

Abstract
Brain-derived neurotrophic factor (BDNF) is reported to enhance synaptic transmission and to play a role in long-term potentiation in hippocampus and neocortex. If so, a shortage or blockade of BDNF might lead to another form of synaptic plasticity, long-term depression (LTD). To test this possibility and to elucidate mechanisms if it is the case, EPSCs evoked by test stimulation of layer IV were recorded from layer II/III neurons in visual cortical slices of young rats in the whole-cell voltage-clamp mode. LTD was induced by low-frequency stimulation (LFS) at 1 Hz for 10–15 min if each pulse of the LFS was paired with depolarization of neurons to −30 mV but was not induced if their membrane potentials were kept at −70 mV. Such an LTD was blocked by exogenously applied BDNF, probably through presynaptic mechanisms. Suppression of endogenous BDNF activity by the anti-BDNF antibody or an inhibitor for BDNF receptors made otherwise ineffective stimuli (LFS without postsynaptic depolarization) effective for LTD induction, suggesting that endogenous BDNF may prevent low-frequency inputs from inducing LTD in the developing visual cortex.
Keywords: long-term depression, brain-derived neurotrophic factor, long-term potentiation, synaptic plasticity, visual cortex, development
Neurotrophins, the family of nerve growth factor (NGF), have been reported to enhance excitatory synaptic transmission (Lohof et al., 1993; Leβmann et al., 1994; Kang and Schuman, 1995; Levine et al., 1995; Stoop and Poo, 1996; Akaneya et al., 1997; Carmignoto et al., 1997; Scharfman, 1997) or to suppress inhibitory transmission (Kim et al., 1994; Tanaka et al., 1997) and further to play a role in a form of synaptic plasticity, long-term potentiation (LTP), in the hippocampus and developing visual cortex (Castrén et al., 1992; Patterson et al., 1992; Kang and Schuman, 1995; Korte et al., 1995; Figurov et al., 1996; Patterson et al., 1996;Akaneya et al., 1997). In field potential recordings from the visual cortex of young rats, it was reported that one of the neurotrophins, brain-derived neurotrophic factor (BDNF), blocks the induction of another form of synaptic plasticity, long-term depression (LTD), which otherwise is induced by low-frequency stimulation (LFS) at 1 Hz for 10–15 min of afferents (Akaneya et al., 1996; Huber et al., 1998). Such an action of BDNF was suggested to play a role in consolidation or protection of immature synapses (Akaneya et al., 1996), which are known to be easily depressed by low-frequency, repetitive inputs (Dudek and Bear, 1993; Dudek and Friedlander, 1996). It is not yet known, however, whether the site of the LTD-blocking action of exogenously applied BDNF is presynaptic or postsynaptic, and more importantly whether endogenous BDNF actually plays such a role in the developing visual cortex.
To examine these questions we recorded EPSCs with whole-cell voltage clamping of layer II/III neurons in visual cortical slices of young rats and found that LTD was induced only when LFS was paired with a certain level of postsynaptic depolarization, and that BDNF may exert its action on presynaptic sites where LTD is reported to be expressed in visual cortex (Torii et al., 1997). Furthermore, the inhibition of activity of endogenous BDNF with the anti-BDNF antibody made the otherwise ineffective LFS effective to induce LTD. This result suggests that endogenous BDNF may prevent low-frequency inputs from inducing LTD and thus play a crucial role in consolidation of immature synapses, which are bombarded by inputs at various frequencies from retinae in the developing visual cortex (Shatz, 1990).
MATERIALS AND METHODS
Preparation of slices. Sprague Dawley rats, aged from 13 to 18 postnatal days, were deeply anesthetized with ketamine (30 mg/kg, i.p.), followed by cervical dislocation. Coronal slices of visual cortex (300 μm thickness) were cut using a rotor slicer (DTY7000, Dosaka, Japan). Procedures for maintaining the slices were essentially the same as described previously (Akaneya et al., 1997). In short, slices were submerged in a stream of the perfusion medium at the rate of 200 ml/h. The composition of incubation and perfusion medium of the slices was as follows (in mm): NaCl 124, KCl 5, KH2PO4 1.2, MgSO4 1.3, CaCl2 2.4, NaHCO3 26, and glucose 10. All of the recordings were performed at 30–31°C.
Stimulation of afferents and recording of synaptic currents.A bipolar stimulating electrode was placed in layer IV of the cortex. EPSCs evoked by test stimulation of layer IV were recorded from pyramidal cell-like neurons in layer II/III of the cortex through whole-cell patch-clamp electrodes (Yoshimura and Tsumoto, 1994). These neurons were identified visually using an upright microscope with Nomarski optics (Axioscope FS, Zeiss, Germany). Patch pipettes (resistance, 8–10 MΩ) were filled with a solution containing (in mm): 130 potassium gluconate, 10 KCl, 10 HEPES, 3 MgATP, 0.5 Na2GTP, and adjusted to pH 7.2 by KOH. The osmolarity of the solution was 270 mOsm. Visually identified neurons were voltage-clamped at −70 mV with a patch-clamp amplifier (Axoclamp 2B, Axon Instruments, Foster City, CA). The fast transient capacitive currents for recording electrodes were canceled. The series resistance was <30 MΩ. Data were digitized at a rate of 10 kHz and fed into an IBM-PC clone computer for analysis. Synaptic currents were filtered at 1 kHz. These data were analyzed mainly with pClamp or AxoScope software (Axon Instruments).
Test pulses of 0.1 msec width were delivered at 0.1 Hz, and the stimulus intensity was adjusted usually to 1.2–1.5 times the threshold for EPSCs. The intensity of test shocks was usually 1.5–3.0 V for 0.1 msec duration. After having confirmed that test shocks given to layer IV at 0.1 Hz induced EPSCs with almost constant amplitude for 10 min, LFS at 1 Hz was applied to the same site in layer IV for 10 or 15 min. The width of each pulse of LFS and its voltage were the same as those of test shocks. EPSCs were further observed for at least 20 min after cessation of the LFS. To see a possible dependence of LTD induction on postsynaptic membrane potential, each pulse of the LFS was paired with depolarization of recorded neurons to −30 or −50 mV for 100 msec (Yoshimura and Tsumoto, 1994). This depolarization was initiated 10 msec before each shock of LFS. The pairing procedure was performed at 1 Hz for 10 or 15 min. To evaluate effects of LFS on EPSCs in various conditions quantitatively, the means of peak amplitude and initial slope of EPSCs for 10 min before LFS were taken as control value. The slope was calculated using the value for 10–60% of the initial, falling phase of EPSCs.
In most of the experiments, recombinant human BDNF (provided from Sumitomo Pharmaceutical Co., Ltd., Japan) or K252a (provided from Kyowa Hakko Co., Ltd., Japan) was applied to slices through the perfusion medium 20 min before the initiation of LFS. BDNF was prepared at the concentration of 500 μg/ml containing 0.1% bovine serum albumin in undiluted Ca2+-, Mg2+-free PBS. Then it was diluted to be 20 ng/ml with the perfusion medium just before recordings. For application of K252a, it was prepared to be the final concentration of 200 or 300 nmwith dimethyl sulfoxide (DMSO) as vehicle, of which the final concentration was 0.01% (v/v). In the experiments in which K252b was injected into cortical neurons, it was prepared with DMSO and patch pipette solution. The concentrations of K252b and DMSO in pipettes were 200 nm and 0.01% (v/v), respectively. In part of the experiments, K252b was mixed with rhodamine (15 μm) to see the spread of K252b into neurons with the fluorescent microscope (Axioscope FS). To prevent a possible adhesion of the neurotrophins to the wall of the perfusion system, the inner walls of the reservoir, recording chamber, and tubes connecting them were coated by silicone 2–3 d before recording.
Preparation and purification of the anti-BDNF antibody.BDNF-specific antibodies were prepared as described previously (Katoh-Semba et al., 1997). In short, BDNF (3 ng/1.5 ml) was treated with formalin (3 μl) at room temperature for 30 min, and then the excess formalin was inactivated with Tris-HCl, pH 8.0. Polyclonal antibodies against BDNF were raised by immunizing rabbits with the formalin-treated BDNF. The BDNF-specific IgGs were purified from the antiserum on a column of Sepharose CL-4B coupled with recombinant human BDNF. The specificity of the resultant polyclonal antibodies against BDNF was checked by the immunoblotting procedure as described previously (Katoh-Semba et al., 1997).
Evaluation of biological activity of the anti-BDNF antibody in neuronal cultures. Biological activity of the polyclonal antibody was assayed in dissociated primary cultures of basal forebrain neurons from embryonic rats, as described previously (Hatanaka and Tsukui, 1986). Briefly, tissue fragments of the basal forebrain area containing the septum and the vertical limb of the diagonal band were dissected out from embryonic day 18 rats (Wistar ST, Shizuoka, Japan), digested with papain (90 U; Worthington, Freehold, NJ) twice for 15 min each time at 37°C, and then resuspended in the medium containing 5% (v/v) precolostrum newborn calf serum (Mitsubishi Kasei), 5% (v/v) horse serum (Life Technologies, Gaithersburg, MD), 1% (v/v) rat serum, and 89% (v/v) DF medium (1:1 mixture of DME and Ham’s F12 medium containing 15 mm HEPES buffer, pH 7.4, 30 nmselenium, and 1.9 mg/ml sodium bicarbonate). Cells were then dissociated by gentle drawing through plastic tips of two sizes (1.2 mm followed by 0.8 mm in diameter). Cells were resuspended in the above serum-containing medium and seeded onto polyethyleneimine-coated 48-well plates (0.65 cm2/well; Sumitomo Bakelite) at a density of 5.8 × 105cells/cm2. Seeded cells were cultured in a humidified 5% CO2 incubator at 37°C. After 1 d of culture, the medium was changed, and then the neurotrophic factors and antibodies were added. Culture was continued for 6 d. Choline acetyltransferase (ChAT) activity was assayed according to the method described previously (Hatanaka and Tsukui, 1986).
RESULTS
Whole-cell patch-clamp recordings were performed from pyramidal cell-like neurons in layer II/III of the visual cortex. To check possible changes in recording conditions and membrane properties of cells under observation, we continuously monitored series and input resistances. If these values changed >20% of the initial control, the recordings were stopped and the data were deleted from the analysis. Eighty-four neurons that were recorded for longer than 40 min satisfied these criteria. In the 61 neurons in which the initial control records were obtained without any drug, single shocks applied to layer IV of the cortex induced inward currents with the mean peak latencies and amplitudes of 7.4 ± 2.0 (SD) msec and 115.1 ± 59.9 pA, respectively, at the membrane potential of −70 mV (see specimen records of Figs. 1, 3, and 7). These currents were judged as monosynaptically elicited EPSCs on the basis of the published criteria, such as very short and constant latency and reversal potential near 0 mV (Yoshimura and Tsumoto, 1994). After test shocks of layer IV at 0.1 Hz were confirmed to elicit EPSCs in the stable condition for 10 min, LFS was given at 1 Hz for 10 or 15 min to the same site in layer IV. In seven neurons, pair-pulse stimulation at the interval of 50 msec was applied to layer IV at 0.1 Hz without LFS, as will be described later.
Fig. 1.
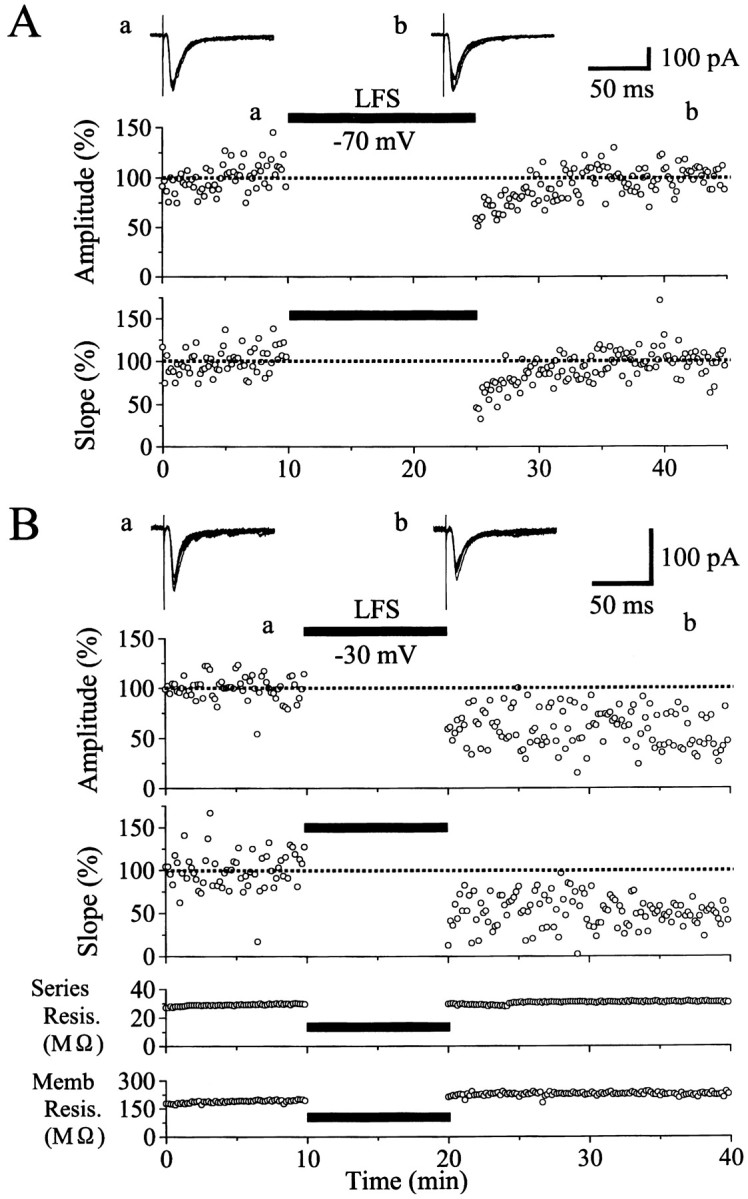
Dependence of LTD induction on postsynaptic membrane potential. A, An example of ineffectiveness for LFS to induce LTD when the membrane potential of a postsynaptic neuron was clamped at −70 mV. At the top are shown examples of EPSCs recorded at the time point indicated by corresponding letters in the bottom graph. Each record is obtained by superimposition of six consecutive sweeps. The peak amplitude and the initial slope of EPSCs are plotted against time in thetop and bottom graphs, respectively. The value is expressed as the percentage of the mean of 60 responses before LFS. The time when LFS was applied to layer IV is indicated by ahorizontal bar. B, An example of LTD induced by LFS that was paired with depolarization of a postsynaptic neuron to −30 mV. The peak amplitude and the initial slope of EPSCs are plotted against time in the top two graphs. In thebottom two graphs, the series resistance of the electrode and the membrane resistance of the cell are plotted against time. Other conventions are the same as in A.
Fig. 3.

Blockade of LTD by BDNF (A), and effective (B) and ineffective (C) antagonism by K252a applied through the medium and K252b injected into a postsynaptic neuron, respectively. The initial slope of EPSCs is plotted against time in the graphs. The value is expressed as the percentage of the mean of 60 responses before LFS. An open, horizontal bar indicates the period during which BDNF was applied to the slice. In B, K252a (300 nm) was applied to a slice, as indicated by ashaded, horizontal bar, simultaneously with BDNF. InC, K252b (200 nm) was injected into a neuron through a patch pipette. Other conventions are the same as in Figure1A.
Fig. 7.
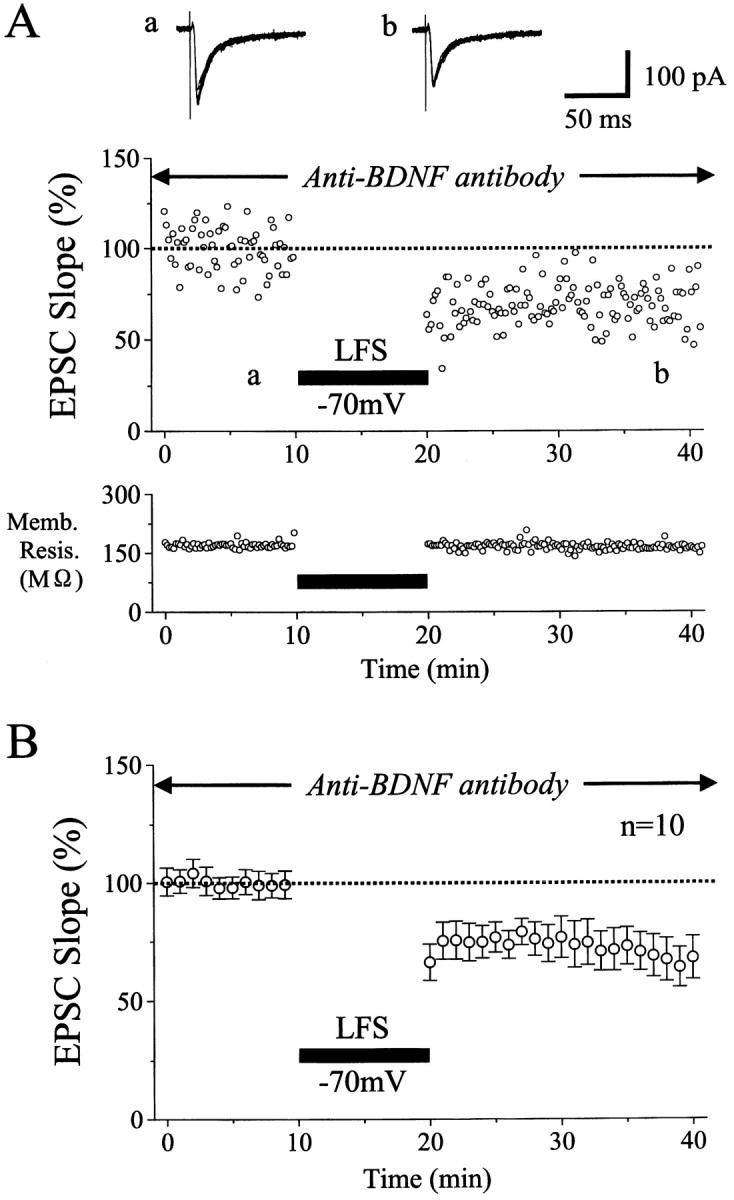
Emergence of LTD during the inhibition of activity of endogenous BDNF. A, An example of LTD induced by LFS without postsynaptic depolarization in a slice that was perfused with the anti-BDNF antibody at the concentration of 1 μg/ml. Other conventions are the same as in Figure 1A. In thebottom graph, the membrane resistance of this neuron is plotted against time. B, Time course of the mean EPSC slope for 10 neurons. Slices containing these neurons had been incubated with the anti-BDNF antibody. Other conventions are the same as in Figure 2.
Dependence of the induction of LTD on postsynaptic depolarization
Initially we found that LFS of layer IV did not induce LTD of EPSCs in most of layer II/III neurons when the membrane potential was kept at −70 mV during the LFS. An example of this finding is shown in Figure 1A. In this case the duration of LFS was set to 15 min to exclude the possibility that the shorter duration of LFS might make the induction of LTD infrequent. As shown in the top graph of Figure 1A, the peak amplitude of EPSCs seemed to be slightly depressed after the LFS with the clamped membrane potential of −70 mV. However, they rapidly recovered to the original control level within 5 min and thereafter remained at the control level until 20 min after cessation of the LFS. The peak amplitude of EPSCs might be contaminated with inhibitory currents, so we also measured the initial slope of EPSCs (Fig. 1A, bottom graph). The initial slope changed in parallel with the peak amplitude of EPSCs after the LFS. Such parallelism between the peak amplitude and the initial slope of EPSCs was seen in other cells also (Fig.1B). In the present study, we used the initial slope of EPSCs for evaluation of changes in excitatory synaptic response to minimize the above-mentioned possibility of contamination of inhibitory currents. Mean values of the initial slope of EPSCs for nine cells were plotted against time in Figure2A. In seven of the nine cells, LFS was applied to layer IV for 10 min, because this duration of LFS, if paired with postsynaptic depolarization to −30 mV, consistently induced LTD, as will be described later. The mean value of ratios of the EPSC slope 15–20 min after cessation of LFS to that before the LFS was 96.0 ± 18.5% (SD) for the seven cells. This value was not significantly different from that (96.6 ± 6.4%) for the other two cells tested with LFS of the 15 min duration. Therefore, these data were combined in Figure 2A and treated as a single group of data in the present study.
Fig. 2.

Time courses of the mean EPSC slope. For each cell, the mean slope of six consecutive EPSCs was calculated as a percentage of that of 30 control EPSCs before LFS. Vertical bars indicate twice the SEM. In A, LFS* indicates that its duration was 10 min for seven cells and 15 min for the other two cells. The values after cessation of the LFS were combined as described in Results. Thus, thenumbers in parentheses along the abscissa indicate the time for the latter two cells. In all nine cells the membrane potential was kept at −70 mV throughout recordings. InB and C, LFS was paired with depolarization of recorded cells to −30 and −50 mV, respectively. The duration of LFS was 10 min, as indicated by horizontal bars. Other conventions are the same as in A. InD, recorded cells were depolarized to −30 mV without layer IV stimulation, as indicated by the horizontal bar. Other conventions are the same as inA.
We then attempted to determine conditions in which LTD of EPSCs of patch-clamped neurons was induced reliably by LFS of layer IV. We found that LTD was induced consistently when LFS was paired with depolarization of recorded neurons to −30 mV. An example of this finding is shown in Figure 1B. In this neuron the mean peak amplitude and initial slope of control EPSCs before LFS was 99.0 ± 12.2 pA and 28.1 ± 10.9 nA/sec, respectively. After LFS was combined with depolarization to −30 mV for 10 min, the amplitude and slope of EPSCs decreased to ∼65 and 60% of the control, respectively, and remained at this level 20 min after cessation of the LFS. This depression of EPSCs was not caused by a deterioration of the recording and neuronal conditions, because the series and membrane resistances were very stable (Fig.1B, bottom two graphs). Mean EPSC slopes for nine cells were plotted in Figure 2B. It is obvious that LTD was induced by LFS that was paired with depolarization of postsynaptic neurons to −30 mV.
Then, we tested to what degree depolarization is necessary to induce LTD. For this, LFS of layer IV was paired with depolarization of the membrane potential to −50 mV in another nine cells. As shown in Figure2C, this pairing procedure did not yield LTD of EPSCs. The mean value of ratios of the EPSC slope 15–20 min after LFS to that before LFS for the nine cells was 91.8 ± 15.9% (SD), which was not significantly (p = 0.16; paired ttest) different from the control. In the present study, we did not systematically change the membrane potential of recorded cells, because we were not interested in the threshold for the postsynaptic depolarization to induce LTD, but rather in the action of BDNF on LTD. There is a possibility that LTD induced by pairing of LFS with the depolarization of the recorded neurons to −30 mV might be attributable to strong postsynaptic depolarization per se. To test this possibility, eight neurons were depolarized to −30 mV (duration, 100 msec) at 1 Hz for 10 min without layer IV stimulation. As a whole, EPSCs were not significantly depressed (Fig. 2D). The mean value of the EPSC slope 15–20 min after cessation of the depolarization procedure for the eight cells (96.3 ± 13.5%) was not significantly different from the control.
Blockade of LTD by exogenous BDNF
As reported previously (Akaneya et al., 1997), BDNF at the concentration of 20 ng/ml did not significantly change EPSCs evoked by test stimulation of layer IV at 0.1 Hz. In the present study, however, we found that BDNF at this concentration did block LTD of EPSCs even when LFS was paired with depolarization of postsynaptic neurons to −30 mV. An example of this finding is shown in Figure3A. Twenty minutes after initiation of the BDNF application, LFS paired with the depolarization of this neuron to −30 mV was applied to layer IV at 1 Hz for 10 min. Such a pairing procedure turned out to be ineffective. After cessation of the LFS, the amplitude and initial slope of EPSCs were not decreased at all (Fig. 3A). Mean EPSC slopes for the eight cells tested in this way were plotted in Figure4A. It is evident that LTD was not induced by LFS. The mean value of ratios of the EPSC slope 15–20 min after LFS to that before the LFS for the eight cells was 98.4 ± 19.4%. Without BDNF, LFS of the same parameters induced significant LTD in all of the nine cells, as mentioned above (Fig.2B). The mean ratio for the nine cells was 55.6 ± 17.4%. The difference between these two values was statistically significant (p < 0.001; unpaired ttest).
Fig. 4.
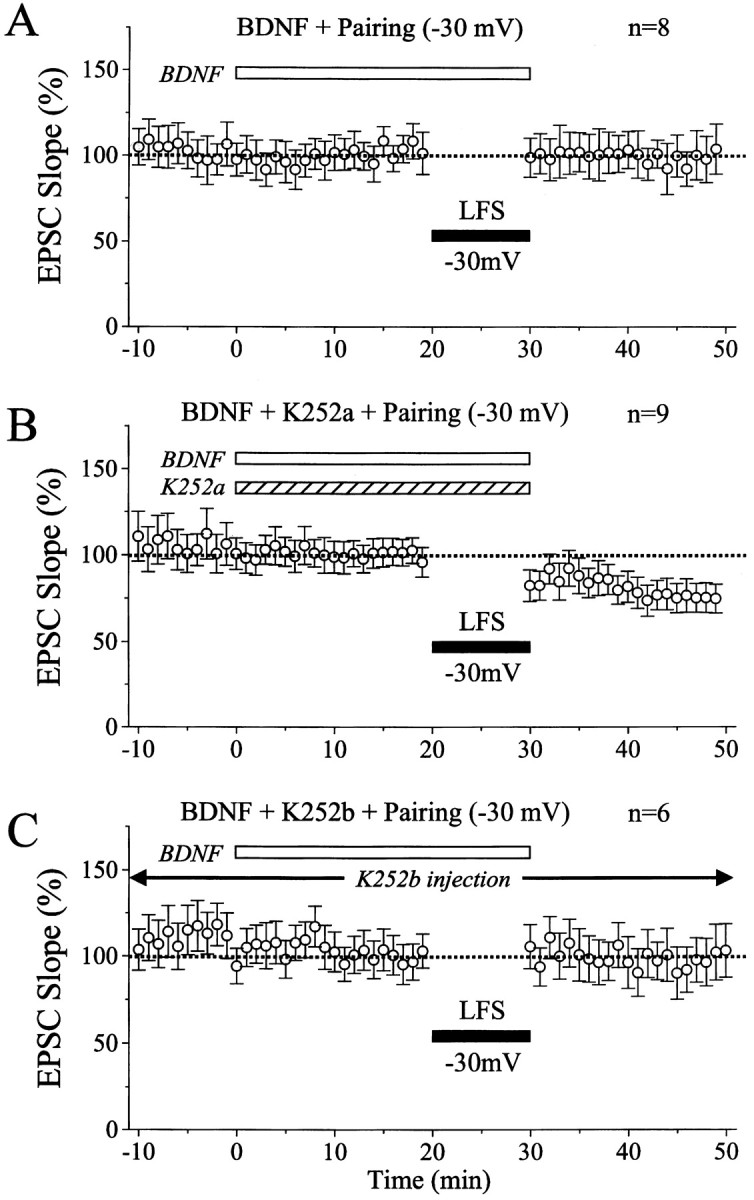
Time courses of the mean EPSC slope in the conditions as indicated. Conventions are the same as in Figure 2. InA, BDNF was applied to slices, as indicated by theopen, horizontal bar, and then LFS was paired with −30 mV depolarization of recorded neurons during the period indicated by the filled horizontal bar. In B, BDNF and K252a were applied to slices through the perfusion medium, as indicated by horizontal bars. In C, K252b was injected into recorded neurons through patch pipettes, and BDNF was applied to slices through the medium, as indicated by the open, horizontal bar.
Next, we tested whether K252a could antagonize such a blocking action of BDNF on the LTD induction. K252a is a membrane-permeable inhibitor for Trk receptor tyrosine kinases without affecting other tyrosine kinases at the concentration of 200–300 nm, and thus is an effective blocking agent on biological actions of BDNF and other neurotrophins (Knüssel and Hefti, 1992; Tapley et al., 1992). As reported previously (Akaneya et al., 1996, 1997), an application of K252a at this concentration did not significantly change synaptic responses of layer II/III neurons evoked by test stimulation of layer IV at 0.1 Hz. When K252a was coapplied with BDNF, LFS paired with the depolarization of postsynaptic neurons to −30 mV induced LTD of EPSCs. An example of this finding is shown in Figure 3B. In this case, the initial slope of EPSCs 15–20 min after the paired LFS decreased to 55.4 ± 17.2% of the control. This result indicates that K252a antagonized the LTD-blocking action of BDNF. Mean EPSC slope for the nine cells tested in this way shows that LFS of layer IV during the coapplication of BDNF and K252a induced LTD of EPSCs (Fig.4B), although the magnitude of LTD (73.5 ± 16.5%, 15–20 min after LFS) was slightly but significantly (p < 0.05; unpaired t test) smaller than that induced by paired LFS alone. This might be attributable to an imperfect action of K252a at 200–300 nm in the slice preparations. Nevertheless, the value of 73.5 ± 16.5% was significantly (p < 0.02; unpaired ttest) smaller from that (98.4 ± 19.4%) of the eight cells without K252a but with BDNF (Fig. 4A). In sum, these results suggest that the blockade of LTD by BDNF is mediated at least in part through an activation of Trk receptor tyrosine kinases.
Then, we examined the question of whether the site of the activation of Trk receptor tyrosine kinases by BDNF is presynaptic or postsynaptic. For this, K252b was injected into postsynaptic neurons through patch pipettes. K252b is a membrane-impermeable inhibitor of Trk receptor tyrosine kinases over a wide range of concentrations without being cytotoxic (Knüssel and Hefti, 1992). To confirm that K252b actually spread to the whole dendritic region of postsynaptic neurons, a mixture of K252b and rhodamine was injected into four neurons through whole-cell patch pipettes. The molecular weights of K252b and the dye are similar (453.5 and 606.7, respectively), and thus they are expected to spread almost equally. The fluorescent dye was observed to spread to distal parts of dendrites of layer II/III pyramidal neurons approximately 20 min after the cell membrane had been ruptured (Fig.5). It is to be noted that the stable recording of EPSCs was usually obtained at least 10 min after the formation of patch-clamp recording configuration, and thus LFS was initiated at least 20 min after the rupture of the membrane. Therefore, K252b is expected to spread to almost the whole dendritic region at a concentration not very different from that in pipettes, when LFS was applied to layer IV.
Fig. 5.

A fluorescent image of a pyramidal neuron into which a mixture of K252b and rhodamine was injected through a patch pipette, which is seen to be attached to the soma. This cell was located in layer II/III of the cortex so that the right side of the figure corresponds to layer I of the cortex. Scale bar, 20 μm.
Figure 3C shows an example of the results obtained from the experiments in which the possibility of postsynaptic action of K252b was tested. About 10 min after having obtained the configuration of whole-cell patch-clamp recording with a pipette containing K252b at 200 nm, EPSCs became stable and then remained almost constant. Ten minutes later, BDNF at 20 ng/ml was applied to this cell through the perfusion medium. EPSCs were not significantly changed by BDNF (Fig. 3C, record b). Then, LFS paired with depolarization of this cell to −30 mV was applied to layer IV, but EPSCs were not depressed at all (Fig. 3C, recordc and graph). Mean EPSC slopes for the six cells tested in this way were plotted in Figure 4C. The mean value of ratios of the EPSC slope 15–20 min after LFS to that before the LFS for this group of cells (95.5 ± 19.5%) was nearly equal to that of the cells with BDNF alone (Fig. 4A), indicating that the injection of K252b into postsynaptic neurons could not antagonize the action of BDNF.
The ineffectiveness of the postsynaptic injection of K252b but the effective blockade of the BDNF action by the bath application of K252a suggests that the site of its action is presynaptic. To confirm this suggestion, we observed EPSCs evoked by pair-pulse stimulation of layer IV and measured the ratio of the initial slope of second EPSC to that of first EPSC [pair-pulse ratio (PPR)], because PPR is thought to be an indicator of a change in presynaptic processes of transmitter release (Zucker, 1989). So, we stimulated layer IV with pair-pulse shocks at the interval of 50 msec and measured PPRs before and after the application of BDNF at 20 ng/ml in seven neurons. Consistent with the previous report (Torii et al., 1997), five of the seven cells showed pair-pulse depression, and the other two cells did not show pair-pulse depression or potentiation. The mean PPR for the seven cells was 0.83 ± 0.16 (SD). After the application of BDNF, EPSCs to second stimuli were relatively enhanced in most of the cells, and consequently the mean PPR of these seven cells was increased to 0.98 ± 0.22. The difference between these two values is statistically significant (p < 0.01; pairedt test). This confirms the suggestion that the site of the BDNF action is presynaptic.
Emergence of LTD during the inhibition of endogenous BDNF
As the next step of analysis, we attempted to determine whether endogenous BDNF has a role in the blockade of LTD similar to that of exogenous BDNF, as mentioned above. In other words, the ineffectiveness of LFS without postsynaptic depolarization might be caused by the LTD-blocking action of endogenous BDNF. To test this possibility, we initially applied K252a to cortical slices, and LFS was given to layer IV without postsynaptic depolarization. In seven of the nine cells tested in this way, LFS effectively induced LTD even when the membrane potential was clamped at −70 mV. The mean value of ratios of the EPSC slope 15–20 min after LFS to that before LFS was 72.4 ± 9.1% for the nine cells.
The action of K252a is not strictly selective for TrkB receptor tyrosine kinases. To block the action of endogenous BDNF more selectively, therefore, we used the polyclonal antibody against BDNF. This antibody reacted with BDNF but not with NGF and neurotrophin-3 (NT-3) (Fig. 6A). Then, we tested whether the antibody could block biological activities of BDNF selectively and if so at what concentration. For this test, dissociated primary cultures of basal forebrain neurons obtained from embryonic rats were prepared, and ChAT activity was assayed according to the method as described previously (Hatanaka and Tsukui, 1986). Results obtained from this assay are shown in Figure6B. The antibody at the concentration of 1 μg/ml clearly antagonized the action of BDNF, whereas it did not block that of NGF. This indicates that the antibody at 1 μg/ml could selectively block the biological activity of BDNF. The antibody might not easily penetrate into slices, even if the slices were incubated with the antibody for ∼1 hr. Initially, therefore, we incubated seven slices with the antibody at the concentration of 5 μg/ml and found a clear antagonizing effect against BDNF. In three other slices, we used the antibody at 1 μg/ml, which again showed the same degree of the antagonizing effect. So, the data obtained with the two concentrations of the antibody were combined in the present analysis. An example of the results obtained with the antibody at 1 μg/ml is shown in Figure7A. As shown in specimen records, test stimulation of layer IV elicited EPSCs with the mean peak latency and amplitude of 6.1 ± 0.4 msec and 147.1 ± 12.4 pA, respectively. These values were within those of control EPSCs without the antibody. For the 10 cells incubated with the antibody, the mean peak latency and amplitude of control EPSCs were 8.4 ± 1.8 msec and 110.1 ± 42.0 pA, respectively, which were not significantly different from those of the other neurons (7.5 ± 1.9 msec and 116.1 ± 57.2 pA, respectively). Thus, the incubation of slices with the anti-BDNF antibody did not affect control EPSCs of layer II/III neurons evoked by layer IV stimulation at 0.1 Hz. In the cell shown in Figure 7A, LFS of layer IV induced a clear LTD even when the cell was clamped at −70 mV. The mean value of the initial slope of EPSCs 15–20 min after LFS was 70.0 ± 9.8% of the control. This LTD was not caused by a deterioration of the cell condition, because the membrane resistance was fairly constant (Fig.7A, bottom histogram). For the 10 cells tested in this way, mean EPSC slopes were plotted in Figure 7B. The mean value of ratios of the EPSC slope 15–20 min after LFS to that before the LFS (69.1 ± 21.8%) was significantly (p < 0.01; unpaired t test) smaller than that (96.1 ± 16.2%) of the cells clamped at −70 mV without any drug or antibody, as shown in Figure 2A.
Fig. 6.

Specificity of the anti-BDNF antibody.A, Western blot. NGF (purified mouse submandibular β-NGF, 2.6 ng), BDNF (recombinant human BDNF, 2.5 ng), and NT-3 (recombinant human NT-3, 2.5 ng) were loaded in respective lanes. The blot was stained with the anti-BDNF antibody. An arrow indicates a band corresponding to a subunit of BDNF with molecular mass of 14 kDa.B, Assay of activity of the anti-BDNF antibody. Action of the antibody on ChAT activity enhanced by BDNF or NGF was examined in the conditions as indicated at the bottom. Eachcolumn with short bars indicates the mean ± SD of the mean for the four determinations.
DISCUSSION
In the present study we found that LFS of layer IV induces LTD of EPSCs of layer II/III neurons in the visual cortex of young rats if each shock of the LFS is paired with depolarization of the neurons to −30 mV, but such a paired LFS becomes ineffective during the application of BDNF. This indicates that exogenously applied BDNF prevents paired LFS from inducing LTD of synaptic transmission. Considering the existence of endogenous BDNF in the cerebral cortex (Wetmore et al., 1991; Dugich-Djordjevic et al., 1995; Nawa et al., 1995; Katoh-Semba et al., 1997; Yan et al., 1997a), it is also possible to state that endogenous BDNF alone cannot block the induction of LTD by paired LFS. The present results also demonstrate that the inhibition of activity of endogenous BDNF by the anti-BDNF antibody or K252a makes the otherwise ineffective LFS effective for the induction of LTD in most of the neurons that were voltage-clamped at −70 mV. This suggests that endogenous BDNF may prevent repetitive, low-frequency inputs to cortical synapses from inducing LTD if postsynaptic neurons are not depolarized or only weakly depolarized. In the physiological condition, however, cortical neurons are not voltage-clamped so that repetitive inputs would generate a strong postsynaptic depolarization. It is known, however, that repetitive synaptic inputs at low frequencies such as 1 Hz do not always generate strong depolarizations or action potentials of neurons in hippocampus (Mulkey and Malenka, 1992) and visual cortex (Dudek and Friedlander, 1996), probably because of no summation of excitatory responses and/or a concomitant activation of inhibitory circuits. Thus, it seems possible that repetitive, low-frequency inputs to cortical synapses could not induce LTD in the presence of endogenous BDNF. In other words, endogenous BDNF is assumed to prevent such inputs from inducing LTD in the physiological condition.
In the developing visual cortex, it is suggested that cortical synapses are bombarded by repetitive inputs originated from retinal spontaneous activity at various frequencies, including the range of LFS (Shatz, 1990). Furthermore, immature synapses in the cortex are known to be easily suppressed by such repetitive inputs (Dudek and Bear, 1993;Dudek and Friedlander, 1996). Taking these and the above-mentioned considerations altogether, it seems reasonable to suggest that endogenous BDNF may prevent immature cortical synapses from being depressed by such spontaneous inputs during postnatal development.
The site in which BDNF exerts its action: tentative models
As mentioned above, a substantial depolarization of cortical neurons in conjunction with repetitive synaptic inputs at 1 Hz for 10–15 min is necessary for the induction of LTD. This result indicates that processes for the induction of LTD may take place at postsynaptic sites. On the other hand, the previous study using the same type of preparations as the present ones suggested that LTD of layer II/III neurons in the visual cortex is expressed at presynaptic sites (Torii et al., 1997). Thus, it seems reasonable to conclude that the locus for the induction of LTD is postsynaptic, but that the expression is presynaptic in visual cortex, as reported in the CA1 area of the hippocampus (Bolshakov and Siegelbaum, 1994; Stevens and Wang, 1994). This suggests that there is a retrograde messenger that might act as an LTD expression factor in the visual cortex (Fig.8A).
Fig. 8.

Schematic diagrams showing the two models for the blocking action of BDNF on LTD. In A, BDNF is hypothesized to be derived from presynaptic sites and to serve as an inhibitor for an LTD expression factor that would be released from postsynaptic sites. In this model, LFS is supposed to induce a mild level of depolarization and a consequent rise of Ca2+ in postsynaptic sites through activation of glutamate receptors (Glu-R) and voltage-dependent Ca2+ channels (VDCC). This rise of Ca2+ would activate an unknown process, which in turn would release or produce the LTD expression factor. This factor would decrease transmitter release from presynaptic terminals for a long time. The activation of TrkB by BDNF would then suppress the action of this factor in presynaptic terminals. In B, BDNF itself is hypothesized to operate as an LTD-blocking or a synaptic transmission-maintaining factor. The LFS-induced rise in postsynaptic Ca2+ would activate an unknown factor, possibly protein phosphatases, which would suppress the release or production of BDNF at postsynaptic sites. This would in turn lead to a shortage of BDNF at presynaptic terminals. Thus, LFS without support of BDNF would lead to a decrease in transmitter release from presynaptic terminals for a long time.
Then, an obvious question is this: in which site BDNF does exert its action? Previous studies with immunohistochemistry reported that high-affinity receptors for BDNF, TrkB receptors, exist both at presynaptic axons and postsynaptic dendrites and soma in the cortex (Cabelli et al., 1996; Fryer et al., 1996; Yan et al., 1997b). The present results—that the injection of K252b into postsynaptic neurons was not effective to block the action of BDNF, whereas the perfusion of K252a to the whole slice was effective—suggest that BDNF exerts its action on the expression mechanism in presynaptic sites. This suggestion was further confirmed by the result that the PPR of EPSCs was changed by the application of BDNF in most of the cases in which pair-pulse stimulation was given to layer IV. Previous studies also suggest that BDNF acts on presynaptic sites to increase the release of transmitters (Lohof et al., 1993; Leβmann et al., 1994; Kang and Schuman, 1995; Stoop and Poo, 1996; Carmignoto et al., 1997; Gottschalk et al., 1998; Leβmann and Heumann, 1998). Then, the next question is this: from which site is BDNF derived? Recently it has been suggested that BDNF is transported anterogradely and localized around vesicles in presynaptic terminals (von Bartheld et al., 1996; Conner et al., 1997;Fawcett et al., 1997). Thus, it is possible to assume that BDNF released from axon terminals activates TrkB receptors at the terminals, which in turn inhibits the action of an LTD expression factor (Fig.8A). However, this possibility seems inconsistent with the present result that LTD was induced by LFS without postsynaptic depolarization during the application of the anti-BDNF antibody. In this situation the postsynaptic process was not activated, so that the LTD expression factor would not be produced or released. Nevertheless the inactivation of BDNF by the antibody resulted in the induction of LTD by LFS.
Thus, we are led to an alternative hypothesis (Fig.8B). In this hypothesis BDNF itself is supposed to act as a retrograde messenger, as suggested previously (Thoenen, 1995;Bonhoeffer, 1996; Katz and Shatz, 1996). A novel point of this hypothesis is that the basal level of BDNF would be released as a transmission-maintaining factor from postsynaptic neurons with the membrane potential at or near the resting level, but its release would be decreased during the moderate level of postsynaptic depolarization induced by LFS. A very strong depolarization that is induced by high-frequency inputs would rather increase the release of BDNF, as suggested in hippocampal neurons that were depolarized very strongly with high K+ solution (Blöchl and Thoenen, 1995; Goodman et al., 1996). The increased BDNF at presynaptic sites then would play a role in the maintenance of LTP, as suggested previously (Kang and Schuman, 1995; Akaneya et al., 1997). On the other hand, the moderate level of depolarization induced by LFS would activate a factor that suppresses the release or production of BDNF. Thus, the decrease in release of BDNF from postsynaptic sites and the consequent shortage of BDNF at presynaptic sites may lead to LTD. In other words, LFS would depress synaptic transmission for a long time if a certain amount of BDNF is not available to presynaptic sites. This hypothesis seems consistent with the results obtained in the present study. For example, the inhibition of activity of BDNF by the antibody would result in LTD without postsynaptic depolarization. The factor that reduces the release or production of BDNF in response to the moderate level of postsynaptic depolarization might be Ca2+/calmodulin-dependent protein phosphatase, whereas the factor with the opposite action might be Ca2+/calmodulin-dependent protein kinases, as suggested previously (Tsumoto, 1992; Bear and Malenka, 1994; Singer, 1995). However, little is known about the intracellular processes that control the release or production of BDNF, and thus more experimental data are necessary for the establishment of this hypothesis.
Footnotes
This study is supported by a Grant-in-Aid for Scientific Research (07279102) from the Ministry of Education, Science, Sports and Culture of Japan. We express many thanks to Sumitomo Pharmaceutical Co., Ltd. and Kyowa Hakko Kogyo Co., Ltd. for kind gifts of recombinant human BDNF and K252a, respectively. We also thank Dr. Yasuhiro Abiru for helpful advice to performing the experiments.
Correspondence should be addressed Dr. Tadaharu Tsumoto, Department of Neurophysiology, Biomedical Research Center, Osaka University Medical School, 2-2 Yamadaoka, Suita City 565-0871 Japan.
REFERENCES
- 1.Akaneya Y, Tsumoto T, Hatanaka H. Long-term depression blocked by brain-derived neurotrophic factor in rat visual cortex. J Neurophysiol. 1996;76:4198–4201. doi: 10.1152/jn.1996.76.6.4198. [DOI] [PubMed] [Google Scholar]
- 2.Akaneya Y, Tsumoto T, Kinoshita S, Hatanaka H. Brain-derived neurotrophic factor enhances long-term potentiation in rat visual cortex. J Neurosci. 1997;17:6707–6716. doi: 10.1523/JNEUROSCI.17-17-06707.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 4.Blöchl A, Thoenen H. Characterization of nerve growth factor (NGF) release from hippocampal neurons: evidence for a constitutive and an unconventional sodium-dependent regulated pathway. Eur J Neurosci. 1995;7:1220–1228. doi: 10.1111/j.1460-9568.1995.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 5.Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- 6.Bonhoeffer T. Neurotrophins and activity-dependent development of the neocortex. Curr Opin Neurobiol. 1996;6:119–126. doi: 10.1016/s0959-4388(96)80017-1. [DOI] [PubMed] [Google Scholar]
- 7.Cabelli RJ, Allendoerfer KL, Radeke MJ, Welcher AA, Feinstein SC, Shatz CJ. Changing patterns of expression and subcellular localization of TrkB in the developing visual system. J Neurosci. 1996;16:7965–7980. doi: 10.1523/JNEUROSCI.16-24-07965.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carmignoto G, Pizzorusso T, Tia S, Vicini S. Brain-derived neurotrophic factor and nerve growth factor potentiate excitatory synaptic transmission in the rat visual cortex. J Physiol (Lond) 1997;498:153–164. doi: 10.1113/jphysiol.1997.sp021848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castrén E, Pitkanen M, Sirviö J, Parsadanian A, Lindholm D, Thoenen H, Riekkinen PJ. The induction of LTP increases BDNF and NGF mRNA but decreases NT-3 mRNA in the dentate gyrus. NeuroReport. 1992;4:895–898. doi: 10.1097/00001756-199307000-00014. [DOI] [PubMed] [Google Scholar]
- 10.Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dudek SM, Bear MF. Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J Neurosci. 1993;13:2910–2918. doi: 10.1523/JNEUROSCI.13-07-02910.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dudek SM, Friedlander MJ. Developmental down-regulation of LTD in cortical layer IV and its independence of modulation by inhibition. Neuron. 1996;16:1097–1106. doi: 10.1016/s0896-6273(00)80136-1. [DOI] [PubMed] [Google Scholar]
- 13.Dugich-Djordjevic MM, Peterson C, Isono F, Ohsawa F, Widmer HR, Denton TL, Bennett GL, Hefti F. Immunohistochemical visualization of brain-derived neurotrophic factor in the rat brain. Eur J Neurosci. 1995;7:1831–1839. doi: 10.1111/j.1460-9568.1995.tb00703.x. [DOI] [PubMed] [Google Scholar]
- 14.Fawcett JP, Aloyz R, McLean JH, Pareek S, Miller FD, McPherson PS, Murphy RA. Detection of brain-derived neurotrophic factor in a vesicular fraction of brain synaptosomes. J Biol Chem. 1997;272:8837–8840. doi: 10.1074/jbc.272.14.8837. [DOI] [PubMed] [Google Scholar]
- 15.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 16.Fryer RH, Kaplan DR, Feinstein SC, Radeke MJ, Grayson DR, Kromer LF. Developmental and mature expression of full-length and truncated TrkB receptors in the rat forebrain. J Comp Neurol. 1996;374:21–40. doi: 10.1002/(SICI)1096-9861(19961007)374:1<21::AID-CNE2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 17.Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AJ, Hefti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996;7:222–238. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- 18.Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hatanaka H, Tsukui I. Differential effects of nerve growth factor and glioma-conditioned medium on neurons cultured from various regions of fetal rat central nervous system. Dev Brain Res. 1986;30:47–56. doi: 10.1016/s0006-8993(86)80007-5. [DOI] [PubMed] [Google Scholar]
- 20.Huber KM, Sawtell NB, Bear MF. Brain-derived neurotrophic factor alters the synaptic modification threshold in visual cortex. Neuropharmacology. 1998;37:571–579. doi: 10.1016/s0028-3908(98)00050-1. [DOI] [PubMed] [Google Scholar]
- 21.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 22.Katoh-Semba R, Takeuchi IK, Semba R, Kato K. Distribution of brain-derived neurotrophic factor in rats and its changes with development in the brain. J Neurochem. 1997;69:34–42. doi: 10.1046/j.1471-4159.1997.69010034.x. [DOI] [PubMed] [Google Scholar]
- 23.Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- 24.Kim HG, Wang T, Olafsson P, Lu B. Neurotrophin 3 potentiates neuronal activity and inhibits γ-aminobutyratergic synaptic transmission in cortical neurons. Proc Natl Acad Sci USA. 1994;91:12341–12345. doi: 10.1073/pnas.91.25.12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knüssel B, Hefti F. K-252 compounds: modulator of neurotrophin signal transduction. J Neurochem. 1992;59:1987–1996. doi: 10.1111/j.1471-4159.1992.tb10085.x. [DOI] [PubMed] [Google Scholar]
- 26.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leßmann V, Heumann R. Modulation of unitary glutamatergic synapses by neurotrophin-4/5 or brain-derived neurotrophic factor in hippocampal microcultures: presynaptic enhancement depends on pre-established paired-pulse facilitation. Neuroscience. 1998;86:399–413. doi: 10.1016/s0306-4522(98)00035-9. [DOI] [PubMed] [Google Scholar]
- 28.Leßmann V, Kottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- 29.Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptor. Proc Natl Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lohof AM, Ip NY, Poo M-M. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- 31.Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- 32.Nawa H, Carnahan J, Gall C. BDNF protein measured by a novel enzyme immunoassay in normal brain and after seizure: partial disagreement with mRNA levels. Eur J Neurosci. 1995;7:1527–1535. doi: 10.1111/j.1460-9568.1995.tb01148.x. [DOI] [PubMed] [Google Scholar]
- 33.Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- 34.Patterson SL, Abel T, Deuel TAS, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 35.Scharfman HE. Hyperexcitability in combined entorhinal/hippocampal slices of adult rat after exposure to brain-derived neurotrophic factor. J Neurophysiol. 1997;78:1082–1095. doi: 10.1152/jn.1997.78.2.1082. [DOI] [PubMed] [Google Scholar]
- 36.Shatz CJ. Impulse activity and the patterning of connections during CNS development. Neuron. 1990;5:745–756. doi: 10.1016/0896-6273(90)90333-b. [DOI] [PubMed] [Google Scholar]
- 37.Singer W. Development and plasticity of cortical processing architectures. Science. 1995;270:758–764. doi: 10.1126/science.270.5237.758. [DOI] [PubMed] [Google Scholar]
- 38.Stevens CF, Wang Y. Changes in reliability of synaptic function as a mechanism for plasticity. Nature. 1994;371:704–707. doi: 10.1038/371704a0. [DOI] [PubMed] [Google Scholar]
- 39.Stoop R, Poo M-M. Synaptic modulation by neurotrophic factors: differential and synergistic effects of brain-derived neurotrophic factor and ciliary neurotrophic factor. J Neurosci. 1996;16:3256–3264. doi: 10.1523/JNEUROSCI.16-10-03256.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tapley P, Lamballe F, Barbacid M. K252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene. 1992;7:371–381. [PubMed] [Google Scholar]
- 42.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 43.Torii N, Tsumoto T, Uno L, Astrelin AV, Voronin LL. Quantal analysis suggests presynaptic involvement in expression of neocortical short- and long-term depression. Neuroscience. 1997;79:317–321. doi: 10.1016/s0306-4522(97)00129-2. [DOI] [PubMed] [Google Scholar]
- 44.Tsumoto T. Long-term potentiation and long-term depression in the neocortex. Prog Neurobiol. 1992;39:209–228. doi: 10.1016/0301-0082(92)90011-3. [DOI] [PubMed] [Google Scholar]
- 45.Von Bartheld CS, Byers MR, Williams R, Bothwell M. Anterograde transport of neurotrophins and axodendritic transfer in the developing visual system. Nature. 1996;379:830–833. doi: 10.1038/379830a0. [DOI] [PubMed] [Google Scholar]
- 46.Wetmore C, Cao Y, Pettersson RF, Olson L. Brain-derived neurotrophic factor: subcellular compartmentalization and interneuronal transfer as visualized with anti-peptide antibodies. Proc Natl Acad Sci USA. 1991;88:9843–9847. doi: 10.1073/pnas.88.21.9843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yan Q, Rosenfeld RD, Matheson CR, Hawkins N, Lopez OT, Bennett L, Welcher AA. Expression of brain-derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience. 1997a;78:431–448. doi: 10.1016/s0306-4522(96)00613-6. [DOI] [PubMed] [Google Scholar]
- 48.Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol. 1997b;378:135–157. [PubMed] [Google Scholar]
- 49.Yoshimura Y, Tsumoto T. Dependence of LTP induction on postsynaptic depolarization: a perforated patch-clamp study in visual cortical slices of young rats. J Neurophysiol. 1994;71:1638–1645. doi: 10.1152/jn.1994.71.5.1638. [DOI] [PubMed] [Google Scholar]
- 50.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]