

Abstract
Precise growth cone guidance is the consequence of a continuous reorganization of actin filament structures within filopodia and lamellipodia in response to inhibitory and promoting cues. The small GTPases rac1, cdc42, and rhoA are critical for regulating distinct actin structures in non-neuronal cells and presumably in growth cones. Collapse, a retraction of filopodia and lamellipodia, is a typical growth cone behavior on contact with inhibitory cues and is associated with depolymerization and redistribution of actin filaments. We examined whether small GTPases mediate the inhibitory properties of CNS myelin or collapsin-1, a soluble semaphorin, in chick embryonic motor neuron cultures. As demonstrated for collapsin-1, CNS myelin-evoked growth cone collapse was accompanied by a reduction of rhodamine–phalloidin staining most prominent in the growth cone periphery, suggesting actin filament disassembly. Specific mutants of small GTPases were capable of desensitizing growth cones to CNS myelin or collapsin-1. Adenoviral-mediated expression of constitutively active rac1 or rhoA abolished CNS myelin-induced collapse and allowed remarkable neurite extension on a CNS myelin substrate. In contrast, expression of dominant negative rac1 or cdc42 negated collapsin-1-induced growth cone collapse and promoted neurite outgrowth on a collapsin-1 substrate. These findings suggest that small GTPases can modulate the signaling pathways of inhibitory stimuli and, consequently, allow the manipulation of growth cone behavior. However, the fact that opposite mutants of rac1 were effective against different inhibitory stimuli speaks against a universal signaling pathway underlying growth cone collapse.
Keywords: growth cone, actin filaments, small GTPases, myelin, collapsin-1, rac1, semaphorin
The function of the adult nervous system depends on the precise guidance of neuronal growth cones to their appropriate target by a myriad of attractive and inhibitory cues (Kolodkin, 1996; Tessier-Lavigne and Goodman, 1996). Sensory capacity, adhesion, and motility of growth cones all reside in lamellipodia and filopodia. The dynamics and morphology of these highly motile structures is almost universally coupled to a coordinated assembly, disassembly, and retrograde flow of actin filaments (Stossel, 1993;Tanaka and Sabry, 1995). The small GTPases rac1, cdc42, and rhoA represent key regulators of distinct actin filament structures in non-neuronal cells in response to extrinsic signals (Nobes and Hall, 1995; for review, see Hall, 1998). Recently, their importance has been demonstrated in neurons as well (for review, see Luo et al., 1997). Rac1 is involved in neurite outgrowth (Luo et al., 1994; Kuhn et al., 1998), correct target innervation (Kaufmann et al., 1998), formation of dendritic arbors (Luo et al., 1996), differentiation of neurites (Threadgill et al., 1997), and growth cone collapse (Jin and Strittmatter, 1997). Depending on the mode of interaction, inhibitory cues typically elicit growth cone repulsion, a local retraction of lamellipodia and filopodia, as opposed to growth cone collapse, a total loss of lamellipodia and filopodia. In the case of collapsin-1, a concomitant disassembly of actin filaments has been demonstrated (Fan et al., 1993). Conversely, an accumulation of actin filaments in filopodia that contact attractive cues precedes turning of growth cones toward the guidance source (O’Connor and Bentley, 1993).
To interfere with the detrimental effects of inhibitory cues, both receptor complexes and associated signaling pathways represent suitable targets, yet are poorly understood both for collapsin-1 and CNS myelin-associated factors. The CNS myelin-derived inhibitors NI35/250 and myelin-associated glycoprotein (MAG) elicit rises in free intracellular Ca2+, whereas collapsin-1 acts via Ca2+-independent pathways (Ivins et al., 1991;Bandtlow et al., 1993; Song et al., 1998). Only recently have neuropilins been identified as receptors for semaphorin family members (Chen at al., 1997; Kolodkin et al., 1997). Because of the actin-dependent changes in growth cone morphology associated with collapse and repulsion, small GTPases represent excellent candidates. It is conceivable that small GTPases directly participate in the intracellular signaling of inhibitory cues as suggested by Jin and Strittmatter (1997). However, their effects on distinct actin filament structures could simply prove beneficial in the presence of inhibitory cues.
The present investigation demonstrates that adenoviral-mediated expression of specific constitutively active and dominant negative mutants of small GTPases in chick motor neurons can compensate for the inhibitory properties of CNS myelin-associated factors and a soluble semaphorin, collapsin-1. In particular, opposing rac1 mutants interfered with growth cone collapse induced by CNS myelin and collapsin-1 and allowed neurite outgrowth in the presence of either growth inhibitor, respectively. These results further support a model in which different inhibitory stimuli elicit distinct intracellular signals rather than a universal response.
MATERIALS AND METHODS
Reagents. Unless specified otherwise, all reagents were purchased from Sigma (St. Louis, MO).
Cell culture. Spinal cords were dissected from 6- to 7-d-old chick embryos (White leghorn). The ventral halves of spinal cords, containing mostly motor neurons, were trypsinized (4% trypsin, 0.2 gm/ml EDTA, 15 min, 37°C) and triturated, and the dissociated cells were preplated (1 hr, 37°C) in high glucose DMEM (Life Technologies, Gaithersburg, MD) and 10% FBS (Hyclone, Logan, UT). Nonadherent cells were resuspended in DMEM, pH 7.3 (325 ± 5 mOsm), 10% FBS, 12 nm fluorodeoxyuridine, and 1% N3 supplement (10 μg/ml bovine serum albumin, 100 μg/ml transferrin, 10 μg/ml insulin, 32 ng/ml putrescine, 20 ng/ml triiodothyronine, 10 ng/ml sodium selenite, 12.6 ng/ml progesterone, 200 ng/ml corticosterone) and plated at either 50,000/ml (collapse assay) or 100,000/ml (stripe assay). Glass coverslips (22 × 22 mm; Carolina Biological Supply, Burlington, NC) or 24-well plates were pretreated with poly-d-lysine (PL) (10 μg/ml, borate buffer, pH 8.4) and coated with 2 μg/cm2 fibronectin (FN) (Boehringer Mannheim, Indianapolis, IN) for 1 hr at 37°C.
Collapse assay and stripe assay. For collapse assays, CNS myelin, enriched collapsin-1, or recombinant collapsin-1 were added in soluble form to motor neuron cultures. Protein concentrations were adjusted such that a 10 μl aliquot of CNS myelin or enriched collapsin-1 was added to 500 μl culture medium. In the case of recombinant collapsin-1, protein concentration was adjusted (15 μg/ml, addition of 75 ng) so that ∼70% of growth cones collapsed. After incubation for 1 hr at 37°C, cultures were washed with PBS (37°C) and fixed with 2% glutaraldehyde in PBS (37°C), and the percentage of collapsed growth cones was determined. In stripe assays, 2 × 5 mm stripes of filter paper (Whatman #50) were UV-sterilized and saturated with CNS myelin (100 μg/ml) or recombinant collapsin-1 (15 μg/ml) supplemented with 15% rhodamine-labeled dextran (Molecular Probes, Eugene, OR). Filter stripes were placed on FN-coated (CNS myelin stripes) or poly-d-lysine-coated glass coverslips (collapsin-1 stripes) for 15 min in a humidified incubator at 37°C and removed before plating of cells. Borders between CNS myelin and FN or collapsin-1 and PL were visible under fluorescence or even under phase (CNS myelin).
Recombinant adenoviruses. Recombinant adenoviruses were generated as described by Becker et al. (1994) using an adenovirus subtype 5 deletion mutant (Ad5 dl309). Briefly, c-myc-tagged constitutively active (V12rac, V12cdc42, V14rhoA) or dominant negative (N17rac1, N17cdc42) mutants of small GTPases (generous gift from Dr. A. Hall, Cambridge, UK) or lacZ (control) were subcloned into the pMLE1A shuttle plasmid (kindly provided by Dr. I. Maxwell, Denver, CO) under the viral E1A promoter. In the shuttle plasmid, the transcription cassettes were flanked by the Ad5 fragment ranging from map unit 0 to 1.3 and the Ad5 fragment ranging from map unit 9.1 to 15.9. Shuttle plasmids were linearized at a unique XhoI restriction site and 10 μg of each was co-transfected with 10 μg of pJM17 (McGrory et al., 1988) (Microbix Biosystems, Toronto, Ontario, Canada) into 293 cells (ATCC CRL 1573). Homologous recombination rescued viral sequences with the expression cassette, containing c-myc-tagged small GTPase mutants, inserted into the left end of the truncated viral genome resulting in replication-deficient adenoviruses (V12rac-AdE1A,V12cdc42-AdE1A,V14rhoA-AdE1A,N17rac1-AdE1A,N17cdc42-AdE1A, lacZ-AdE1A). Routinely, titers were 1 × 108 to 1 × 109, and aliquots of viral stocks were stored at −80°C. Long-term storage and repetitive freeze-thawing had no effect on virus titers. Motor neuron cultures were infected 4 hr after plating with 200 plaque-forming units per neuron [multiplicity of infection (moi)] of recombinant adenovirus in 300 μl of culture medium. Cultures were supplemented with 700 μl of fresh medium 16 hr after infection. We were unable to generate recombinant adenoviruses for expression of small GTPases under the cytomegalovirus (CMV) immediate early promoter. Mutants of small GTPases were toxic for proliferating cells, even bacteria, possibly because of leaky expression under the CMV promoter. However, using the E1A promoter we successfully generated recombinant adenovirus vectors expressing the small GTPases. Expression of genes under the E1A promoter is downregulated in 293 cells because the presence of the E1A transgene product in these cells represses E1A promoter activity (Schaack et al., 1998). Therefore, the E1A promoter was suitable for use to construct adenovirus vectors to express proteins that were toxic for cell proliferation.
Immunocytochemistry and histochemistry. Cultured motor neurons were fixed in 4% paraformaldehyde, 0.1% glutaraldehyde, 100 mm Na-phosphate, pH 7.4, 120 mm sucrose for 30 min at room temperature, washed three times with PBS, and permeabilized with 0.5% Triton X-100 in PBS. Primary antibodies or rhodamine–phalloidin were added for 2 hr at room temperature. Secondary antibodies diluted in PBS/2%BSA were incubated for 1 hr at room temperature. Cultures were stored in 90% glycerol/PBS at −20°C. The following primary antibodies were used: a monoclonal mouse anti-c-myc IgG (9E10) (1 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA), a rabbit polyclonal anti-c-myc IgG (0.5 μg/ml; Upstate Biotechnology, Lake Placid, NY), rhodamine–phalloidin (1:50; Molecular Probes, Eugene, OR), and a monoclonal mouse anti-tubulin (1:250; Sigma, St. Louis, MO). A Texas Red-labeled polyclonal goat anti-mouse IgG (Molecular Probes) and a fluorescein-labeled polyclonal goat anti-mouse IgG (Molecular Probes) were used as secondary antibodies (1:500). Alternatively for histochemical staining, cultures were fixed, permeabilized, and incubated with anti-c-myc IgG (0.5 μg/ml) followed by a biotinylated goat anti-mouse IgG (Pierce, Rockford, IL). β-Galactosidase coupled to avidin (0.2 U/ml; Pierce) was added (1 hr, 37°C), and cultures were incubated overnight with 0.5 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactoside (Life Technologies, Gaithersburg, MD), 5 mm potassium ferricyanide, 5 mm potassium ferrocyanide, 2 mmMgCl2, 150 mm NaCl, and 15 mm sodium phosphate, pH 7.3.
Preparation and characterization of growth cone particles.Highly enriched preparations of growth cone particles (GCPs) were obtained as described in Pfenninger et al. (1983) with modifications according to Lockerbie et al. (1991). Briefly, fresh brains from 10- to 14-d-old chick embryos were homogenized (on ice) in 6–8 vol (wet weight to volume) of 5 mm HEPES, pH 7.3, 1 mmMgCl2, and 0.32 m sucrose. After a low-speed centrifugation of the crude homogenate (1500–1660 ×gmax, 15 min, 4°C), the resulting supernatant was overlaid onto 5 mm HEPES, pH 7.3, 1 mm MgCl2, and 0.75 msucrose and centrifuged at 150,000 × gmax(1 hr, 4°C). Material at the interface was collected, diluted six- to sevenfold with 5 mm HEPES, pH 7.3, 1 mmMgCl2, and 0.32 m sucrose (added dropwise on ice), overlaid onto 2 ml of Maxidense (Sigma), and centrifuged at 40,000 × gmax (1 hr, 4°C). Sealed GCPs were collected from the interface between load and Maxidense cushion, gently resuspended in Krebs’ buffer (145 mm NaCl, 5 mm KCl, 1.2 mmNaH2PO4, 1.2 mmMgCl2, 5 mm HEPES, pH 7.3), and stored at −80°C. Freshly prepared GCPs (100 μg total protein) were treated with various conditions for 1 hr at 37°C. Samples were solubilized in 1% Triton X-100/0.02% saponin and separated by high-speed centrifugation (1 hr, 4°C, 100,000 ×gmax) into a cytoskeletal (pellet) and a cytosol (supernatant) fraction. Proteins in the supernatant were precipitated with chloroform/methanol (Wessel and Flügge, 1984), resuspended in SDS sample preparation buffer (250 mmTris-Cl, pH 6.8, 10% glycerol, 1% SDS, 10% 2-mercaptoethanol, 0.01% bromophenol blue), and boiled for 5 min. Protein pellets were directly resuspended in this buffer.
Gel electrophoresis and Western blotting. Cell cultures were extracted with 2% SDS, 10 mm Tris-Cl, pH 7.5, 10 mm NaF, 5 mm dithiothreitol, and 2 mm EGTA. Soluble protein was boiled (5 min), precipitated with chloroform/methanol, and resuspended in SDS sample preparation buffer. Protein from GCPs was prepared as detailed above. All protein concentrations were determined as described by Minamide and Bamburg (1990). SDS-PAGE was performed according to Laemmli (1970), and gels were stained with Coomassie blue or silver (Oakley et al., 1980). Western blotting onto polyvinylidene difluoride (PVDF) membranes (Gelman Sciences, Ann Arbor, MI) was performed as detailed in Towbin et al. (1979). Membranes were blocked with 5% nonfat dry milk in TBS (10 mm Tris-Cl, pH 8.0, 150 mm NaCl), washed with TBS/0.05% Tween 20, and incubated for 1 hr with primary antibody diluted in TBS/0.05% Tween 20/1% BSA. The primary antibody used was a monoclonal mouse anti-Rac1 (1 μg/ml; Upstate Biotechnology) or a monoclonal mouse anti-actin (ICN Biomedicals, Aurora, OH). Membranes were washed with TBS/0.05% Tween-20 and incubated with secondary antibodies for 1 hr (alkaline phosphatase conjugated to goat anti-rabbit IgG or anti-mouse IgG, 1:10,000), and bands were visualized with Nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-indolylphosphate p-toluidine salt (Life Technologies).
Preparation of CNS myelin and enriched collapsin-1. CNS myelin was prepared from adult rat brain as reported by Norton and Poduslo (1973). Briefly, adult rat brain was homogenized in 0.32m sucrose, and the homogenate was overlaid onto 0.85m sucrose and centrifuged for 30 min (75,000 ×gmax). Crude CNS myelin was collected from the interface and washed twice in ice-cold water (25,000 ×gmax, 30 min). The resulting pellet was resuspended in 0.32 m sucrose, overlaid onto 0.85m sucrose, and centrifuged for 1 hr (75,000 ×gmax). Purified CNS myelin was collected from the interface, washed twice in ice-cold water, and stored at −80°C. Before addition to cell cultures, purified CNS myelin was resuspended in PBS. Protein extracts were prepared from purified CNS myelin by solubilizing in 2% octylglucoside followed by centrifugation (1 hr, 4°C, 100,000 × gmax) and dialysis of the resulting supernatant overnight against PBS. Enriched collapsin-1 was obtained according to Fan et al. (1993). Briefly, purified chick brain membranes from 10- to 14-d-old chick embryos were solubilized in 2% 3-[(3-cholamidopropyl)dimethylammonio]-1-propane sulfonate and centrifuged (1 hr, 4°C, 100,000 × gmax), and the resulting supernatant was dialyzed overnight against PBS. This protein extract was used immediately thereafter or stored for a maximum of 3 d at 4°C. Dialyzed extraction buffer was used as control condition. Purified recombinant collapsin-1 was prepared as described previously (Luo et al., 1995) and used at a concentration to collapse 70% of motor neuron growth cones as was obtained with enriched collapsin-1 or purified CNS myelin.
Image analysis and statistics. Analysis of growth cone collapse was performed on a Nikon inverted microscope (Diaphot 300) using a 40× oil objective (Nikon) and a Dage MTI tube video camera (Vidicon VT-1000). Quantitative analysis of rhodamine–phalloidin was performed on a Nikon inverted microscope (Diaphot 200) equipped with a computer controlled CCD camera (PXL Photometrics, Tucson, AZ) using a 60× oil objective (Nikon). Images were captured with Metamorph imaging software run on a Pentium PC (Universal Imaging Corporation, West Chester, PA). Images were acquired using identical parameters, and total fluorescence intensity was measured on a pixel-by-pixel basis after background subtraction. Growth cones were subdivided into a peripheral region, a central region (defined by microtubule bundles), and the proximal neurite. All measurements were normalized to the average of the peripheral region under control conditions. One-way ANOVA analysis and a Kruskal–Wallis test were used when comparing multiple samples. Dunnett’s test was used when comparing the means of multiple conditions with a single control. In both cases, ap value < 0.01 was considered significant.
RESULTS
Adenoviral-mediated expression of small GTPases in primary chick motor neurons
We used recombinant, replication-deficient adenoviruses to express c-myc-tagged constitutively active and dominant negative mutants of rac1 (V12rac1, N17rac1), cdc42 (V12cdc42, N17cdc42), and rhoA (V14rhoA) in dissociated, embryonic chick motor neurons. Two to three days after infection with a recombinant adenovirus carrying V12rac1 orV12cdc42 under the viral E1A promoter (V12rac1-AdE1A orV12cdc42-AdE1A), V12rac1 andV12cdc42 proteins were present in motor neuron growth cones as revealed by indirect immunofluorescence against c-myc. We measured a 2.7-fold increase in the total fluorescence intensity per square micrometer in growth cones ofV12rac1-AdE1A-infected motor neurons (4.96 ± 0.5, n = 36, p < 0.01) compared with lacZ-AdE1A-infected cultures (1.86 ± 0.9, n = 24), which was our control. A 1.83-fold increase in total fluorescence intensity per square micrometer was measured in growth cones of neurons infected withV12cdc42-AdE1A (3.30 ± 0.31,n = 27, p < 0.01) compared with uninfected controls (1.80 ± 0.24, n = 20). Quantitative Western blotting revealed a total rac1 immunoreactivity proportional to the moi. There was a 20–50% increase in immunoreactivity inV12rac1-AdE1A-infected cultures compared with controls (Fig.1B). An moi-dependent increase in expression was also measured for V12cdc42 andN17cdc42. Adenoviral expression of small GTPase mutants was maximal 3 d after infection, increasing from 35 ± 5% (n = 208) c-myc-positive motor neurons at 1 d after infection to 88 ± 7% (n = 176,p < 0.05) at 3 d after infection, as illustrated for V12rac1 (Fig.2A). Over a time period of 3 d, the percentage of neurite-bearing neurons remained constant regardless of infection with recombinant adenovirus (200 moi) and expression of small GTPase mutants, suggesting that motor neuron survival was not affected. However, neuronal survival was diminished 3 d after infection with higher mois, in particular for cdc42 mutants.
Fig. 1.

Adenoviral-mediated expression of c-myc-tagged mutants of small GTPases in chick motor neurons. A, Motor neurons from 6-d-old chick embryos were plated on fibronectin and infected with a recombinant adenovirus carrying c-myc-tagged constitutively active rac1 under the viral E1A promoter (V12rac1-AdE1A). Three days after infection, c-myc immunoreactivity is present in growth cones and cell bodies (data not shown) of motor neurons. B, In lacZ-AdE1A-infected cultures, c-myc immunostaining was very faint, indicating very little unspecific staining using the anti-c-myc antibody. C, Motor neuron cultures were infected with V12rac1-AdE1A using a multiplicity of infection (moi or ratio of virus particles to neurons) of 20, 50, and 100. Three days after infection, cultures were solubilized, and extracted proteins were separated by 15% SDS-PAGE. After transfer to PVDF membranes, the presence ofV12rac1 was demonstrated using a rac1-specific antibody.D, Rac1-immunoreactive bands were quantified by densitometric analysis and normalized to the values at 20 moi. Total rac1 immunoreactivity was proportional to the moi.
Fig. 2.

Mutants of small GTPases expressed via recombinant adenoviruses are functional. A, Motor neurons were infected with c-myc V12rac1-AdE1A (200 moi) and stained for c-myc using β-galactosidase-enhanced histochemistry. The percentage of c-myc-positive motor neurons is plotted against days after infection in c-mycV12rac1-AdE1A-infected cultures (▪) compared with controls (▵). More than 90% of c-mycV12rac1-AdE1A-infected motor neurons were c-myc positive 3 d after infection. B, Motor neurons were grown on FN and infected withV12rac1-AdE1A (▪, 200 moi) or lacZ-AdE1A (○, 200 moi) at the time of plating. The longest neurite per neurons was measured, and their distribution in a motor neuron population was plotted. Only neuronal processes >10 μm long were considered neurites. Expression of V12rac1 attenuates β1 integrin-mediated neurite outgrowth on the FN substrate (2 day postinfection), which is consistent with previous findings using trituration loading of motor neurons with purified recombinant V12rac1 protein (Kuhn et al., 1998).
Adenoviral-mediated expression of rac1 mutants affected growth cone advance and actin filament distribution of growth cones as shown previously by trituration loading of chick motor neurons with purified, recombinant rac1 mutants (Kuhn et al., 1998). Expression ofV12rac1 attenuated β1 integrin-mediated neurite outgrowth of motor neurons on FN (Fig. 2B). Average neurite length was significantly reduced to 29 ± 2 μm (n = 107, p < 0.001) at 1 d after infection and 38 ± 2 μm (n = 111,p < 0.001) at 2 d after infection compared with lacZ-AdE1A-infected cultures 1 d after infection (39 ± 2 μm, n = 64) or 2 d after infection (76 ± 4 μm, n = 74), respectively. Accumulation of actin filaments occurred in the presence ofV12rac1 as suggested by a significant increase of the total rhodamine–phalloidin fluorescence per growth cone (1.25 + 0.06,n = 32, p < 0.01; normalized to control) in V12rac1-AdE1A-infected cultures. Infection of motor neuron cultures with mutants of cdc42 and rhoA also resulted in changes in growth cone morphology, neurite outgrowth, and actin filament distribution (data not shown). In summary, adenoviral-mediated gene transfer allows the effective expression of constitutively active and dominant negative mutants of small GTPases in dissociated, embryonic chick motor neurons. Behavioral and morphological changes of growth cones in conjunction with a redistribution of actin filaments provide strong evidence that small GTPases are functional.
CNS myelin induces a loss of actin filaments in growth cones
Growth cone collapse, a loss of lamellipodia and filopodia, is inherently connected to severe alterations in the distribution of actin filaments as has been demonstrated for collapsin-1 (Fan et al., 1993). To test whether CNS myelin-associated inhibitors evoke similar effects, we visualized actin filament distribution in motor neurons exposed to CNS myelin using rhodamine-labeled phalloidin.
In the presence of purified CNS myelin (100 μg/ml), motor neuron growth cones displayed a collapsed morphology. A decrease in rhodamine–phalloidin fluorescence suggested a CNS myelin-induced rearrangement of actin filaments (Fig.3). We quantified changes in rhodamine–phalloidin staining by integrating the fluorescence intensity on a pixel-by-pixel basis over the area of three growth cone regions: a central region defined by microtubule bundles, a peripheral region containing filopodia and lamellipodia, and 15 μm of the proximal neurite. All values were normalized to the average integrated fluorescence intensity of the peripheral region under control conditions. CNS myelin caused a substantial loss of actin filaments throughout the entire growth cone but was most pronounced in the periphery (Fig. 4A). We detected a large reduction of rhodamine fluorescence intensity in the peripheral region of CNS myelin-treated growth cones (0.17 ± 0.03, n = 23, *p < 0.001) and, to a lesser degree, in the central region (0.35 ± 0.06,n = 23, *p < 0.001) and in the proximal neurite (0.18 ± 0.03, n = 23, *p < 0.001) compared with the same regions in controls (1.0 ± 0.11, n = 20, peripheral region; 0.69 ± 0.11, n = 20, central region; 0.45 ± 0.06,n = 20, proximal neurite).
Fig. 3.

CNS myelin-associated growth inhibitors cause a rearrangement of actin filaments in motor neuron growth cones. Motor neurons were grown on FN for 2 d and then treated with PBS (A, B) or 100 μg/ml CNS myelin (C–F). Cultures were fixed and stained for actin filaments with rhodamine-labeled phalloidin (A, C, E) or for microtubules using a monoclonal anti-tubulin antibody followed by a fluorescein-conjugated secondary antibody (B, D, F). In controls, growth cones displayed many actin filament-rich filopodia and lamellipodia (A) with a dense bundle of microtubules defining the central region of the growth cones (B). On exposure to CNS myelin, growth cones retracted both lamellipodia and filopodia concomitant with a decrease in rhodamine fluorescence predominantly in the periphery.C, Often motor neuron growth cones responded to CNS myelin by retracting the entire peripheral region using the tubulin staining as our criteria. E, In some cases, motor neuron growth cones displayed stubby filopodial remnants with decreased rhodamine fluorescence in the peripheral region. Images were acquired using identical parameters. Scale bar, 10 μm.
Fig. 4.
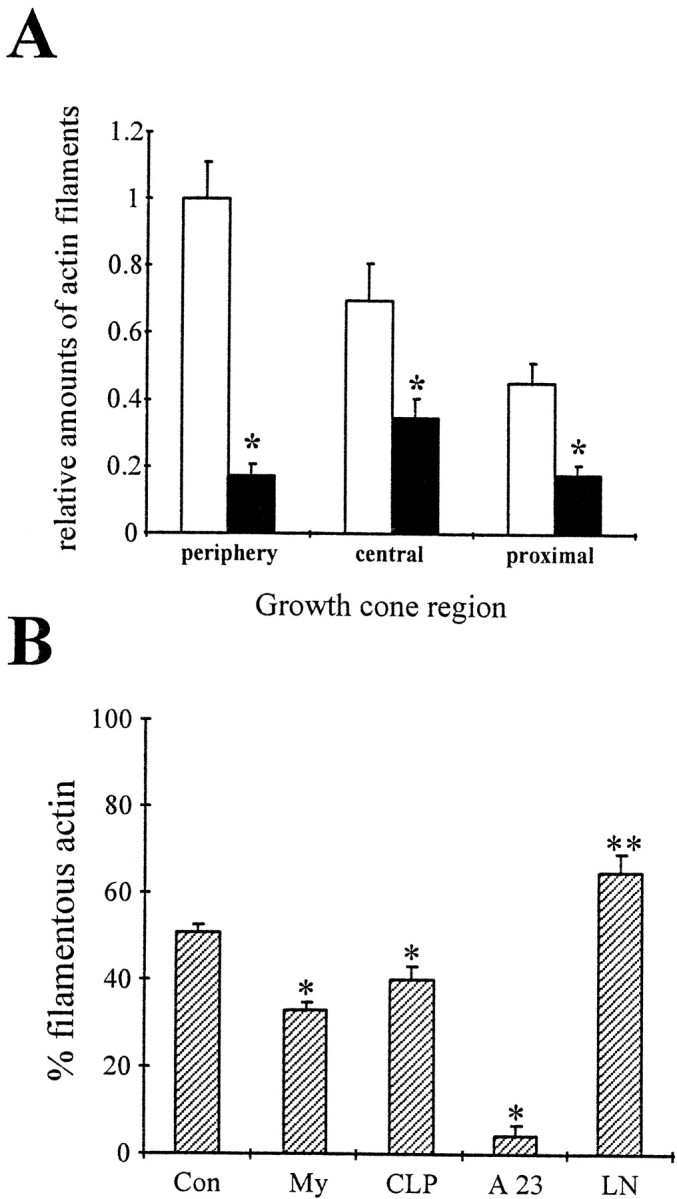
CNS myelin-associated growth inhibitors signal a disassembly of actin filaments. A, Motor neurons were grown on FN for 2 d, treated with PBS (open bars) or 100 μg/ml CNS myelin (filled bars), and actin filaments were visualized with rhodamine-labeled phalloidin. Growth cones were divided into a peripheral region, a central region defined by intense microtubule staining, and 15 μm of the proximal neurite. Images were acquired using identical parameters, and the total fluorescence intensity was analyzed in each growth cone region on a pixel-by-pixel basis after background subtraction. All values were normalized to the total fluorescence intensity of the peripheral region in control growth cones. CNS myelin caused a significant reduction of rhodamine fluorescence in the peripheral region (*p< 0.001) and also in the central region (*p < 0.001) and the proximal neurite (*p < 0.001). The loss of rhodamine fluorescence suggests a net disassembly of actin filaments signaled by CNS myelin as reported for collapsin-1 (Fan et al., 1993). B, Freshly prepared, intact growth cone particles were incubated with 100 μg/ml CNS myelin (My), 15 μg/ml enriched collapsin-1 (CLP), a mixture of 2 mm CaCl2and 10 μm A23187 (A23), 50 μg/ml laminin (LN), or PBS (Con). After 1 hr, samples were solubilized and separated into a cytoskeletal fraction and cytosol fraction by high-speed centrifugation (100,000 ×gmax). Proteins were separated on 10% SDS-PAGE and blotted onto PVDF membranes. The percentage of total actin in each fraction was determined by densitometry. Plotted is the percentage of total actin immunoreactivity in the cytoskeletal fraction, most likely actin filaments of various lengths, for each treatment. A significant decrease in filamentous actin occurred during incubation with CNS myelin (*p < 0.0001) or enriched collapsin-1 (*p < 0.01). As our control, large increases in the free intracellular Ca2+concentration results in an almost complete depletion of actin filaments (*p < 0.0001). In contrast, incubation with LN caused an increase in actin immunoreactivity in the cytoskeletal fraction, suggesting a net polymerization of actin filaments (**p < 0.01).
A biochemical assay further supported a CNS myelin-induced loss of actin filaments. Highly enriched, sealed GCPs (100 μg of total protein) were treated with CNS myelin (100 μg/ml), purified recombinant collapsin (750 ng/ml), laminin (50 μg/ml), CaCl2 (2 mm)/A23187 (10 μm), or buffer. A cytoskeletal fraction, containing insoluble actin, was separated from a cytosol fraction, containing soluble actin, by high-speed centrifugation (Helmke and Pfenninger, 1995). Relative actin content in each fraction was determined by quantitative Western blotting (Fig. 4B). Incubation of GCPs with CNS myelin reduced the relative actin content in the cytoskeletal fraction (33 ± 2%, n = 6, *p < 0.0001) compared with control (51 ± 2%, n = 6), implying a depolymerization of actin filaments. Also, treatment with 2 mm CaCl2 and 10 μm A23187 resulted in an almost complete depletion of actin in the cytoskeletal fraction (4 + 2%, n = 3, *p < 0.0001). In contrast, laminin increased actin in the cytoskeletal fraction (65 + 4%, n = 3, p < 0.01). Taken together, these results suggest that changes in growth cone morphology and behavior induced by CNS myelin originate, at least partially, from a loss of actin filament structures.
V12Rac1 and V14rhoA protect motor neurons from CNS myelin-induced growth cone collapse
CNS myelin as well as a protein extract obtained from CNS myelin caused a dose-dependent growth cone collapse when added in soluble form to motor neuron cultures (Fig.5A). In the presence of 80 μg/ml CNS myelin, a maximum of 72 ± 2% (open bars;n = 100, *p < 0.01) of growth cones investigated displayed a collapsed morphology compared with PBS, our control (26 ± 4% collapsed growth cones; n = 70). Similarly, 80 μg/ml of CNS myelin-derived protein extract collapsed 69 ± 7% of growth cones (n = 103, **p < 0.01) compared with 24 ± 5% collapsed growth cones (n = 127) in controls (dialyzed extraction buffer). It is noteworthy that a fair number of motor neuron growth cones exhibited a collapsed morphology under normal culture conditions.
Fig. 5.
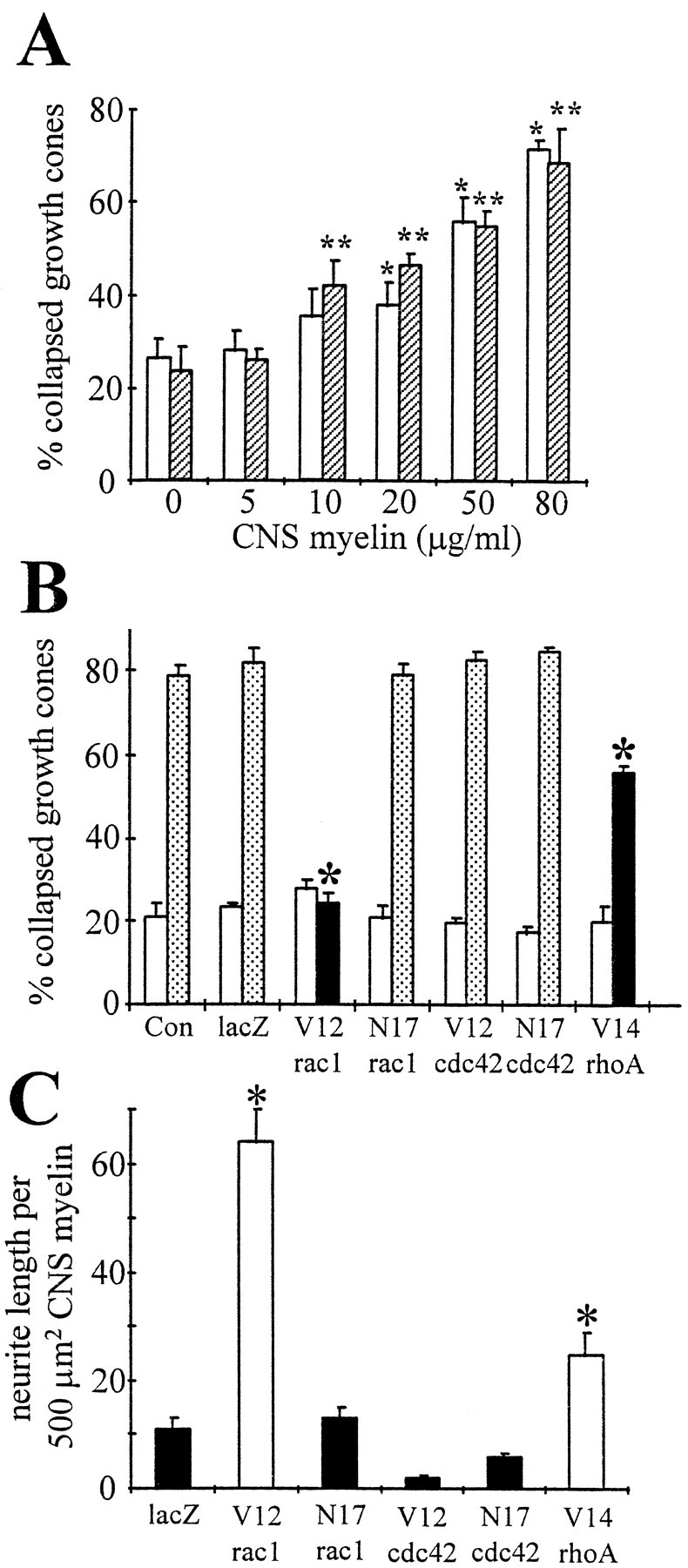
Dose-dependent CNS myelin-induced collapse of motor neuron growth cones is abolished by expressingV12rac1 or V14rhoA. A, Motor neurons were grown on FN for 2 d and then treated with CNS myelin (open bars) or a protein extract obtained from CNS myelin (hatched bars). In the case of CNS myelin, concentration of 20 μg/ml or higher achieved significant numbers of collapsed growth cones (*p < 0.01), whereas as little as 10 μg/ml CNS myelin protein extract induced significant growth cone collapse (**p < 0.01). Importantly, a basal level of collapsed growth cones existed in these motor neuron cultures. B, Motor neurons were infected at the time of plating with recombinant adenovirus carrying mutants of small GTPases and grown for 3 d. Cultures were treated with 100 μg/ml CNS myelin (stippled and filled bars) or with PBS (open bars). The percentage of collapsed growth cones was determined as a function of small GTPase mutants that were expressed. Only expression of constitutively active rac1 (V12rac1, filled bar) and rhoA (V14rhoA, filled bar) inhibited CNS myelin-induced growth cone collapse (*p < 0.01). Expression of other small GTPase mutants or lacZ was ineffective (stippled bars). It is noteworthy that a fraction of motor neuron growth cones exhibited a collapsed morphology regardless of proteins expressed (open bars). C, Motor neurons were plated on FN-coated dishes containing stripes of CNS myelin (100 μg/ml), infected with recombinant adenovirus carrying mutants of small GTPases, and grown for 3–4 d. The length of neurites grown entirely on CNS myelin stripes or grown into CNS myelin stripes was measured. Neurite length per 500 μm2 CNS myelin is plotted as a function of small GTPase mutants expressed. Expression of either V12rac1 or V14rhoA (open bars) resulted in considerable neurite outgrowth on CNS myelin (*p < 0.01) compared with lacZ or the other small GTPase mutants (filled bars). Con, PBS; lacZ, reporter gene coding β-galactosidase; V12rac1, constitutively active rac1; N17rac1, dominant negative rac1; V12cdc42, constitutively active cdc42; N17cdc42, dominant negative cdc42;V14rhoA, constitutively active rhoA.
Motor neurons expressing mutants of small GTPases were exposed to soluble CNS myelin to elucidate their potential role in CNS myelin-induced growth cone collapse. Dissociated motor neurons were infected with V12rac1-AdE1A,N17rac1-AdE1A,V12cdc42-AdE1A,N17cdc42-AdE1A,V14rhoA-AdE1A, or lacZ-AdE1A (200 moi) and treated with 100 μg/ml CNS myelin 3 d after infection, and the percentage of collapsed growth cones was determined (Fig. 5B). Importantly, expression of V12rac1 (24 ± 2% collapsed growth cones; n = 75, *p < 0.01) andV14rhoA (55 ± 4% collapsed growth cones;n = 113, *p < 0.01) protected motor neuron growth cones from CNS myelin-induced collapse compared with lacZ-AdE1A-infected cultures (82 ± 1% collapsed growth cones; n = 82) or uninfected cultures (79 ± 3% collapsed growth cones; n = 95), respectively. An identical correlation was evident when distinguishing between motor neurons expressing the c-myc tag and noninfected motor neurons using β-galactosidase-enhanced histochemistry. In the case ofV12rac1, 12 ± 5% (n = 100,p < 0.05) of c-myc-positive motor neurons (i.e., those expressing the viral-encoded rac1 mutants) had collapsed growth cones as opposed to 78 ± 8% (n = 100) of c-myc-negative motor neurons. InN17rac1-AdE1A-infected cultures, the fraction of collapsed growth cones of c-myc-positive motor neurons (90 ± 4%, n = 90) was indistinguishable from that of c-myc-negative motor neurons (85 ± 6%, n= 100) or lacZ-AdE1A-infected cultures (80 ± 5%, n = 100). Because β-galactosidase-enhanced histochemistry is a nonlinear amplification, and given the tight correlation of both analyses, evaluation of collapse of all growth cones is a much less biased approach. None of the other small GTPase mutants tested interfered with CNS myelin-induced collapse or affected the basal level of collapsed growth cones (Fig. 5A, open bars). Taken together, constitutively active mutants of rac1 and rhoA specifically compensated for the collapsing activity of CNS myelin. These findings suggest either that rac1 and rhoA participate in CNS myelin signaling through their inactivation or their constitutively active mutants preferentially alter growth cone behavior to resist collapse, probably through their effects on actin organization.
Expression of V12rac1 or V14rhoA enables motor neurons to extend neurites in the presence of CNS myelin
Because V12rac1 and V14rhoA protected growth cones against the effects of CNS myelin, we tested whether these mutants would permit neurite outgrowth in the presence of CNS myelin. Motor neurons were plated on FN-coated dishes containing stripes of CNS myelin (100 μg/ml) and infected withV12rac1-AdE1A,N17rac1-AdE1A,V12cdc42-AdE1A,N17cdc42-AdE1A,V14rhoA-AdE1A, or lacZ-AdE1A (200 moi). As illustrated in Figure6A,V12rac1-expressing motor neuron cultures and alsoV14rhoA-expressing motor neurons (data not shown) exhibited considerable neurite outgrowth 3 d after infection in the presence of CNS myelin (55 and 33% of fields investigated, respectively). In contrast, only marginal neurite outgrowth was achieved by expressingN17rac1 (Fig. 6C), V12cdc42,N17cdc42, or lacZ (17% of fields investigated). Neurite outgrowth on FN-coated areas was comparable among cultures infected with mutants of small GTPases or lacZ (Fig. 6B,D). To quantify these effects, we determined the length of neuronal process either grown entirely on or extending into CNS myelin-coated regions (area surveyed, >100,000 μm2) (Fig.5C). Expression of V12rac1 achieved an average neurite length per 500 μm2 of CNS myelin of 64 ± 6 μm (*p < 0.01) or an almost sixfold increase compared with controls (lacZ-AdE1A, 11 ± 2 μm/500 μm2 CNS myelin). The presence ofV14rhoA also achieved considerable neurite outgrowth on CNS myelin (25 ± 4 μm/500 μm2 CNS myelin). On FN, motor neurons established a rather elaborate network of processes after 3 d in culture (100,000 cells/ml); thus, length of individual neurites, grown entirely on FN, could not be determined. Taken together, motor neurons extend neurites in the presence of CNS myelin only when expressing V12rac1 or V14rhoA, supporting our findings with regard to CNS myelin-induced growth cone collapse.
Fig. 6.
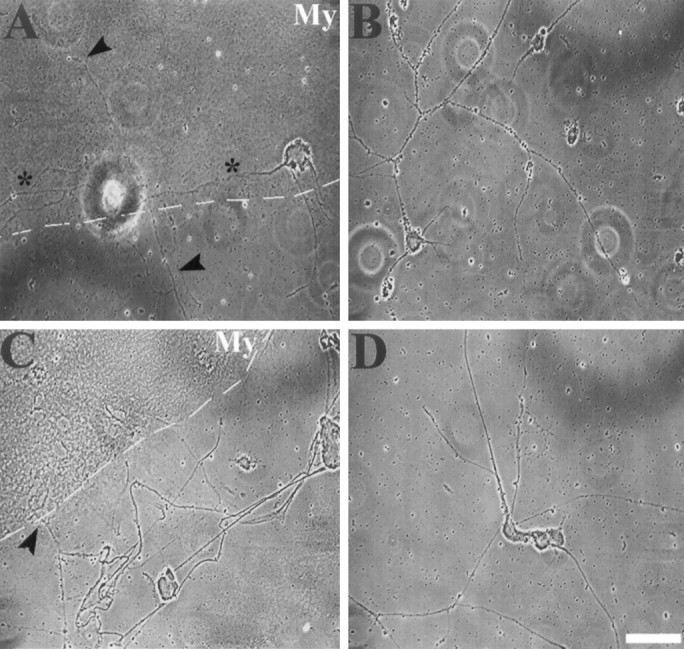
Motor neurons only establish neurites on CNS myelin when expressing V12rac1. Motor neurons were plated on FN-coated dishes containing stripes of CNS myelin (100 μg/ml) and infected (200 moi) with V12rac1-AdE1A(A, B) or N17rac1-AdE1A(C, D). A, Three days after infection, motor neurons expressing V12rac1 exhibit neurites grown entirely (asterisk) on CNS myelin-coated areas (My) as well as crossing into CNS myelin stripes (arrowheads). Dotted line marks the FN–CNS myelin border. B, Neurites grown exclusively on FN, however, are much longer and more branched (same cultures as inA), forming a rather intricate network.C, Despite N17rac1-expressing motor neurons extending neurites toward CNS myelin stripes (My), neurite growth into or entirely on CNS myelin stripes was only marginal (arrowhead) or even absent. The dashed line indicates the border between CNS myelin and FN.D, Nevertheless, on FN (same culture as inC), N17rac1-expressing motor neurons established a network of neurites comparable to that inV12rac1-infected cultures. Scale bar, 20 μm.
N17Rac1 and N17cdc42 protect motor neuron growth cones from the inhibitory effects of collapsin-1
In dorsal root ganglion (DRG) neuron cultures, rac1 mediates collapsin-1-induced growth cone collapse (Jin and Strittmatter, 1997). Therefore, we tested whether, in motor neuron cultures, collapsin-1-induced growth cone collapse was also associated with rac1 and possibly cdc42 and rhoA.
Enriched collapsin-1 (200 μg/ml) caused a dose-dependent collapse of motor neurons reaching 62 ± 3% (n = 124,p < 0.01) collapsed growth cones (Fig.7A). Incubation of GCPs with enriched collapsin-1 reduced the relative actin content in the cytoskeletal fraction (40 ± 4%, n = 3,p < 0.01; control = 51 ± 2%,n = 6) supporting a depolymerization of actin filaments as described in DRG growth cones (Fan et al., 1993). To investigate the role of small GTPases in mediating the action of collapsin-1, motor neurons expressing mutants of small GTPases were treated with 200 μg/ml enriched collapsin-1 or 150 ng/ml purified recombinant collapsin-1 3 d after infection (Fig. 7B). Expression of N17rac1 drastically reduced the fraction of collapsed growth cones (32 ± 3%, n = 134, *p < 0.01) compared with lacZ-AdE1A-infected cultures (71 ± 2% collapsed growth cones; n = 152), confirming previous observations by Jin and Strittmatter (1997). In our study, expression of N17cdc42 also effectively abolished growth cone collapse (39 ± 3%, n = 145, *p < 0.01). Furthermore, we coated collapsin-1 in small stripes onto poly-d-lysine to assess a potential stimulation of neurite outgrowth by N17rac1 orN17cdc42 or both. In accordance with our results in the collapse assay, motor neurons expressing N17rac1 orN17cdc42 displayed significant neurite outgrowth in the presence of collapsin-1 (Figs. 7C,8). Outgrowth on collapsin-1 (95 ± 7 μm, n = 61, *p < 0.001 in the case of N17rac1 and 91 ± 5 μm, n = 75, *p < 0.001 in the case of N17cdc42, respectively) was comparable to that on PL in the same cultures (N17rac1: 98 ± 7 μm, n = 59 andN17cdc42: 92 ± 6 μm, n = 68). However, in our culture system, a basal level of neurite outgrowth was observed on collapsin-1 stripes regardless of the expression of mutants of small GTPases or lacZ (57 ± 5 μm, n = 72). In summary, only N17rac1 or N17cdc42 negate collapsin-1-induced collapse and support growth on collapsin-1. This finding suggests either that cdc42 and rac1 are constituents of a signaling cascade activated by collapsin-1 or their dominant negative mutants alter actin filament organization favorably to protect against collapse.
Fig. 7.
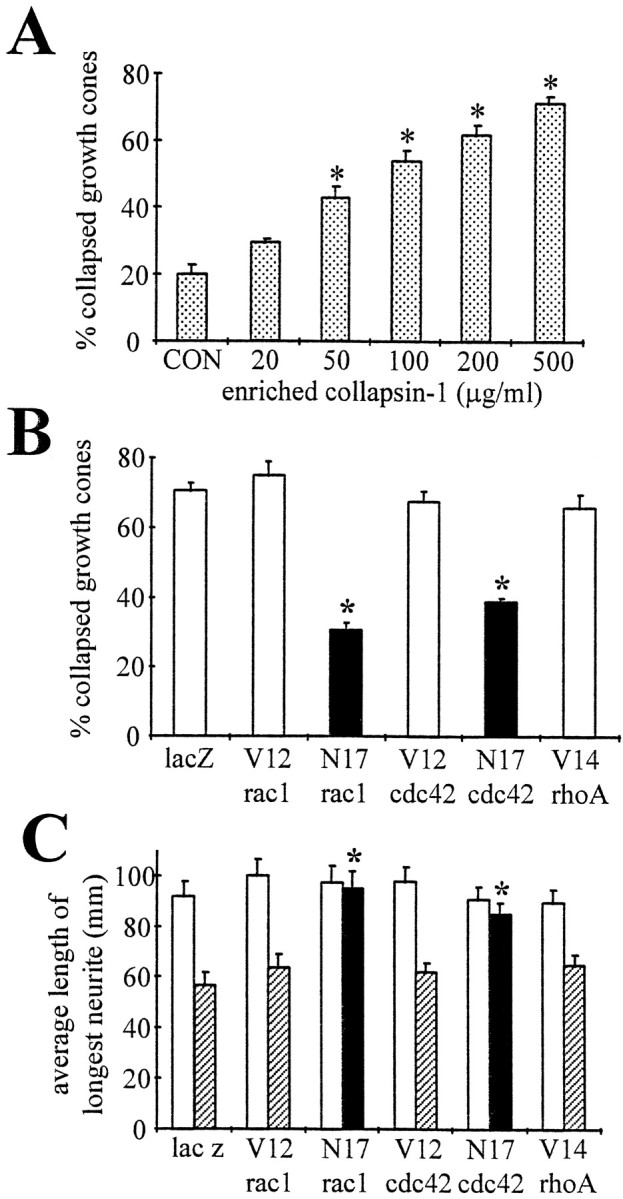
N17rac1 andN17cdc42 protect motor neuron growth cones from collapsin-1-induced collapse and support neurite outgrowth on a collapsin-1 substrate. A, Motor neurons grown on FN for 2 d were treated with increasing concentrations of enriched collapsin-1, and the number of collapsed growth cones was determined. Growth cone collapse was dose-dependent. A minimal concentration of 50 μg/ml was required for a significant number of collapsed growth cones (*p < 0.01). B, Motor neurons were plated on FN and infected with recombinant adenovirus carrying mutants of small GTPases. Three days after infection, cultures were treated with purified recombinant collapsin-1 (75 ng, 150 ng/ml). The percentage of collapsed growth cones is plotted as a function of the small GTPase expressed. Only N17rac1 orN17cdc42 reduced the fraction of collapsed growth cones (*p < 0.01) (filled bars), whereas other small GTPase mutants and lacZ expression were ineffective (open bars). In particular, the protective effect ofN17rac1 has been reported previously in DRG neurons (Jin and Strittmatter, 1997). Even in the absence of collapsin-1, a fraction of motor neuron growth cones was collapsed, independent of proteins expressed as shown for CNS myelin in Figure 5B.C, In a similar experiment, motor neurons were plated on poly-d-lysine-coated dishes containing collapsin-1 stripes and infected with recombinant adenovirus carrying mutants of small GTPases. The neurite length of the longest neurite per neuron was measured on polylysine alone (open bars) or on collapsin-1 (hatched bars and filled bars), and the average length was plotted as a function of expressed proteins. Neurite outgrowth on polylysine was comparable among conditions (open bars). In particular, expression of N17rac1 or N17cdc42 supported neurite outgrowth on collapsin-1 (filled bars) that was indistinguishable from growth on polylysine alone (*p < 0.001). Neither of the other small GTPase mutants that were tested significantly increased neurite length on collapsin-1 (hatched bars). It is noteworthy that motor neurons exhibited a basal outgrowth on collapsin-1.lacZ, Control; V12rac1, constitutively active rac1; N17rac1, dominant negative rac1; V12cdc42, constitutively active cdc42; N17cdc42, dominant negative cdc42;V14rhoA, constitutively active rhoA.
Fig. 8.

Dominant negative mutants of rac1 and cdc42 abolish the growth inhibitory effect of collapsin-1. Motor neurons expressing V12rac1 (A, D),N17rac1 (B, E), or N17cdc42 (C, F) were plated on polylysine-coated dishes containing collapsin-1 stripes. A–C, On polylysine, motor neurons formed neurites (arrows) regardless of expressing V12rac1 (A),N17rac1 (B), or N17cdc42 (C). D–F, On collapsin-1, expression of V12rac1 (D) achieved only basal outgrowth (asterisk), whereas bothN17rac1 (E) and N17cdc42 (F) supported neurite outgrowth (arrowheads) comparable to levels observed on polylysine alone. Scale bar, 30 μm.
DISCUSSION
During development, inhibitory cues direct advancing growth cones via repulsion and comprise one fundamental guidance force (Tessier-Lavigne and Goodman, 1996; Kolodkin et al., 1997). In the adult organism, an excessive presence of similar inhibitory cues is suspected of impairing regeneration of nerve fibers by inducing the collapse of growth cones. CNS myelin-associated factors, including NI35/250 and MAG, are potent growth inhibitors both in vitroand in vivo (Caroni and Schwab, 1988; Schnell and Schwab, 1990; McKerracher et al., 1994; Mukhopadhyay et al., 1994; Tang et al., 1997). Retraction of lamellipodia and filopodia are typical growth cone behaviors on contact with inhibitory cues, reflecting a substantial rearrangement of actin filaments (Kapfhammer et al., 1987; for review, see Luo and Raper, 1994). CNS myelin-induced growth cone collapse is associated with a reduction of rhodamine–phalloidin staining that is most prominent in the growth cone periphery, suggesting a loss of actin filaments (this report). A similar correlation has been demonstrated in growth cones of sensory neurons exposed to collapsin-1 (Fan et al., 1993). Nevertheless, partial loss of actin filaments is not a universal prerequisite of collapse. The Eph-ligand AL-1 induces growth cone collapse attributable to redistribution of actin filaments with no overall loss (Meima et al., 1997).
We tested whether modulating the activity of small GTPases could compensate for actin rearrangements in growth cones exposed to inhibitory cues. We used recombinant, replication-deficient adenoviruses to express constitutively active or dominant negative mutants of epitope-tagged small GTPases in embryonic chick motor neurons. The expression of virally encoded small GTPases was revealed by immunocytochemistry and Western blotting (Fig. 1). The function of small GTPases requires isoprenylation in the C-terminal region (Adamson et al., 1992). Attenuation of neurite outgrowth on fibronectin and changes in the actin filament distribution in motor neuron growth cones indicated that virally expressed small GTPases were functional. Identical observations have been obtained previously using trituration loading with recombinant V12rac1 protein (Kuhn et al., 1998). In support of these interpretations, microinjection of small GTPases in the form of protein or DNA into fibroblasts induced changes in cell behavior and morphology, suggesting successful isoprenylation (Paterson et al., 1990; Ridley et al., 1992). Furthermore, isoprenylation of small GTPases has been directly demonstrated in vitro and in vivo (Birchmeier et al., 1985; Kinsella and Maltese, 1992; Lang et al., 1996).
Inhibitory stimuli mediated by small GTPases
Our results in combination with previous reports provide evidence that small GTPases participate in mediating growth cone collapse. Expression of V12rac1 and V14rhoA but notV12cdc42 compensated for CNS myelin-induced growth cone collapse and supported neurite extension in the presence of CNS myelin. Motor neurons expressing dominant negative mutants of rac1 and cdc42 remained sensitive to CNS myelin. In sensory neurons, neitherN17rac1 nor wild-type rac1 impaired CNS myelin-dependent collapse, whereas V12rac1 and V14rhoA have not been tested (Jin and Strittmatter; 1997). Only recently, LIM-kinase 1 has been identified as a rac1 target. LIM-kinase 1 phosphorylates cofilin and probably actin depolymerizing factor (ADF), thereby inactivating these proteins (Arber et al., 1998; Yang et al., 1998). Increases in the activity of cofilin/ADF support neurite outgrowth (Meberg et al., 1998). Presently, the role of rhoA in growth cone collapse is at best controversial. Inhibition of rhoA blocked lysophosphatidic acid-mediated growth cone collapse in neuronal cell lines but also caused massive collapse of sensory neuron growth cones (Jalink et al., 1994; Jin and Strittmatter, 1997). The following processes are thought to support recruitment/activation of rhoA to the plasma membrane and cell motility. Phosphorylation of rhoA by protein kinase A, caused by increases in cAMP, enables a GDP dissociation inhibitor (rho-GDI) to bind the active, membrane-bound form of rhoA (Lang et al., 1996; Dong et al., 1998). Association of rho-GDI with radixin, an ezrin family protein, facilitates rhoA activation, possibly in a cAMP- and/or Ca2+-dependent manner (Takahashi et al., 1997). Interestingly, a depletion of radixin in the growth cone periphery has been observed in collapsing growth cones, and MAG and NI35/250 stimulate decreases in cAMP and increases in Ca2+ (Bandtlow et al., 1993; Gonzalez-Agosti and Solomon, 1996; Song et al., 1998).
Collapsin-1-dependent growth cone collapse and inhibition of neurite outgrowth was compensated by expressing N17rac1 andN17cdc42. Similar findings have been reported in sensory neurons using trituration loading of purified recombinant mutants (Jin and Strittmatter, 1997). N17Rac1 but notN17cdc42 blocked collapsin-1-induced growth cone collapse. This discrepancy might be attributable to the rather broad range of loading efficiency by trituration compared with viral expression, which achieves more constant levels in the majority of infected cells. Nevertheless, these results suggest that collapsin-1 causes growth cone collapse in a rac1/cdc42-dependent manner via increased activity of small GTPases. This is intriguing because increased GTPase activity usually relates to actin filament formation, whereas collapse usually correlates with actin filament reduction. However, altering the coordinated assembly and disassembly of actin filaments by disrupting either process could contribute to collapse. Small GTPases modulate both protein kinases (including LIM-kinase 1) and protein phosphatases such as calcineurin and phosphatase 1, which could influence actin filament dynamics via the degree of phosphorylation of cofilin/ADF (Meberg et al., 1998). Indeed, it has been shown that myosin light-chain phosphatase is inactivated in a rhoA-dependent manner (Kimura et al., 1996). Thus, in different neuronal cell types, the relative amounts and localization within the growth cones of different components of the same signaling pathway could bring about opposite affects on actin filament dynamics or myosin activation.
Inhibitory stimuli simply compensated by small GTPase
Our findings demonstrated that only certain small GTPase mutants abolished inhibitory effects of CNS myelin or collapsin-1, whereas their corresponding opposite mutants neither mimicked nor enhanced the inhibitory stimuli. There are two explanations for this phenomenon, both of which are likely occurring. First, the regulation of the GTP/GDP cycling of small GTPases is tightly regulated by multiple factors, including GDP/GTP exchangers (GEFs), GDIs, and GTPase-activating proteins (GAPs) (Boguski and McCormick, 1993). Constitutively active mutants elicit their effects in the absence of GEFs, whereas dominant negative mutants act by sequestering endogenous GEFs. Furthermore, there are regulatory factors that interact with subsets of small GTPases. Pertinent to our finding, rac1 and cdc42 share a GEF, TIAM-1, which has been implicated in neurite outgrowth, and rac1 and rhoA share yet another GEF, Trio (Ehler et al., 1997;Bellanger et al., 1998). Thus, signals that modulate TIAM-1 or Trio could simultaneously target both rac1 and cdc42 or rac1 and rhoA, respectively. Second, multiple parallel pathways may be stimulated by inhibitory cues. Thus, a single small GTPase mutant could abolish an inhibitory signal, whereas the opposite mutant might have no effect. For instance, MAG and NI-35/250 alter levels of cAMP and Ca2+ but collapsin-1 affects cGMP levels and heterotrimeric G-proteins (Song et al., 1998). These data suggest that different inhibitory cues elicit distinct intracellular signaling pathways and are supported by our finding that opposite rac1 mutants impaired CNS myelin- versus collapsin-1-induced collapse. However, our data do not exclude the alternative that small GTPase mutants indirectly interfere with collapse via a beneficial reorganization of the actin cytoskeleton.
Downstream targets of rac1 and cdc42 include N-WASP and IQGAP, two actin-associated components, and several protein kinases such as p65PAK, p120ACK, PI-3-kinase, and S6 kinase (Tapon and Hall, 1997). RhoA-dependent inactivation of myosin phosphatase strengthens myosin–actin interactions that could alter retrograde actin filament flow and filopodial motility (Lin et al., 1996; Wang et al., 1996). Also, GAP-43-induced filopodial formation and stabilization of microtubules requires rhoA (Aarts et al., 1998; Cook et al., 1998). Finally, collapsin-1 stimulates anterograde and retrograde axoplasmic transport, and inhibition of vesicle fusion at the tip of growth cones induces collapse, suggesting that axonal transport represents another aspect of collapse (Igarashi et al., 1996,Goshima et al., 1997).
Conversion of an inhibitory into a permissive environment
Our results provide evidence that small GTPases mediate the inhibitory effects of CNS myelin and collapsin-1, a soluble semaphorin. As a consequence, inhibitory properties of CNS myelin and collapsin-1 are switched to a permissive environment. An analogous situation exists in netrin-dependent growth cone guidance. Commissural neurons are attracted by netrin, whereas thoracic neurons are repelled (Kennedy et al., 1994; Colamarino and Tessier-Lavigne, 1995). Such a switch in the nature of the guidance stimuli can be mimicked simply by inhibiting protein kinase A (Ming et al., 1997). Song et al. (1998) demonstrated that growth cones, in the presence of a cGMP agonist, are attracted by collapsin-1. Similarly, growth cones become attracted to MAG by elevating cAMP levels. In conclusion, modulating signaling pathways linked to inhibitory stimuli might represent an alternative to achieving neurite outgrowth in an inhibitory environment. In particular, small GTPases are ideal candidates because of their regulation by multiple upstream factors and their influence on actin filament dynamics.
Footnotes
This work was supported by Grant 1643 from the Paralyzed Veterans of America Spinal Cord Research Foundation to T.B.K. and National Institutes of Health Grants GM35126 and GM54004 to J.R.B. We gratefully acknowledge Dr. Alan Hall (Cambridge, UK) for his generous contribution of G-protein expression plasmids. We also thank Don Traul and Dr. Peter Meberg for critical advice and help in the production of recombinant adenovirus, and Laurie Minamide for technical advice and assistance.
Correspondence should be addressed to Dr. James R. Bamburg, Department of Biochemistry and Molecular Biology, Colorado State University, Fort Collins, CO 80523.
REFERENCES
- 1.Aarts LH, Schrama LH, Hage WJ, Bos JL, Gispen WH, Schotman P. B-50/GAP-43-induced formation of filopodia depends on rho-GTPase. Mol Biol Cell. 1998;9:1279–1292. doi: 10.1091/mbc.9.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adamson P, Marshall CJ, Hall A, Tilbrook PA. Posttranslational modification of p21 rho proteins. J Biol Chem. 1992;267:20033–20038. [PubMed] [Google Scholar]
- 3.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 4.Bantdlow CE, Schmidt MF, Hassinger TD, Schwab ME, Kater SB. Role of intracellular calcium in NI-35 evoked collapse of neuronal growth cones. Science. 1993;259:80–83. doi: 10.1126/science.8418499. [DOI] [PubMed] [Google Scholar]
- 5.Becker TC, Noel RJ, Coats WS, Gomez-Foix AM, Alam T, Gerard RD, Newgrad CB. Use of recombinant adenovirus for metabolic engineering of mammalian cells. Methods Cell Biol. 1994;43:161–189. doi: 10.1016/s0091-679x(08)60603-2. [DOI] [PubMed] [Google Scholar]
- 6.Bellanger JM, Lazaro JB, Diriong S, Fernandez A, Lamb N, Debant A. The two guanine nucleotide exchange factor domains of Trio link the rac1 and rhoA pathway in vivo. Oncogene. 1998;16:147–152. doi: 10.1038/sj.onc.1201532. [DOI] [PubMed] [Google Scholar]
- 7.Birchmeier C, Broek D, Wigler M. Ras proteins can induce meiosis in Xenopus oocytes. Cell. 1985;43:615–621. doi: 10.1016/0092-8674(85)90233-8. [DOI] [PubMed] [Google Scholar]
- 8.Boguski MS, McCormick F. Proteins regulating ras and its relatives. Nature. 1993;366:643–654. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- 9.Caroni P, Schwab ME. Two membrane protein fractions from rat central myelin with inhibitory properties for neurite growth and fibroblast spreading. J Cell Biol. 1988;106:1281–1288. doi: 10.1083/jcb.106.4.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Chedotal A, He Z, Goodman CS, Tessier-Lavigne M. Neuropilin-2, a novel member of the neuropilin family, is a high affinity receptor for the semaphorins Sema E and Sema IV but not Sema III. Neuron. 1997;19:547–559. doi: 10.1016/s0896-6273(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 11.Colamarino SA, Tessier-Lavigne M. The axonal chemoattractant netrin-1 is also a chemorepellent for trochlear axons. Cell. 1995;81:621–629. doi: 10.1016/0092-8674(95)90083-7. [DOI] [PubMed] [Google Scholar]
- 12.Cook TA, Nagasaki T, Gundersen GG. Rho guanosine triphosphatase mediates the selective stabilization of microtubules induced by lysophosphatidic acid. J Cell Biol. 1998;141:175–185. doi: 10.1083/jcb.141.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong JM, Leung T, Manser E, Lim L. cAMP-induced morphological changes are counteracted by the activated rhoA small GTPase and the rho kinase ROKalpha. J Biol Chem. 1998;273:22554–22562. doi: 10.1074/jbc.273.35.22554. [DOI] [PubMed] [Google Scholar]
- 14.Ehler E, van Leeuwen F, Collard JG, Salinas PC. Expression of Tiam-1 in the developing brain suggests a role for the Tiam-1-Rac signaling pathway in cell migration and neurite outgrowth. Mol Cell Neurosci. 1997;9:1–12. doi: 10.1006/mcne.1997.0602. [DOI] [PubMed] [Google Scholar]
- 15.Fan J, Mansfield SG, Redmond T, Gordon-Weeks PR, Raper JA. The organization of F-actin and microtubules in growth cones exposed to a brain-derived collapsing factor. J Cell Biol. 1993;121:867–878. doi: 10.1083/jcb.121.4.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez-Agosti C, Solomon F. Response of radixin to perturbations of growth cone morphology and motility in chick sympathetic neurons in vitro. Cell Motil Cytoskeleton. 1996;34:122–136. doi: 10.1002/(SICI)1097-0169(1996)34:2<122::AID-CM4>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 17.Goshima Y, Kawakami T, Hori H, Sugiyama Y, Takasawa S, Hashimoto Y, Kagoshima-Maezono M, Takenaka T, Misu Y, Strittmatter SM. A novel action for collapsin: collapsin-1 increases antero- and retrograde axoplasmic transport independently of growth cone collapse. J Neurobiol. 1997;33:316–328. doi: 10.1002/(sici)1097-4695(199709)33:3<316::aid-neu9>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 18.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 19.Helmke S, Pfenninger KH. Growth cone enrichment and cytoskeletal association of non-receptor tyrosine kinases. Cell Motil Cytoskeleton. 1995;30:194–207. doi: 10.1002/cm.970300304. [DOI] [PubMed] [Google Scholar]
- 20.Igarashi M, Kozaki S, Terakawa S, Kawano S, Ide C, Komiya Y. Growth cone collapse and inhibition of neurite neurite growth by botulinum neurotoxin C1: a t-SNARE is involved in axonal growth. J Cell Biol. 1996;134:205–215. doi: 10.1083/jcb.134.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivins JK, Raper JA, Pittman RN. Intracellular calcium levels do not change during contact-mediated collapse of chick DRG growth cone structure. J Neurosci. 1991;11:1597–1608. doi: 10.1523/JNEUROSCI.11-06-01597.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jalink K, van Corven EJ, Hengeveld T, Morii N, Narumiya S, Moolenaar WH. Inhibition of lysophosphatidate- and thrombin-induced neurite retraction and neuronal cell rounding by ADP ribosylation of the small GTP-binding protein rho. J Cell Biol. 1994;126:801–810. doi: 10.1083/jcb.126.3.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin Z, Strittmatter SM. Rac1 mediates collapsin-1-induced growth cone collapse. J Neurosci. 1997;17:6256–6263. doi: 10.1523/JNEUROSCI.17-16-06256.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kapfhammer JP, Grunewald BE, Raper JA. The selective inhibition of growth cone extension by specific neurites in culture. J Neurosci. 1987;6:2527–2534. doi: 10.1523/JNEUROSCI.06-09-02527.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaufmann N, Wills ZP, Van Vactor D. Drosophila Rac1 controls motor axon guidance. Development. 1998;125:453–461. doi: 10.1242/dev.125.3.453. [DOI] [PubMed] [Google Scholar]
- 26.Kennedy TE, Serafini T, de la Torre JR, Tessier-Lavigne M. Netrins are diffusible chemotropic factors for commissural axons in the embryonic spinal cord. Cell. 1994;78:425–435. doi: 10.1016/0092-8674(94)90421-9. [DOI] [PubMed] [Google Scholar]
- 27.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by rho and rho-associated kinase (rho-kinase). Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 28.Kinsella BT, Maltese WA. rab GTP-binding proteins with three different carboxyl-terminal cysteine motifs are modified in vivo by 20-carbon isoprenoids. J Biol Chem. 1992;267:3940–3945. [PubMed] [Google Scholar]
- 29.Kolodkin AL. Growth cones and the cues that repel them. Trends Neurosci. 1996;19:507–513. doi: 10.1016/S0166-2236(96)10057-6. [DOI] [PubMed] [Google Scholar]
- 30.Kolodkin AL, Levengood DV, Rowe EG, Tai YT, Giger RJ, Ginty DD. Neuropilin is a semaphorin III receptor. Cell. 1997;90:753–762. doi: 10.1016/s0092-8674(00)80535-8. [DOI] [PubMed] [Google Scholar]
- 31.Kuhn TB, Brown MD, Bamburg JR. Rac1-dependent actin filament organization in growth cones is necessary for β1 integrin-mediated advance but not for growth on poly-d-lysine. J Neurobiol. 1998;37:524–540. doi: 10.1002/(sici)1097-4695(199812)37:4<524::aid-neu3>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 32.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 33.Lang P, Gesbert F, Delespine-Carmagnat M, Stancou R, Pouchelet M, Bertoglio J. Protein kinase A phosphorylation of rhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 1996;15:510–519. [PMC free article] [PubMed] [Google Scholar]
- 34.Lin CH, Espreafico EM, Mooseker MS, Forscher P. Myosin drives retrograde F-actin flow in neuronal growth cones. Neuron. 1996;16:769–782. doi: 10.1016/s0896-6273(00)80097-5. [DOI] [PubMed] [Google Scholar]
- 35.Lockerbie RO, Miller VE, Pfenninger KH. Regulated plasmalemmal expansion in nerve growth cones. J Cell Biol. 1991;112:1215–1227. doi: 10.1083/jcb.112.6.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo L, Liao YJ, Jan LY, Jan YN. Distinct morphogenetic functions of similar small GTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. Genes Dev. 1994;8:1787–1802. doi: 10.1101/gad.8.15.1787. [DOI] [PubMed] [Google Scholar]
- 37.Luo L, Hensch TK, Ackerman L, Barbel S, Jan LY, Jan YN. Differential effects of the rac GTPase on Purkinje cell axons and dendritic trunks and spines. Nature. 1996;379:837–840. doi: 10.1038/379837a0. [DOI] [PubMed] [Google Scholar]
- 38.Luo L, Jan LY, Jan YN. Rho family small GTP-binding proteins in growth cone signaling. Curr Opin Neurobiol. 1997;7:81–86. doi: 10.1016/s0959-4388(97)80124-9. [DOI] [PubMed] [Google Scholar]
- 39.Luo Y, Raper JA. Inhibitory factors controlling growth cone motility and guidance. Curr Opin Neurobiol. 1994;4:648–654. doi: 10.1016/0959-4388(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 40.Luo Y, Raible D, Raper JA. Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell. 1995;75:217–227. doi: 10.1016/0092-8674(93)80064-l. [DOI] [PubMed] [Google Scholar]
- 41.McGrory WJ, Bautista DS, Graham FL. A simple technique for the rescue of early region I mutations into infectious human adenovirus type 5. Virology. 1988;163:614–617. doi: 10.1016/0042-6822(88)90302-9. [DOI] [PubMed] [Google Scholar]
- 42.McKerracher L, David S, Jackson DL, Kottis V, Dunn RJ, Braun PE. Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron. 1994;13:805–811. doi: 10.1016/0896-6273(94)90247-x. [DOI] [PubMed] [Google Scholar]
- 43.Meberg PJ, Ono S, Minamide LS, Takahashi M, Bamburg JR. Actin depolymerizing factor and cofilin phosphorylation dynamics: response to signals that regulate neurite extension. Cell Motil Cytoskeleton. 1998;39:172–190. doi: 10.1002/(SICI)1097-0169(1998)39:2<172::AID-CM8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 44.Meima L, Kljavin IJ, Moran P, Shih A, Winslow JW, Caras IW. AL-1-induced growth cone collapse of rat cortical neurons is correlated with REK7 expression and rearrangement of the actin cytoskeleton. Eur J Neurosci. 1997;9:177–188. doi: 10.1111/j.1460-9568.1997.tb01365.x. [DOI] [PubMed] [Google Scholar]
- 45.Minamide LS, Bamburg JR. A filter paper dye-binding assay for quantitative determination of protein without interference from reducing agents or detergents. Anal Biochem. 1990;190:66–70. doi: 10.1016/0003-2697(90)90134-u. [DOI] [PubMed] [Google Scholar]
- 46.Ming GL, Song HJ, Berninger B, Holt CE, Tessier-Lavigne M, Poo MM. cAMP-dependent growth cone guidance by netrin-1. Neuron. 1997;19:1225–1235. doi: 10.1016/s0896-6273(00)80414-6. [DOI] [PubMed] [Google Scholar]
- 47.Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT. A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron. 1994;13:757–767. doi: 10.1016/0896-6273(94)90042-6. [DOI] [PubMed] [Google Scholar]
- 48.Nobes CD, Hall A. Rho, rac and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 49.Norton WT, Poduslo SE. Myelination in the rat brain: method of myelin isolation. J Neurochem. 1973;21:749–757. doi: 10.1111/j.1471-4159.1973.tb07519.x. [DOI] [PubMed] [Google Scholar]
- 50.Oakley BR, Kirsch DR, Norris NR. A simplified ultrasensitive silver stain for detecting proteins in polyacrylamide gels. Anal Biochem. 1980;105:361–363. doi: 10.1016/0003-2697(80)90470-4. [DOI] [PubMed] [Google Scholar]
- 51.O’Connor TP, Bentley D. Accumulation of actin in subsets of pioneer growth cone filopodia in response to neural and epithelial guidance cues in situ. J Cell Biol. 1993;123:935–948. doi: 10.1083/jcb.123.4.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paterson HF, Self AJ, Garrett MD, Just I, Actories K, Hall A. Microinjection of recombinant p21rho induces rapid changes in cell morphology. J Cell Biol. 1990;111:1001–1007. doi: 10.1083/jcb.111.3.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pfenninger KH, Ellis L, Johnson MP, Friedman LB, Somlo ST. Nerve growth cones isolated from fetal rat brain: subcellular fractionation and characterization. Cell. 1983;35:573–584. doi: 10.1016/0092-8674(83)90191-5. [DOI] [PubMed] [Google Scholar]
- 54.Ridley AJ, Paterson HF, Johnston CL, Diekman D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 55.Schaack J, Allen B, Maxwell IH, Smith RL (1998) Promoter strength in adenovirus transducing vectors: down-regulation of the adenovirus E1A promoter in 293 cells facilitates vector construction. J Virol, in press. [DOI] [PubMed]
- 56.Schnell L, Schwab ME. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature. 1990;343:269–272. doi: 10.1038/343269a0. [DOI] [PubMed] [Google Scholar]
- 57.Song H, Ming G, He Z, Lehmann M, Tessier-Lavigne M, Poo M. Conversion of neuronal growth cone responses from repulsion to attraction by cyclic nucleotides. Science. 1998;281:1515–1518. doi: 10.1126/science.281.5382.1515. [DOI] [PubMed] [Google Scholar]
- 58.Stossel TP. On the crawling of animal cells. Science. 1993;260:1086–1094. doi: 10.1126/science.8493552. [DOI] [PubMed] [Google Scholar]
- 59.Takahashi K, Sasaki T, Mammoto A, Takaishi K, Kameyama T, Tsukita S, Takai Y. Direct interaction of the rho GDP dissociation inhibitor with ezrin/radixin/moesin initiates the activation of the rho small G protein. J Biol Chem. 1997;272:23371–23375. doi: 10.1074/jbc.272.37.23371. [DOI] [PubMed] [Google Scholar]
- 60.Tanaka E, Sabry J. Making the connection: cytoskeletal rearrangements during growth cone guidance. Cell. 1995;83:171–176. doi: 10.1016/0092-8674(95)90158-2. [DOI] [PubMed] [Google Scholar]
- 61.Tang S, Woodhall RW, Shen YJ, deBellard ME, Saffell JL, Doherty P, Walsh FS, Filbin MT. Soluble myelin-associated glycoprotein (MAG) found in vivo inhibits axonal regeneration. Mol Cell Neurosci. 1997;9:333–346. doi: 10.1006/mcne.1997.0633. [DOI] [PubMed] [Google Scholar]
- 62.Tapon N, Hall A. Rho, rac and cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr Opin Cell Biol. 1997;9:86–92. doi: 10.1016/s0955-0674(97)80156-1. [DOI] [PubMed] [Google Scholar]
- 63.Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- 64.Threadgill R, Bobb K, Ghosh A. Regulation of dendritic growth and remodeling by rho, rac and cdc42. Neuron. 1997;19:625–634. doi: 10.1016/s0896-6273(00)80376-1. [DOI] [PubMed] [Google Scholar]
- 65.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang FS, Wolenski JS, Cheney RE, Mooseker MS, Jay DG. Function of myosin-V in filopodial extension of neuronal growth cones. Science. 1996;273:660–663. doi: 10.1126/science.273.5275.660. [DOI] [PubMed] [Google Scholar]
- 67.Wessel D, Flügge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 68.Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393:809–812. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]