

Abstract
Mnemonic deficits resulting from excitotoxic lesion of the basal forebrain have been classically attributed to the resulting depletion of cortical acetylcholine activity. It has been demonstrated that in spite of the strong cholinergic depletion after injections into the basal forebrain of the immunotoxin 192IgG–saporin, no detectable deficit can be found in the acquisition of several learning tasks, including conditioned taste aversion. Conversely, NMDA-induced lesions of the basal forebrain strongly impair taste aversion learning. In this study we show that 192IgG–saporin produces an efficient and selective cholinergic deafferentation of the rat neocortex but not the amygdala. Furthermore, a stronger relationship between severity of memory impairment after NMDA lesions and basoamygdaloid cholinergic deafferentation was found. Therefore, in a second experiment, we show that combining NMDA-induced lesions into the basolateral amygdala with 192IgG–saporin injections into the basal forebrain results in a strong disruption of taste aversion learning, whereas none of these treatments were by themselves capable of producing any detectable impairment in this learning task. The double lesion effect was only paralleled by simple NMDA lesions into the basal forebrain, suggesting that the learning deficits associated to excitotoxic lesions of the basal forebrain are the result of the simultaneous destruction of the corticopetal and basoamygdaloid interaction. A model is proposed, according to which the modulation of learning processes exerted by the basal forebrain can be redundantly performed by both the basocortical and basoamygdaloid pathway.
Keywords: conditioned taste aversion, learning, cholinergic basal forebrain, ChAT, insular cortex, amygdala
The cholinergic basal forebrain complex, including the nucleus basalis magnocellularis (NBM), provides widespread, topologically organized afferent cholinergic innervation to many brain regions, including the whole cortical mantle, hippocampus, and amygdala (Bigl et al., 1982). Many studies have implicated these cholinergic neurons in the mediation of learning and memory processes (Hepler et al., 1985; Etherington et al., 1987; Everitt et al., 1987;Dunnett and Fibiger, 1993; Sinden et al., 1995). Lesions of the NBM using injections of excitatory amino acid agonists have been associated with learning deficits in a great variety of tasks (Sinden et al., 1995; Wenk, 1997). Mnemonic deficits resulting from this type of lesions have been classically attributed to the depletion of cortical acetylcholine activity. However, those excitotoxins previously reported to produce the greatest mnemonic deficits also produce the largest decreases in amygdaloid choline acetyltransferase (ChAT) (Dunnet et al., 1987; Beninger et al., 1994; Mallet et al., 1995).
It has been demonstrated that the immunotoxin 192IgG–saporin, injected into the NBM, induces a selective loss of cortically projecting cholinergic nerve growth factor receptor-positive neu- rons. However, this treatment spares basolateral amygdaloid (BLA)-projecting fibers (Heckers and Mesulam, 1994). Interestingly, in spite of the massive reduction of cortical cholinergic input (Wiley, 1992; Book et al., 1994), this immunotoxin has repeatedly failed to reproduce the kind of memory deficits normally found as a result of less selective excitotoxic lesions (Berger-Sweeney et al., 1994; Torres et al., 1994;Wenk et al., 1994, 1996; Baxter et al., 1995; Waite and Thal, 1996). Taken together, these data support the view that learning deficits associated to excitotoxic lesions are the result of basoamygdaloid deafferentation rather than the destruction of basocortical cholinergic projection. This interpretation, however, challenges the extensive evidence supporting a direct role for the cortical cholinergic input in memory formation.
Here we use a cortically mediated learning paradigm, conditioned taste aversion (CTA), to study the modulation exerted by the basal forebrain on memory formation. CTA is a very robust model for the study of learning and memory processes (Bermúdez-Rattoni and Yamamoto, 1998). In this model, an animal acquires an aversion to a novel taste when it is followed by digestive malaise. It has been shown that bilateral lesions of the insular cortex (IC) disrupt acquisition and retention of CTA (Kiefer and Brown, 1979; Yamamoto et al., 1980;Aggleton et al., 1981; Braun et al., 1982; Kiefer, 1985;Bermúdez-Rattoni and Yamamoto, 1998). In addition, the participation of the basolateral and central amygdaloid nucleus in associative processing of gustatory stimuli has been described (Bermúdez-Rattoni and Yamamoto, 1998). The amygdaloid complex receives gustatory, visceral, and olfactory afferents (Norgren, 1974). Furthermore, the insular cortex and amygdaloid system are reciprocally and functionally interconnected (Kapp et al., 1985;Lasiter and Glanzman, 1985; Escobar et al., 1998).
Excitotoxic lesions of the NBM disrupt the ability to acquire taste aversion learning, implying the involvement of the basal cholinergic projecting system in memory processing of CTA (Lopez-García et al., 1993). However, it remains to be tested whether the behavioral effects of this kind of lesion are exclusively caused by the basocortical cholinergic component rather than by the basoamygdaloid pathway. The CTA thus offers an appropriate model for the analysis of the possible functional role of the ascending cholinergic pathway in cortically mediated learning mechanisms.
The following experiments were designed to reveal the relative contribution of both the basocortical and basoamygdaloid projection in the mediation of taste aversion learning. In the first experiment, we used comparisons between 192IgG–saporin and the less specific NMDA-induced lesions directly into the NBM as a valuable tool for assessing the possible differential involvement of both ascending pathways. In the second experiment, by means of a combined lesion strategy, we further inspected the possible interplay between the NBM–amygdala and NBM–cortex interaction on this behavioral paradigm.
EXPERIMENT 1
In this experiment we explore the possible differential contributions of either the basocortical or basoamygdaloid projection in the mediation of taste aversion learning. To this end, we relate the behavioral effects with the extent of cholinergic deafferentation in both the insular cortex and amygdala after either bilateral injections of 192IgG–saporin or NMDA directly into the NBM.
Materials and methods
Subjects. Forty-nine male Wistar rats weighing 250–300 gm were used in this experiment. They were individually caged and kept in a 12 hr light/dark cycle. All behavioral and biochemical manipulations were performed in the light cycle phase.
Surgical procedure. Animals were anesthetized with sodium pentobarbital (65 mg/kg) and placed in a head holder. Lesions were made by stereotaxic infusion of the toxin via a 30 gauge stainless steel cannula connected via teflon tubing to a 10 μl glass microsyringe mounted in a microdrive pump. The stereotaxic coordinates used were anteroposterior, −0.8 mm from bregma; lateral, ±2.5 mm; and dorsoventral −7.5 mm. A total volume of 0.8 μl of a solution of either 0.2 μg/μl of 192IgG–saporin (Chemicon, Temecula, CA) or NMDA (10 μg/μl; Sigma, St. Louis, MO) in PBS was bilaterally infused at a constant rate of 0.5 μl/min. The injector was left in place for 3 min to allow proper diffusion.
Following the aforementioned surgical procedure, 18 animals received bilateral microinjections of 192IgG–saporin directly into the NBM (N-I). Another group of 15 animals received bilateral intra-NBM injections of NMDA (N-E). An additional group of 16 animals remained unoperated during the whole procedure as an intact control group (CTR).
CTA procedure. Two weeks after the surgery all the rats were prevented to drink and left without water for the following 24 hr. After that, they were given water in their home cages every 12 hr for 15 min, and consumption was measured. When they reached an asymptotic consumption level, they received an acquisition trial. The presentation of a novel taste was done by adding saccharin to the water (1 gm/l). Fifteen minutes later, in accordance with our standard procedure, a malaise-inducing drug (LiCl, 0.15 m; 7.5 ml/kg) was administered intraperitoneally (Gutiérrez et al., 1997). Subsequent drinking trials were performed with water only. After three drinking trials the subjects were presented with the saccharin-flavored water for the second time, and their consumption was used as a measure of strength of aversion.
Enzymatic assay. One day after the behavioral study, randomly sampled animals obtained from each group were killed by decapitation (N-I, n = 10; N-E, n = 7; and CTR, n = 8). Samples of insular cortex, amygdaloid complex were dissected under a stereoscopic microscope and stored at −70°C before analysis of ChAT activity. Dorsal striatum tissue samples were included as a nonbasal dependent cholinergic control. Each sample was sonicated in 1 ml of 25 mm phosphate buffer containing 0.5% Triton X-100 and maintained on ice to avoid overheating. The homogenate was centrifuged at 20,000 ×g for 15 min. ChAT activity in 200 μl of the supernatant was assayed by adding 50 μl of a substrate solution containing (in mm): 10 choline chloride, 0.2 neostigmine, 20 EDTA, and 0.4 acetyl coenzyme A in 0.1 m sodium phosphate buffer, pH 7.4, and incubated for 5 min at 37°C. Reaction was stopped by adding 10 μl of 1 m perchloric acid on ice and 1 ml of distilled water. The mixture was passed through a 30,000 MW ultraspin filter (Cole-Parmer) and stored at −70°C before chromatographic analysis.
Analysis of ACh levels. Samples were assayed for ACh levels using HPLC with electrochemical detection, using a mobile phase, pH 8.5, containing 50 mm sodium phosphate buffer and 0.5% Kathon reagent (BAS) microbicide. All samples were injected on a polymeric reversed phase column (BAS ACh-choline assay kit), ACh and choline were then converted into hydrogen peroxide and betaine in a postcolumn reactor containing immobilized acetylcholinesterase and choline oxidase (BAS). A choline oxidase–catalase reactor (BAS) was added to avoid choline detection in the substrate solution. The hydrogen peroxide was detected electrochemically with a platinum electrode set at 500 mV (vs Ag/AgCl). The detection limit, defined as the amount of ACh producing a peak twice the basal noise, was ∼0.2 pmol.
Histology. Immediately after the behavioral study, five animals were randomly selected for each of the N-I, N-E, and CTR groups, respectively. Animals were perfused, and 40 μm sections were obtained. Acetylcholinesterase histochemistry was then performed according to a protocol modified from Paxinos and Watson (1986). Briefly, sections were incubated overnight in 50 mm sodium acetate buffer, pH 5.0, 4 mm copper sulfate, 16 mm glycine, 4 mm acetylthiocholine iodide, and 0.1 mm ethopropazine. After incubation, the slides were immersed into a developing solution (1% sodium sulfide, pH 7.5) for 10 min. After cholinesterase histochemistry, to obtain a quantitative estimation of the effects of intrabasal NMDA or 192IgG–saporin-induced lesions on AChE-positive fibers in the basolateral amygdala (BLA), computer images of the basolateral amygdala were directly acquired using a DDC camera coupled to a light microscope. After the automatic segmentation of the BLA, mean color density of this nucleus was obtained using the standard 256 level gray density scale. Six sections throughout the BLA nucleus were bilaterally analyzed per brain. The average value of mean gray level among sections was determined for each brain.
The basal forebrain cholinergic cells have been shown to stain intensely for acetylcholinesterase (AChE) and show a particularly rapid recovery of enzyme activity after systemic administration of the irreversible inhibitor di-isopropylfluorophosphate (DFP). This procedure easily reveals the cholinergic somata of the basal forebrain and their proximal processes to an appreciable extent. Therefore, three animals from each of the N-I, N-E, and CTR groups, respectively, were subjected to this analysis. DFP-cholinesterase pharmacohistochemical regimen was performed according to the protocol of Bigl et al. (1982). Briefly, animals were injected intramuscularly with 1.8 mg/kg DFP (Calbiochem, La Jolla, CA) 2 hr before perfusion. Once the brains were obtained and cut, mounted sections were subjected to the normal acetylcholinesterase histochemistry described above.
Results
CTA. Simple ANOVA was done on the test day consumption volume for all groups. No differences were found in the baseline water intake among groups (F(2,46) = 0.325;p > 0.05). Mean baseline water intake was 15.08 ± 0.64, 15.74 ± 0.85, and 14.95 ± 1.13 for each of the CTR, N-I , and N-E groups, respectively. As can be seen in Figure1, during the acquisition trial, no differences in saccharine consumption (the novel gustatory stimulus) were found among groups (F(2,46) = 0.24; p > 0.05). During the test presentation of saccharine solution, significant differences were found among groups (F(2,46) = 43.94; p < 0.01). A post hoc pairwise Fisher test showed that only the NMDA-induced lesions resulted in a significant disruption in the acquisition of taste aversion, as indicated by the increased saccharine consumption when compared with the control and immunotoxin-lesioned groups (p values < 0.01). The 192IgG–saporin-treated group showed no disruption of taste aversion as compared with the intact control group. The behavioral difference observed after either treatment further implies a neurotoxin-specific effect.
Fig. 1.

Effects of intrabasal 192IgG–saporin or NMDA microinjections on taste aversion retention test. Aversion is expressed as mean (± SEM) percentage of baseline consumption during the retention trial. CTR, Intact control;N-I, 192IgG–saporin-lesioned group; N-E, NMDA-induced lesioned group. **p < 0.01 versus intact control. Comparisons between saccharin consumption values during the acquisition trial expressed as men (± SEM) percentage of baseline consumption are also shown.
ChAT activity. Simple ANOVAs were used for comparisons of ChAT activity among groups and post hoc pairwise Fisher test where appropriate. As can be seen in Figure2, both NMDA and 192IgG–saporin-induced lesions displayed markedly reduced ChAT activity in the insular cortex relative to the control group (F(2,22)= 18.46; p < 0.01). However, the strongest reduction in enzymatic activity was apparent in the immunotoxin-lesioned group. Subsequent post hoc tests showed statistically significant differences between the intact control and both lesioned groups (p values < 0.01). Additionally, significant differences were found between both treated groups (p < 0.05) implying that, at the used dose, 192IgG–saporin treatments result in a significantly stronger cholinergic deafferentation of the cortex as compared with that resulting from intrabasal microinjections of NMDA. As shown in Figure2, an inverse pattern of ChAT activity was apparent in the amygdala. Significant differences among groups were found (F(2,22) = 6.36; p < 0.01). The corresponding post hoc tests showed that only the NMDA-treated group resulted in a significant reduction in the amygdaloid ChAT activity as compared with the intact control (p < 0.01), whereas no significant reduction in ChAT activity in the amygdala was detected in the immunotoxin-treated group. No effect was found in the dorsal striatum after either treatment (F(2,22) = 0.38;p > 0.05).
Fig. 2.

ChAT activity analysis in the insular cortex, amygdala, and dorsal striatum for each of the groups used in experiment 1. CTR, Intact control; N-I, 192IgG–saporin-lesioned group; N-E, NMDA-induced lesioned group. Activity is expressed as mean picomoles of acetylcholine formed per minute per milligram of protein ± SEM. **p < 0.01 against the intact control group. φp < 0.05 against the immunotoxin-lesioned group (only comparisons between both lesioned groups are shown, see Results).
Confirmation of the lesions. The location and extent of the cholinergic lesions were confirmed in all experimental conditions used by means of cholinesterase and the DFP-cholinesterase pharmacohistochemical regimen. Figure 3shows the cholinesterase fiber staining in the cortex for intact controls (A), as well as for both 192IgG–saporin (B) and NMDA-induced intrabasal lesions (C). Note the relative difference in AChE staining in both lesioned groups when compared with the control staining. As can be seen in Figure 4A–C, both immunotoxic lesions and NMDA-induced lesions into the NBM resulted in a strong reduction of AChE-positive somata in the basal forebrain as compared with the control staining. Although the above described enzymatic assay of ChAT activity was restricted to a tissue sample of the amygdaloid complex, it can be argued, given the close proximity of the amygdaloid nuclei, that the cholinergic effects do not necessarily reflect the biochemical status of the basolateral nucleus itself. Therefore, in Figure 4D–F we compared the effects of intrabasal NMDA-induced excitotoxic lesions and 192IgG–saporin injections on cholinesterase fiber staining in the BLA. Subsequent color density analysis showed a strong significant difference between the BLA staining in the NMDA-lesioned animals when compared with the control group (F(2,12) = 8.83;p < 0.01; Table 1). In contrast, 192IgG–saporin lesions into the NBM did not produce any detectable effect in the cholinergic marker when compared with the control group. These data confirm previous findings regarding the effects of intrabasal excitotoxic and immunotoxic lesions on the NBM–BLA pathway (Heckers and Mesulam, 1994; Heckers et al., 1994;Mallet et al., 1995).
Fig. 3.

Photomicrographs of AChE histochemistry of coronal sections taken from a control brain (A), an immunolesioned brain (192IgG–saporin; B), and NMDA-lesioned brain (C). Note the reduction of cortical AChE staining in both lesioned brains as compared to the control brain.
Fig. 4.

A–C, Photomicrographs taken at the level of the anterior nucleus basalis following DFP–cholinesterase pharmacohistochemical regimen. AChE-positive somata in the basal forebrain in a control brain (A), 192IgG–saporin-treated brain (B), and NMDA-induced intrabasal lesion (C). Scale bar, 1 mm. D–F, Photomicrographs taken at the level of the basolateral amygdaloid nucleus showing cholinesterase fiber staining in either control (D), intrabasal 192IgG–saporin-treated (E), or intrabasal NMDA-induced lesioned brains (F). Scale bar, 200 μm.
Table 1.
Color density analysis of AChE-positive fibers in the BLA
| CTR | N-I | N-E |
|---|---|---|
| 198.08 ± 9.75 | 196.304 ± 5.67 | 155.93 ± 8.11** |
CTR, Intact control group; N-I, intrabasal 1921gG-saporin-lesioned group; N-E, intrabasal NMDA-induced lesioned group.
Values are mean gray scale levels ± SEM; **p < 0.01.
EXPERIMENT 2
As shown in the previous experiments, in spite of an even strong cholinergic deafferentation of the IC, immunotoxic lesions into the NBM do not affect CTA learning. Conversely, excitotoxic lesions of the NBM impair taste aversion learning. Similar discrepant results with respect to both excitotoxic and immunotoxic lesions have been repeatedly found in other learning tasks as spatial water maze, radial maze, inhibitory avoidance, etc. (Berger-Sweeney et al., 1994; Torres et al., 1994; Wenk et al., 1994, 1996; Baxter et al., 1995; Waite and Thal, 1996) Here, we have found a better correlation between severity of memory impairment and simultaneous deafferentation of the cortex and amygdala. These results are consistent with previous findings according to which those excitotoxins reported to produce the greatest mnemonic deficits, also produced the largest decreases in amygdaloid ChAT (Beninger et al., 1994; Mallet et al., 1995). Additionally, in agreement with Heckers and Mesulam (1994) and Heckers et al. (1994), we have found that 192IgG–saporin lesions produce an efficient and selective deafferentation of the rat neocortex but selectively spare an important population of basolateral amygdala projecting fibers. Taken together, these data might support the view of a primary role for the basoamygdaloid interaction on the regulation of memory formation exerted by the basal forebrain. Should this be the case, one would expect BLA lesions to disrupt CTA learning. However, excitotoxic lesions applied into the BLA do not disrupt taste aversion learning (Dunn and Everitt, 1988; Bermúdez-Rattoni and McGaugh, 1991). A possible solution for this paradox is that the modulation of learning processes exerted by the basal forebrain might be simultaneously performed by both the basocortical and basoamygdaloid pathway.
To test this hypothesis, we assessed the effects of (1) NMDA-induced lesions into the NBM, (2) 192IgG–saporin injections into the NBM, or (3) combined NMDA lesions into the BLA with 192IgG–saporin injections into the NBM, using the experimental groups described in Table2.
Table 2.
Experimental groups used in the behavioral study of Experiment 2
| Group | ||||||
|---|---|---|---|---|---|---|
| CTR (n = 9) | N-I (n = 8) | A-E (n = 8) | N-I/A-E (n = 8) | N-E (n = 8) | N-V/A-V (n = 7) | |
| NBM | 192-IgG Saporin | 192-IgG Saporin | NMDA | Vehicle | ||
| Amygdala | NMDA | NMDA | Vehicle | |||
Materials and methods
Subjects. Forty-eight male rats weighing 250–300 gm were used in this experiment. They were kept and maintained as described in experiment 1.
One group of animals received bilateral injections of 192IgG–saporin into the NBM following the same procedure and coordinates indicated in experiment 1 (N-I; n = 8). Another group of animals (A-E; n = 8) received NMDA-induced lesions into the BLA using the following stereotaxic coordinates from bregma: anteroposterior, −1.8 mm; lateral, ±4.7 mm, and dorsoventral, −8.3 mm. A total volume of 0.8 μl of NMDA (10 μg/μl) was bilaterally infused at a constant rate of 0.5 μl/min. A third group (N-I/A-E;n = 8) received both intra-amygdala NMDA-induced lesions and intrabasal 192IgG–saporin microinjections in the same dose and coordinates indicated in the previous experiment. The fourth group of animals (N-E; n = 8) received bilateral intra-NBM injections of NMDA. Another group (N-V/A-V; n = 7) received bilateral vehicle injections (0.8 μl of PBS) in both BLA and NBM. An additional group remained unoperated as an intact control group (CTR; n = 9). After a postoperative period of 2 weeks, all the animals were subjected to taste aversion training according to the same procedure described in the previous experiment.
Histology. Brains were processed for standard acetylcholinesterase histochemistry and DFP-cholinesterase pharmacohistochemical regimen (data not shown), as described in the previous experiment. Immunohistochemistry for p75 NGF receptor detection was performed using the standard avidin–biotin ABC procedure (Hsu et al., 1981). Anti-p75 monoclonal antibodies (1:500) were obtained from Boehringer Mannheim (Mannheim, Germany).
Results
CTA. Simple ANOVA was done on the test day consumption volume for all groups. No differences were found in the baseline water intake among groups (F(5,42) = 0.57;p > 0.05). Mean baseline water intake was 14.94 ± 0.69, 15.94 ± 1.23, 14.87 ± 0.61, 17.25 ± 1.0, 17.55 ± 1.21, and 17.47 ± 0.52 for each of the CTR, N-I, A-E, N-I/A-E, N-E, and N-V/A-V groups, respectively. Again, during the acquisition trial, no differences in saccharine consumption were found among groups (F(5,42) = 0.66;p > 0.05). As shown in Figure5, during the test presentation of the saccharin solution, significant differences were found among the six groups (F(5,42) = 19.368;p < 0.01). A post hoc pairwise Fisher test showed that NMDA-induced lesions into the NBM (N-E) as well as the combined NMDA lesions into the BLA with 192IgG–saporin injections into the basal forebrain (N-I/A-E) had a significant disruption in the acquisition of taste aversion as indicated by the increased saccharine consumption when compared with the control and vehicle-treated groups on the retention trial (p values < 0.01). Neither the amygdala-lesioned group (A-E), the group that only received immunotoxin lesions into the NBM (N-I), or the vehicle-treated group (N-V/A-V) showed any impairment in the ability to acquire CTA as compared with the intact control group.
Fig. 5.

Effects of intrabasal 192IgG–saporin, intra-amygdala NMDA-induced lesions, and combined intrabasal immunotoxin and amygdala excitotoxic lesions on CTA retention test (see Table 2). CTR, Intact control; N-I, 192IgG–saporin-lesioned group; N-E, NMDA lesioned group; N-I/A-E, Combination of intrabasal immunotoxic lesion with intra-amygdala NMDA-induced lesion;N-V/A-V, combined vehicle administration (see Materials and Methods).**p < 0.01 versus intact control.
Confirmation of the lesions. All intrabasal NMDA and 192IgG—saporin-induced lesions were again verified by means of DFP-cholinesterase pharmacohistochemical regimen (to avoid repetition, these photomicrographs are not shown). The specific destruction of the low-affinity NGF receptor-positive cholinergic cells in the basal forebrain, caused by 192IgG–saporin microinjections, was verified by means of p75 immunohistochemistry. Samples are shown in Figure6, A and D. The correct placement of NMDA lesions into the NBM and BLA was verified by means of cholinesterase histochemistry (Fig.6B,C,E,F).
Fig. 6.
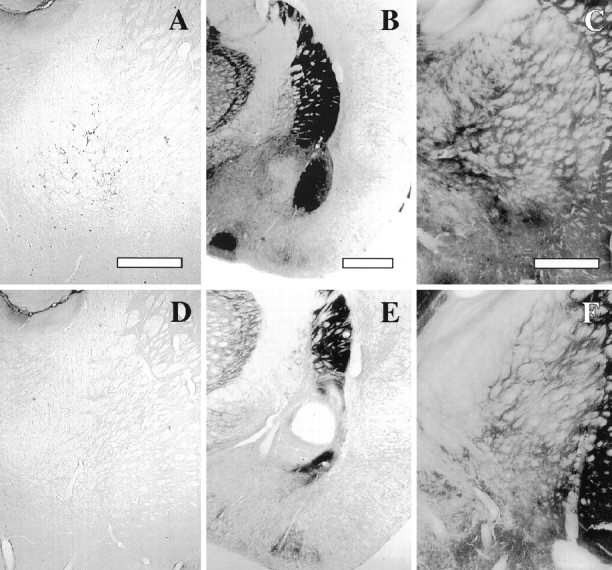
Confirmation of the location and extent of the lesions used in Experiment 2. A, D, p75 immunostaining of NBM magnocellular neurons in a control and immunolesioned brain, respectively. Scale bar, 600 μm. B, E, AChE histochemistry at the level of the basolateral amygdala in a control and NMDA-induced intra-amygdala lesion, respectively. Scale bar, 1 mm.C, F, AChE histochemistry in the basal forebrain in a control and NMDA-induced intrabasal lesion, respectively. Scale bar, 600 μm.
DISCUSSION
In the present experiments, we started from the fact that NMDA injections into the NBM result in a strong disruption of CTA learning, whereas 192IgG–saporin lesions into the NBM do not. The existing discrepancy between both kinds of lesions can be explained in two ways. First, cholinergic neurons in the NBM are interspersed with other neurons in the basal forebrain, including GABAergic neurons of the dorsal and ventral pallidum and noncholinergic magnocellular corticopetal neurons. The use of excitotoxins, especially those acting through NMDA receptors such as NMDA and ibotenic acid, can also destroy noncholinergic pallidal and other neurons in the substantia innominata, whereas there seems to be little doubt that 192IgG–saporin is a powerful and selective lesioning tool for basal cholinergic neurons, which are enriched in the low-affinity nerve growth factor receptor. Second, different excitotoxins, when injected into the NBM, produce differential effects on cholinergic projections to the cortex and amygdala (Boegman et al., 1992). That is, those excitotoxins previously reported to produce the greatest mnemonic deficits also produced the largest decreases in the basolateral amygdaloid ChAT (Beninger et al., 1994; Mallet et al., 1995). In contrast, virtually all of the cortically projecting cholinergic cells within the NBM are vulnerable to the immunotoxin, with the exception being those cholinergic neurons that send an efferent projection to the basolateral amygdala (Heckers and Mesulam, 1994; Heckers et al., 1994).
After intrabasal microinjections of 192IgG–saporin, no detectable disruption in taste aversion learning was found in the acquisition and performance of this well known cortically mediated learning paradigm. Previous results from our laboratory (Gutiérrez et al., 1999) demonstrate that IC-mediated associative processes involved in taste aversion learning can be performed in spite of up to 86% reduction in mean ChAT activity and at least 97% reduction in extracellular acetylcholine release as assessed by intracortical in vivo microdialysis. This result is consistent with previous reports according to which, in spite of the massive reduction of cortical cholinergic input, intraparenchymal treatments performed using this immunotoxin have repeatedly failed to reproduce the kind of memory deficits normally found as a result of the less selective excitotoxic lesions (Berger-Sweeney et al., 1994; Torres et al., 1994; Wenk et al., 1994, 1996; Baxter et al., 1995; Waite and Thal, 1996; Dornan et al., 1997). In our present study, subsequent monitoring of cholinergic markers in the basolateral amygdala confirms the sparing of amygdaloid projecting fibers after the 192IgG–saporin treatment. In contrast, NMDA-induced lesion into to basal forebrain do affect cholinergic projections to both the cortex and basolateral amygdala. However, consistent with previous findings comparing several excitotoxins versus 192IgG–saporin (Waite and Thal, 1996), NMDA microinjections into the basal forebrain resulted in significantly less cholinergic loss in the cortex than that obtained by the administration of 192IgG–saporin. This result directly implies that learning deficits obtained after NMDA-induced lesions into the basal forebrain cannot be the sole result of cortical cholinergic hypofunction, since a much stronger and selective reduction in cortical acetylcholine does not result in aversion learning deficits. Taken together, these results prompted us to hypothesize an additional involvement of the basoamygdaloid projection in the regulation of memory processes exerted by the basal forebrain.
There seems to be little doubt concerning the involvement of the amygdaloid complex (particularly the basolateral and central nucleus) in associative processing of gustatory stimuli (Bermúdez-Rattoni and Yamamoto, 1998). However, NMDA and ibotenic acid-induced lesions in the BLA alone, in spite of the extensive destruction of this nucleus, fail to produce any detectable deficit in CTA acquisition and retention (Bermúdez-Rattoni and McGaugh, 1991; Chambers, 1990; Dunn and Everitt, 1988). Given the differential effects of 192IgG–saporin and NMDA-induced lesions into the basal forebrain on CTA acquisition, we suggest that the cholinergic modulation of learning processes exerted by the basal forebrain might be redundantly performed by both the basocortical and the basoamygdaloid pathway. Should this be the case, one would expect the combined deafferentation of the cortex and the excitotoxic ablation of BLA (hence the disruption of the putative NBM-BLA function) to result in the same learning deficits normally found as a result of the less selective excitotoxic destruction of the NBM.
By combining NMDA lesions into the basolateral amygdala with 192IgG–saporin injections into the basal forebrain, we have found a strong disruption of taste aversion learning, even when none of these treatments were by themselves capable of producing any detectable impairment in this learning task. This behavioral deficit was in fact only paralleled by the effect of intrabasal NMDA microinjections. The cooperative effects of both lesions suggest the possibility of a redundant scheme according to which the modulatory function exerted by the basal forebrain can be, to a given extent, facultatively performed by both the NBM–amygdala and NBM–cortex pathway. Relevant cellular mechanisms have been documented to take place in both the insular cortex and amygdala during the early processing of a novel gustatory stimulus including cholinergic-dependent thyrosine phosphorylation of NMDA receptors (Rosenblum et al., 1995, 1997), cAMP-mediated gene transcription (Dudai, 1987; Lamprecht et al., 1997), and PKC activity (Yasoshima and Yamamoto, 1997). Interestingly, combined ibotenic acid-induced lesions aimed at the basolateral amygdala and insular cortex have stronger disruptive effects on CTA learning than either treatment alone (Yamamoto et al., 1990; Yamamoto, 1993). These data further support the concept of a complementary and/or redundant role for both structures in the processing of gustatory information. Moreover, the fact that NMDA-induced lesions of the basal forebrain result in a strong learning disruption implies that the basal system is also involved in this CTA processing circuit. Because the insular cortex is ultimately needed for the taste aversion conditioning to be learned and expressed, the NBM-meditated associative processing of the gustative stimulus should at the end reach this cortical area.
Figure 7 shows a model of the proposed modulation exerted by the basal forebrain during taste aversion processing. The only two structures functionally linked to the cholinergic basal forebrain so far known to be involved in taste aversion conditioning are the amygdaloid complex and the insular cortex (Bermúdez-Rattoni and Yamamoto, 1998). Therefore, the disruptive effects of the basal excitotoxic lesion can be explained in terms of both ascending pathways. Taking into account that even a strong cortical deafferentation does not per se destroy the cortical function, and being on the other hand that the NMDA-induced lesion into the NBM also reduces cholinergic markers in the basolateral amygdala, our combined lesion study suggests that both ascending pathways are, to some extent, redundantly participating in CTA memory processing. It remains, however, to be directly tested whether the basal forebrain-amygdala interaction is in fact being performed through the basoamygdaloid cholinergic projection fibers, rather than through an alternative indirect and/or noncholinergic pathway. A proper answer to these questions demands a detailed characterization of the basal forebrain–amygdala communication pathways, whether or not they are cholinergic.
Fig. 7.

Schematic representation of the proposed model of the modulation of learning processes exerted by the basal forebrain (NBM), according to which this regulation can be redundantly performed by both the basocortical, and basoamygdaloid interaction. BLA, IC, andGS represent the basolateral amygdala, insular cortex, and gustative stimulus, respectively. A, Effective modulation of taste aversion learning (CTA) after a strong reduction of basocortical cholinergic input. B,CTA is still acquired after BLA excitotoxic lesions and the subsequent disruption of the NBM–BLA–IC circuit. C,Defective basal forebrain modulation of CTA learning caused by excitotoxic lesioning of the NBM and subsequent simultaneous (although partial) deafferentation of both the cortex and BLA. D,Combination of A and B conditions resulting in the functional inactivation of both modulatory pathways.
It should be pointed out that these data do not necessarily imply a roughly equivalent role for the basal forebrain cholinergic modulation on cortex and amygdala functions. Differences in the relative weight of both ascending projections are to be expected. Indeed, direct infusions of several muscarinic antagonists into the insular cortex have been shown to disrupt taste aversion learning (Naor and Dudai, 1996), suggesting that the sole blockade of the cortical cholinergic input can by itself disrupt the cortical learning function. The fact that an almost complete cortical cholinergic depletion does not result in detectable learning deficits suggests that some residual cortical cholinergic activity is still sufficient for mnemonic function. It can be argued that there might be a significant difference between permanent or long-term lesion (i.e., anatomical of cytotoxic ablations) inflicted long before the effect on behavior is tested, and transient metabolic lesions, inflicted a few minutes before the physiological test. However, in agreement with our own interpretation, recent findings in our laboratory show that intracortical infusion of scopolamine in animals that previously received intrabasal microinjections of 192IgG–saporin still result in a strong disruption of taste aversion learning (Gutiérrez et al., 1999). Taken together this data suggest a highly critical role for the basocortical cholinergic pathway. On the other hand, the ablation of the BLA nucleus (and the corresponding disruption of the NBM–BLA–IC circuit) does not by itself disrupt aversion learning, thereby implying a less critical role for this pathway. According to our model, the involvement of the NBM–BLA interaction becomes evident as soon as both the basocortical and basoamygdaloid pathways are compromised. In fact both ascending pathways might be participating in different processes cooperatively involved in learning mechanisms. It has been suggested that the NBM–amygdala projections have a role in retention of affective aspects of conditioning processes, whereas the basocortical projection might be contributing to attentional processes (Everitt and Robins, 1997). In this regard, recent findings have shown that although classical learning paradigms (i.e., radial maze, water maze, and taste aversion learning) are not sensitive to even a strong basocortical deafferentation (after intraparenchymal 192IgG–saporin treatments), more delicate attention-sensitive tasks show a contrasting critical dependence on intact cortical cholinergic function (Chiba et al., 1995;Baxter et al., 1997). In the present study, we show evidence for an additional functional overlapping attained by both ascending projections, because functional deficits associated to experimentally induced cortical cholinergic hypofunction can seemingly be overcome by the NBM–BLA–cortex circuit and vice versa.
The results of this study extend previous work that investigated the effect of 192IgG–saporin and excitatory neurotoxins treatments into the NBM and provide support to the notion that the basal forebrain–amygdala interaction participates in aversive conditionings. They further provide a framework that may help to solve the well documented discrepancies regarding the effects of different neurotoxins in basal forebrain lesion studies. Moreover, they suggest that the regulation exerted by the basal forebrain can be redundantly and additively performed by both the basocortical and the basoamygdaloid projection systems in adult normal rat.
Footnotes
This work was supported in part by Consejo Nacional de Ciencia y Tecnologia Grant 3260p-N9607. We acknowledge the technical assistance of Jimena Estrada, Oreste Carbajal, and Federico Jandete and give thanks to Enrique Espinosa for reviewing the text and to Yolanda Díaz de Castro for the preparation of this manuscript.
Correspondence should be addressed to F. Bermúdez-Rattoni, Instituto de Fisiología Celular, Universidad Nacional Autónoma de México, Apartado postal 70–253, 04510 México, D.F., México.
REFERENCES
- 1.Aggleton JP, Petrides M, Iversen SD. Differential effects of amygdaloid lesion on conditioned taste aversion learning by rats. Physiol Behav. 1981;27:397–403. doi: 10.1016/0031-9384(81)90322-x. [DOI] [PubMed] [Google Scholar]
- 2.Baxter MG, Bucci DJ, Gorman LK, Wiley RG, Gallagher M. Selective immunotoxic lesions of basal forebrain cholinergic cells: effects on learning and memory in rats. Behav Neurosci. 1995;109:714–722. doi: 10.1037//0735-7044.109.4.714. [DOI] [PubMed] [Google Scholar]
- 3.Baxter MG, Holland PC, Gallagher M. Disruption of decrements in conditioned stimulus processing by selective removal of hippocampal cholinergic input. J Neurosci. 1997;17:5230–5236. doi: 10.1523/JNEUROSCI.17-13-05230.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beninger RJ, Kuhnemann S, Ingles JL, Jhmandas K, Boegman RJ. Mnemonic deficits in the double Y-maze are related to the effects of nucleus basalis injections of ibotenic and quisqualic acids on choline acetyltransferase in the rat amygdala. Brain Res Bull. 1994;35:147–152. doi: 10.1016/0361-9230(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 5.Berger-Sweeney J, Heckers S, Mesulam MM, Wiley RG. Differential effects of spatial navigation of immunotoxin-induced cholinergic lesions of the medial septal area and nucleus basalis magnocellularis. J Neurosci. 1994;14:4507–4519. doi: 10.1523/JNEUROSCI.14-07-04507.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bermúdez-Rattoni F, McGaugh JL. Insular cortex and amygdala lesions differentially affect acquisition on inhibitory avoidance and conditioned taste aversion. Brain Res. 1991;549:165–170. doi: 10.1016/0006-8993(91)90616-4. [DOI] [PubMed] [Google Scholar]
- 7.Bermúdez-Rattoni F, Yamamoto T. Neuroanatomy of CTA: lesion studies. In: Bures J, Bermúdez-Rattoni F, Yamamoto T, editors. Conditioned taste aversion. Memory of a special kind. Oxford UP; New York: 1998. pp. 28–44. [Google Scholar]
- 8.Bigl V, Woolf NJ, Butcher LL. Cholinergic projection from the basal forebrain to frontal, parietal, temporal, occipital, and cingulate cortices: a combined fluorescent tracer and acetylcholinesterase analysis. Brain Res Bull. 1982;8:727–749. doi: 10.1016/0361-9230(82)90101-0. [DOI] [PubMed] [Google Scholar]
- 9.Boegman RJ, Cockhill J, Jhamandas K, Beninger RJ. Excitotoxic of the rat basal forebrain: differential effects on choline acetyltransferase in the cortex an amygdala. Neuroscience. 1992;51:129–135. doi: 10.1016/0306-4522(92)90477-j. [DOI] [PubMed] [Google Scholar]
- 10.Book AA, Wiley RG, Shweitzer JB. 192IgG saporin: Specific lethality for cholinergic neurons in the basal forebrain of the rat. J Neuropathol Exp Neurol. 1994;53:95–102. doi: 10.1097/00005072-199401000-00012. [DOI] [PubMed] [Google Scholar]
- 11.Braun JJ, Lasiter PS, Kiefer SW. The gustatory neocortex of the rat. Physiol Psychol. 1982;10:13–45. [Google Scholar]
- 12.Chambers KC. A neural model for conditioned taste aversion. Annu Rev Neurosci. 1990;13:373–385. doi: 10.1146/annurev.ne.13.030190.002105. [DOI] [PubMed] [Google Scholar]
- 13.Chiba AA, Bucci DJ, Holland PC, Gallagher M. Basal forebrain cholinergic lesions disrupts increments but not decrements in conditioned stimulus processing. J Neurosci. 1995;15:7315–7322. doi: 10.1523/JNEUROSCI.15-11-07315.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dornan WA, McCambell AR, Tinkler GP, Hickman LJ, Bannon AW, Decker MW, Gunther KL. Comparisons of site specific injections into the basal forebrain on water maze and radial maze performance in the male rat after immunolesioning with 192IgG saporin. Behav Brain Res. 1997;86:181–189. doi: 10.1016/s0166-4328(96)02264-4. [DOI] [PubMed] [Google Scholar]
- 15.Dudai Y. The cAMP cascade in the nervous system: molecular sites of action and possible relevance to neural plasticity. Crit Rev Biochem. 1987;22:221–281. doi: 10.3109/10409238709101484. [DOI] [PubMed] [Google Scholar]
- 16.Dunn LT, Everitt BJ. Double dissociation of the effects of amygdala and insular cortex lesions on conditioned taste aversion, passive avoidance, and neophobia in the rat using the excitotoxin ibotenic acid. Behav Neurosci. 1988;102:3–23. doi: 10.1037//0735-7044.102.1.3. [DOI] [PubMed] [Google Scholar]
- 17.Dunnett SB, Fibiger HC. Role of forebrain cholinergic system in learning and memory: Relevance to cognitive deficits of aging and Alzheimer’s dementia. Prog Brain Res. 1993;98:413–420. doi: 10.1016/s0079-6123(08)62425-5. [DOI] [PubMed] [Google Scholar]
- 18.Dunnet SB, Whishaw IQ, Jones GH, Bunch ST. Behavioral, biochemical, and histological effects of different neurotoxic amino acids injected into nucleus basalis magnocellularis of rats. Neuroscience. 1987;20:653–669. doi: 10.1016/0306-4522(87)90117-5. [DOI] [PubMed] [Google Scholar]
- 19.Escobar ML, Chao V, Bermúdez-Rattoni F. In vivo long-term potentiation in the insular cortex: NMDA receptor dependence. Brain Res. 1998;779:314–319. doi: 10.1016/s0006-8993(97)01175-x. [DOI] [PubMed] [Google Scholar]
- 20.Etherington RE, Mittleman G, Robbins TW. Comparative effects of nucleus basalis and fimbria-fornix lesions on delayed matching and alteration tests memory. Neurosci Res Commun. 1987;22:441–469. [Google Scholar]
- 21.Everitt BJ, Robins TW. Central cholinergic system and cognition. Annu Rev Psychol. 1997;48:649–684. doi: 10.1146/annurev.psych.48.1.649. [DOI] [PubMed] [Google Scholar]
- 22.Everitt BJ, Robbins TW, Evenden JL, Marston HM, Jones GH, Sirkia TE. The effects of excitotoxic lesions of the substancia innominata, ventral and dorsal globus pallidus on the acquisition and retention of a conditional visual discrimination: implication for cholinergic hypothesis of learning and memory. Neuroscience. 1987;22:441–469. doi: 10.1016/0306-4522(87)90346-0. [DOI] [PubMed] [Google Scholar]
- 23.Gutiérrez H, Miranda M, Bermúdez-Rattoni F. Learning impairments and cholinergic deafferentation after cortical nerve growth factor deprivation. J Neurosci. 1997;17:3796–3803. doi: 10.1523/JNEUROSCI.17-10-03796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gutiérrez H, Gutiérrez R, Silva-Gandarias R, Estrada J, Miranda MI, Bermúdez-Rattoni F. Differential effects of 192IgG-saporin and NMDA-induced lesions into the basal forebrain on cholinergic activity and taste aversion memory formation. Brain Res. 1999;834:136–141. doi: 10.1016/s0006-8993(99)01519-x. [DOI] [PubMed] [Google Scholar]
- 25.Heckers S, Mesulam M. Two types of cholinergic projections to the rat amygdala. Neuroscience. 1994;14:383–97. doi: 10.1016/0306-4522(94)90252-6. [DOI] [PubMed] [Google Scholar]
- 26.Heckers S, Ohtake T, Wiley RG, Lappi DA, Geula C, Mesulam MM. Complete and selective cholinergic denervation of rat neocortex and hippocampus but not amygdala by an immunotoxin against the p75 NGF receptor. J Neurosci. 1994;14:1271–1289. doi: 10.1523/JNEUROSCI.14-03-01271.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hepler DJ, Wenk GL, Cribbs BL, Olton DS, Coyle JT. Lesions in nucleus basalis magnocellularis and medial septal area of rats produce qualitatively similar memory impairments. J Neurosci. 1985;5:866–873. doi: 10.1523/JNEUROSCI.05-04-00866.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu SM, Raine L, Fanger H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedure. J Histochem Cytochem. 1981;29:577–580. doi: 10.1177/29.4.6166661. [DOI] [PubMed] [Google Scholar]
- 29.Kapp BS, Schwaber JS, Driscoll PA. Frontal cortex projections to the amygdaloid central nucleus in the rabbit. Neuroscience. 1985;15:327–346. doi: 10.1016/0306-4522(85)90216-7. [DOI] [PubMed] [Google Scholar]
- 30.Kiefer SW. Neural mediation of conditioning food aversions. Ann NY Acad of Sci. 1985;443:100–109. doi: 10.1111/j.1749-6632.1985.tb27066.x. [DOI] [PubMed] [Google Scholar]
- 31.Kiefer SW, Brown JJ. Acquisition of taste avoidance habits in rats lacking gustatory neocortex. Physiol Psychol. 1979;7:245–250. [Google Scholar]
- 32.Lamprecht R, Hazvi S, Dudai Y. cAMP response element-binding protein in the amygdala is required for long- but not short-term conditioned taste aversion memory. J Neurosci. 1997;17:8443–50. doi: 10.1523/JNEUROSCI.17-21-08443.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lasiter PS, Glanzman DL. Cortical substrates of taste aversion learning: Involvement of the dorsolateral amygdaloid nuclei and the temporal neocortex in taste aversion learning. Behav Neurosci. 1985;99:257–276. doi: 10.1037//0735-7044.99.2.257. [DOI] [PubMed] [Google Scholar]
- 34.Lopez-García JC, Ruiz JF, Escobar ML, Rattoni FB, Tapia R. Effects of excitotoxic lesions of the nucleus basalis magnocellularis on conditioned taste aversion and inhibitory avoidance in the rat. Pharmacol Biochem Behav. 1993;45:147–152. doi: 10.1016/0091-3057(93)90098-e. [DOI] [PubMed] [Google Scholar]
- 35.Mallet PE, Beninger RJ, Flescher SN, Jhamandas K, Boegman RJ. Nucleus basalis lesions: implication of basoamygdaloid cholinergic pathways in memory. Brain Res Bull. 1995;36:51–56. doi: 10.1016/0361-9230(94)00162-t. [DOI] [PubMed] [Google Scholar]
- 36.Naor C, Dudai Y. Transient impairment of cholinergic function in the rat insular cortex disrupts the encoding of taste in conditioned taste aversion. Behav Brain Res. 1996;79:61–67. doi: 10.1016/0166-4328(95)00262-6. [DOI] [PubMed] [Google Scholar]
- 37.Norgren R. Gustatory afferents to ventral forebrain. Brain Res. 1974;81:285–295. doi: 10.1016/0006-8993(74)90942-1. [DOI] [PubMed] [Google Scholar]
- 38.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; San Diego: 1986. [DOI] [PubMed] [Google Scholar]
- 39.Rosenblum K, Schul R, Meiri N, Hadari YR, Zick Y, Dudai Y. Modulation or protein tyrosine phosphorylation in rat insular cortex after conditioned taste aversion. Proc Natl Acad Sci USA. 1995;92:1157–1161. doi: 10.1073/pnas.92.4.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosemblum K, Berman DE, Hazvi S, Lamprecht R, Dudai Y. NMDA receptor and the tyrosine phosphorylation of its 2B subunit in taste learning in the rat insular cortex. J Neurosci. 1997;17:5129–5135. doi: 10.1523/JNEUROSCI.17-13-05129.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sinden JD, Hodges H, Gray JA. Neural transplantation and recovery of cognitive function. Behav Brain Sci. 1995;18:10–35. [Google Scholar]
- 42.Torres EM, Perry TA, Blockland A, Wilkinson LS, Wiley RG, Lappi DA, Dunnet SB. Behavioural, histochemical and biochemical consequences oƒ 83 selective immunolesions in discrete regions of the basal forebrain cholinergic system. Neuroscience. 1994;63:95–122. doi: 10.1016/0306-4522(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 43.Waite JJ, Thal LJ. Lesions of the cholinergic nuclei in the rat basal forebrain: excitotoxins vs an immunotoxin. Life Sci. 1996;58:1947–1953. doi: 10.1016/0024-3205(96)00184-1. [DOI] [PubMed] [Google Scholar]
- 44.Wenk GL. The nucleus basalis magnocellularis cholinergic system: one hundred years of progress. Neurobiol Learn Mem. 1997;67:85–95. doi: 10.1006/nlme.1996.3757. [DOI] [PubMed] [Google Scholar]
- 45.Wenk GL, Stoehr JD, Quintana G, Mobley S, Wiley RG. Behavioral, biochemical, histological, and electrophysiological effects of 192IgG-saporin injections into the basal forebrain of rats. J Neurosci. 1994;14:5986–5995. doi: 10.1523/JNEUROSCI.14-10-05986.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wenk GL, Stoehr JD, Mobley SL, Gurney J, Morris RJ. Age-related decrease in vulnerability to excitatory amino acids in the nucleus basalis. Neurobiol Aging. 1996;17:1–7. doi: 10.1016/0197-4580(95)02025-x. [DOI] [PubMed] [Google Scholar]
- 47.Wiley RG. Neural lesioning with ribosome-inactivating proteins: suicide transport and immunolesioning. Trends Neurosci. 1992;15:285–290. doi: 10.1016/0166-2236(92)90078-m. [DOI] [PubMed] [Google Scholar]
- 48.Yamamoto T (1993) Neuronal mechanisms of taste aversion learning. 16:181–185. [DOI] [PubMed]
- 49.Yamamoto T, Matsuo R, Kawamura Y. Localization of cortical gustatory area in rats and its role in taste discrimination. J Neurophysiol. 1980;44:440–454. doi: 10.1152/jn.1980.44.3.440. [DOI] [PubMed] [Google Scholar]
- 50.Yamamoto T, Matsuo R, Ichikawa H, Wakisaka S, Akai M, Imai Y, Yonehera N, Inoki R. Functional relations between the cortical gustatory area and the amygdala: electrophysiological and behavioral studies in rats. Neurosci Lett. 1990;112:167–172. doi: 10.1016/0304-3940(90)90197-h. [DOI] [PubMed] [Google Scholar]
- 51.Yasoshima Y, Yamamoto T. Rat gustatory memory requires protein kinase C activity in the amygdala and cortical gustatory areas. NeuroReport. 1997;8:1363–1367. doi: 10.1097/00001756-199704140-00009. [DOI] [PubMed] [Google Scholar]