

Abstract
Peripheral myelin protein 22 (PMP22) is an integral membrane protein that is essential for the normal formation and maintenance of peripheral myelin. Duplications, deletions, or mutations in the PMP22 gene account for a set of dominantly inherited peripheral neuropathies. The heterozygous Trembler-J (TrJ) genotype in mice is similar genetically to a Charcot–Marie–Tooth disease type 1A pedigree in humans, whereas the homozygousTrJ condition leads to the most severe form of PMP22-associated neuropathies. To characterize the consequences of theTrJ mutation, we labeled wild-type (wt-) and TrJ-PMP22 in the third loop of the protein with different epitope tags and expressed them separately or together in COS7 cells and primary Schwann cells. Here we show that the transport of the mutant TrJ-PMP22 is interrupted in the intermediate compartment, preventing its insertion into the plasma membrane and affecting the morphology of the endoplasmic reticulum. In addition, TrJ-PMP22 forms a heterodimer with the wt-PMP22. This interaction causes a fraction of the wt-PMP22 to be retained with TrJ-PMP22 in the intermediate compartment of COS7 and Schwann cells. The relative stability of a wt-mutant PMP22 heterodimer as compared with the wt-wt PMP22 homodimer may determine whether a particular mutation is semidominant or dominant. The neuropathy itself appears to result both from decreased trafficking of wt-PMP22 to the plasma membrane and from a toxic gain of function via the accumulation of wt- and TrJ-PMP22 in the intermediate compartment.
Keywords: PMP22, peripheral neuropathy, myelin, Schwann cells, intermediate compartment, dimerization, protein trafficking, Trembler-J mouse, epitope tag
Peripheral myelin protein 22 (PMP22), a 22 kDa glycoprotein, is found mainly in compact peripheral nervous system (PNS) myelin (Snipes et al., 1992). The protein is highly conserved among species, and hydrophobicity plots suggest the presence of four hydrophobic membrane-associated domains (Manfioletti et al., 1990; Patel et al., 1992). Studies of animal models expressing altered levels of mutant PMP22 indicate that PMP22 is required for the formation and maintenance of PNS myelin (Adlkofer et al., 1995, 1997a,b; Huxley et al., 1996; Magyar et al., 1996; Sereda et al., 1996). Point mutations in the gene for PMP22 and, especially, duplication or deletion of the PMP22 gene are responsible for a set of dominantly inherited peripheral neuropathies in humans and mice (for review, see Muller et al., 1997).
In the Trembler-J (TrJ) andTrembler (Tr) mice the neuropathies result from missense mutations in different membrane-associated regions of the protein (Suter et al., 1992a,b). In the heterozygous TrJmice the peripheral nerve myelination occurs during development, although not to the level seen in normal mice (Notterpek et al., 1997). This myelin is progressively lost as the animals age, mediated by degradation in lysosomes, as indicated by the significant upregulation of the lysosomal pathway in the sciatic nerves of affected animals (Notterpek et al., 1997).
Despite many similarities the TrJ and Trphenotypes are not identical. The progression of the disease is less severe in heterozygous TrJ than Tr mice (Henry et al., 1983). However, the homozygous TrJ genotype leads to the most severe known peripheral neuropathy, and the animals die early in postnatal life (Henry and Sidman, 1983). In the major human neuropathy, the Charcot–Marie–Tooth disease type 1A (CMT1A), ∼20 different point mutations have been identified in PMP22, and almost all are located in the membrane-associated domains (Naef and Suter, 1998). The severity of the disease varies according to the type and location of the amino acid changes and is usually more severe than that caused by PMP22 gene duplication. Mutations identical to those in theTrJ and Tr mice have been found in CMT1A and the more severe Déjérine–Sottas Syndrome (Valentijn et al., 1992; Ionasescu et al., 1997). Comparison of the phenotypes of PMP22-deficient mice and mice expressing mutated forms of PMP22 suggests that the mutations act by a toxic gain of function mechanism rather than via a loss of function (Adlkofer et al., 1997a,b; Naef et al., 1997; D’Urso et al., 1998). Various degradation pathways exist to avoid the accumulation of mutated or potentially toxic proteins. These pathways include the lysosomes and the proteasome complex as well as endoplasmic reticulum (ER)-specific mechanisms, and all have been shown to be involved in the degradation of membrane proteins (Jensen et al., 1995).
To explore the molecular mechanisms underlying the TrJneuropathy, we have examined the effects of the TrJ mutation on the cellular localization and trafficking of PMP22 in COS7 cells and primary rat Schwann cells (SCs) in culture. The wild-type (wt-) and TrJ-PMP22 were tagged with two different epitopes to distinguish the proteins when they were expressed in the same cell. The results show that the wt-PMP22, but not the TrJ-PMP22 protein, is transported to the cell membrane. The TrJ protein accumulated in vesicles of the intermediate compartment (IC). When the proteins were coexpressed, wt-PMP22 partially colocalized with TrJ-PMP22 in the IC, but its transport to the cell surface was not blocked completely. Our data suggest that the colocalization of wt- and TrJ-PMP22 results from the formation and trafficking of wt-PMP22 and TrJ-PMP22 as heterodimers.
MATERIALS AND METHODS
Molecular cloning. The mouse wild-type or the TrJ-PMP22 open reading frame (ORF) (Bsu36I–NheI fragment) in pSP72 (Promega, Madison, WI) was used for inserting the tag epitopes. By patch PCR (Squinto et al., 1991), the c-Myc or the hemagglutinin (HA) tag was cloned downstream of the sitesStyI (nucleotide 138 of ORF) and EagI (nucleus 280 of ORF) and upstream of the HincII site (nucleus 379 of ORF) (see Fig. 1A). After the PCR-amplified DNA fragments were sequenced, the three differentially tagged ORFs (BanII–BanI fragment) were cloned into the eukaryotic expression vector pCMX (17).
Fig. 1.
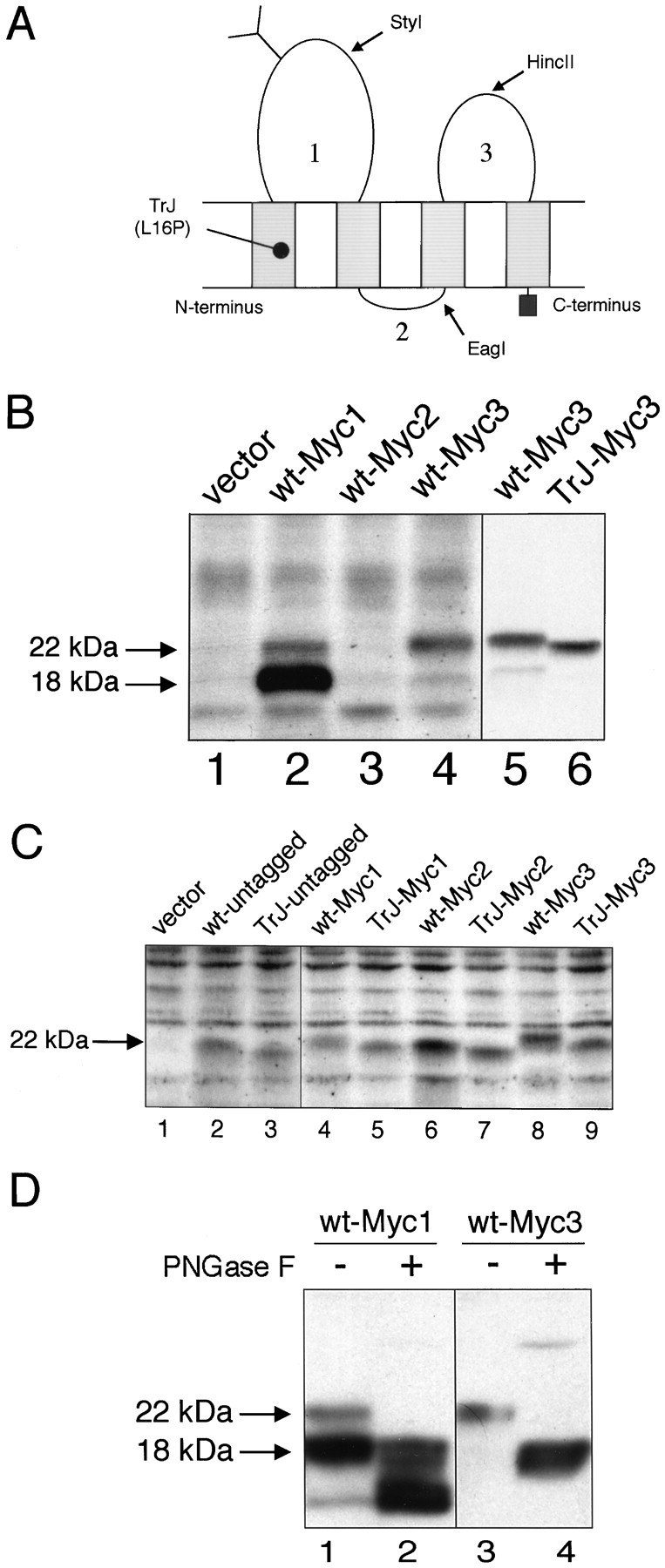
Characterization of the c-Myc epitope-tagged PMP22s. A, Expression of the c-Myc epitope in the three loops of PMP22. Shown is a schematic representation of PMP22. The position of the TrJ mutation is shown near the N terminus. The restriction sites used for inserting the tags are marked by arrows. Numbers represent the position of proposed loops 1, 2, or3. B, Immunodetection of c-Myc tagged wt- and TrJ-PMP22 in COS7 cells. The Myc epitope was inserted into loop 1 (Myc1), loop 2 (Myc2), or loop 3 (Myc3) of wt- or TrJ-PMP22. The expression vector without the cDNA insert was used as a control (vector). Cell lysates were prepared 36 hr after transfection and separated on 12.5% SDS gels; after blotting, the membranes were incubated with a polyclonal rabbit antibody against the c-Myc epitope. The positions of the arrows are derived from molecular weight markers that identify the N-glycosylated PMP22 (22 kDa) and the deglycosylated PMP22 (18 kDa). C, The expression of the c-Myc epitope in the three loops of wt- or TrJ-PMP22 monitored by an anti-PMP22 antibody. The Myc epitope was inserted into loop 1 (Myc1), loop 2 (Myc2), or loop 3 (Myc3) of wt- or TrJ-PMP22, as described in Materials and Methods. The expression vector without a cDNA insert (vector) was used as a control. Cell lysates were prepared 36 hr after transfection and separated on 12.5% SDS gels; after blotting, the membranes were analyzed with a polyclonal rabbit antibody against loop 3 of PMP22. The position of thearrow is derived from molecular weight markers and indicates the migration position of N-glycosylated PMP22 (22 kDa). D, Carbohydrate analysis of wt-Myc1 and wt-Myc3 shows the expected pattern for wt-Myc3, but not for wt-Myc1. Wt-PMP22 with the c-Myc epitope in loop 1 (Myc1) or loop 3 (Myc3) was expressed in COS7 cells. Cell extracts were prepared 36 hr after transfection, incubated with PNGaseF (+), and separated on 12.5% SDS gels. After blotting, the membranes were incubated with a polyclonal rabbit antibody against the c-Myc epitope. Untreated cell extracts were used as controls (−). The positions of the arrows are derived from molecular weight markers and indicate the position of N-glycosylated (22 kDa) and deglycosylated (18 kDa) forms of PMP22.
Cell culture. COS7 cells (American Type Culture Collection, Rockville, MD) were maintained at 37°C in 5% CO2 on untreated tissue culture dishes in DMEM containing 6% bovine calf serum, 6% equine serum (HyClone, Logan, UT), and penicillin/streptomycin. For passaging or before electroporation, the plates were washed once with HBSS and detached with 0.05% Trypsin/EDTA. SC cultures were prepared from neonatal rat sciatic nerves according to the methods of Brockes et al. (1979) and modified as described previously (Pareek et al., 1997). SCs were maintained in DMEM supplemented with 200 μg/ml pituitary extract (Biomedical Technologies, Stoughton, MA) and 2 μm forskolin.
Transfection and immunofluorescent staining. Plates at 80% confluency were trypsinized, washed twice in PBS, and resuspended in PBS. PBS (180 μl) containing 1.5 × 106 cells was mixed with plasmid DNA (30 μg for Cos7 cells; 60 μg for SCs) and incubated for 5 min on ice. After electroporation (125 μF, 0.3 kV; Bio-Rad Gene Pulser, Hercules, CA) the cells were incubated on ice for 5 min and gently resuspended in culture medium. COS7 cells were maintained in 10 cm Petri dishes or on untreated glass coverslips (50,000 cells/12 mm coverslip). SCs were cultured on poly-l-lysine-coated coverslips (100,000 cells/12 mm coverslip). The transfection efficiency of COS7 cells was in the range of 5–15% and <1% for SC. Immunostaining was performed 36 hr after transfection. All steps were performed at room temperature, except for the incubation with antibodies (4°C). Cells were washed twice in DMEM, fixed for 10 min in 4% paraformaldehyde/DMEM, washed twice in Tris-buffered saline (TBS; 25 mm Tris, pH 7.5, 136 mm NaCl, and 2.6 mm KCl), washed once in 1 mm glycine/TBS for 10 min, and washed again in TBS. Then the cells were permeabilized in methanol at −20°C (except for rbet1 and ERGIC53 staining), washed in TBS, and blocked for 30 min in blocking buffer (TBS containing 2% BSA, 0.1% porcine skin gelatin type A, 2% goat serum, and 1% Triton X-100); for rbet1 and ERGIC53 staining only 0.2% Triton X-100 was used. Samples were incubated with the following primary antibodies, diluted according to the manufacturer’s instructions in blocking buffer for 1 hr at 4°C: monoclonal mouse antibodies against c-Myc (Santa Cruz Biotechnology, Santa Cruz, CA), HA (Babco, Hercules, CA), immunoglobulin heavy-chain binding protein (BiP) (StressGen, Victoria, Canada), and ERGIC53 (1:200) (Schweizer et al., 1988). Monoclonal mouse anti-rbet1 hybridoma supernatant (diluted 1:5) was a gift from R. H. Scheller (Stanford University, Stanford, CA), and mouse monoclonal anti-P0 was provided by A. A. Archelos (Archelos et al., 1993). Then the samples were washed three times in TBS (in blocking buffer for rbet1 and ERGIC53 staining) and again for 15 min in blocking buffer. FITC- and Texas Red-conjugated secondary goat anti-mouse and goat anti-rabbit antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) were diluted according to the manufacturer’s instructions in blocking buffer containing 0.5 μg/ml Hoechst dye (Boehringer Mannheim, Indianapolis, IN) and incubated for 1 hr at 4°C with the samples, followed by three washes in TBS. After being mounted in Mowiol (Calbiochem, San Diego, CA) on glass slides, the coverslips were examined and photographed on a Nikon Microphot FXA microscope.
Enzymatic treatments and Western blot analysis. Cells on 10 cm culture plates were washed in PBS, lysed in 0.5 ml of SDS sample buffer (62.5 mm Tris, pH 6.8, 10% glycerol, and 3% SDS), and boiled for 15 min at 95°C. For glycosylation studies the cells were lysed in 500 μl of radioimmunoprecipitation assay (RIPA) buffer (150 mm NaCl, 0.2% SDS, 1% NP-40, 1% deoxycholate, and 50 mm Tris, pH 7.4), rotated in a microcentrifuge tube for 40 min at 4°C, and centrifuged (100,000 × g at 4°C) for 1 hr. Enzymatic treatments with N-glycosidase F (PNGaseF) and endoglycosidase H (EndoH) (both from New England Biolabs, Beverly, MA) were performed overnight according to the manufacturer’s instructions. After 0.2 vol of 6× concentrated SDS sample buffer was added to the reaction mixture, the samples were boiled for 5 min at 95°C. All samples were separated on 12.5% gels and transferred to polyvinylidene difluoride membranes (Immobilon-P, Millipore, Marlborough, MA). Membranes were blocked for 1 hr at 4°C in blocking buffer: TBS containing 1% blocking reagent (Boehringer Mannheim) and 2% goat serum. The polyclonal rabbit antibodies, anti-Myc and anti-PMP22 (Pareek et al., 1997), were diluted 1:500 in antibody buffer (TBS containing 0.5% blocking reagent and 2% goat serum) and incubated for 1 hr at 4°C. Membranes were washed twice in TBS containing 0.1% Tween 20, twice in antibody buffer, and once in blocking buffer and then were incubated with horseradish peroxidase-conjugated anti-rabbit secondary antibodies for 1 hr at 4°C. The membranes were washed three times in TBS/0.1% Tween 20, and the immunoreactive proteins were identified with an enhanced chemiluminescence detection kit (NEN Life Science Products, Boston, MA), according to the manufacturer’s instructions, using Kodak X-OMAT AR film (Kodak, Rochester, NY).
Antibody coimmunoprecipitation. After being washed in PBS, the cells in 10 cm plates were lysed in 0.5 ml of RIPA buffer, rotated in a microcentrifuge tube for 40 min at 4°C, and centrifuged for 1 hr (100,000 × g at 4°C). Cell lysates were rotated for 1 hr at 4°C with 4 μl of polyclonal rabbit anti-c-Myc antibody and then for 2 hr with protein A/G Plus-Agarose (Santa Cruz Biotechnology). The agarose beads were washed four times in RIPA buffer and then boiled for 5 min in 1× SDS sample buffer before separation on 12.5% denaturing gels. Western blot analysis with monoclonal mouse anti-c-Myc or mouse anti-HA antibodies was performed as described above.
Extraction of adult mouse sciatic nerve. Sciatic nerves were collected from adult mice and were frozen immediately in liquid nitrogen. Nerves pooled from two to four animals were pulverized under liquid nitrogen, and total tissue homogenates were prepared in 0.5% SDS/1% β-mercaptoethanol. Digestion with PNGaseF and EndoH was performed according to the manufacturer’s suggestions for 16 hr at 37°C. Protein samples were separated on 12.5% SDS gels under reducing conditions, and Western blot analysis with polyclonal rabbit anti-PMP22 antibody at 1:1000 dilution (Notterpek et al., 1997) was performed as described above.
RESULTS
An epitope tag in loop 3 of PMP22 does not alter its properties
The localization of wt- and TrJ-PMP22 in COS7 and Schwann cells was assessed by overexpressing epitope-tagged proteins and staining with antibodies against the epitope tags. Analysis of the wt-PMP22 protein with the c-Myc epitope in loop 1 (Fig.1A, wt-Myc1) by Western blotting with anti-c-Myc antibody showed two main bands (Fig.1B, lane 2). The upper of these two bands migrated close to wt-PMP22 isolated from rat sciatic nerve (as indicated by the position of the top arrow in Fig. 1B) (Notterpek et al., 1997). Polyclonal anti-PMP22 antibodies raised against a peptide from loop 3 of PMP22 detected the upper, but not the lower band of wt-Myc1 (Fig. 1C, lane 4). The upper band again migrated close to the position of wt-PMP22 (Fig. 1C, lane 2, wt-untagged). These results show that the wt-Myc1 is not suitable for further cellular localization studies. The c-Myc epitope in loop 2 (Fig. 1A, wt-Myc2) was not recognized by anti-c-Myc antibodies (Fig. 1B, lane 3) despite the fact that the polyclonal PMP22 antibody was able to detect the protein (Fig. 1C, lane 6). Thus wt-Myc2 was also not appropriate for further studies.
Analysis of the wt protein with the c-Myc epitope in loop 3 (Fig.1A, wt-Myc3) showed a single main band with either the anti-c-Myc (Fig. 1B, lanes 4, 5) or the anti-PMP22 (Fig. 1C, lane 8) antibodies migrating close to wt-PMP22 from sciatic nerve (Fig. 1B, top arrow;C, arrow) (Notterpek et al., 1997). Insertion of c-Myc into loop 3 of wt-PMP22 appeared to have no obvious deleterious effect on the processing and conformation of the parent protein.
As a further test of the integrity of the epitope-tagged PMP22 proteins, the effect of tagging on the N-linked glycosylation of PMP22 was determined. Wt-Myc1 and wt-Myc3 were treated with PNGaseF, an enzyme that removes all N-linked oligosaccharides from glycoproteins regardless of their carbohydrate composition (Maley et al., 1989). Both species of wt-Myc1 (Fig. 1B, lane 2) identified with the anti-c-Myc antibody were reduced by ∼4 kDa, the weight of the N-linked oligosaccharide in PMP22 (Snipes et al., 1993) (Fig.1D, lanes 1, 2), suggesting that both forms were glycosylated normally. More importantly, the deglycosylation of the single component of wt-Myc3 was identical to that of wt-PMP22 (Fig.1D, lanes 3, 4) (Pareek et al., 1997). Taken together, these results show that loop 3 is an appropriate site for a tag insertion because it does not interfere with the normal biosynthesis of PMP22 nor, apparently, does it affect the integrity or conformation of the resultant tagged protein.
Analysis of the Myc-tagged TrJ-PMP22 expressed in COS7 cells yielded results similar to those obtained with wt-PMP22. TrJ-Myc1 showed a similar major component of unknown composition as wt-Myc1, whereas TrJ-Myc2 was not recognized by the anti-c-Myc antibody (data not shown). Both tagged proteins, however, as well as TrJ-Myc3 were recognized by anti-PMP22 antibody (Fig. 1C). All of the untagged and tagged TrJ-PMP22 proteins migrated at a slightly faster rate than their wild-type counterparts (Fig. 1B,C). N-terminal sequence analysis of wt- and TrJ-PMP22 showed that both N termini were identical (MLLLLLGI), precluding a proteolytic cleavage at this end of the molecule. Thus, the differences in mobility probably reflect the single amino acid change between wt- and TrJ-PMP22, as also has been reported for rat and human ciliary neurotrophic factor (Panayotatos et al., 1993).
The TrJ mutant PMP22 alters the trafficking of wt-PMP22 in COS7 and Schwann cells
Expression in COS7 cells
A possible explanation for the demyelinating phenotype produced by the TrJ mutation is interference with the intracellular processing and trafficking of PMP22. To determine the intracellular localization of wt and mutated PMP22, we expressed wt-Myc3 and TrJ-Myc3 separately in COS7 cells. Because nontransfected cells (Fig. 2, asterisks) or cells transfected only with the expression vector (data not shown) did not stain with anti-c-Myc antibody, the nontransfected cells served as an internal control for antibody specificity. Immunolabeling with polyclonal anti-c-Myc antibody indicated that, in cells expressing relatively high amounts of wt-Myc3, most of the protein remained intracellular although a fraction was transported to the cell membrane (Fig. 2A). Similar results have been reported in COS7 cells (Naef et al., 1997) and in HeLa cells (D’Urso et al., 1998). Double immunolabeling with a monoclonal anti-BiP antibody revealed colocalization of much of the intracellular protein with the ER (Fig.2A,B). The presence of wt-Myc3 in the cell membrane was confirmed by surface staining of transfected, unpermeabilized COS7 cells with polyclonal anti-c-Myc or monoclonal anti-HA antibodies (data not shown). The ratio between membrane-localized and intracellularly retained wt-Myc3 varied. Some cells showed mainly membrane staining with relatively little accumulation in the ER, whereas in other cells wt-Myc3 was found in both the cell membrane and the ER (Fig.2A).
Fig. 2.
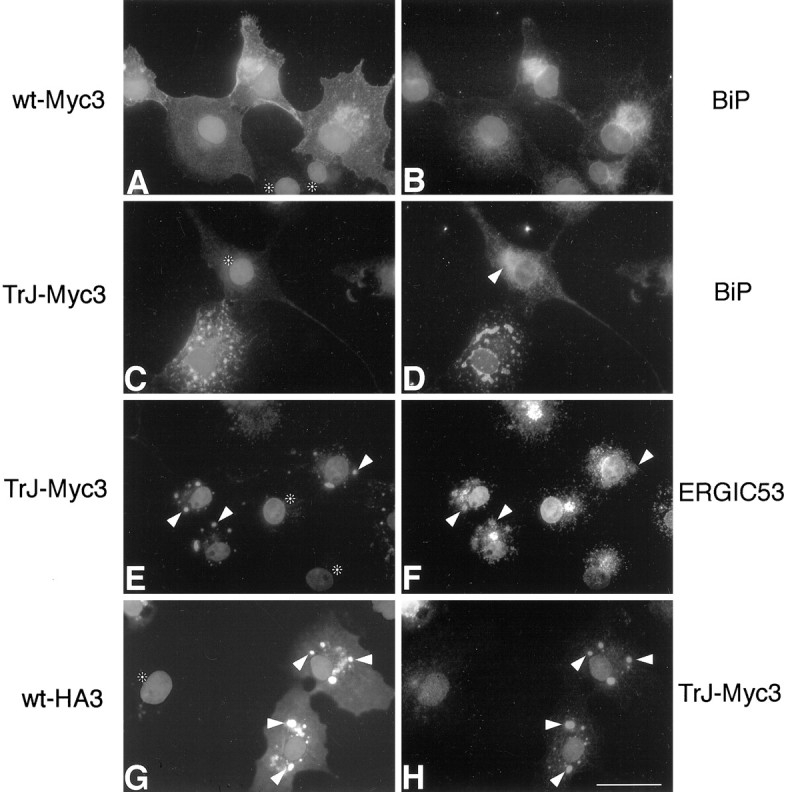
The localization of TrJ-Myc3 is different from that of wt-Myc3 in COS7 cells; coexpression results in the partial colocalization of wt-Myc3 with TrJ-Myc3. Cells were transfected, as described in Materials and Methods, with wt-Myc3 (A, B) and TrJ-Myc3 (C–F) or cotransfected with wt-HA3 and TrJ-Myc3 (G, H). Cells were immunostained 36 hr after transfection. Immunostained proteins are indicated on eachside of the panels, and nuclei are visualized with Hoechst dye. Shown is coimmunodetection of wt-Myc3 (A) with the ER marker BiP (B). TrJ-Myc3 (C, E) did not show colocalization with the ER marker BiP (D) but costained with the intermediate compartment (IC) marker ERGIC53 (F). The arrowhead inD indicates the BiP staining of normal ER morphology for comparison with the TrJ-Myc3-expressing cell below. Thearrowheads in E and Fpoint to vesicular structures that show immunostaining both with antibodies against c-Myc (E) and the IC marker ERGIC53 (F). When wt-HA3 (G) and TrJ-Myc3 (H) were coexpressed, the two proteins partially colocalized in intracellular structures in which TrJ-Myc3 accumulates (arrowheads in G andH). Untransfected cells in A, C, E, and G are marked withasterisks. Scale bar (in H), 33 μm.
The localization of TrJ-Myc3 in COS7 cells differed significantly from that of wt-Myc3 (compare Fig.2A,C). The mutated protein was retained within the cell, with no TrJ-Myc3 observed in the cell membrane (Fig. 2C). In addition, the intracellular TrJ-Myc3 accumulated in large vesicular-like structures (Fig. 2C) that did not show labeling with the ER marker BiP (Fig.2D). However, the ER of the cells expressing the mutated protein (Fig. 2D) showed an altered, condensed morphology as compared with cells expressing wt-Myc3 (Fig.2B) or untransfected cells (Fig. 2D, arrowhead). The morphology of the Golgi compartment was unaltered. Double-immunolabeling studies with various organelle markers revealed that the vesicular-like structures containing TrJ-Myc3 (Fig.2E) stained positive for ERGIC53 (Fig.2F), an IC protein that functions as a mannose-selective lectin (Itin et al., 1996). The colocalization of TrJ-Myc3 and ERGIC53 was confirmed by confocal microscopy (data not shown). The IC is a dynamic collection of vesicles involved in cargo transport from the ER to the Golgi (Hay et al., 1996). It was noted that not all of the ERGIC53-positive IC contained TrJ-Myc3 (Fig.2F). The expression of TrJ-Myc3 therefore is distinct from wt-Myc3 in two ways: it alters the appearance of the ER, and it accumulates in a unique part of the cell, the IC.
To determine whether the expression of TrJ-PMP22 with wt-PMP22 affects the intracellular localization of the latter, we coexpressed both proteins in the same cell. A HA epitope was inserted into loop 3 of the wt-PMP22 to produce the tagged wt-HA3. Coexpression of wt-HA3 and wt-Myc3 showed that the localization of wt-HA3 in COS7 cells was identical to wt-Myc3 (data not shown), indicating that the nature of the tag had no effect on localization. In cells expressing both wt-HA3 and TrJ-Myc3, we detected both proteins in the vesicular-like structures identified as the IC (Fig. 2G,H, arrowheads). Interestingly, some of the wt-HA3 was still observed in the cell membrane (Fig. 2G), whereas TrJ-Myc3 was not (Fig.2H). The intracellular fate of wt-HA3 therefore is altered, in part, when coexpressed with TrJ-Myc3. These findings were confirmed by confocal microscopy (data not shown).
An alternative, biochemical approach to assess the subcellular localization of wt-Myc3 and TrJ-Myc3 was used also. Cell lysates from transfected COS7 cells were treated with EndoH or PNGaseF (Fig.3). EndoH degrades only high-mannose-containing carbohydrates that have not been cleaved by mannosidase II, an enzyme found in the medial- and trans-Golgi (Kornfeld and Kornfeld, 1985). For this reason EndoH-sensitive proteins are localized in the ER or cis-Golgi compartments, whereas EndoH-resistant proteins are found in post-ER compartments, such as the trans-Golgi. In contrast, PNGaseF cleaves all of the carbohydrate moiety from the single N-linked site, leaving only the core PMP22 protein (18 kDa) (Snipes et al., 1993). Analysis of the enzyme-treated lysates showed that, as previously observed for PMP22 (Pareek et al., 1997), a fraction of wt-Myc3 was resistant to EndoH digestion (Fig. 3,lane 2), consistent with a trans-Golgi or plasma membrane localization. On the other hand, all of the TrJ-Myc3 was sensitive to EndoH digestion (Fig. 3, lane 5), indicating that the mutated protein accumulates in a compartment before the trans-Golgi, presumably in the IC, as noted above. The failure of the TrJ-Myc3 protein to pass to the trans-Golgi and to become EndoH-resistant is consistent with the finding that no TrJ-Myc3 was detectable in the cell membrane.
Fig. 3.

Wt-Myc3 expressed in COS7 cells becomes partially resistant to EndoH digestion, but TrJ-Myc3 does not. The wt-Myc3 and TrJ-Myc3 proteins were tagged in loop 3 and expressed in transiently transfected COS7 cells. Cell extracts were prepared 36 hr after transfection, incubated with PNGaseF or EndoH, and separated on 12.5% SDS gels. Untreated extracts were used as a control (−). After blotting, the membranes were incubated with a polyclonal antibody against the c-Myc epitope. The arrow is derived from molecular weight markers and indicates the position of glycosylated (22 kDa) PMP22.
Expression in Schwann cells
Because PMP22 is expressed predominantly in SCs, we performed a similar series of localization studies in these cells, using the constructs described above. As observed in COS7 cells, a fraction of wt-Myc3 was transported to the cell membrane of the SCs while a significant amount was retained intracellularly (Fig.4A) (D’Urso et al., 1998). The same differences between low and high expression of wt-Myc3 as seen in COS7 cells also were observed in SCs, with the exception that at the highest expression levels wt-Myc3 accumulated not only in BiP-positive ER organelles (Fig. 4B) but also in a perinuclear compartment that was not labeled with ER (BiP, Fig. 4B), Golgi, or IC markers (data not shown). The ER marker BiP is localized predominantly in the rough ER, whereas the smooth ER is characterized by a much lower concentration of this protein (Bole et al., 1989). TrJ-Myc3 was retained intracellularly and, as in COS7 cells, was not detected in the cell membrane (Fig.4C). Likewise, the vesicle-like structures in which TrJ-Myc3 accumulated (Fig. 4C, arrowheads) stained positive for the IC marker protein rbet1 (Zhang et al., 1997) (Fig. 4D, arrowheads). The TrJ-Myc3 containing IC compartments were, however, smaller in SCs than in COS7 cells (compare Fig.4G,H). The morphology of the ER in SCs also was affected by expression of TrJ-Myc3 (data not shown), as observed in COS7 cells (see Fig. 2D).
Fig. 4.
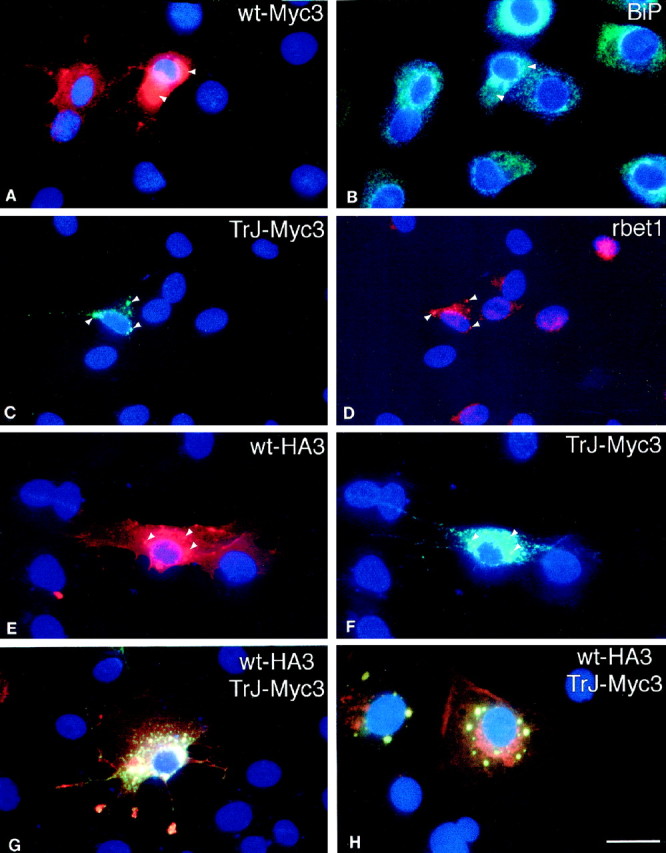
The localization of TrJ-Myc3 is different from that of wt-Myc3 in Schwann cells; coexpression results in partial colocalization of wt-Myc with TrJ-Myc3. Cells were transfected, as described in Materials and Methods, with wt-Myc3 (A, B) and TrJ-Myc3 (C, D) or cotransfected with wt-HA3 and TrJ-Myc3 (E–H). Cells were immunostained 36 hr after transfection. The stained proteins are indicated in thetop right corner of each panel. Nuclei are visualized with Hoechst dye. Wt-Myc3 (A) partially colocalized with the ER marker BiP (B).Arrowheads in A and Bpoint to areas with accumulated wt-Myc3 but weak BiP staining response. TrJ-Myc3 (C) shows partial costaining with the IC marker rbet1 (D). Arrowheads inC and D indicate colocalization of intracellularly accumulated TrJ-Myc3 (C) with rbet1 (D). When wt-HA3 (E) and TrJ-Myc3 (F) are coexpressed, the two proteins colocalize in intracellular structures in which TrJ-Myc3 accumulates (see the arrowsin E and F). If the wt-HA3 (red) and the TrJ-Myc3 staining (green) is superimposed in SCs (G) or COS7 cells (H), only wt-Myc3 (red staining in G and H) can be detected in the cell membrane, whereas yellowvesicles indicate the intracellular colocalization of the two proteins. Scale bar (in H), 33 μm.
Coexpression of wt-HA3 and TrJ-Myc3 in SCs gave similar results to those observed in COS7 cells. Wt-HA3 was observed in the IC (Fig.4E, arrowheads) colocalized with TrJ-Myc3 (Fig.4F, arrowheads). Superimposition of the wt-HA3 (red) and TrJ-Myc3 (green) labeling confirmed that most of the IC contained both proteins and appeared yellow (Fig. 4G). A COS7 cell in which the two proteins were coexpressed is shown in Figure4H for comparison. A fraction of the wt-HA3 appeared in the SC (Fig. 4G) or COS7 cell membrane (Fig.4H), and some typical ER staining with wt-HA3 was observed, particularly in the COS7 cells. The SCs showed an unusual accumulation of wt-HA3 (red), and some colocalized wt-HA3 and TrJ-Myc3 (yellow) at the tips of the processes (Fig. 4G). The expression of the mutant TrJ-Myc3 in SCs therefore affects the intracellular trafficking of wt-HA3 as it does in COS7 cells. The influence of TrJ-Myc3 on wt-HA3 trafficking is unlikely to be explained by nonspecific protein interactions because the trafficking of the endogenous P0 is not affected by the expression of TrJ-Myc3 (Fig.5). Although the TrJ-Myc3 accumulated in the IC in these SCs (Fig. 5A), the intracellular and membrane localization of P0 was normal (Fig. 5B) (Cheng and Mudge, 1996).
Fig. 5.

The localization of endogenous P0 in SCs is not affected by the expression of TrJ-Myc3. SCs were transfected with TrJ-Myc3, as described in Materials and Methods, and immunostained with anti-Myc antibody (A) or anti-P0 antibody (B). The immunostained proteins are indicated in the top right corner of each panel. P0 was not detected in the IC vesicles containing TrJ-Myc3. An untransfected cell inA is marked with an asterisk. Scale bar (in B), 33 μm.
PMP22 heterodimers form in cells expressing both wt- and TrJ-PMP22
The ability of TrJ-PMP22 to alter partially the trafficking pattern of wt-PMP22 suggests that the two proteins interact directly, or indirectly via chaperone proteins. Evidence in favor of a direct interaction is provided by the analysis of PMP22 extracted from mouse sciatic nerve. The Western blot that used polyclonal anti-PMP22 antibody showed not only a major PMP22 component (22 kDa) but also a second component for which the molecular mass (40–45 kDa) is consistent with that of a PMP22 dimer (Fig.6, lane 1). Significantly, removal of the N-linked carbohydrate with PNGaseF before analysis not only reduced the molecular mass of the PMP22 by ∼4 kDa, as anticipated, but it also completely disrupted the putative PMP22 dimer (Fig. 6, lane 3). Treatment with EndoH had an intermediate effect (see Fig. 6, lane 2), disrupting some but not all of the putative PMP22 dimers. Therefore, the stability of the proposed PMP22 dimers seems to depend on the presence and molecular composition of the N-linked carbohydrate moiety. The same results were obtained after the extraction of rat sciatic nerve (data not shown). Whether higher aggregates also form, such as the tetramers observed with P0 in crystals (Shapiro et al., 1996), cannot be determined from these data.
Fig. 6.
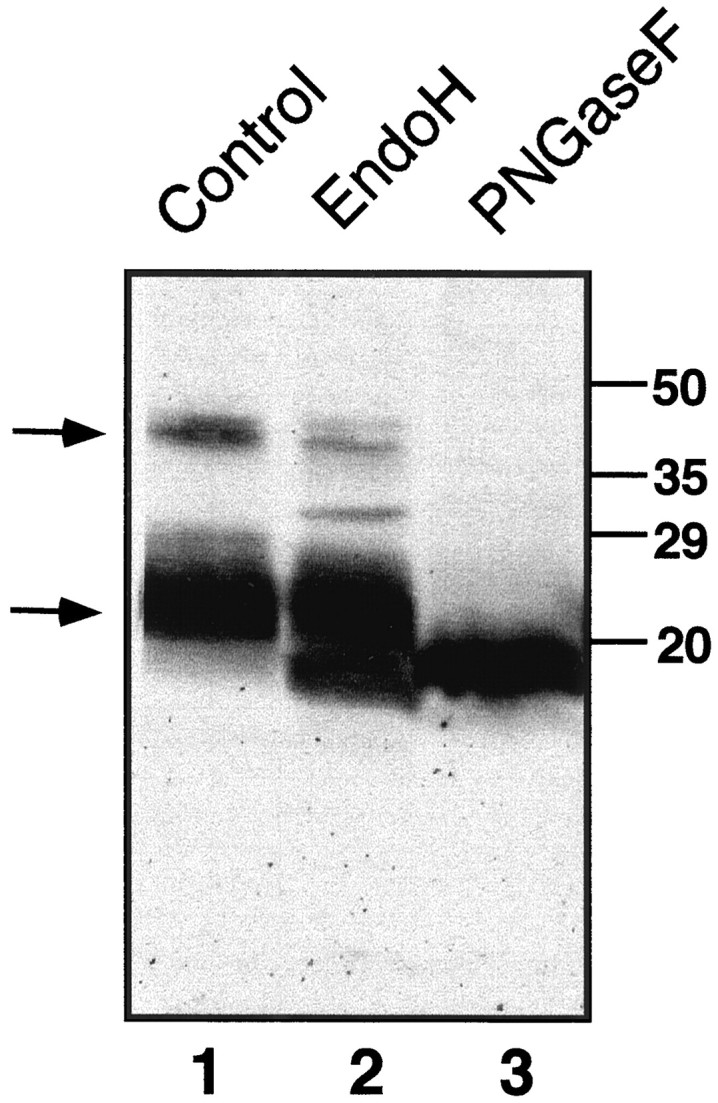
PMP22 dimers are observed in extracts of adult mouse sciatic nerve. Dimerization is stabilized by the N-linked carbohydrate domain. Mouse sciatic nerves were extracted as described in Materials and Methods and treated with EndoH (lane 2) or PNGaseF (lane 3). An extract incubated without the addition of an enzyme is shown in lane 1. Thebottom arrow on the left indicates N-glycosylated PMP22, and the top arrow indicates the position of the putative PMP22 dimer. All lanes contain 12.5 μg of total protein. Molecular markers are indicated on theright (in kDa).
If PMP22 does, indeed, form dimers during trafficking in the cell, coexpression of TrJ- and wt-PMP22s could lead to the formation of heterodimers. The existence of heterodimers in this situation has been confirmed by coimmunoprecipitation experiments (Fig.7). Extracts of COS7 cells transfected with wt-HA3 alone or wt-HA3 together with wt-Myc3 or TrJ-myc3 were precipitated with a polyclonal anti-c-Myc antibody; the immunoprecipitates were probed, after electrophoretic separation, either with a monoclonal anti-HA antibody (Fig. 7, top) or the monoclonal anti-c-Myc-antibody (Fig. 7, bottom). Whereas cells expressing only wt-HA3 showed no immunoprecipitate with the initial anti-c-Myc antibody (Fig. 7, wt-HA3 lane), cells expressing both forms showed coimmunoprecipitation of wt-HA3 with either wt-Myc3 or TrJ-Myc3. A similar experiment showed that TrJ-PMP22 also can form homodimers (data not shown). Heterodimer formation was not, however, observed when COS7 cells, separately transfected with wt-HA3 or wt-Myc3, were mixed and lysed together, showing that dimer formation was not the result of detergent-induced aggregation. The ability of PMP22 to form dimers may explain why TrJ-PMP22 partially can exert its own unique trafficking pattern on wt-PMP22.
Fig. 7.

Epitope-tagged wt- and TrJ-PMP22 coimmunoprecipitate from transfected COS7 cells. COS7 cells were transfected with wt-HA3 or were cotransfected with either wt-Myc3 and wt-HA3 or TrJ-Myc3 and wt-HA3, as indicated at the topof the figure. The cells were lysed and precipitated with a polyclonal anti-c-Myc antibody and protein A-agarose. Washed agarose beads were boiled with SDS sample buffer, and the samples were separated on 12.5% denaturing gels. Western blot analysis was performed with mouse monoclonal anti-c-Myc or anti-HA antibodies, as indicated on theleft side of the figure. Proteins precipitated from cotransfected COS7 cells with anti-c-Myc antibody reacted with anti-HA antibody (top panel) and anti-c-Myc antibody (bottom panel). No protein was precipitated with the anti-c-Myc antibody from COS7 cells transfected with wt-HA3 alone. The position of the arrows is derived from molecular weight markers and indicates the migration position of N-glycosylated PMP22.
DISCUSSION
The use of two different epitope tags in PMP22 permitted the localization of wt-PMP22 and TrJ-PMP22 to be compared in cells in which either one or both forms of PMP22 were expressed. This analysis revealed significant differences in trafficking between wild-type and mutant protein. The main conclusions are that TrJ-PMP22, unlike wt-PMP22, does not reach the cell membrane but accumulates in the IC. In addition, the expression of TrJ-PMP22 causes part, but not all, of the wt-PMP22 to be diverted from its normal trafficking pathway and to be retained in the IC. The mechanism behind this effect probably is the ability of TrJ-PMP22 to form heterodimers with wt-PMP22.
An epitope tag in loop 3 of PMP22 does not affect trafficking
In these experiments it is obviously important that the location of the epitope tag in PMP22 does not disturb its function(s). Because point mutations in the putative transmembrane domains of PMP22 lead to hereditary neuropathies, it is likely that these domains are unsuitable for epitope insertion. Similar, the N terminus, another commonly used site for epitope tagging, is located possibly in the membrane in PMP22 and again may not be a suitable location for a tag. Attaching an epitope to the highly charged intracellular C terminus that has sequence similarity to ER retrieval signals (Goepfert et al., 1995;Naef and Suter, 1998) possibly could alter the trafficking of PMP22 in the cell, although Naef et al. (1997) and D’Urso et al. (1998) have added vesicular stomatitis virus (VSV) G-protein and Flag tags, respectively, at this position with no apparent deleterious effect.
Only the insertion of the Myc tag into loop 3, as opposed to loops 1 or 2 of PMP22, exposed the tag in an accessible way without interfering with the processing of PMP22. Wt-Myc3 reacted with antibodies to both Myc and PMP22, had the appropriate molecular mass, and was deglycosylated completely by PNGaseF. The TrJ-Myc3 protein displayed similar properties to wt-Myc3. Loop 3 of PMP22 is, therefore, a suitable site for insertion of an epitope tag, and two such epitopes, Myc and HA, were used in this study. The trafficking of wt-PMP22 modified with these two different tags was indistinguishable.
Differences in the cellular localization of wild-type and TrJ-PMP22
When wt-Myc3 was expressed in COS7 cells, a fraction of the protein appeared in the cell membrane. In the cells expressing high amounts of wt-Myc3 the protein also accumulated in intracellular ER compartments that reacted with the BiP antibody. Consistent with these findings was the observation that a proportion of the wt-Myc3 was EndoH-resistant, indicating that its N-linked carbohydrate had been modified in the Golgi before transport to the membrane. In contrast, no TrJ-Myc3 was observed in COS7 cell membranes, and the glycosylation analysis supported this observation. All of the expressed TrJ-Myc3 was sensitive to EndoH, showing that none of it had reached the medial-Golgi compartment through which it would have to pass en route to the cell membrane. The intracellular localization of TrJ-Myc3 was also in marked contrast to that of wt-Myc3 in that it was not accumulated in the ER like wt-Myc3, but in some, although not all, of the IC. When both wt and TrJ proteins were coexpressed with different epitope tags, only wt-HA3 reached the membrane. However, both proteins colocalized to the IC. Thus the expression of the mutant TrJ-Myc3 influenced the trafficking of the wt-HA3 by retaining some of the latter to the IC. The remainder of the wt-HA3 moved normally through the intracellular compartments to the membrane (see Fig.4H).
Essentially the same results were obtained when the epitope-tagged proteins were expressed in SCs although, because of the low efficiency of SC transfection, it was not possible to perform the same biochemical analyses that were performed on the transfected COS7 cells. Moreover, with one exception, the data obtained in this study with wt-Myc3 are similar to those previously observed with endogenous untagged wt-PMP22 in SCs in culture, with or without neurons, or in whole peripheral nerve (Pareek et al., 1997). The conclusion from these earlier studies was that a major fraction of wt-PMP22 is degraded in the ER and only a smaller fraction passes to the Golgi. The level of endogenous wt-PMP22 that reaches the SC membrane under nonmyelinating culture conditions, if any, is too low to be detected with the available PMP22 antibodies. The fact that wt-Myc3 and wt-HA3 were observed in both COS7 and SC membranes suggests either that the expression levels of the tagged forms of PMP22 are higher than those of endogenous wt-PMP22 and/or that the anti-Myc and anti-HA antibodies are more sensitive.
Whereas wt-PMP22 or wt-Myc3 are natural substrates for ER resident proteases (Pareek et al., 1997), TrJ-Myc3 appears less accessible to them because it is not strongly accumulated in the distorted ER of COS7 or SCs. TrJ-Myc3 appears to escape the quality control mechanisms in the ER by accumulating in the IC. How this is achieved is not yet known. Because both wt- and TrJ-PMP22 form homodimers, it is unlikely that dimer formation is responsible for the aberrant trafficking of TrJ-PMP22. It is clear, however, that the ability of TrJ-Myc3 to sequester wt-HA3 into the IC stems from its ability to form a wt-TrJ PMP22 heterodimer. Dimer formation probably occurs during biosynthesis in the ER. Moreover, how much wt-PMP22 is diverted from its normal pathway will depend on the relative stabilities of the wt-wt and wt-TrJ dimers. Although the current data do not permit quantitative estimates of dimer stability, the observation that the wt-PMP22 appears in both normal and abnormal trafficking pathways suggests that the stability of the wt-wt and wt-TrJ PMP22 dimers may be similar. The ability of PMP22 to form homodimers or heterodimers with TrJ-PMP22 also provides a potential explanation for the semidominant nature of this particular mutation (Henry and Sidman, 1983).
A comparison of TrJ-PMP22 and Tr-PMP22 localization
It is of interest to compare the cellular localization of the TrJ protein with that of the Tr protein. The Tr protein, like the TrJ protein, is not transported to the COS7 cell membrane (Naef et al., 1997). However, the Tr protein accumulates in an expanded ER and not in the IC as observed for the TrJ protein (Naef et al., 1997). Moreover, coexpression of the Tr protein with wt-PMP22, both tagged with a VSV epitope, allowed the transport of wt-PMP22 to the cell membrane in only ∼15% of transfected cells. Thus the altered processing and transport of the Tr protein are essentially dominant over the usual trafficking of wt-PMP22, accounting for the dominant nature of the Trmutation (Falconer, 1951). At the molecular level this could arise because the stability of the wt-Tr heterodimer is significantly greater than that of the wt-wt homodimer. Although the relative stabilities of the mutant and wild-type PMP22 heterodimers help to explain, in part, the different phenotypes of TrJ and Tr mice, the dissimilar trafficking patterns of the TrJ and Tr proteins also must play a significant role in determining the severity of the defects imposed on the SCs. Information on these aspects of the peripheral neuropathies awaits a full understanding of the protein interactions that dictate the trafficking of PMP22 and how mutations in PMP22 alter these interactions.
Many examples of mutations leading to protein misfolding and ultimately to disease have been described (for review, see Muller et al., 1997). Among them are the mutations in PLP, a CNS tetraspan myelin protein, that lead to Pelizaeus–Merzbacher disease (Hodes et al., 1993;Griffiths et al., 1995). A number of missense mutations in PLP/DM20 cause one or both of these proteins to accumulate in the ER of COS7 cells, preventing them from being transported to the membrane (Gow et al., 1998). Of interest is the finding that transport-competent wild-type or mutated DM20 proteins facilitate the transfer of mutant PLP proteins that do not usually traffic to the cell membrane (Gow and Lazzarini, 1996). It is plausible that, as with PMP22, this facilitation could involve the direct interaction of these two related proteins.
Although the conclusions on the trafficking of TrJ-PMP22 reported here and of Tr-PMP22 (Naef et al., 1997) agree in part with those described by D’Urso et al. (1998), there are some discrepancies. All studies show that the mutant TrJ- and Tr-PMP22 proteins are not transported to the membrane of COS7, HeLa, or SCs. The discrepancies in experimental observations are in the effects of coexpressing the wild-type and mutant proteins together. D’Urso et al. (1998) reported that although the wt- and TrJ- or Tr-PMP22 proteins coexisted in some intracellular compartments, neither TrJ- nor Tr-PMP22 affected the trafficking of wt-PMP22 in HeLa or SCs. In the present study the demonstration by immunohistochemistry of the inability of TrJ-Myc3 to move to the cell membrane was supported by the biochemical analysis that showed that none of the TrJ-Myc3 protein was complex-glycosylated in the Golgi, a prerequisite for trafficking to the cell membrane. Moreover, the identification of a specific compartment, the IC, in which TrJ-Myc3 accumulated allowed us to show that some wt-Myc3 also accumulated with TrJ-Myc3 in the IC when they were coexpressed. At the same time it was possible to demonstrate that the remainder of the wt-Myc3 resided in the ER and the plasma membrane, indicative of normal trafficking. The finding that PMP22 is able to form homo- and heterodimers provides a molecular basis for all of these phenomena.
If these results are applicable to the in vivo situation, it would suggest that the severe neuropathy in the TrJ mouse and in humans is not attributable to the insertion of the mutant TrJ-PMP22 into myelin but rather to a decrease in the amount of the wt-PMP22 trafficking to the membrane as well as the deleterious effect of the accumulation of wt- and TrJ-PMP22 in the intermediate compartment of the SCs.
Footnotes
This work was supported by Grants from National Institutes of Health (NS04270 to E.M.S. and NS09694 to L.N.), the American Paralysis Association (SA2-9606 to E.M.S.), the Muscular Dystrophy Association (to E.M.S.), the Swiss National Science Foundation and the Novartis Jubilaumsstiftung (to A.T.), the National Multiple Sclerosis Society (FG 1120-A-1 to L.N.), and the American Philosophical Society (to L.N.). U.S. is supported by the Swiss National Science Foundation, the Swiss National Research Program for Neurodegenerative Disorders (NFP38), the European Union project “Clinical, Genetic, and Functional Analysis of Peripheral Neuropathies: An Integrated Approach,” and the Swiss Bundesamt for Science.
Correspondence should be addressed to Dr. Eric M. Shooter, Department of Neurobiology, Stanford University School of Medicine, Stanford, CA 94305-5125.
REFERENCES
- 1.Adlkofer K, Martini R, Aguzzi A, Zielasek J, Toyka KV, Suter U. Hypermyelination and demyelinating peripheral neuropathy in PMP22-deficient mice. Nat Genet. 1995;11:274–280. doi: 10.1038/ng1195-274. [DOI] [PubMed] [Google Scholar]
- 2.Adlkofer K, Frei R, Neuberg DH, Zielasek J, Toyka KV, Suter U. Heterozygous peripheral myelin protein 22-deficient mice are affected by a progressive demyelinating tomaculous neuropathy. J Neurosci. 1997a;17:4662–4671. doi: 10.1523/JNEUROSCI.17-12-04662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adlkofer K, Naef R, Suter U. Analysis of compound heterozygous mice reveals that the Trembler mutation can behave as a gain-of-function allele. J Neurosci Res. 1997b;49:671–680. doi: 10.1002/(SICI)1097-4547(19970915)49:6<671::AID-JNR2>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 4.Archelos JJ, Roggenbuck K, Schneider-Schaulies J, Linington C, Toyka KV, Hartung HP. Production and characterization of monoclonal antibodies to the extracellular domain of P0. J Neurosci Res. 1993;35:46–53. doi: 10.1002/jnr.490350107. [DOI] [PubMed] [Google Scholar]
- 5. Bole DG, Dowin R, Doriaux M, Jamieson JD. Immunocytochemical localization of BiP to the rough endoplasmic reticulum: evidence for protein sorting by selective retention. J Histochem Cytochem 37 1989. 1817 1823 Referenced on page 2 os m.s. [DOI] [PubMed] [Google Scholar]
- 6.Brockes JP, Fields KL, Raff MC. Studies on cultured rat Schwann cells. I. Establishment of purified populations from cultures of peripheral nerve. Brain Res. 1979;165:105–118. doi: 10.1016/0006-8993(79)90048-9. [DOI] [PubMed] [Google Scholar]
- 7.Cheng L, Mudge AW. Cultured Schwann cells constitutively express the myelin protein P0. Neuron. 1996;16:309–319. doi: 10.1016/s0896-6273(00)80049-5. [DOI] [PubMed] [Google Scholar]
- 8.D’Urso D, Prior R, Greiner-Petter R, Gabreels-Festen AA, Muller HW. Overloaded endoplasmic reticulum–Golgi compartments, a possible pathomechanism of peripheral neuropathies caused by mutations of the peripheral myelin protein PMP22. J Neurosci. 1998;18:731–740. doi: 10.1523/JNEUROSCI.18-02-00731.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falconer DS. Two new mutants, “trembler” and “reeler,” with neurological actions in the house mouse. Genetics. 1951;50:192–201. doi: 10.1007/BF02996215. [DOI] [PubMed] [Google Scholar]
- 10.Goepfert PA, Wang G, Mulligan MJ. Identification of an ER retrieval signal in a retroviral glycoprotein [letter]. Cell. 1995;82:543–544. doi: 10.1016/0092-8674(95)90026-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gow A, Lazzarini RA. A cellular mechanism governing the severity of Pelizaeus–Merzbacher disease. Nat Genet. 1996;13:422–428. doi: 10.1038/ng0896-422. [DOI] [PubMed] [Google Scholar]
- 12.Gow A, Southwood CM, Lazzarini RA. Disrupted proteolipid protein trafficking results in oligodendrocyte apoptosis in an animal model of Pelizaeus–Merzbacher disease. J Cell Biol. 1998;140:925–934. doi: 10.1083/jcb.140.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffiths IR, Schneider A, Anderson J, Nave KA. Transgenic and natural mouse models of proteolipid protein (PLP)-related dysmyelination and demyelination. Brain Pathol. 1995;5:275–281. doi: 10.1111/j.1750-3639.1995.tb00604.x. [DOI] [PubMed] [Google Scholar]
- 14.Hay JC, Hirling H, Scheller RH. Mammalian vesicle trafficking proteins of the endoplasmic reticulum and Golgi apparatus. J Biol Chem. 1996;271:5671–5679. doi: 10.1074/jbc.271.10.5671. [DOI] [PubMed] [Google Scholar]
- 15.Henry EW, Sidman RL. The murine mutation Trembler-J: proof of semidominant expression by use of the linked vestigial tail marker. J Neurogenet. 1983;1:39–52. doi: 10.3109/01677068309107071. [DOI] [PubMed] [Google Scholar]
- 16.Henry EW, Cowen JS, Sidman RL. Comparison of Trembler and Trembler-J mouse phenotypes: varying severity of peripheral hypomyelination. J Neuropathol Exp Neurol. 1983;42:688–706. doi: 10.1097/00005072-198311000-00008. [DOI] [PubMed] [Google Scholar]
- 17.Hodes ME, Pratt VM, Dlouhy SR. Genetics of Pelizaeus–Merzbacher disease. Dev Neurosci. 1993;15:383–394. doi: 10.1159/000111361. [DOI] [PubMed] [Google Scholar]
- 18.Huxley C, Passage E, Manson A, Putzu G, Figarella-Branger D, Pellissier JF, Fontes M. Construction of a mouse model of Charcot–Marie–Tooth disease type 1A by pronuclear injection of human YAC DNA. Hum Mol Genet. 1996;5:563–569. doi: 10.1093/hmg/5.5.563. [DOI] [PubMed] [Google Scholar]
- 19.Ionasescu VV, Searby CC, Ionasescu R, Chatkupt S, Patel N, Koenigsberger R. Dejerine–Sottas neuropathy in mother and son with same point mutation of PMP22 gene. Muscle Nerve. 1997;20:97–99. doi: 10.1002/(sici)1097-4598(199701)20:1<97::aid-mus13>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 20.Itin C, Roche AC, Monsigny M, Hauri HP. ERGIC-53 is a functional mannose-selective and calcium-dependent human homologue of leguminous lectins. Mol Biol Cell. 1996;7:483–493. doi: 10.1091/mbc.7.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 22.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 23.Magyar JP, Martini R, Ruelicke T, Aguzzi A, Adlkofer K, Dembic Z, Zielasek J, Toyka KV, Suter U. Impaired differentiation of Schwann cells in transgenic mice with increased PMP22 gene dosage. J Neurosci. 1996;16:5351–5360. doi: 10.1523/JNEUROSCI.16-17-05351.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maley F, Trimble RB, Tarentino AL, Plummer TH., Jr Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal Biochem. 1989;180:195–204. doi: 10.1016/0003-2697(89)90115-2. [DOI] [PubMed] [Google Scholar]
- 25.Manfioletti G, Ruaro ME, Del Sal G, Philipson L, Schneider C. A growth arrest-specific (gas) gene codes for a membrane protein. Mol Cell Biol. 1990;10:2924–2930. doi: 10.1128/mcb.10.6.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller HW, Suter U, Van Broeckhoven C. Advances in Charcot–Marie–Tooth disease research: cellular function of CMT-related proteins, transgenic animal models, and pathomechanisms. Neurobiol Dis. 1997;4:215–220. doi: 10.1006/nbdi.1997.0148. [DOI] [PubMed] [Google Scholar]
- 27.Naef R, Suter U. Many facets of the peripheral myelin protein PMP22 in myelination and disease. Microsc Res Tech. 1998;41:359–371. doi: 10.1002/(SICI)1097-0029(19980601)41:5<359::AID-JEMT3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 28.Naef R, Adlkofer K, Lescher B, Suter U. Aberrant protein trafficking in Trembler suggests a disease mechanism for hereditary human peripheral neuropathies. Mol Cell Neurosci. 1997;9:13–25. doi: 10.1006/mcne.1997.0604. [DOI] [PubMed] [Google Scholar]
- 29.Notterpek L, Shooter EM, Snipes GJ. Upregulation of the endosomal–lysosomal pathway in the Trembler-J neuropathy. J Neurosci. 1997;17:4190–4200. doi: 10.1523/JNEUROSCI.17-11-04190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Panayotatos N, Radziejewska E, Acheson A, Pearsall D, Thadani A, Wong V. Exchange of a single amino acid interconverts the specific activity and gel mobility of human and rat ciliary neurotrophic factors. J Biol Chem. 1993;268:19000–19003. [PubMed] [Google Scholar]
- 31.Pareek S, Notterpek L, Snipes GJ, Naef R, Sossin W, Laliberte J, Iacampo S, Suter U, Shooter EM, Murphy RA. Neurons promote the translocation of peripheral myelin protein 22 into myelin. J Neurosci. 1997;17:7754–7762. doi: 10.1523/JNEUROSCI.17-20-07754.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel PI, Roa BB, Welcher AA, Schoener SR, Trask BJ, Pentao L, Snipes GJ, Garcia CA, Francke U, Shooter EM. The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot–Marie–Tooth disease type 1A. Nat Genet. 1992;1:159–165. doi: 10.1038/ng0692-159. [DOI] [PubMed] [Google Scholar]
- 33.Schweizer A, Fransen JA, Bachi T, Ginsel L, Hauri HP. Identification, by a monoclonal antibody, of a 53-kDa protein associated with a tubulovesicular compartment at the cis-side of the Golgi apparatus. J Cell Biol. 1988;107:1643–1653. doi: 10.1083/jcb.107.5.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sereda M, Griffiths I, Pühlhofer A, Stewart H, Rossner MJ, Zimmerman F, Magyar JP, Schneider A, Hund E, Meinck H-M, Suter U, Nave K-A. A transgenic rat model of Charcot–Marie–Tooth disease. Neuron. 1996;16:1049–1060. doi: 10.1016/s0896-6273(00)80128-2. [DOI] [PubMed] [Google Scholar]
- 35.Shapiro L, Doyle JP, Hensley P, Colman DR, Hendrickson WA. Crystal structure of the extracellular domain from P0, the major structural protein of peripheral nerve myelin. Neuron. 1996;17:435–449. doi: 10.1016/s0896-6273(00)80176-2. [DOI] [PubMed] [Google Scholar]
- 36.Snipes GJ, Suter U, Welcher AA, Shooter EM. Characterization of a novel peripheral nervous system myelin protein (PMP-22/SR13). J Cell Biol. 1992;117:225–238. doi: 10.1083/jcb.117.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Snipes GJ, Suter U, Shooter EM. Human peripheral myelin protein-22 carries the L2/HNK-1 carbohydrate adhesion epitope. J Neurochem. 1993;61:1961–1964. doi: 10.1111/j.1471-4159.1993.tb09840.x. [DOI] [PubMed] [Google Scholar]
- 38.Squinto SP, Stitt TN, Aldrich TH, Davis S, Bianco SM, Radziejewski C, Glass DJ, Masiakowski P, Furth ME, Valenzuela DM, DiStefano PS, Yancopoulos GD. trkB encodes a functional receptor for brain-derived neurotrophic factor and neurotrophin-3 but not nerve growth factor. Cell. 1991;65:885–893. doi: 10.1016/0092-8674(91)90395-F. [DOI] [PubMed] [Google Scholar]
- 39.Suter U, Moskow JJ, Welcher AA, Snipes GJ, Kosaras B, Sidman RL, Buchberg AM, Shooter EM. A leucine-to-proline mutation in the putative first transmembrane domain of the 22-kDa peripheral myelin protein in the Trembler-J mouse. Proc Natl Acad Sci USA. 1992a;89:4382–4386. doi: 10.1073/pnas.89.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suter U, Welcher AA, Ozcelik T, Snipes GJ, Kosaras B, Francke U, Billings GS, Sidman RL, Shooter EM. Trembler mouse carries a point mutation in a myelin gene. Nature. 1992b;356:241–244. doi: 10.1038/356241a0. [DOI] [PubMed] [Google Scholar]
- 41.Valentijn LJ, Baas F, Wolterman RA, Hoogendijk JE, van den Bosch NH, Zorn I, Gabreels-Festen AW, de Visser M, Bolhuis PA. Identical point mutations of PMP-22 in Trembler-J mouse and Charcot–Marie–Tooth disease type 1A. Nat Genet. 1992;2:288–291. doi: 10.1038/ng1292-288. [DOI] [PubMed] [Google Scholar]
- 42.Zhang T, Wong SH, Tang BL, Xu Y, Peter F, Subramaniam VN, Hong W. The mammalian protein (rbet1) homologous to yeast Bet1p is primarily associated with the pre-Golgi intermediate compartment and is involved in vesicular transport from the endoplasmic reticulum to the Golgi apparatus. J Cell Biol. 1997;139:1157–1168. doi: 10.1083/jcb.139.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]