 1990;346:374–376. doi: 10.1038/346374a0. [DOI] [PubMed] [Google Scholar]
- 64.Schmitz Y, Witkovsky P. Glutamate release by the intact light-responsive photoreceptor layer of the Xenopus retina. J Neurosci Methods. 1996;68:55–60. doi: 10.1016/0165-0270(96)00070-2. [DOI] [PubMed] [Google Scholar]
- 65.Schmitz Y, Witkovsky P. Dependence of photoreceptor glutamate release on a dihydropyridine-sensitive calcium channel. Neuroscience. 1997;78:1209–1216. doi: 10.1016/s0306-4522(96)00678-1. [DOI] [PubMed] [Google Scholar]
- 66.Schumann K, Romanin C, Baumgartner W, Groschner K. Intracellular Ca2+ inhibits smooth muscle L-type Ca2+ channels by activation of protein phosphatase type 2B and by direct interaction with the channel. J Gen Physiol. 1997;110:503–513. doi: 10.1085/jgp.110.5.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sham JSK. Ca2+ release-induced inactivation of Ca2+ current in rat ventricular myocytes: evidence for local Ca2+ signalling. J Physiol (Lond) 1997;500:285–295. doi: 10.1113/jphysiol.1997.sp022020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sitsapesan R, Williams AJ. Mechanisms of caffeine activation of single calcium-release channels of sheep cardiac sarcoplasmic reticulum. J Physiol (Lond) 1990;423:425–439. doi: 10.1113/jphysiol.1990.sp018031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Somlyo AP, Walz B. Elemental distribution in Rana pipiens retinal rods: quantitative electron probe analysis. J Physiol (Lond) 1985;358:183–195. doi: 10.1113/jphysiol.1985.sp015547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sridhar TS, Brown MC, Sewell WF. Unique postsynaptic signaling at the hair cell efferent synapses permits calcium to evoke changes on two time scales. J Neurosci. 1996;17:428–437. doi: 10.1523/JNEUROSCI.17-01-00428.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tse FW, Tse A, Hille B, Horstmann H, Almers W. Local Ca2+ release from internal stores controls exocytosis in pituitary gonadotrophs. Neuron. 1997;18:121–132. doi: 10.1016/s0896-6273(01)80051-9. [DOI] [PubMed] [Google Scholar]
- 72.Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- 73.Tsien RY. New calcium indicators and buffers with high selectivity against magnesium and protons: design, synthesis and properties of prototype structures. Biochemistry. 1980;19:2396–2404. doi: 10.1021/bi00552a018. [DOI] [PubMed] [Google Scholar]
- 74.Ungar F, Piscopo I, Letizia J, Holtzman E. Uptake of calcium by the endoplasmic reticulum of the frog photoreceptor. J Cell Biol. 1984;98:1645–1655. doi: 10.1083/jcb.98.5.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walton PD, Airey JA, Sutko JL, Beck CF, Mignery GA, Südhof TC, Deerinck TJ, Ellisman MH. Ryanodine and inositol triphosphate receptors coexist in avian cerebellar Purkinje neurons. J Cell Biol. 1991;113:1145–1157. doi: 10.1083/jcb.113.5.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Witkovsky P, Schmitz Y, Akopian A, Krizaj D, Tranchina D. Gain of rod to horizontal cell synaptic transfer: relation to glutamate release and a dihydropyridine-sensitive calcium current. J Neurosci. 1997;17:7297–7306. doi: 10.1523/JNEUROSCI.17-19-07297.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zorzato F, Scutari E, Tegazzin V, Clementi E, Treves S. Chlorocresol: an activator of ryanodine receptor-mediated Ca2+ release. Mol Pharmacol. 1993;44:1192–1201. [PubMed] [Google Scholar]