

Abstract
Mitochondria are sites of cellular energy production but may also influence life and death decisions by initiating or inhibiting cell death. Mitochondrial depolarization and the subsequent release of pro-apoptotic factors have been suggested to be required for the activation of a cell death program in some forms of neuronal apoptosis. We induced apoptosis in cultured rat hippocampal neurons by exposure to the protein kinase inhibitor staurosporine (STS) (300 nm). The time course of mitochondrial membrane potential (ΔΨm) during apoptosis was examined using the probe tetramethylrhodamine ethyl ester (TMRE). Cells exhibited no decrease in TMRE fluorescence, indicative of mitochondrial depolarization, up to 8 hr after STS exposure. Rather, baseline TMRE fluorescence remained unchanged up to 2 hr and thereafter actually increased significantly. Throughout this time period, the mitochondria could also be depolarized with the protonophore carbonyl cyanidep-trifluoromethoxy-phenylhydrazone (FCCP, 0.1 μm), exhibiting the same relative magnitude of fluorescence release (unquenching) as controls. Even after 16 hr of staurosporine treatment, neurons that showed signs of nuclear apoptosis maintained ΔΨm and could be depolarized with FCCP. In contrast, caspase-3-like activity had increased roughly sevenfold by 2 hr and >20-fold by 8 hr. Double-labeling of hippocampal neurons with the potential-sensitive probe Mitotracker Red Chloromethyl X-Rosamine and an antibody to cytochrome c demonstrated at the subcellular level that mitochondrial cytochrome c release also occurred in the absence of mitochondrial depolarization. These data suggest that mitochondrial depolarization is not a decisive event in neuronal apoptosis.
Keywords: programmed cell death, mitochondrial membrane potential (ΔΨm), cytochrome c, permeability transition pore (PTP), caspases, staurosporine (STS), tetramethylrhodamine ethyl ester (TMRE), carbonyl cyanidep-trifluoromethoxy-phenylhydrazone (FCCP)
It has become increasingly clear that the activation of a cell death program may be responsible for pathophysiological cell death in neurodegenerative disorders, including Alzheimer’s disease (Thompson, 1995). Known elements of this program include members of the bcl-2 gene family, Apaf-1, and proteases of the caspase family (Cohen, 1997; Reed, 1997a; Salvesen and Dixit, 1997;Zou et al., 1997).
Once thought to be merely the center of cellular ATP production, mitochondria are increasingly implicated as sensors and executioners in the cell’s decision to live or die (Kroemer et al., 1997; Reed, 1997b;Murphy et al., 1999) and may even influence the mode of cell death chosen, necrosis or apoptosis, depending on their functional state (Nicotera and Leist, 1997; Tsujimoto, 1997). It has been suggested that mitochondria are capable of releasing pro-apoptotic factors into the cytosol, including apoptosis-inducing factor (AIF), which has been shown to induce chromatin condensation and oligonucleosomal DNA fragmentation in isolated nuclei (Susin et al., 1996, 1999; Zamzami et al., 1996a), and cytochrome c, which can activate, with the help of Apaf-1 and (d)ATP, procaspase-9 and the caspase cascade (Li et al., 1997; Zou et al., 1997).
What remains as yet unresolved is the mechanism by which pro-apoptotic factors are released into the cytosol. One candidate is the so-called mitochondrial permeability transition pore (PTP) (Kroemer et al., 1997;Marzo et al., 1998). The PTP is triggered by an increase in matrix Ca2+, pro-oxidants, and mitochondrial depolarization and appears to be nonspecific and permeable to solutes under 1.5 kDa (Zoratti and Szabo, 1995). Because it results in inner membrane permeability to protons, PTP is associated with a decrease in mitochondrial transmembrane potential (ΔΨm) (Zoratti and Szabo, 1994; Zamzami et al., 1996a). Because cytochrome c has a molecular weight of 15 kDa, the opening of a PTP is probably not sufficient to release cytochrome c directly but could trigger cytochrome c release by causing mitochondrial swelling and/or rupture of the outer mitochondrial membrane (Vander Heiden et al., 1997).
A reduction in ΔΨm has been observed in a number of models of apoptosis (Ankarcrona et al., 1995; Zamzami et al., 1996b; Boise and Thompson, 1997; Wadia et al., 1998; Heiskanen et al., 1999). On the other hand, however, studies in non-neuronal cells have suggested that this is not a primary event in apoptosis (Kluck et al., 1997; Vander Heiden et al., 1997; Yang et al., 1997; Bossy-Wetzel et al., 1998; Yoshida et al., 1998). Thus, although the importance of mitochondria in programmed cell death (PCD) seems fairly clear with respect to the release of pro-apoptotic factors, the events preceding this remain unresolved. Staurosporine (STS), a potent inhibitor of survival protein kinases (Bertrand et al., 1994), is widely used to activate PCD in neuronal and non-neuronal cells (Falcieri et al., 1993; Koh et al., 1995; Prehn et al., 1997). We examined the importance of mitochondrial depolarization in STS-induced apoptosis of cultured rat hippocampal neurons using the potential-dependent probes tetramethylrhodamine ethyl ester (TMRE) and Mitotracker Red Chloromethyl X-Rosamine (CMXRos) in conjunction with digital video and immunofluorescence microscopy.
MATERIALS AND METHODS
Cell culture. Primary cultures of hippocampal neurons were prepared from neonatal (P1) Fischer 344 rats according to Prehn (1998). Dissected hippocampi were incubated for 20 min at 37°C in Leibovitz L-15 medium (Life Technologies, Eggenstein, Germany) containing 0.1% papain. Afterward, the medium was removed, and the cells were suspended by gentle trituration in MEM supplemented with 10% NU-serum, 2% B-27 supplement (50× concentrate), 2 mm l-glutamine, 20 mmd-glucose, 26.2 mm sodium bicarbonate, 100 U/ml penicillin, and 100 μg/ml streptomycin (Life Technologies). The suspension was layered over medium containing 10 mg/ml trypsin inhibitor and centrifuged for 10 min at 600 rpm. The cells were then resuspended, plated, and incubated in MEM at 37°C in an atmosphere of 95% air and 5% carbon dioxide. For imaging studies, cells were grown on poly-l-lysine-coated glass coverslips that had been placed into 35 mm Petri dishes (Falcon, Heidelberg, Germany). For immunofluorescence microscopy experiments, cells were plated onto eight-well tissue culture slides (Becton-Dickinson, Heidelberg, Germany). In all other cases, neurons were plated onto poly-l-lysine-coated 24-well plates (Nunc, Hamburg, Germany). After 24 hr in vitro, cultures were treated with the anti-proliferation agent cytosine β-arabinofuranoside (CAF) (1 μm; Sigma, Deisenhofen, Germany). After an additional 24 hr, the cells were given fresh CAF-containing medium. Experiments were performed on 8- to 10-d-old cultures. Animal care followed official government guidelines.
Induction of apoptosis. We induced apoptosis in rat hippocampal neurons by exposure to the protein kinase inhibitor STS. STS-induced cell death is a commonly used model in the study of apoptosis in both neuronal and non-neuronal cell types (Falcieri et al., 1993; Bertrand et al., 1994; Koh et al., 1995; Prehn et al., 1997); is characterized by shrinkage of the cell body, membrane blebbing, chromatin condensation, nuclear pyknosis, and positive labeling of 3′-OH-DNA ends using the terminal deoxynucleotidyl transferase-based dUTP-digoxigenin nick-end labeling (TUNEL) technique; and can be reduced by 24 hr pretreatment with the protein synthesis inhibitor cycloheximide or the G1/S cell cycle inhibitor mimosine, as well as by caspase inhibition (Koh et al., 1995;Prehn et al., 1997; Krohn et al., 1998). To induce apoptosis, staurosporine (Sigma; 1 mm stock in DMSO) was added to the culture medium to a final concentration of 300 nm, a concentration that has been shown to induce rapid apoptotic cell death in >75% of cultured rat hippocampal neurons (Prehn et al., 1997). Controls were exposed to an equal volume of the vehicle.
Estimation of mitochondrial membrane potential (ΔΨm). TMRE is a cationic, lipophilic dye that accumulates in the negatively charged mitochondrial matrix according to the Nernst equation potential (Ehrenberg et al., 1988). A TMRE (Molecular Probes, Leiden, The Netherlands) stock was prepared at a concentration of 10 mg/ml in DMSO and stored at −20°C. Working stocks of 1 mg/ml were made up fresh in distilled water. For estimation of ΔΨm, cells were incubated with 100 nm TMRE for 15 min at room temperature in HEPES-buffered saline (HBS) consisting of (in mm): 144 NaCl, 10 HEPES, 2 CaCl2, 1 MgCl2, 5 KCl, 10d-glucose; 320 mOsm; pH 7.4. The dye was present in the buffer during the entire course of the experiment. TMRE fluorescence was then measured using a fluorescence microscope (Axiovert 100 inverted-stage microscope; Zeiss, Oberkochen, Germany) with a 40× fluorescence objective and attenuated UV illumination from a 75 W xenon arc. Optics were as follows: excitation, 490 nm; dichroic mirror, 505 nm; and emission, >510 nm. Under these conditions, autofluorescence was negligible. Images were collected every 15 sec using an intensified CCD camera (C 2400–87, Hamamatsu, Herrsching, Germany) with the camera controller gain set to 2.5. Sixteen frames were averaged for each image, which was digitized as 256 × 256 eight-bit pixels. A background image was taken before each experiment and was later subtracted from the images. Data were analyzed using Argus-50 software (Hamamatsu). Neurons were recognized by morphology as well as by their position in a higher plane of focus than astrocytes; they generally showed considerably lower basal TMRE fluorescence. Fluorescence data, which reflect the average pixel intensity obtained from the neuronal soma excluding the nucleus, are expressed in arbitrary fluorescence units (FlU).
Baseline TMRE fluorescence was measured for the first 5 min of each experiment. At that point, the cells were exposed to the protonophore carbonyl cyanide p-trifluoromethoxy-phenylhydrazone (FCCP) (0.1 μm in TMRE-containing HBS; Sigma). FCCP depolarizes mitochondria by abolishing the proton gradient across the inner mitochondrial membrane (Gunter and Pfeiffer, 1990; Prehn et al., 1994). After 5 min of exposure, the FCCP-containing buffer was replaced with fresh TMRE-containing HBS, allowing the cells to recover ΔΨm.
For some experiments, the nuclei of cells were imaged after estimation of ΔΨm. The culture medium was returned, and cultures were incubated with the cell-permeant, live-cell nucleic acid stain SYTO 16 (1 mm stock in DMSO; Molecular Probes) at a concentration of 1 μm for 30 min at 37°C (Frey, 1995). The medium was then aspirated, the cultures were washed in HBS, and the nuclear fluorescence was acquired using the above-mentioned imaging system. Apoptotic nuclei were distinguished by increased SYTO 16 fluorescence, indicative of chromatin condensation (Frey, 1995).
Mitotracker Red CMXRos and cytochrome c double-labeling.After the indicated length of exposure to 300 nm STS, hippocampal cultures were incubated with 50 nm Mitotracker Red CMXRos (Molecular Probes) for 30 min at 37°C in culture medium. Similarly to TMRE, this dye is taken up by mitochondria as a result of ΔΨm (Susin et al., 1999). After uptake, CMXRos forms thiol conjugates with cell peptides, inhibiting release of the dye from mitochondria and enabling fixation of cells while retaining the dye. After incubation with CMXRos, cultures were washed with PBS, fixed for 20 min at 37°C with freshly made 4% formaldehyde in PBS, and permeabilized with 0.1% Triton X-100 for 3 min at 4°C. After permeabilization, the cultures were washed again, then incubated in blocking buffer (2% low-fat milk powder, 2% bovine serum albumin, 0.1% Tween 20, in PBS, pH 7.4) for 1 hr at room temperature. The primary antibody (anti-cytochrome c, clone 6H2.B4, PharMingen, San Diego, CA), which recognizes the native form of cytochrome c, was then added at a concentration of 10 μg/ml for 2 hr at room temperature. After washing, biotin-conjugated anti-mouse IgG (1:1000; Vector Laboratories, Burlingame, CA) was added for 1 hr at room temperature, followed by a streptavidin–Oregon Green conjugate (1 μg/ml, 20 min, Molecular Probes). CMXRos and Oregon Green fluorescence were observed using an Eclipse TE300 inverted microscope and either a 20× dry or 100× oil immersion objective (Nikon, Düsseldorf, Germany) with the following optics: for CMXRos, excitation, 510–560 nm; dichroic mirror, 575 nm; emission, >590 nm; for Oregon Green, excitation, 465–495 nm; dichroic mirror, 505 nm; emission, 515–555 nm. Digital images of equal exposure for control and STS-treated cultures were acquired with a SPOT-2 camera (Diagnostic Instruments, Sterling Heights, MI) using Metamorph software (Universal Imaging Corporation, West Chester, PA). The images were deconvoluted using No Neighbor Deblurring software (kindly provided by Dr. B. Lindemann, University of Saarbrücken, Germany), which applies the algorithm of Monck et al. (1992) to reduce image background haze attributable to light originating from unsharp areas of the specimen.
Assessment of mitochondrial mass: quantitation of Mitotracker Green FM and nonyl acridine orange fluorescence. Mitotracker Green FM (MTGFM) is a mitochondrion-selective probe that becomes fluorescent in the lipid environment of mitochondria. MTGFM contains a thiol-reactive chloromethyl moiety, resulting in stable peptide and protein conjugates after accumulation in mitochondria. Unlike TMRE and CMXRos, uptake of this probe is less dependent on ΔΨm (see Table 2 and manufacturer’s information), thus allowing estimation of mitochondrial mass in both live and fixed cells (Metivier et al., 1998). A 1 mm stock solution of Mitotracker Green FM (Molecular Probes) was made in DMSO. At the indicated time points after addition of STS, the stock solution was diluted in the culture medium to a final concentration of 200 nm, and the cultures were incubated for 30 min at 37°C. The cultures were then rinsed twice with PBS and extracted with 200 μl lysis buffer (10% SDS, 0.1 m Tris, pH 7.5). Extract fluorescence was then measured using the fluorescent plate reader described below, with an excitation wavelength of 485 nm and an emission wavelength of 530 nm. Nonyl acridine orange (NAO) accumulates in mitochondria by binding to cardiolipin and has been used previously to measure changes in mitochondrial mass (Cossarizza et al., 1995;Mancini et al., 1997; Metivier et al., 1998). Experiments with NAO were performed as with MTGFM, using a dye concentration of 1 μg/ml. Values are expressed as fluorescence units per microgram protein. Protein content was determined using a Pierce BCA Micro Protein Assay Kit.
Table 2.
Cellular uptake of Mitotracker Green FM is independent of ΔΨm
| Probe | FlU/μg protein |
|---|---|
| TMRE | |
| Control | 65.8 ± 6.8 |
| FCCP | 12.1 ± 0.7* |
| Mitotracker Green FM | |
| Control | 9.9 ± 0.9 |
| FCCP | 9.5 ± 1.0 |
Hippocampal neurons were treated for 30 min with FCCP (0.1 μm) or vehicle (ethanol). During FCCP exposure, cells were loaded with either TMRE (100 nm) or Mitotracker Green FM (200 nm). Fluorescence of cellular extracts was then quantified using a fluorescence plate reader. Data are means ± SEM from n = 8–12 cultures. *p < 0.05 with respect to control.
Modulation of plasma membrane potential. The influence of plasma membrane potential on mitochondrial TMRE uptake was abolished by incubation in high K+ buffer. Hippocampal neurons were first treated with 300 nm STS or vehicle for 6 hr. After this, the medium was exchanged for a Ca2+-free HBS containing either 5 or 50 mm KCl, in which the cells were incubated for 5 min before the addition of 100 nm TMRE. After a 10 min loading period, cellular TMRE fluorescence was acquired with the aforementioned SPOT-2 camera system, and fluorescence intensities were analyzed using Metamorph software.
Measurement of caspase-3-like activity. After the indicated lengths of exposure to staurosporine, the culture medium was aspirated, the cultures were washed with PBS, and the cells were lysed with 200 μl of lysis buffer [10 mm HEPES, pH 7.4, 42 mm KCl, 5 mm MgCl2, 1 mm PMSF, 0.1 mm EDTA, 0.1 mm EGTA, 1 mm dithiothreitol (DTT), 1 μg/ml pepstatin A, 1 μg/ml leupeptin, 5 μg/ml aprotinin, 0.5% 3-(3-cholamidopropyldimethylammonio)-1-propane sulfonate (CHAPS)]. Fifty microliters of this extract were added to 150 μl of reaction buffer (25 mm HEPES, 1 mm EDTA, 0.1% CHAPS, 10% sucrose, 3 mm DTT, pH 7.5), which was supplemented with 10 μm acetyl-DEVD-aminomethylcoumarin (Ac-DEVD-AMC) (Alexis, Grünstetten, Germany), a fluorogenic substrate for caspase-3 and related proteases, including caspase-1, -4, -6, -7, and -8 (Talanian et al., 1997; Garcia-Calvo et al., 1999). Production of fluorescent AMC was monitored for 120 min at room temperature using a fluorescent plate reader (FL 500, Biotek, Hamburg, Germany) (excitation 380 nm, emission 460 nm). Fluorescence of blanks containing no cellular extracts were subtracted from the values. Caspase-3-like activity is expressed as change in fluorescence units per 120 min per microgram protein. Total protein content, determined as above, did not change significantly up to 12 hr into STS exposure.
Nuclear changes. To observe nuclear changes occurring during apoptosis, the chromatin-specific dye Hoechst 33258 (Sigma) was used. Cultures were fixed for 10 min with 4% formaldehyde in PBS at 37°C, then permeabilized by treatment with a 19:1 mixture of ethanol/acetic acid for 15 min at −20°C. After being washed with PBS, the cells were stained with 1 μg/ml Hoechst 33258 in PBS for 20 min at room temperature and then washed again. Hoechst staining was viewed with an Axiovert 135 fluorescence microscope (Zeiss) using a 40× fluorescence objective, with UV illumination from a 50 W xenon arc. Optics were as follows: excitation, 365 nm; dichroic mirror, 395 nm; and emission, 420 nm. Nuclei of control cells appeared oval, with a septate pattern of blue fluorescence. During STS-induced apoptosis, nuclei displayed increased brightness, indicative of chromatin condensation, or fragmented into smaller bodies. Apoptotic and nonapoptotic nuclei were counted in 10 randomly chosen subfields per culture well.
Inhibitors of mitochondrial permeability transition. The immune suppressant cyclosporine A inhibits the opening of the mitochondrial PTP by binding to cyclophilin D and bongkrekic acid by stabilizing the adenine nucleotide translocator in the matrix-facing conformation (Zoratti and Szabo, 1995; Murphy et al., 1999). Hippocampal neurons were pretreated for 1 hr with cyclosporine A (10 μm) or bongkrekic acid (50 μm), and 300 nm STS was added to the culture medium. After 24 hr incubation, apoptotic degeneration was assessed by Hoechst staining (see above).
Statistics. Where not noted otherwise, data are presented as means ± SEM. For statistical comparison of viability and caspase activity data, one-way ANOVA followed by Tukey’s test were used. Imaging data were analyzed using the nonparametric Kruskal–Wallis test, followed by pair-wise comparisons using the Mann—WhitneyU test. p values <0.05 were considered to be statistically significant.
RESULTS
Estimation of mitochondrial membrane potential
Exposure of rat hippocampal neurons to STS in a concentration range from 10 to 1000 nm induces apoptosis (Prehn et al., 1997; Krohn et al., 1998). In this study, we used an STS concentration of 300 nm, which results in rapid and extensive neuronal cell death in the cultures (Fig. 1) (Prehn et al., 1997). To relate changes in ΔΨm to these apoptosis-specific events, we established a time course of nuclear changes using Hoechst 33258. As assessed with the Hoechst dye, control cultures showed a low level of nuclear condensation and fragmentation (2.4 ± 1.6%) (Fig.1C). After 2 hr of staurosporine exposure, this increased significantly to 17.7 ± 4.3%, and by 4 hr it increased to 28.2 ± 7.2%. By 8 hr, slightly more than half of the neurons showed a condensed or fragmented nuclear morphology. There was a further increase up to 24 hr, the end point of observation, by which time 76.8 ± 5.5% of the neurons were affected.
Fig. 1.
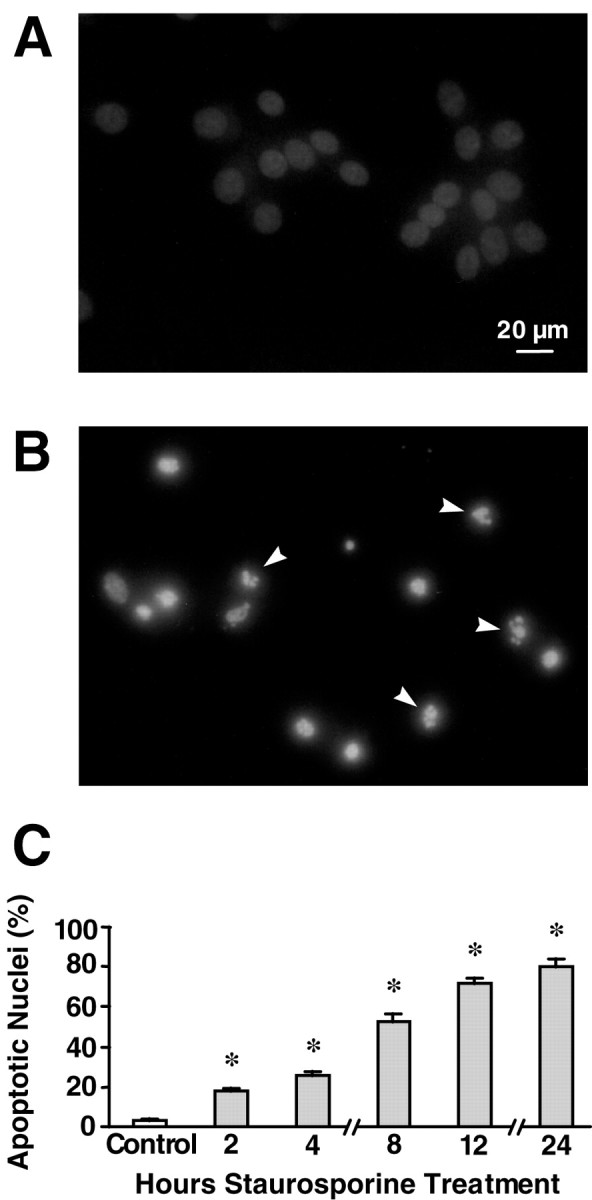
Nuclear apoptotic changes in hippocampal neurons exposed to staurosporine. A, B, Hoechst 33258 staining of nuclear chromatin. Cultured rat hippocampal neurons were treated with 300 nm staurosporine or vehicle for 24 hr. Note the condensation and increased fluorescence of nuclei from STS-treated cells as well as the appearance of nuclear fragmentation (arrows). Scale bar, 20 μm. C, Time course of nuclear apoptosis. Cultured rat hippocampal neurons were treated with 300 nm staurosporine for the indicated periods of time (0–24 hr). Nuclei were considered apoptotic if the nucleus became increasingly bright and decreased in size or fragmented into apoptotic bodies. Data are means ± SEM from n= 4–8 cultures in three separate experiments. *p< 0.05 with respect to control.
TMRE, a cationic, lipophilic dye was used for the estimation of mitochondrial membrane potential (ΔΨm) after STS exposure. TMRE enters cells and reversibly accumulates in the highly negatively charged mitochondrial matrix according to the Nernst equation, so that potential can be measured dynamically; release of TMRE after mitochondrial depolarization and its reuptake after repolarization can be quantified. Cultures that had been treated with 300 nm staurosporine for the indicated times were loaded with 100 nm TMRE, which remained present in the buffer solution, and measured by digital video microscopy. The basal fluorescence of individual cells remained stable over a period of 5 min, indicating that TMRE had fully equilibrated. Control cells had a mean basal fluorescence of 18 ± 1 FlU. Subsequent statistical analysis of control and STS-treated neurons revealed that the data were not normally distributed and were heterogeneous with respect to variance. Kruskal–Wallis ANOVA, however, revealed a significant difference between groups. Thus the nonparametric Mann—Whitney test was used for comparisons between groups, and the data are presented for clarity in histogram form (Fig. 2). In comparison with controls, there was no decline in TMRE fluorescence, indicative of mitochondrial depolarization, up to 8 hr after the onset of STS exposure, at which point more than half of the neurons showed a condensed or fragmented nuclear morphology. On the contrary, STS-treated cells showed a clear shift to higher fluorescence values, which was already significant at 2 hr and increased further during the course of STS treatment. The total pixel number of neuronal somata remained unchanged up to 12 hr after addition of STS (334 ± 11 pixels for the 12 hr STS-treated cells vs 305 ± 10 for the controls; p > 0.05; n = 69 and 77 cells, respectively), suggesting that cell shrinkage occurred only later (Fig.3A,E).
Fig. 2.
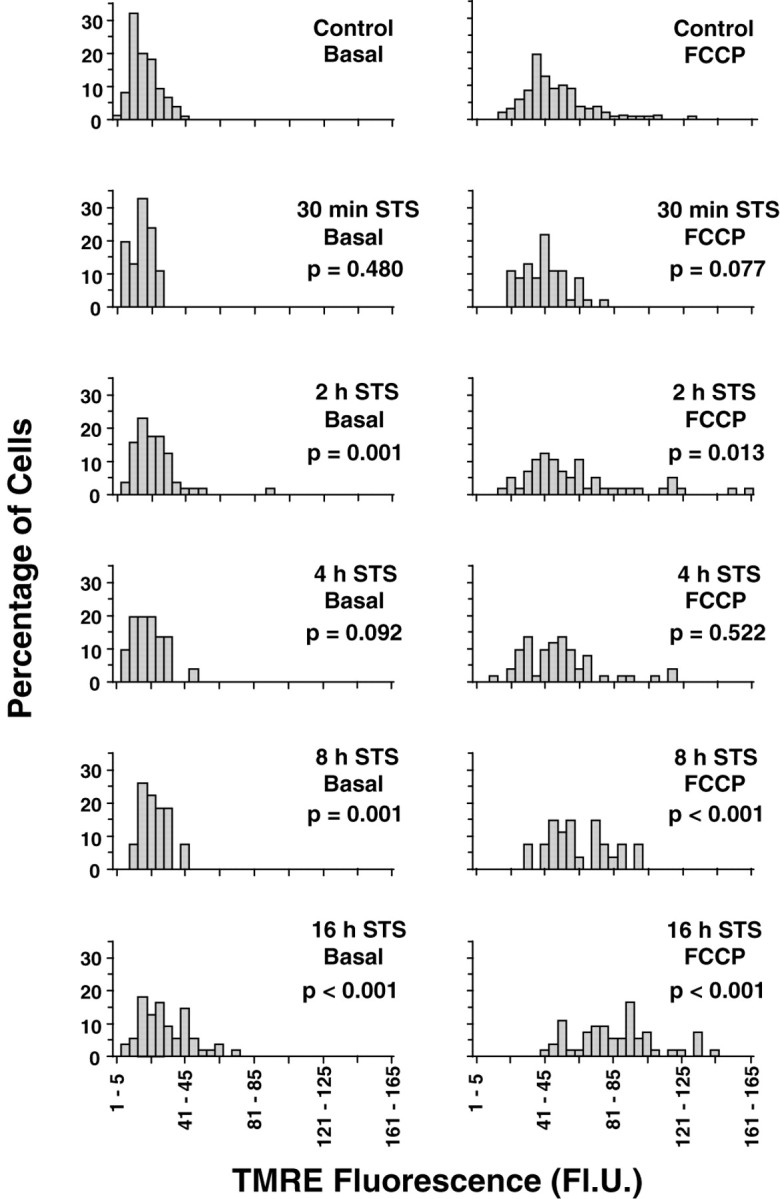
Histogram representation of cellular TMRE fluorescence during the course of staurosporine-induced apoptosis. After the indicated lengths of exposure to 300 nmstaurosporine, hippocampal cultures were incubated with 100 nm TMRE, and fluorescence was acquired using digital video microscopy. The x-axis represents fluorescence intervals into which the data were grouped, whereas the y-axis shows the percentage of cells from each treatment with fluorescence values within a given interval. Values in the left column represent baseline TMRE fluorescence (Basal). Values in the right column represent the peak fluorescence after exposure to 0.1 μm FCCP, when the dye is released from mitochondria after depolarization (“fluorescence unquenching”). In both cases, there is a progressive shift toward the right, corresponding to higher TMRE fluorescence. p values from Mann—WhitneyU test comparisons with the corresponding controls are shown under the treatment conditions. Data are fromn = 27–150 cells (see Table 1).
Fig. 3.

Representative TMRE images from vehicle-treated and 300 nm staurosporine-treated rat hippocampal cultures.A, E, Phase-contrast (PC) images. B, F, Baseline TMRE fluorescence. Images were taken after loading cells with 100 nm TMRE.C, G, Peak TMRE fluorescence after treatment with 0.1 μm FCCP. The higher fluorescence of staurosporine-treated cells is clearly visible. D,H, Corresponding traces of the two experiments. After loading with TMRE, baseline fluorescence was monitored for 5 min. Note the stability of the values, indicating that the dye distribution was fully equilibrated. At 5 min, the cultures were exposed to 0.1 μm FCCP, which depolarizes the mitochondria, resulting in a characteristic increase in fluorescence (“unquenching”) as the dye concentration equilibrates with that outside the mitochondria. The mitochondria of both control and staurosporine-treated cells could be depolarized with FCCP. After 5 min of FCCP exposure, the buffer was exchanged, and cells were allowed to recover ΔΨm. Scale bar, 20 μm.
STS-treated neurons respond to FCCP
To prove that STS-treated cells functionally respond to pharmacological alteration of ΔΨm and to ensure that the increased TMRE fluorescence values were not a result of TMRE release from the mitochondria through depolarization during dye loading and consequent “unquenching” of the dye (see below), we next examined the reaction of TMRE-loaded neurons to the protonophore FCCP (0.1 μm). FCCP depolarizes mitochondria, releasing TMRE into the cytoplasm, which is accompanied by a transient increase in fluorescence (“unquenching”) (Fig. 3) (Duchen and Biscoe, 1992;Prehn et al., 1994). We first characterized the neuronal response to various concentrations of FCCP and found that 0.1 μm was sufficient to depolarize mitochondria from all cells, resulting in a fluorescence increase. Concentrations higher than this (0.3 or 1.0 μm) did not lead to a greater TMRE release from mitochondria (data not shown). Fluorescence values collected after 0.1 μm FCCP exposure (total exposure time: 5 min) showed the same trend as the basal values: a gradual increase in mean TMRE fluorescence, which first became significant at 2 hr and continued up to 16 hr, the latest time point at which cells were imaged (Fig. 2), thus indicating that the mitochondria of STS-treated neurons could still be depolarized. Of note, unquenching values, calculated by dividing TMRE fluorescence after FCCP exposure by the basal fluorescence, were not significantly different between groups (Table1), suggesting the absence of mitochondrial depolarization during dye loading.
Table 1.
TMRE fluorescence unquenching by FCCP
| Ratio peak/baseline | n | p | |
|---|---|---|---|
| Control | 2.86 ± 0.07 | 150 | |
| 30 min | 2.70 ± 0.08 | 46 | 0.91 |
| 2 hr | 2.62 ± 0.10 | 57 | 0.57 |
| 4 hr | 2.82 ± 0.22 | 51 | 1.00 |
| 8 hr | 2.64 ± 0.07 | 27 | 0.88 |
| 16 hr | 3.14 ± 0.13 | 55 | 0.37 |
Cultured rat hippocampal neurons were exposed to 300 nm STS for the indicated periods. Unquenching ratios were calculated as the ratio of peak TMRE fluorescence after 0.1 μm FCCP treatment to baseline fluorescence. Values are expressed as means ± SEM from n neurons.
To exclude the possibility that damaged cells from the later time points were washed out in preparation for imaging, and to ensure that the cells whose TMRE fluorescence we measured were representative of the entire population, we stained selected cultures with SYTO 16, a live-cell nucleic acid probe, subsequent to imaging with TMRE. Cells that had been treated with staurosporine for 16 hr and showed signs of chromatin condensation, indicated by an increased SYTO 16 fluorescence, still maintained ΔΨm and could be depolarized with FCCP (Fig. 4).
Fig. 4.

ΔΨm remains as nuclei condense. Representative images of control (A–D) and 16 hr staurosporine-treated (E–H) rat hippocampal cultures. A, E, Phase-contrast images. Neuronal damage is clearly visible in the 16 hr staurosporine-treated culture. B, F, SYTO 16 staining. After their TMRE fluorescence was measured, cells were returned to the culture medium and incubated with 1 μmSYTO 16, a live-cell nucleic acid stain (30 min, 37°C). Nuclei of staurosporine-treated cells show increased fluorescence, as well as a more condensed and irregular morphology. C,G, Baseline TMRE fluorescence. Cells that show evidence of nuclear condensation in the SYTO 16 image (arrows) maintain ΔΨm. TMRE fluorescence of the staurosporine-treated cells is actually higher than that of controls.D, H, Peak TMRE fluorescence after 0.1 μm FCCP treatment. Here the difference between the control and treated cells is even more pronounced. Cells that show signs of nuclear apoptosis can still be depolarized by FCCP. Scale bar, 20 μm.
Mitochondrial mass remains stabile during STS-induced neuronal apoptosis
Increases in both basal and FCCP-induced TMRE fluorescence could indicate an increase in ΔΨm, mitochondrial volume, mitochondrial mass, or even plasma membrane potential. In an attempt to differentiate between these possibilities, we first used the probe Mitotracker Green FM, an indicator of mitochondrial mass (Metivier et al., 1998). A preliminary experiment in hippocampal neurons showed that uptake of this dye, in contrast to TMRE, was indeed not dependent on ΔΨm (Table2). Additional studies performed in PC12 pheochromocytoma cells indicated that uptake of this probe was also not influenced by exposure to FCCP (10 μm, 30 min), oligomycin (5 μg/ml, 30 min), the Ca2+ionophore A-23187 in combination with thapsigargin (5 and 1 μm, 4 hr), or mastoparan (50 μm, 4 hr) (data not shown). In subsequent experiments, we found that Mitotracker Green FM fluorescence of the hippocampal neuron cultures remained unchanged up to 12 hr after the start of staurosporine exposure (Fig.5A). In addition, we found that uptake of another probe used to determine changes in mitochondrial mass, nonyl acridine orange (Cossarizza et al., 1995; Mancini et al., 1997; Metivier et al., 1998), also remained unchanged up to 12 hr after STS exposure (Fig. 5B).
Fig. 5.
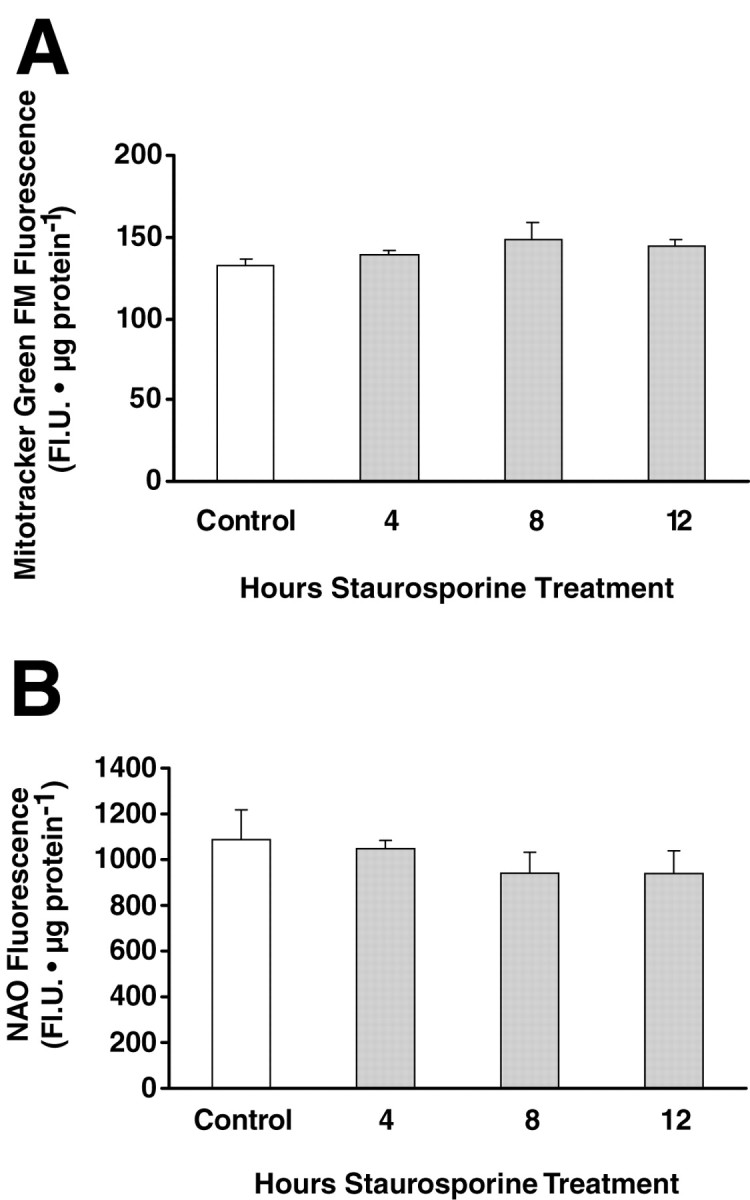
Measurement of mitochondrial mass using Mitotracker Green FM (MTGFM) and nonyl acridine orange (NAO). Cultured rat hippocampal neurons exposed to STS for the indicated times were loaded with MTGFM (200 nm, 30 min) or NAO (1 μg/ml) in medium for 30 min at 37°C, and the fluorescence of cell extracts was measured using a fluorescence plate reader. No significant differences were found with respect to the controls. Data are the means ± SEM from n = 6 cultures. The experiments were repeated with similar results.
Incubation in high K+ buffer abolishes the STS-induced increase in TMRE fluorescence
Because mitochondrial mass remained unchanged during STS-induced apoptosis, we next examined the possibility that an increase in plasma membrane potential was in fact responsible for the increased uptake of TMRE after STS treatment. The Nernstian behavior of this cationic probe would predict a concomitant increase in uptake as either mitochondrial or plasma potential increases (Ehrenberg et al., 1988). Plasma membrane hyperpolarization attributable to enhancement of outward potassium current has been observed in several models of neuronal apoptosis (Yu et al., 1997). To eliminate this influence, a high K+ buffer containing 50 mm KCl was used to depolarize the plasma membrane. Hippocampal neurons exposed to 300 nm STS for 6 hr and loaded with TMRE again showed an increase in fluorescence compared with controls (Fig. 6). In contrast, incubation in high K+ buffer decreased mitochondrial TMRE uptake in control cells but also prevented the increase in TMRE uptake seen after STS treatment. A reduction of the STS-induced increase in mitochondrial TMRE uptake was also observed in cultures treated with the potassium channel blockers tetraethylammonium (25 mm) and clofilium (1 μm) 15 min before TMRE loading (data not shown).
Fig. 6.

The STS-induced increase in cellular TMRE fluorescence is abolished by incubation in high K+buffer. Hippocampal neurons were treated with 300 nm STS for 6 hr. After this, the medium was exchanged for Ca2+-free HBS containing either 5 mm(Control) or 50 mm (High K+) KCl, in which the cells were incubated for 5 min before the addition of 100 nm TMRE. After a 10 min loading period, cellular TMRE fluorescence was acquired. Data are presented in median/quartile form and represent values fromn = 100–137 cells in two experiments per treatment. *p < 0.05 with respect to control.
Cytochrome c release proceeds without loss of ΔΨm
Because we found no evidence of early mitochondrial depolarization in our experiments with TMRE, we next examined one possible consequence of mitochondrial depolarization—–release of cytochrome c, which has been implicated in apoptotic cell death—–with the aid of immunofluorescence microscopy in con- junction with monitoring of ΔΨm using the potential-sensitive probe Mitotracker Red CMXRos. Unlike TMRE, CMXRos forms stable thiol conjugates with cellular peptides, inhibiting release of the dye from mitochondria after loading. In addition, the formation of thiol conjugates allows fixation and double-labeling of cells with an antibody to cytochrome c. Comparable to the results seen with TMRE, we saw no decrease in neuronal CMXRos fluorescence after addition of STS but rather an increase by 8 hr (Fig.7A,C). In contrast, cytochrome c staining of STS-treated cells was clearly lower than that of the respective controls by this time (Fig.7B,D). In fact, cells that had already lost a considerable amount of cytochrome c by 8 hr exhibited increased CMXRos fluorescence (Fig. 7, arrows). Furthermore, in many cells we observed a transition from a punctate to a diffuse staining pattern. Our observation of a decrease in cytochrome c staining could be indicative of cytochrome c modification or degradation after its release into the cytoplasm (Neame et al., 1998;Varkey et al., 1999).
Fig. 7.

Cytochrome c is released while ΔΨm remains stabile. Cultured rat hippocampal neurons were exposed to either vehicle (A, B) or 300 nm STS (C, D) for 8 hr, then incubated with 50 nm CMXRos for 30 min at 37°C. After fixation and permeabilization, cultures were subjected to immunocytochemistry using an antibody specific to native cytochrome c in conjunction with biotinylated anti-mouse IgG and streptavidin-conjugated Oregon Green.A, C, CMXRos staining to visualize changes in ΔΨm. Note that in the majority of neurons, CMXRos fluorescence, indicative of ΔΨm, increases during STS exposure. B, D, Cytochrome c immunofluorescence observed with Oregon Green. In contrast to that seen with CMXRos, Oregon Green fluorescence decreases considerably during STS exposure. Arrows inC and D indicate cells with particularly pronounced increases in CMXRos fluorescence and decreases in Oregon Green fluorescence compared with controls. Scale bar, 20 μm.
A similar pattern was seen when the cultures were examined with a 100× oil objective, allowing resolution of individual mitochrondria and mitochondria-rich regions (Fig. 8). Mitochondria from STS-treated cells showed only a small level of staining for cytochrome c compared with controls; this was even the case in mitochondria and mitochondria-rich regions that showed a high level of staining with CMXRos (Fig.8B,E).
Fig. 8.

High-resolution imaging of cytochrome c release. Control (A–C) and STS-treated (D–F) hippocampal cultures were treated as in Figure 7. CMXRos (ΔΨm) and Oregon Green (cytochrome c) staining were then observed with a 100× oil objective.A, D, CMXRos staining for ΔΨm. Mitochondria from STS-treated cells show no loss of ΔΨm 8 hr after addition of the drug.B, E, Cytochrome c immunofluorescence staining with Oregon Green. Mitochondria from control cells show a high level of staining for cytochrome c. In comparison, mitochondria from STS-treated cells show extensive loss of cytochrome c staining encompassing most of the mitochondrial population. C,F, Superimposed images of CMXRos (red) and Oregon Green staining. Yellow areas are indicative of colocalized CMXRos and Oregon Green staining. Arrowsindicate areas with especially pronounced loss of staining for cytochrome c without a corresponding decrease in ΔΨm. Scale bar, 5 μm.
Time course of caspase-3-like activity
Cytochrome c release from mitochondria can lead to activation of the caspase cascade through consecutive activation of caspase-9 and caspase-3 (Li et al., 1997). To assess whether caspase activation could proceed without mitochondrial depolarization, we measured caspase-3-like activity by monitoring the cleavage of a fluorogenic caspase substrate by extracts from staurosporine-treated neurons. Ac-DEVD-AMC is cleaved efficiently by caspase-3 but also by caspase-1, -4, -6, -7, and -8 (Talanian et al., 1997; Garcia-Calvo et al., 1999). Production of the fluorescent cleavage product AMC was negligible with extracts from control cultures (1.1 ± 0.6 FlU · 120 min−1 · μg−1 protein) (Fig.9). By 2 hr of staurosporine exposure, cleavage activity had increased significantly to 7.6 ± 1.1 FlU · 120 min−1 · μg−1 protein. By 8 hr, at which point there had been no decrease in ΔΨm, caspase-3-like activity reached a maximum, 23.6 ± 0.7 FlU · 120 min−1 · μg−1 protein, a >20-fold increase compared with the control. Cleavage activity then decreased by approximately one-half between 8 and 12 hr. Overall protein content remained unchanged up to 12 hr after addition of STS (data not shown). We also obtained evidence for the activation of caspases by Western blotting of caspase substrates. We observed cleavage of both poly-(ADP-ribose) polymerase, which is cleaved most efficiently by caspase-3 but also by other caspases (Margolin et al., 1997), as well as α-spectrin, both reaching a maximum by 6 hr after STS addition (data not shown). Thus, cytochrome c release was accompanied by activation of caspase-3-like proteases, without a corresponding decrease in ΔΨm.
Fig. 9.

Time course of caspase-3-like protease activity in cytosolic protein extracts. Cultured rat hippocampal neurons were treated with staurosporine (300 nm) for the indicated periods of time. Caspase-3-like protease activity was measured by cleavage of the fluorogenic substrate Ac-DEVD-AMC (10 μm). Activities are represented as increase in AMC fluorescence [in arbitrary fluorescence units (Fl.U.)] over 120 min per microgram protein. Data are means ± SEM from n = 6 cultures. A duplicate experiment yielded similar results. Different from controls, *p < 0.05.
No prevention of staurosporine-induced neuronal death by cyclosporine A or bongkrekic acid, inhibitors of the mitochondrial permeability transition
We pretreated hippocampal cultures with cyclosporine A (10 μm) or bongkrekic acid (50 μm), two potent inhibitors of PTP with distinct modes of action, 1 hr before exposure to 300 nm staurosporine. The percentage of apoptotic nuclei was assayed 24 hr after the start of staurosporine exposure by Hoechst 33258 staining. Cyclosporine A and bongkrekic acid both showed no significant toxicity when added alone (Fig.10). Neither PTP inhibitor reduced the level of apoptotic nuclear morphology after STS exposure. Cell viability, assessed by trypan blue staining, was also unaffected (data not shown). In addition, loss of mitochondrial cytochrome c was not influenced by treatment with cyclosporine A (45.3 ± 2.5% neurons showing loss of mitochondrial cytochrome c immunofluorescence 6 hr after STS treatment vs 52.5 ± 6.4% in STS-treated and cyclosporine A-treated cultures; n = 4 cultures per treatment; p > 0.1).
Fig. 10.

Inhibitors of the mitochondrial PTP do not prevent STS-induced apoptosis. Hippocampal neurons were pretreated for 1 hr with cyclosporine A (CsA, 10 μm) or bongkrekic acid (BA, 50 μm), then 300 nm STS was added to the culture medium. After 24 hr of STS treatment, apoptotic cell death was assessed by Hoechst 33258 staining and determined as in Figure 1. Data are means ± SEM fromn = 4 cultures. *p < 0.05 with respect to control. A duplicate experiment yielded comparable results.
DISCUSSION
In an attempt to clarify the importance of mitochondrial membrane potential in PCD of rat hippocampal neurons, we investigated the time courses of ΔΨm, cytochrome c release, caspase activation, and apoptotic nuclear changes during staurosporine-induced apoptosis of cultured rat hippocampal neurons. Treatment with 300 nm staurosporine led to rapid, apoptotic cell death, which was characterized by the appearance of nuclear apoptotic morphology visualized with the chromatin stain Hoechst 33258. The percentage of nuclei showing condensation and fragmentation was already significantly higher than the controls by 2 hr after staurosporine treatment and by 8 hr had increased to more than half the cell population (Fig.1C). Caspase-3-like activity increased significantly (by nearly sevenfold) as early as 2 hr after staurosporine exposure and peaked 8 hr after the start of treatment (Fig. 9), at which point mitochondrial release of cytochrome c, an inducer of caspase activity, was readily apparent (Figs. 7, 8).
In this context it is especially interesting that we observed no mitochondrial depolarization during the course of these staurosporine-induced apoptotic changes using either TMRE or CMXRos, probes that accumulate reversibly and irreversibly, respectively, in mitochondria according to the magnitude of ΔΨm. Particularly important, cells could be depolarized with FCCP throughout the length of staurosporine treatment, as shown with the reversible probe TMRE. Interestingly, even individual cells that were in the later stages of apoptosis (showing apoptotic nuclear changes as seen with SYTO 16) could be shown to maintain mitochondrial function (Fig. 4).
We cannot fully discount the possibility that only a small portion of mitochondria depolarized, which might be sufficient to trigger events downstream. In our high-magnification observations with the probe CMXRos, however, we found no such evidence at the subcellular level (Fig. 8). It is indeed conceivable that such a strategy would be risky for the organism, because a small number of mitochondria could determine the fate of entire cells (Murphy et al., 1999). Our results are in agreement with the thought that apoptosis is an active process during which cells must maintain sufficient ATP levels and thus, presumably, mitochondrial function (Nicotera and Leist, 1997).
We also examined the possibility that the mitochondrial PTP might be involved in staurosporine-induced PCD. Evidence for involvement of PTP in apoptosis includes the observation that known inhibitors of PTP such as cyclosporine A and bongkrekic acid prevent a number of apoptosis-associated events (Marchetti et al., 1996; Pastorino et al., 1996; Zamzami et al., 1996b; Marzo et al., 1998). A 1 hr pretreatment with cyclosporine A or bongkrekic acid (10 and 50 μm, respectively) failed to prevent apoptosis after subsequent treatment with staurosporine (Fig. 10), suggesting that PTP is not necessary for the initiation of STS-induced apoptosis. The TMRE fluorescence data are further evidence against the participation of a PTP in this model. The opening of a PTP leads to loss of ΔΨm, because the mitochondrion is no longer able to maintain a proton gradient across the inner membrane (Zoratti and Szabo, 1994; Zamzami et al., 1996a). We observed no such loss of ΔΨmduring STS-induced apoptosis.
By 8 hr, when more than one-half the cell population showed signs of nuclear apoptosis, cytochrome c release was evident, and caspase-3-like activity had peaked, TMRE baseline fluorescence had actually increased by roughly 30% (Fig. 2). A similar increase in CMXRos fluorescence was also seen at this time point. By 16 hr after treatment, the increase in TMRE fluorescence was roughly 70%. This increase in TMRE fluorescence seen during the course of neuronal apoptosis could be caused by mitochondrial hyperpolarization/swelling (Vander Heiden et al., 1997), an increase in mitochondrial mass (Mancini et al., 1997), or plasma membrane hyperpolarization (Ehrenberg et al., 1988). Our results support the latter alternative. We examined cellular uptake of Mitotracker Green FM and nonyl acridine orange, two probes used to evaluate changes in mitochondrial mass, and found no change in fluorescence up to 12 hr after the start of staurosporine treatment (Fig. 5). On the other hand, incubation of cells in high K+ buffer was sufficient to prevent the increase in TMRE uptake seen after STS treatment (Fig. 6), suggesting that this increase was caused at least partly by an increase in plasma membrane potential. Interestingly, Yu et al. (1997) found an increase in outward K+ current during neuronal apoptosis induced by STS or serum deprivation. Blocking this increase with high K+ or tetraethylammonium reduced the level of apoptosis, which suggests the potential importance of the plasma membrane potential in apoptotic cell death processes.
On the other hand, there is support for the idea that mitochondrial volume homeostasis could be involved in apoptosis and that anti-apoptotic Bcl-2-family proteins might function by stabilizing ΔΨm and preventing swelling [Vander Heiden et al. (1997, 1999); but see Shimizu et al. (1998)]. It has been suggested that mitochondrial swelling could result in rupture of the outer membrane, a possible mechanism of release of pro-apoptotic factors (Vander Heiden et al., 1997). Our observations of neurons incubated in high K+ buffer notwithstanding, it is possible that the increase in TMRE and CMXRos fluorescence that we observed could reflect an actual increase in ΔΨm or mitochondrial volume secondary to a defect in mitochondrial ADP/ATP exchange (Vander Heiden et al., 1999). Moreover, mitochondrial hyperpolarization may increase the likelihood of mitochondrial generation of superoxide (Korshunov et al., 1997), an event that significantly contributes to STS-induced neuronal cell death (Krohn et al., 1998). Alternatively, the release of pro-apoptotic factors could be controlled by specific channels or contact points located in the mitochondrial membrane (Reed, 1997b), could be subject to specific proteolytic processes, or could be caused by a reversal of protein import processes.
Although it is becoming clearer that mitochondria play an important role in PCD, especially in light of evidence that the release of pro-apoptotic factors from the mitochondria can initiate the caspase cascade, there is much conflicting data concerning the role of mitochondria upstream of this event. Using STS-induced cell death in cultured rat hippocampal neurons, we demonstrate that cytochrome c release, caspase activation, and nuclear apoptosis may proceed independently of loss of mitochondrial potential. We also show that mitochondria of neurons displaying morphological signs of apoptosis functionally respond to a depolarizing agent. Taken together, our data suggest that mitochondrial depolarization is not required for STS-induced neuronal apoptosis. This model of neuronal cell death may have broad biological significance, because it involves inhibition of survival kinases, increased oxidant stress, and a disturbance of neuronal Ca2+ homeostasis (Bertrand et al., 1994; Prehn et al., 1997; Krohn et al., 1998; Kumar and Mattson, 1999), findings that have been suggested to be involved in neuronal cell death in neurodegenerative disorders.
Footnotes
This work was supported by a grant from Alzheimer Forschung Initiative e.V. to J.H.M.P. and IZKF Universität Münster. We thank Elke Bauerbach for technical assistance and Professor J. Krieglstein for his support.
Correspondence should be addressed to Dr. Jochen H. M. Prehn, Interdisciplinary Center for Clinical Research (IZKF), Junior Research Group “Apoptosis and Cell Death,” Faculty of Medicine, Westphalian Wilhelms-University, Röntgenstrasse 21, D-48149 Münster, Germany.
REFERENCES
- 1.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 2.Bertrand R, Solary E, O’Connor P, Kohn KW, Pommier Y. Induction of a common pathway of apoptosis by staurosporine. Exp Cell Res. 1994;211:314–321. doi: 10.1006/excr.1994.1093. [DOI] [PubMed] [Google Scholar]
- 3.Boise LH, Thompson CB. Bcl-xl can inhibit apoptosis in cells that have undergone Fas-induced protease activation. Proc Natl Acad Sci USA. 1997;94:3759–3764. doi: 10.1073/pnas.94.8.3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bossy-Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cossarizza A, Cooper EL, Quaglino D, Salvioli S, Kalachnikova G, Franceschi C. Mitochondrial mass and membrane potential in coelomocytes from the earthworm Eisenia foetida: studies with fluorescent probes in single intact cells. Biochem Biophys Res Commun. 1995;214:503–510. doi: 10.1006/bbrc.1995.2315. [DOI] [PubMed] [Google Scholar]
- 7.Duchen MR, Biscoe TJ. Relative mitochondrial membrane potential and [Ca2+] in type I cells isolated from the rabbit carotid body. J Physiol (Lond) 1992;450:33–61. doi: 10.1113/jphysiol.1992.sp019115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehrenberg B, Montana V, Wie MD, Wuskell JP, Loew LM. Membrane potential can be determined in individual cells from the nernstian distribution of cationic dyes. Biophys J. 1988;53:785–794. doi: 10.1016/S0006-3495(88)83158-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falcieri E, Martelli AM, Bareggi R, Cataldi A, Cocco L. The protein kinase inhibitor staurosporine induces morphological changes typical of apoptosis in MOLT-4 cells without concomitant DNA fragmentation. Biochem Biophys Res Commun. 1993;193:19–25. doi: 10.1006/bbrc.1993.1584. [DOI] [PubMed] [Google Scholar]
- 10.Frey T. Nucleic acid dyes for detection of apoptosis in live cells. Cytometry. 1995;21:265–274. doi: 10.1002/cyto.990210307. [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Calvo M, Peterson EP, Rasper DM, Vaillancourt JP, Zamboni R, Nicholson DW, Thornberry NA. Purification and catalytic properties of human caspase family members. Cell Death Differ. 1999;6:362–369. doi: 10.1038/sj.cdd.4400497. [DOI] [PubMed] [Google Scholar]
- 12.Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- 13.Heiskanen KM, Bhat MB, Wang H-W, Ma J, Nieminen A-L. Mitochondrial depolarization accompanies cytochrome c release during apoptosis in PC6 cells. J Biol Chem. 1999;274:5654–5658. doi: 10.1074/jbc.274.9.5654. [DOI] [PubMed] [Google Scholar]
- 14.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 15.Koh J-Y, Wie MB, Gwag BJ, Sensi SL, Canzoniero LMT, Demaro J, Csernansky C, Choi DW. Staurosporine-induced neuronal apoptosis. Exp Neurol. 1995;135:153–159. doi: 10.1006/exnr.1995.1074. [DOI] [PubMed] [Google Scholar]
- 16.Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 17.Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immun Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- 18.Krohn AJ, Preis E, Prehn JHM. Staurosporine-induced apoptosis of cultured rat hippocampal neurons involves caspase-1-like proteases as upstream initiators and increased production of superoxide as a main downstream effector. J Neurosci. 1998;18:8186–8197. doi: 10.1523/JNEUROSCI.18-20-08186.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar I, Mattson MP. Pivotal role of mitochondrial calcium uptake in neural cell apoptosis and necrosis. J Neurochem. 1999;72:529–540. doi: 10.1046/j.1471-4159.1999.0720529.x. [DOI] [PubMed] [Google Scholar]
- 20.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 21.Mancini M, Anderson BO, Caldwell E, Sedghinasab M, Paty PB, Hockenbery DM. Mitochondrial proliferation and paradoxical membrane depolarization during terminal differentiation and apoptosis in a human colon carcinoma cell line. J Cell Biol. 1997;138:449–469. doi: 10.1083/jcb.138.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marchetti P, Castedo M, Susin SA, Zamzami N, Hirsch T, Macho A, Haeffner A, Hirsch F, Geuskens M, Kroemer G. Mitochondrial permeability transition is a central coordinating event of apoptosis. J Exp Med. 1996;184:1155–1160. doi: 10.1084/jem.184.3.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Margolin N, Raybuck SA, Wilson KP, Chen W, Fox T, Gu Y, Livingston DJ. Substrate and inhibitor specificity of interleukin-1β-converting enzyme and related caspases. J Biol Chem. 1997;272:7223–7228. doi: 10.1074/jbc.272.11.7223. [DOI] [PubMed] [Google Scholar]
- 24.Marzo I, Brenner C, Zamzami N, Jürgensmeier JM, Susin SA, Vieira HLA, Prévost M-C, Xie Y, Matsuyama S, Reed JC, Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 25.Metivier D, Dallaporta B, Zamzami N, Larochette N, Susin SA, Marzo I, Kroemer G. Cytofluorometric detection of mitochondrial alterations in early CD95/Fas/APO-1-triggered apoptosis of Jurkat T lymphoma cells: comparison of seven mitochondrion-specific fluorochromes. Immunol Lett. 1998;61:157–163. doi: 10.1016/s0165-2478(98)00013-3. [DOI] [PubMed] [Google Scholar]
- 26.Monck JR, Oberhauser AF, Keating TJ, Fernandez JM. Thin-section ratiometric Ca2+ images obtained by optical sectioning of fura-2 loaded mast cells. J Cell Biol. 1992;116:745–759. doi: 10.1083/jcb.116.3.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murphy AN, Fiskum G, Flint Beal M. Mitochondria in neurodegeneration: bioenergetic function in cell life and death. J Cereb Bood Flow Metab. 1999;19:231–245. doi: 10.1097/00004647-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Neame SJ, Rubin LL, Philpott KL. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J Cell Biol. 1998;142:1583–1593. doi: 10.1083/jcb.142.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicotera P, Leist M. Energy supply and the shape of cell death in neurons and lymphoid cells. Cell Death Differ. 1997;4:435–442. doi: 10.1038/sj.cdd.4400265. [DOI] [PubMed] [Google Scholar]
- 30.Pastorino JG, Simbula G, Yamamoto K, Glascott PA, Jr, Rothman RL, Farber JL. The cytotoxicity of tumor necrosis factor depends on induction of the mitochondrial permeability transition. J Biol Chem. 1996;271:29792–29798. doi: 10.1074/jbc.271.47.29792. [DOI] [PubMed] [Google Scholar]
- 31.Prehn JHM. Mitochondrial transmembrane potential and free radical production in excitotoxic neurodegeneration. Naunyn Schmiedebergs Arch Pharmacol. 1998;357:316–322. doi: 10.1007/pl00005173. [DOI] [PubMed] [Google Scholar]
- 32.Prehn JHM, Bindokas VP, Marcuccilli CJ, Krajewski S, Reed JC, Miller RJ. Regulation of neuronal Bcl2 protein expression and calcium homeostasis by transforming growth factor type β confers wide-ranging protection on rat hippocampal neurons. Proc Natl Acad Sci USA. 1994;91:12599–12603. doi: 10.1073/pnas.91.26.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prehn JHM, Jordan J, Ghadge GD, Preis E, Galindo MF, Roos RP, Krieglstein J, Miller RJ. Ca2+ and reactive oxygen species in staurosporine-induced neuronal apoptosis. J Neurochem. 1997;68:1679–1685. doi: 10.1046/j.1471-4159.1997.68041679.x. [DOI] [PubMed] [Google Scholar]
- 34.Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997a;387:773–776. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- 35.Reed JC. Cytochrome c: can’t live with it—–can’t live without it. Cell. 1997b;91:559–562. doi: 10.1016/s0092-8674(00)80442-0. [DOI] [PubMed] [Google Scholar]
- 36.Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 37.Shimizu S, Eguchi Y, Kamiike W, Funahashi Y, Mignon A, Lacronique V, Matsuda H, Tsujimoto Y. Bcl-2 prevents apoptotic mitochondrial dysfunction by regulating proton flux. Proc Natl Acad Sci USA. 1998;95:1455–1459. doi: 10.1073/pnas.95.4.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996;184:1331–1342. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlet DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 40.Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW. Substrate specificity of caspase family proteases. J Biol Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- 41.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 42.Tsujimoto Y. Apoptosis and necrosis: intracellular ATP level as a determinant for cell death modes. Cell Death Differ. 1997;4:429–434. doi: 10.1038/sj.cdd.4400262. [DOI] [PubMed] [Google Scholar]
- 43.Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xl regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- 44.Vander Heiden MG, Chandel NS, Schumacker PT, Thompson CB. Bcl-xl prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol Cell. 1999;3:159–167. doi: 10.1016/s1097-2765(00)80307-x. [DOI] [PubMed] [Google Scholar]
- 45.Varkey J, Chen P, Jemmerson R, Abrams JM. Altered cytochrome c display precedes apoptotic cell death in Drosophila. J Cell Biol. 1999;144:701–710. doi: 10.1083/jcb.144.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wadia JS, Chalmers-Redman RME, Ju WJH, Carlile GW, Phillips JL, Fraser AD, Tatton WG. Mitochondrial membrane potential and nuclear changes in apoptosis caused by serum and nerve growth factor withdrawal: time course and modification by (−)-deprenyl. J Neurosci. 1998;18:932–947. doi: 10.1523/JNEUROSCI.18-03-00932.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 48.Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- 49.Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LMT, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1997;278:114–118. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- 50.Zamzami N, Susin SA, Marchetti P, Hirsch T, Gomez-Monterrey I, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996a;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zamzami N, Marchetti P, Castedo M, Hirsch T, Susin SA, Masse B, Kroemer G. Inhibitors of permeability transition interfere with the disruption of the mitochondrial transmembrane potential during apoptosis. FEBS Lett. 1996b;384:53–57. doi: 10.1016/0014-5793(96)00280-3. [DOI] [PubMed] [Google Scholar]
- 52.Zoratti M, Szabo I. Electrophysiology of the inner mitochondrial membrane. J Bioenerg Biomembr. 1994;26:543–553. doi: 10.1007/BF00762739. [DOI] [PubMed] [Google Scholar]
- 53.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 54.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4: participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]