

Abstract
The insulin-like growth factor-1 (IGF-1)/receptor tyrosine kinase recently has been shown to mediate neuronal survival and potentiate the activity of specific calcium channel subtypes; survival requires Akt, a serine/threonine kinase. We demonstrate here that Akt mediates the IGF-1-induced potentiation of L channel currents, but not that of N channels. Transient expression of wild-type, dominant–negative, and constitutively active forms of Akt in cerebellar granule neurons causes, respectively, no change in IGF-1/L channel potentiation, complete inhibition of potentiation, and a dramatic increase in basal L currents accompanied by the loss of ability to induce further increases. In no case is the IGF-1 potentiation of N currents affected. We additionally find that IGF-1 partially mediates granule neuron survival via L channel activity and that Akt-dependent L channel modulation is a necessary component. Interestingly, very brief exposure (1 min) to IGF-1 triggers nearly complete survival and requires L channel activity. These results strongly suggest that neuronal receptor tyrosine kinases can control long-term calcium-dependent processes via the rapid control of voltage-sensitive channels.
Keywords: IGF-1, RTK, L channel, modulation, granule neurons, apoptosis, survival
Receptor tyrosine kinases (RTKs) increasingly are being described as rapid modulators of neuronal ion channels, suggesting mechanisms for regulating a variety neuronal processes, including action potential firing, neurotransmitter release, RTK-dependent differentiation, and neuronal survival. RTK ligands such as insulin-like growth factor-1 (IGF-1) have been described to regulate ion channel behavior within minutes of application (Peppelenbosch et al., 1991, 1992; Lovisolo et al., 1992; Hinkle et al., 1993; Naumov et al., 1993; Jonas et al., 1996; Delbono et al., 1997) and sometimes within seconds (Selinfreund and Blair, 1994; Blair and Marshall, 1997;Hilborn et al., 1998).
IGF-1 is of particular interest. Both it and its receptor are highly expressed in the mature nervous system (Kar et al., 1993; LeRoith et al., 1993). Although its role on postmitotic neurons is not fully clear, IGF-1 has been shown to be an essential mediator of neuronal survival (D’Mello et al., 1993). The IGF-1-induced neuroprotective mechanisms are beginning to be elucidated and include such signaling intermediates as phosphatidylinositol 3-OH kinase (PI 3-kinase) and Akt (Galli et al., 1995; Datta et al., 1997; D’Mello et al., 1997; Dudek et al., 1997; Miller et al., 1997; Párrizas et al., 1997).
Akt, a serine/threonine kinase also known as PKB or RAC, recently has been shown to be a direct downstream target of lipid products of PI 3-kinase (Datta et al., 1996; Franke et al., 1997). Stimulation of cells by IGF-1 increases PI 3-kinase activity. The activated kinase then phosphorylates the membrane lipid PI(4,5)P2 to produce PI(3,4,5)P3 (PIP3). In granule neurons, PIP3 can be converted to PI(3,4)P2 (Franke et al., 1997), which binds the Akt PH domain, leading to dimerization and membrane attachment (Datta et al., 1996); it is then activated by phosphorylation by the membrane-associated serine/threonine kinase, PIP3-dependent protein kinase-1 (Alessi et al., 1997). Membrane translocation is an essential step in Akt activation; the addition of an N-terminal myristoylation sequence results in a constitutively active enzyme (Kohn et al., 1996; Andjelkovic et al., 1997). Although the mechanisms mediating neuronal survival appear to involve multiple pathways (Galli et al., 1995; Courtney et al., 1997;Datta et al., 1997; D’Mello et al., 1997; Mattson, 1997; Soler et al., 1998), a role for Akt has been clearly established. Akt is required for NGF-dependent survival in sympathetic neurons (Crowder and Freeman, 1998) and for IGF-1- and PI 3-kinase-dependent survival in granule neurons (Dudek et al., 1997).
A second or overlapping pathway involves L channel-mediated calcium influx. To promote survival, cerebellar granule neurons are standardly grown in elevated potassium to depolarize the cells, increasing L channel activity. Although it has long been clear that L channel activity is important in neuronal survival (Gallo et al., 1987), the underlying mechanism or mechanisms are still under debate. It is, for instance, unclear whether survival requires a sustained or transient elevation of cytosolic calcium (Gallo et al., 1987; Marchetti and Usai, 1996; Schmidt et al., 1996; Ono et al., 1997) or whether the signaling pathway involves IGF-1 and PI 3-kinase (Galli et al., 1995; D’Mello et al., 1997; Miller et al., 1997).
We previously have shown rapid and strongly voltage-dependent potentiation of N and L calcium channels in cerebellar granule neurons by physiological levels of IGF-1. This potentiation is blocked by PI 3-kinase inhibitors and by the transfection of an inactive mutant of the p85 regulatory subunit of the kinase (Blair and Marshall, 1997). Given that Akt has been shown to be a downstream target of PI 3-kinase and that Akt and calcium influx through L channels have been implicated independently in neuronal survival (D’Mello et al., 1993; Datta et al., 1997; Dudek et al., 1997; Mattson, 1997; Ono et al., 1997), we determined whether Akt plays a role in the IGF-1/PI 3-kinase-dependent potentiation of calcium channel currents in cerebellar granule neurons and whether the Akt-mediated survival of these neurons depends on rapid IGF-1 channel modulation.
MATERIALS AND METHODS
Cell culture. Cerebellar granule neurons from P5 rats were cultured by standard means (Messer, 1972; Blair et al., 1998) and tested after 3–6 d. Standard full-serum medium contained 10% fetal bovine serum (Life Technologies, Gaithersburg, MD, or HyClone, Logan, UT) in DMEM, with penicillin/streptomycin and elevated potassium and glucose (25 mm KCl and 6 gm/l glucose). To inhibit proliferation of non-neuronal cells, we added either cytosine arabinofuranoside (10 μm; Sigma, St. Louis, MO) or 5-fluoro-2′-deoxyuridine (20 μm; Calbiochem, La Jolla, CA) after 1 d in culture. To eliminate potential preexposure to IGF-1 and because serum contains a large number of growth factors, we switched cells to media without serum before electrophysiological testing. Test media for the survival assays were the same as the standard medium but were without serum and contained 5 mm KCl (see below).
Expression vectors and transfection of neurons in culture.Three forms of Akt were expressed in granule neurons: normal, wild-type Akt (Franke et al., 1995), and kinase-dead Akt (Franke et al., 1995) as well as Akt containing the src myristoylation signal that confers membrane localization and the ability to maintain the kinase in an elevated state of activity (Kohn et al., 1996). All cDNAs were in pcDNA3 expression vectors (Invitrogen, San Diego, CA) under CMV promoter control.
Granule neurons in primary culture were transfected by using procedures that were modified from those developed for cell lines (Blair and Marshall, 1997; Blair et al., 1998). Briefly, 1 d cultures (2 ml of medium per culture) were cotransfected by calcium phosphate precipitation with 4–8 μg of cDNAs encoding Akt and SuperGlo (sg25)-GFP (Quantum Biotechnologies, Durham, NC) to allow for identification of the transfected cells. Control transfections used only the GFP-containing vector, but the amount of GFP cDNA was increased so that the same total amount of DNA was used as for the Akt plus GFP transfections. Electrophysiological recordings were done as soon as possible (typically, 12–20 hr) after transfection to minimize long-term survival effects on the quality of recordings.
Electrophysiology. The permeabilized patch variation of the standard whole-cell technique (Hamill et al., 1981; Rae et al., 1991) was used to eliminate the loss of soluble cytoplasmic components. Calcium channel currents were evoked and recorded with a List EPC-9 patch-clamp amplifier in conjunction with Macintosh-based data acquisition and analysis software (HEKA, Instrutech, Great Neck, NY). Typically, data were low-pass-filtered at 3 kHz (−3 dB, digital Gaussian filter) and acquired at a sample interval of 50 μsec, using P/4 leak subtraction. Currents were evoked by depolarizing voltage pulses (−40 to +40 mV) from a holding potential of −80 mV. For each cell the currents were recorded immediately before and 10–500 sec after IGF-1 addition. Frozen stocks of human recombinant IGF-1 (Becton Dickinson, Mountain View, CA) were diluted into the extracellular recording saline (final concentration of 20 ng/ml) and superfused over the cells. The extracellular saline contained (in mm): 20 BaCl2 (as the charge carrier), 100 NaCl, 20 tetraethylammonium chloride (TEA), 5 4-aminopyridine (4-AP), 1 μm tetrodotoxin (TTX), and 10 Na-HEPES, pH 7.4. Patch electrodes (Corning 8161 glass) were filled with (in mm): 150 CsCl, 5 BAPTA, and 10 Na-HEPES, pH 7.4, as well as amphotericin B (final concentration, 0.25 mg/ml) to permeabilize the patch and allow low-resistance electrical access without breaking the patch membrane; the electrode resistance was ∼3 MΩ. Under these conditions the calcium channel currents could be recorded for ≥5 min without displaying significant “rundown.” Improperly clamped cells were eliminated; however, because granule neurons are relatively small (soma diameter, ∼5 μm), space-clamp problems generally were not encountered. For a comparison of currents obtained from different cells, peak currents were normalized to the cell surface area as estimated from the membrane capacitance (i.e., current density).
The estimate of errors associated with determining the fold change in basal L currents when Akt isoforms are overexpressed (see Fig. 2) requires factoring in both the errors associated with each reference point (mean level at each potential in the untransfected cells) and the errors associated with each test condition (mean levels at each potential in the wild-type Akt- or constitutively active Akt-transfected cells). First, to normalize for differences in cell size, we calculated the current densities of L currents at each potential for each of the three conditions (untransfected cells and cells transfected with wild-type and constitutively active Akt) from the peak currents before IGF-1 addition and the membrane capacitance. Then, for each condition and membrane potential the mean current densities were determined. To obtain the fold change in basal L current levels at each potential, we divided the mean current densities obtained with wild-type and constitutively active Akt (the two test conditions) by the mean obtained from untransfected cells (the reference condition). Because each reference mean is an experimental value containing error, it was necessary to consider this error, as well as the errors associated with the mean values for each test condition, into each error estimate of fold change. Therefore, final errors were estimated from the variances around the means of each reference and test condition by the two-tailed Student’s tformulation of the SE of differences, taking into account the respective SDs and n values and the fact that no a priori assumption could be made as to whether currents in the test condition would be greater or less than in the reference condition. The theory of this method is identical to that of the Student’s tformulation, which allows for determining the probability that two populations (test vs reference) are different. The cumulative error thus obtained for a test condition at a given potential is, depending on the n values, similar to or greater than the arithmetic sum of the test and reference SEMs at that potential.
Fig. 2.
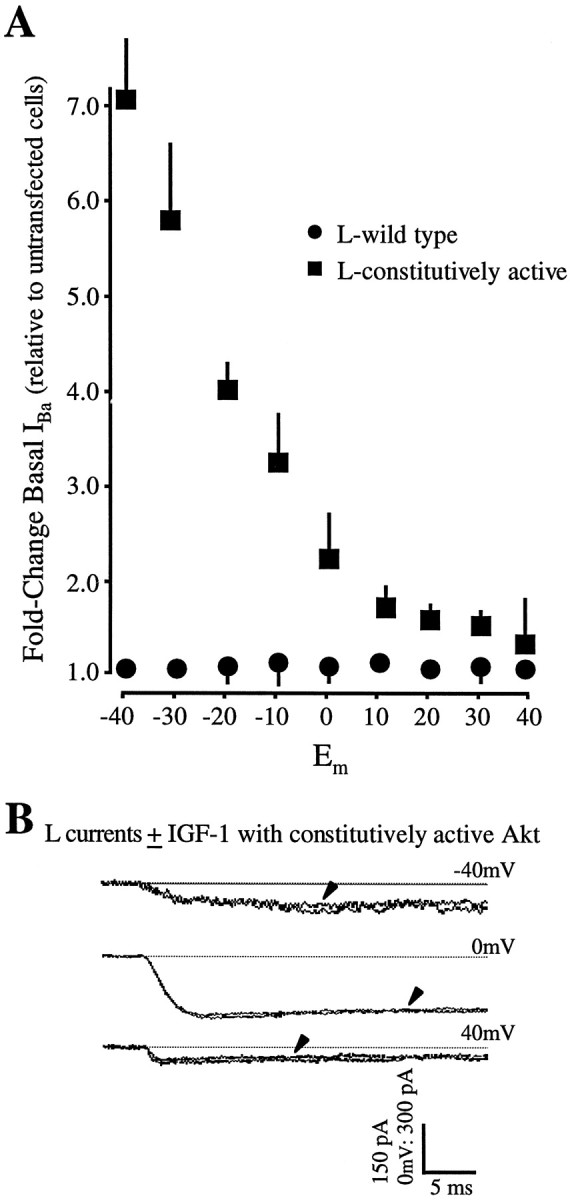
Overexpression of a constitutively active Akt mimics the IGF-1 effect. A, Basal L currents are increased dramatically in neurons transfected with Akt containing a myristoylation sequence that renders it constitutively active. Moreover, superfusion of IGF-1 is incapable of further potentiating L currents. Recordings were performed as in Figure 1; note, however, the change in scale. Arrowheads indicate currents recorded 30 sec after IGF-1 (20 ng/ml) addition. The test pulse protocol is identical to that used in Figure 1. B, Plots of the voltage-dependent changes in basal L currents show that the increase is particularly prominent at hyperpolarized membrane potentials in neurons expressing myristoylated Akt. Current levels before IGF-1 addition in transfected cells were normalized to cell size as estimated by membrane capacitance and were compared with the values obtained in untransfected cells. Values are means ± SEM; where no bars are shown, the errors are smaller than the symbols denoting their means. Errors were calculated as described in Materials and Methods. n= 27 cells transfected with myristoylated Akt; n = 32 cells with wild-type Akt.
Separation of ionic currents. L and N channel currents were studied in isolation by standard techniques of ion substitution and pharmacological blocking agents. Sodium channel activity was blocked with TTX (1 μm; Sigma), and potassium currents were blocked with TEA (20 mm; Sigma) and 4-AP (5 mm; Sigma), as well as by substitution of intracellular (pipette) potassium with cesium. P and Q calcium channels were blocked with ω-agatoxin-IVA (300 nm; gift of Dr. N. A. Saccomano (Pfizer, New York, NY), N channels with ω-conotoxin-GVIA (500 nm; Sigma), and L channels with the dihydropyridine (DHP) inhibitor nimodipine (5–10 μm; Calbiochem).
Survival assays. Before they were switched to the test media, the neurons were cultured under standard full-serum conditions for 3 d, then rinsed three times in 0 serum medium, and cultured for 1–3 d more in the test medium indicated above each panel. Neurons grown in the standard full-serum medium were used as a reference to establish the maximal attainable level of survival. The primary survival test media were the basic survival medium with 0 serum and 5 mm KCl (labeled on the figures as No addition), or 0 serum/5 mm KCl medium with 50 ng/ml IGF-1 (IGF-1), with 10 μm nimodipine (nim), or with both 50 ng/ml IGF-1 and 10 μmnimodipine (IGF-1 + nim). For each experiment all survival media were assayed either in triplicate or quadruplicate and in parallel on sister cultures. Other survival test media included 1 or 10 μm nifedipine ± IGF-1, 1 μmnimodipine ± IGF-1, 80 mm KCl ± nimodipine, and 0.1–50 μm (−)-Bay K8644. Because DHPs are use-dependent blockers, the inhibitor was added 10 min before the IGF-1 for media containing both DHP inhibitors and IGF-1. Depolarization test medium (80 mm KCl ± nimodipine) also contained 1 μm TTX to inhibit sodium-dependent action potential activity. For assessing the effect of overexpressing wild-type or inactive Akt, we used untransfected neurons from the same plates as controls.
Cells then were processed according to the particular assay. For propidium iodide staining to reveal DNA, the cultures were fixed with 2% paraformaldehyde, permeabilized with 0.1% Triton X-100 in PBS, and stained with 2 μg/ml propidium iodide in 0.1% Triton X-100 in PBS. Chromatin staining patterns then were analyzed for individual cells by using rhodamine filters and either a Zeiss Axiovert 100 laser scanning confocal microscope (LSM) or a Nikon Eclipse E800 microscope equipped with a CCD camera; codetection of GFP used the fluorescein settings. For each experiment all neurons in four randomly chosen fields were counted and separated into two groups: those with dispersed chromatin (healthy) or those with condensed or condensed and fragmented nuclei (apoptotic).
For the direct detection of chromatin cleavage, two assays, the terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling reaction (TUNEL; also called in situ end labeling) and DNA laddering, were used. For the TUNEL assay the cultures were processed as per the manufacturer’s instructions, using the In SituCell Death Detection kit (Boehringer Mannheim, Indianapolis, IN), and the labeled (i.e., cleaved) DNA of individual cells was visualized under fluorescein optics on a Zeiss Axiovert 100 LSM. To detect chromatin cleavage via the formation of a DNA ladder (Miller et al., 1988), cells, plated at equal density and grown in the test media as described above, were lysed in 10 mm Tris, pH 8.0, and 10 mm EDTA buffer (TE) with 200 μg/ml proteinase K and 0.5% SDS; proteins were precipitated with 1 m NaCl, RNA was eliminated by treatment with 25 μg/ml RNase A, and any remaining proteins or lipids were removed by phenol/chloroform extraction. DNA, precipitated in ethanol and resuspended in TE, then was electrophoresed on 1.2% agarose gels and detected by ethidium bromide labeling.
RESULTS
IGF-1 rapidly modulates neuronal L channel activity via Akt
We first determined the potential role of Akt/PKB in the rapid voltage-dependent modulation of neuronal N and/or L channels by IGF-1. To assess this, we employed the “dominant–negative” approach (Blair and Marshall, 1997), transiently cotransfecting granule neurons with cDNAs encoding a catalytically inactive mutant of Akt (dn-Akt;Franke et al., 1995) and the jellyfish green fluorescent protein (GFP;Marshall et al., 1995), an autofluorescent protein that enables identification of transfected neurons. Kinase-dead Akt was constructed by introducing a lysine-methionine substitution (amino acid position 179) within the ATP-binding domain (Franke et al., 1995). To control for possible nonspecific effects that might result simply from heterologous overexpression of the Akt protein in neurons, we transfected sister cultures with wild-type Akt, and their ability to respond rapidly to IGF-1 was tested.
We found that overexpression of the catalytically inactive Akt prevented the rapid IGF-1-induced potentiation of pharmacologically isolated L, but not N, calcium channel currents (Figs.1B–D, 3). Conversely, overexpression of wild-type Akt failed to affect L channel potentiation by IGF-1 (Fig. 1A; n = 35 cells), which occurred as previously described (Blair and Marshall, 1997). IGF-1-induced increases in L currents were most prominent at hyperpolarized membrane potentials; the primary kinetic effect appeared to be an increase in the rate of activation, and no differences in deactivation, as indicated by the tail currents, were detected. However, when L currents were tested in neurons overexpressing the inactive dn-Akt, the strong IGF-1 potentiation typically observed at hyperpolarized potentials was fully blocked (Figs.1B,D, 3; n = 21 cells). Nonetheless, when N currents were examined in sister neurons also expressing dn-Akt, IGF-1 induced a rapid (within 10 sec) increase (Figs. 1C, 3;n = 19 cells). Similar increases in N currents were observed in wild-type Akt expressing neurons (data not shown) (n = 11 cells). As expected (Blair and Marshall, 1997), the N channel potentiation in both wild-type and dn-Akt-expressing neurons was particularly prominent at depolarized membrane potentials. These results strongly imply that the signaling pathway mediating the rapid neuromodulatory effects of IGF-1 on L, but not N, channels requires Akt.
Fig. 1.
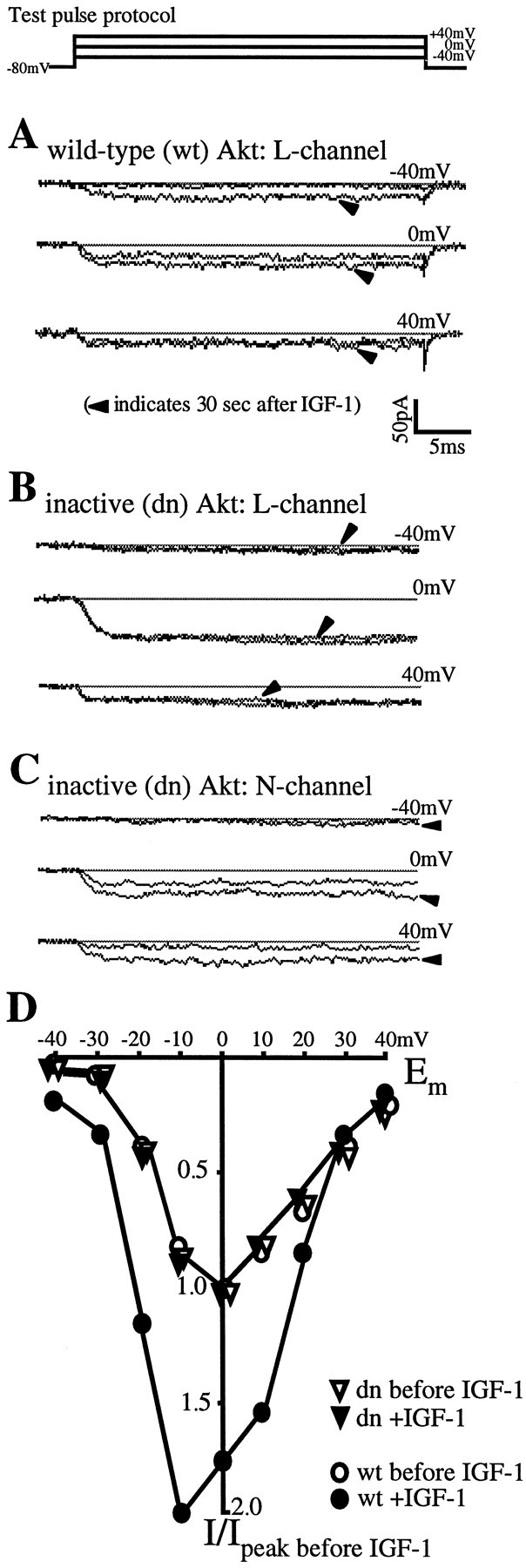
Rapid IGF-1 potentiation of cerebellar L channels requires the serine/threonine kinase Akt. A, Overexpression of wild-type Akt allows the normal, hyperpolarization-dependent IGF-1 potentiation of L channels.A–C, Each panel shows an individual neuron transfected with either wild-type or kinase-dead Akt. Recordings are of the barium currents evoked at three membrane potentials (−40, 0, and +40 mV) before and 30 sec after IGF-1 (20 ng/ml) addition (arrowheads). The test pulse protocol is shownabove A. Calcium channel subtypes were isolated as described in Materials and Methods. B, A K179M mutation in the ATP-binding site of the kinase renders Akt inactive and blocks IGF-1/L channel potentiation. C, Inactive Akt has no effect on the normal depolarization-dependent potentiation of N channels by IGF-1. D, IGF-1 responses in two neurons, one transfected with wild-type Akt (open circles) and one with inactive dn-Akt (inverted open triangles); shown are membrane L channel current–voltage relationships before (open symbols) and 30 sec after the addition of 20 ng/ml IGF-1 (filled symbols). L currents for each cell were normalized to the peak currents measured before IGF-1. Note that, although the currents are relatively small at hyperpolarized potentials, the fold increase in the wild-type Akt-expressing neuron is much greater at −30 and −40 mV than at potentials ≥0 mV.
Fig. 3.

Fold potentiation of N and L currents in wild-type, inactive, and constitutively active Akt-expressing neurons. For each cell the peak current measured after 20 ng/ml IGF-1 was divided by that measured before. Values are means ± SEM; where no bars are shown, the errors are smaller than the symbols denoting their means. Filled circles, inverted open triangles, filled squares, The potentiation of L-currents by 20 ng/ml IGF-1. Transfection conditions: wild-type Akt (L-wt),n = 32 cells; dominant–negative Akt (L-dn), n = 21 cells; constitutively active Akt (L-con-active), n = 27 cells. Open circles, open triangles, open squares, N-current potentiation by IGF-1. Transfection conditions: wild-type Akt (N-wt), n = 11 cells; dominant–negative Akt (N-dn), n = 17 cells; constitutively active Akt (N-con-active),n = 8 cells.
We next tested whether the expression of a constitutively active form of Akt would alter L currents or their ability to respond rapidly to IGF-1. For these experiments a construct encoding an Akt tagged at the N terminus with the src myristoylation signal and conferring membrane localization was used (Kohn et al., 1996). Because the normal activation of wild-type Akt requires translocation to the plasmalemma (Andjelkovic et al., 1997), where it is activated by the membrane-associated PIP3-dependent protein kinase-1 (Alessi et al., 1997), myristoylated Akt displays elevated activity. We found that expression of the constitutively active Akt mimicked the IGF-1 effect on L currents (Fig. 2;n = 27 cells). In the absence of IGF-1 a dramatic increase in basal L currents was observed at hyperpolarized membrane potentials (Fig. 2A). Moreover, superfusion of IGF-1 was unable to evoke further increases (Figs. 2B,3), suggesting that the constitutively active enzyme induced full potentiation. To ensure that any delayed responses were not overlooked, L currents were recorded for up to 8 min after IGF-1 addition. N currents were examined also. As expected from the failure of the dominant–negative Akt to block IGF-1–N channel potentiation, the constitutively active Akt similarly failed to affect either N channel potentiation or basal current levels (n = 8 cells).
Interestingly, the increase in basal L currents in neurons expressing the constitutively active Akt (Fig. 2A) is greater than the IGF-1 potentiation observed in untransfected neurons 30 sec after IGF-1 addition (Blair and Marshall, 1997). One possible explanation is that increased activity of the Akt kinase drives potentiation to (or toward) the maximum. Our results would favor this hypothesis. When IGF-1 responses were recorded from untransfected neurons for periods >30 sec (i.e., for 5–10 min) or from neurons overexpressing rate-limiting intermediates, IGF-1 potentiation increased (Blair and Marshall, 1997; our unpublished data), suggesting that, although strong potentiation can be observed within seconds of exposure to IGF-1, maximal levels are obtained over minutes or, potentially, hours (Selinfreund and Blair, 1994). The inability of IGF-1 to induce further increases in the neurons overexpressing constitutively active Akt also implies that L channel activity was maximally increased in these cells. Alternatively or in addition, Akt-driven increases in calcium influx may increase L channel synthesis, which is known to be regulated by L channel activity (Murphy et al., 1991).
IGF-1 increases neuronal survival via L channel potentiation
To identify potential cellular effects of the rapid channel modulation, we examined whether IGF-1 potentiation of L channels contributes to neuronal survival. For these experiments, cultured cerebellar granule neurons were grown in the presence or absence of IGF-1 and the presence or absence of dihydropyridine (DHP) inhibitors of L channel activity. Additionally, because the normal medium contains 10% serum, a rich source of growth factors, and elevated (25 mm) KCl to promote survival via increased L channel activity, the survival test media eliminated serum and reduced the KCl to normal (5 mm) levels to eliminate exposure to uncontrolled levels of growth factors and exogenous sources of L channel stimulation. Survival was assessed by three technically unrelated assays designed to detect endonucleosis, a key event specifically linked to apoptosis.
All assays indicate that approximately one-third of the IGF-1-mediated neuronal survival is L channel-dependent. Granule neurons, first cultured in a standard (10% serum, 25 mm KCl) medium, were rinsed extensively and subsequently maintained in the basic survival test medium (no serum, 5 mm KCl) or in the survival test medium supplemented with IGF-1 (50 ng/ml), the DHP inhibitor nimodipine (10 μm), or both IGF-1 and nimodipine (50 ng/ml and 10 μm, respectively). To maximize the difference in survival between the basic and IGF-1-containing test media, 50 ng/ml IGF-1 was used. This concentration will potentiate L channels to a slightly greater level than 20 ng/ml (L. A. C. Blair, unpublished results) but will not cross-activate insulin receptors (Cohick and Clemmons, 1993). When survival after 1 or 2 d in the basic test medium was assessed by DNA staining with propidium iodide, it was found that most neurons were apoptotic (Fig.4B,F). By day 3, no neurons survived in this medium, making it impossible to factor in the effect of nimodipine alone (see below). In contrast, neurons maintained in either full-serum medium or survival test medium supplemented with IGF-1 mainly displayed the dispersed chromatin characteristic of healthy cells, with only a tiny fraction exhibiting the condensed and fragmented nuclei indicative of apoptotic cells (Fig.4A,D,F). Neurons maintained in test media with nimodipine showed poorer survival than those maintained in the basic test medium alone (Fig. 4C,F). Because L channel activity has been implicated extensively in granule neuron survival (Gallo et al., 1987; Galli et al., 1995), this could be attributable to the inhibition of basal L channel activity. In addition, partial block of other channel types (e.g., potassium channels; Bean, 1992) also might be a factor. As described below, the decrease in survival due to nimodipine alone was compensated.
Fig. 4.

L channel potentiation by IGF-1 promotes neuronal survival. Granule neurons, cultured for 1 d in the indicated test media, were stained with propidium iodide to reveal DNA; anarrowhead indicates one example/panel of the bright, condensed chromatin characteristic of apoptotic cells.A, In full (10%) serum medium the nuclei are healthy, showing dispersed chromatin. B, At 1 d after serum withdrawal (No addition) most neurons are apoptotic.C, The addition of 10 μm nimodipine, an L channel inhibitor, to block neuronal L channel activity reduces survival over 0 serum alone. D, Conversely, the addition of 50 ng/ml IGF-1 promotes survival almost to the levels observed in full serum. E, Exposure to both IGF-1 and nimodipine partially promotes survival. F, Quantitation of survival: data (mean ± SEM) are from seven (day 1) and four independent (day 2) experiments in which each condition was tested in triplicate. The total number of neurons scored is shownwithin each bar; the percentage of survival is indicatedabove each bar. Shaded bars, Survival in full-serum medium. IGF-1*, Because nimodipine by itself decreases survival beyond that of the basic survival, the effect of nimodipine alone [11% = (survival in No add) − (survival in nim)] was subtracted from the survival in IGF-1 to obtain an estimate (IGF*) that can be compared directly with the survival in the L channel inhibition medium (IGF + nim, nim). IGF + 1 μm nif, IGF + 10 μm nif, IGF + 10 μm nim, Survival in DHP-containing media suggests that 10, but not 1, μm is effective. G, H, DNA laddering (G) and the TUNEL assay (H) for nicked DNA similarly indicate that L channel potentiation by IGF-1 is a component of IGF-1-mediated survival. G, IGF-1 (50 ng/ml) protects chromatin from the cleavage into low molecular weight fragments that occurs in the presence of 10 μm nimodipine. Only partial protection occurs when cells are grown in IGF-1 plus nimodipine. Lane 4, DNA standards. H, Cells with cleaved chromatin are brightly labeled. Unlabeled cells can be identified from the dim autofluorescence. All cells are shown at 1 d in test media. I, The DHP L channel agonist, (−)-Bay K8644, can mimic the IGF-1-dependent and L channel-dependent survival, but only at optimal concentrations. Sister cultures of neurons were grown for 24 hr in the basic survival medium supplemented with the indicated (−)-Bay K8644 concentrations, and survival was assessed by the proportion of cells displaying dispersed chromatin after propidium iodide labeling; the values are expressed as means ± SEM.
Neurons maintained in IGF-1 in the presence of nimodipine survived well but to a considerably lower degree than those maintained in IGF-1 without the L channel inhibitor (Fig. 4E,F), implying L channel involvement. Importantly, however, survival above that observed in the basic test medium cannot be used for direct comparison to other test conditions because nimodipine itself reduces survival, independent of the presence of IGF-1 (Fig.4C,F). Therefore, to remove the nonspecific effects of nimodipine on survival (i.e., effects on basal L channel activity in the absence of IGF-1 and any other nonspecific effects), we subtracted the reduction in survival due to nimodipine alone (see Fig.4F, IGF*). The results indicate that L channels mediated a part of the IGF-1-dependent survival. Namely, if IGF-1 was exerting its effects independently of L channel activity, the expected survival in IGF-1 plus nimodipine would be that of IGF*, that is, IGF-1 alone corrected for the IGF-1-independent decrease observed in nimodipine alone. Because the survival in nimodipine alone was 11% lower than that obtained in the basic test medium (Fig.4F), the level of survival corrected for IGF-1-independent effects on L channels (Fig. 4F, IGF*) would be ∼83%. Significantly, survival in IGF-1 plus nimodipine was only ∼63%. Therefore, using the 32% survival in nimodipine as the base survival that occurs without IGF-1 or active L channels, the data suggest that IGF-1 potentiation of L channel activity accounts for approximately one-third of the IGF-1-induced survival. Similar results were obtained when the presence of cleaved DNA was assessed by DNA laddering and the TUNEL assay (Fig.4G,H) and strongly indicate that L channel potentiation by IGF-1 is a component of IGF-1-mediated survival.
We next compared the effectiveness of different dihydropyridine antagonists. This was of particular interest because studies using 1 μm nifedipine had reported that IGF-1-dependent survival was L channel-independent (Galli et al., 1995). To determine whether different efficacies of the different DHP antagonists or concentration-dependent differences were the source of the differences in results, we assessed the relative abilities of 1 and 10 μm nifedipine and 1 and 10 μm nimodipine to reduce IGF-1-induced survival in the basic survival media. As assayed by DNA condensation detected by propidium iodide staining, 1 μm nifedipine was only poorly effective, 1 μm nimodipine was partially effective, and 10 μm nifedipine was approximately as effective as 10 μm nimodipine (Fig. 4F). Specifically, survival after 1 d in 10 μm nimodipine plus IGF-1 was 63 ± 5%, compared with 75 ± 2% in 1 μmnimodipine plus IGF-1, 89 ± 3% in 1 μm nifedipine plus IGF-1, and 67 ± 4% in 10 μm nifedipine plus IGF-1 (mean ± SEM; n = 3 independent experiments, with all test conditions assayed in parallel and in quadruplicate). Given that survival in IGF-1 alone was 94 ± 2%, our results would suggest that, in the absence of exogenous electrical stimulation, a 10 μm concentration of either DHP is more effective on granule neurons than 1 μm, and 1 μmnimodipine is more effective than 1 μm nifedipine.
The ability of the L channel agonist, (−)-Bay K8644, to induce survival was determined also (Fig. 4I). When granule neurons were cultured for 24 hr in survival medium supplemented with (−)-Bay K8644, we found a very sharp concentration dependence. No concentration was able to induce complete survival and, in concentrations ≤0.5 μm, survival was indistinguishable from the basal survival observed in the absence of any additions. However, exposure to optimal concentrations (1–5 μm) increased survival ∼30%, precisely mimicking the portion of IGF-1-dependent survival attributable to L channel modulation. Interestingly, concentrations ≥10 μm decreased survival below that observed in the basic survival medium alone.
Akt-dependent survival is also partially L channel-dependent
Akt recently has been shown to be an essential link between the IGF-1/RTK and inhibition of apoptosis in granule neurons (Dudek et al., 1997). In addition, L channel activity also has been linked to the survival of various cell types, including granule neurons (Gallo et al., 1987; Galli et al., 1995). We therefore investigated the potential role of Akt-dependent L channel modulation in IGF-1-mediated survival.
Granule neurons were transiently transfected in sister cultures with either wild-type or kinase-dead Akt. We then compared the survival of the transfected and untransfected neurons in the same dish, using GFP fluorescence to identify successful transfectants and propidium iodide labeling to assess the state of the chromatin in both transfected and untransfected cells. Because all neurons in each culture were treated identically, comparing transfected and untransfected cells within each culture optimally controlled for any slight differences in handling between cultures. As a further control, sister cultures were transfected with only the GFP-containing vector to determine whether survival was altered by the transfection procedure or the overexpression of exogenous protein.
We found that expression of dominant–negative Akt resulted in the death of all detectably transfected neurons, even in the presence of IGF-1 (Fig. 5A,E) or 10% serum medium, whereas overexpressed wild-type Akt increased survival (Fig. 5B,E). Similar results have been reported previously for Akt-transfected granule neurons (Dudek et al., 1997). Overexpression of GFP alone slightly but significantly decreased survival (Fig. 5E; p < 0.05; two-tailed Student’s t test). We further evaluated whether the increased survival of neurons overexpressing wt-Akt could be attributable in part to an Akt-dependent increase in L channel activity.
Fig. 5.

Survival via IGF-1 modulation of L channels is mediated by Akt. Granule neurons were cultured for 1 d in the indicated survival media and transfection conditions.A–D, Left, Akt transfectants as indicated by GFP fluorescence. Right, The same fields showing propidium iodide labeling of all neurons to reveal DNA;arrowheads indicate transfectants with the condensed chromatin characteristic of apoptotic cells, and arrowsindicate healthy transfectants; open arrowheads indicate examples of untransfected apoptotic cells. A, In IGF-1-containing (50 ng/ml) medium, neurons expressing dominant–negative (dn) Akt are apoptotic although all surrounding untransfected neurons are healthy. B–E, Neurons expressing wild-type (wt) Akt show increased, but L channel-dependent, survival. B, In IGF-1-containing test medium, wt-Akt-transfected neurons are healthy.C, In the basic test medium (No addition), wt-Akt-transfected neurons survive much better than their untransfected neighbors. D, In nimodipine-containing (10 μm) medium, the survival of transfected and untransfected neurons is similar. E, Quantitation of survival: data (mean ± SEM) are from three independent experiments (except GFP alone,n = 2), with each condition tested in triplicate. In each bar pair the left bar (shaded) represents transfectants, and the right bar represents untransfected cells from the same cultures. Overexpression of wt-Akt increases survival but to a much lesser degree when L channels are blocked (filled arrowheads) than when they are active (open arrowheads). The total number of neurons scored is shown within each bar; the percentage of survival is indicated above each bar.
As summarized in Figure 5E, survival in all test media was significantly greater for wt-Akt-transfected over nontransfected neurons in the same cultures (Fig. 5B–E; p< 0.001 for basic, IGF-1, IGF-1 plus nimodipine, and plus nimodipine test media). Importantly, however, the increased survival was susceptible to L channel block; in the basic 0 serum test medium, survival of the wt-Akt-transfected neurons was 48 ± 1% (mean ± SEM; Fig. 5C,E), 8% greater than for the untransfected sister neurons. However, the addition of 10 μm nimodipine halved the increase because of wt-Akt overexpression (Fig. 5D,E; 33 ± 1% wt-Akt vs 30 ± 1% untransfected cells). In medium containing both IGF-1 and nimodipine, wt-Akt-transfected neurons survived better than their untransfected sisters (Fig. 5E; ∼4% higher survival), but not to the degree observed in medium supplemented with IGF-1 alone (Fig. 5E; ∼7% higher survival). Together, these results imply that Akt potentiation of L channel activity is integral to Akt-dependent survival.
Rapid modulation of L channels triggers IGF-1-dependent survival
Neuronal survival has long been associated with regulating cytosolic calcium levels. Limited increases appear to increase survival (Mattson, 1997), whereas excessive influx is associated with necrotic cell death (Choi, 1995; Gwag et al., 1997). Because recent evidence suggests that transient increases may be sufficient to improve survival (Schmidt et al., 1996; Ono et al., 1997), we performed a time course to determine whether relatively brief exposure to IGF-1 also promoted survival.
We found that applications of IGF-1 for as little as 1 min increased survival to levels almost indistinguishable from those obtained with a 24 hr application (Fig.6A,B,E). IGF-1 applications up to 1 hr were tested, and all were nearly identical to each other and the 24 hr time point (Fig. 6E). Similarly, brief (1 min–1 hr) coapplication of IGF-1 and nimodipine resulted in survival nearly identical to that obtained with 24 hr coapplications of IGF-1 and nimodipine (Fig. 6D,E), implying that IGF-1-induced potentiation of L channels is an element required to achieve full survival. Crucially, the subsequent block of L channel activity after brief exposure to IGF-1 failed to decrease the survival (Fig. 6C,E). Similar results were obtained when chromatin cleavage was assessed by the presence of low molecular weight DNA (DNA laddering, Fig. 6F); brief pulses of IGF-1 strongly retarded chromatin cleavage for up to 2 d; by 3 d the DNA patterns in IGF-1-treated cells were indistinguishable from untreated cells.
Fig. 6.

A brief pulse of IGF-1 promotes neuronal survival via L channel potentiation. Granule neurons were kept in survival test media for a total of 24 hr; IGF-1-containing (50 ng/ml) test media (IGF-1, IGF + Nim) were applied for the indicated times, and then the cultures were rinsed three times and placed in either basic survival medium (No add) or in 10 μmnimodipine-containing medium (Nim) for the remainder of the 24 hr period. DNA was labeled as in Figures 4 and 5; anarrowhead indicates one example/panel of apoptotic cells. A, In IGF-1 medium the nuclei are healthy.B, C, A 1 min pulse of IGF-1 strongly promotes survival, even when L channels subsequently are blocked for the remainder of the test duration (C). D, Conversely, the number of apoptotic nuclei increases after a 1 min pulse of IGF-1 in the presence of nimodipine. E, Quantitation of survival: data (mean ± SEM) are from three independent experiments, with each condition tested in triplicate. The total number of neurons scored is shown within each bar; the percentage of survival is indicated above each bar.Shaded bars, Survival in full-serum medium.F, Brief exposure (1 or 10 min) to IGF-1 (50 ng/ml), followed by continuous exposure to nimodipine (10 μm), reduces chromatin cleavage as determined by DNA laddering. Partial protection can be detected for up to 2 d. Lane 1, DNA standards. G, The activation of L channels by depolarization also inhibits apoptosis. Neurons depolarized with 80 mm KCl for the same periods (1 or 10 min) survive much better than those exposed either to the basic survival medium, which contains 5 mm KCl (i.e., the “No add”medium), or to the 80 mm KCl-containing medium in the presence of 10 μm nimodipine (nim + 80 mm KCl). The 80 mm KCl is expected to drive the membrane potential to approximately −15 mV. After the brief application of 80 mm KCl ± nimodipine or basic (5 mm KCl) medium, all neurons were maintained for 1 d in the basic medium; apoptotic versus nonapoptotic cells were scored as above after propidium iodide labeling.
L channel involvement in rapidly promoting survival also was assessed by briefly depolarizing neurons with elevated potassium. We found that activation of the voltage-sensitive L channels with 80 mmKCl, which would be expected to drive the membrane potential to approximately −15 mV, for relatively short periods also promotes survival and that the increased survival was inhibited fully by simultaneous exposure to specific L channel blockers (Fig.6G). Neurons, depolarized with 80 mm KCl for 1 or 10 min, survived much better than either those exposed to the basic survival medium, which contains 5 mm KCl (i.e., theNo addition medium), or to the 80 mmKCl-containing medium in the presence of 10 μm nimodipine (nim + 80 mm KCl). After the brief (1 or 10 min) exposure, all neurons were maintained for 1 d in the basic medium; apoptotic versus nonapoptotic cells then were scored as above after propidium iodide labeling. Note that nimodipine, when added, was present for only very brief periods and that, at 1 d, survival after brief exposure to 80 mm KCl plus nimodipine was indistinguishable from that observed in the basic medium. Interestingly, however, neurons continuously depolarized to −15 mV for 1 min survived better than those depolarized for 10 min (Fig. 6G; p < 0.001). Moreover, although accurate cell counts were not possible because many cells had detached, the survival of neurons maintained for a full day in 80 mm KCl appeared to be <5%. The decreased survival in extended depolarization is consistent with the observations with high (−)-Bay K8644 and implies that moderate calcium influx promotes survival but that excessive influx induces cell death. Moreover, together with the data on IGF-1- and L channel-dependent potentiation and survival, our results suggest that rapid Akt-mediated potentiation of L-type calcium channels is an essential component of IGF-1-induced neuronal survival.
DISCUSSION
Our results demonstrate that Akt mediates the rapid IGF-1-induced potentiation of L channel currents in cerebellar granule neurons. Specifically, both the inability of IGF-1 to potentiate L channels in the presence of inactive Akt and the ability of constitutively active Akt to mimic IGF-1-induced increases in L currents strongly implicate Akt as a signaling intermediate. Together with previous studies demonstrating the involvement of PI 3-kinase in both N and L channel modulation (Blair and Marshall, 1997) and demonstrating that Akt acts downstream of PI 3-kinase (Franke et al., 1997), the data suggest that the RTK-dependent signaling pathway mediating rapid L channel modulation proceeds via IGF-1/RTK–PI 3-kinase–phosphoinositide intermediates PIP3 and PI(3,4)P2–Akt. Subsequent steps remain to be shown. However, Akt, a serine/threonine kinase, potentially could activate succeeding components or directly phosphorylate one or more of the channel subunits.
Interestingly, the rapid IGF-1-dependent potentiation of N channel activity was unaffected by the presence of inactive Akt. This suggests that the signal transduction pathways responsible for IGF-1 potentiation of L and N channels diverge after PI 3-kinase. Given the differing voltage dependencies of the L and N channel modulation (Blair and Marshall, 1997) and the differing cellular functions and subcellular localizations of L and N channels (Westenbroek et al., 1992; Spitzer, 1994; Sheng et al., 1996), it could be predicted that the pathways, although sharing initial steps, eventually might diverge. Functionally, the IGF-1 potentiation of N channels, which are expressed preferentially in presynaptic terminals, would be expected to increase neurotransmitter release, whereas potentiating somally localized L channels might regulate a variety of cellular processes, including calcium-dependent transcriptional events, neuronal survival, and differentiation (Murphy et al., 1991; Ghosh et al., 1994; Misra et al., 1994; Rosen et al., 1994; Rosen and Greenberg, 1996) (for review, see Spitzer, 1994; Finkbeiner and Greenberg, 1996).
Neuronal survival is promoted by IGF-1 (D’Mello et al., 1993; Dudek et al., 1997; Tagami et al., 1997). Moreover, accumulating evidence suggests that the neuroprotective mechanism of growth factors may be via a transient or sustained elevation of cytosolic calcium, activating signaling pathways protective against excitotoxic insults (Mattson, 1997). Depolarization also promotes survival via L channel-mediated calcium influx (Gallo et al., 1987; Galli et al., 1995). Our recent work demonstrating that IGF-1 rapidly potentiates L channel activity in cerebellar granule neurons (Blair and Marshall, 1997) suggested a connection between the IGF-1 and depolarization/L channel-mediated survival. Nevertheless, it also is well established that excessive intracellular calcium can induce necrotic cell death (Choi, 1995; Gwag et al., 1997), and it has been reported that the inhibition of L channels failed to block the survival-promoting activity of IGF-1 in granule neurons (Galli et al., 1995).
Our work implies that the IGF-1 potentiation of L channel activity is a small but significant component of IGF-1-mediated survival, accounting for approximately one-third of that seen in low-serum conditions. Our experiments further suggest that the earlier conclusion of noninvolvement may have arisen from the voltage dependence of the potency of dihydropyridines. DHP inhibition of L channel activity is strongly use-dependent, a phenomenon that arises from changes in affinity with membrane potential. At strongly depolarized potentials, DHPs typically have affinities in the low nanomolar range. However, at hyperpolarized potentials, up to 10 μm DHP is required to achieve complete block (for review, see Bean, 1992). Consequently, the efficacy of inhibition should vary with the level of spontaneous action potential activity in the granule neurons, an unknown. In addition, the survival media all contain lower potassium than the growth medium, which has 25 mm KCl; 25 mm KCl would, if the membrane behaved as a pure potassium electrode, depolarize the cells to −46 mV. However, to eliminate depolarization-induced L channel activity, survival media typically contain 5 mmKCl; this would be expected to reduce spontaneous action potential activity by driving the membrane to strongly hyperpolarized potentials (maximum of −89 mV).
We found here that L channel effects on IGF-1-mediated survival could be detected easily with 10 μm DHPs but that 1 μm DHPs, particularly 1 μm nifedipine, were less effective. This is consistent with neurophysiological studies. A 10 μm concentration of nimodipine is commonly used to block fully the L currents in granule neurons (Randall and Tsien, 1995), whereas 10 μm nifedipine mainly (60–85%), but incompletely, blocked L currents evoked by long-duration (≥200 msec) test pulses in sensory neurons and in heterologous systems expressing rat brain α1C (Fox et al., 1987; Tomlinson et al., 1993). Moreover, when the holding potential was −90 mV and L currents were evoked by depolarizing pulses resembling action potentials (+30 mV for 10 msec), 1 μm DHPs blocked only 25% of the L channel activity (Cohen and McCarthy, 1987). Because DHPs are rapidly reversible inhibitors, spontaneous action potential activity would need to be very high to achieve complete or near-complete inhibition. However, it also is known that DHPs at 10 μm are not completely selective for L channels (Bean, 1992). Importantly, our studies determined the effects of exposure to 10 μmnimodipine alone and subtracted the nonspecific effects on survival to obtain our estimate of IGF-1/L channel-dependent survival. Earlier survival studies using 1 μm nifedipine (Galli et al., 1995) may have been unable to detect L channel involvement, because nifedipine may be slightly less effective than nimodipine and/or because the spontaneous action potential activity might have been too low for complete block. Interestingly, IGF-1 potentiation of L channel activity is most prominent at hyperpolarized potentials (Blair and Marshall, 1997), raising the possibility that, after exposure to IGF-1, subthreshold membrane activity might be sufficient to induce calcium influx.
Significantly, our results also suggest that a brief exposure to IGF-1 is neuroprotective for an extended period and that L channels must be functional during the IGF-1 exposure, although not after it. We also found that direct, but moderate, depolarization for brief periods also promoted survival in an L channel-dependent manner. This is consistent with recently published studies demonstrating that a relatively brief depolarizing pulse promotes fibroblast growth factor-dependent survival of ciliary neurons via L channel activity (Schmidt et al., 1996), that a 1 min pulse of NGF is sufficient to activate immediate-early genes and engage specific genetic programs in neural crest-, adrenal chromaffin-derivative PC12 cells (Toledo et al., 1995), and that maintenance of granule neurons in depolarizing conditions does not lead to long-term elevation of intracellular calcium levels (Ono et al., 1997). In contrast, other studies have concluded that influx through L channels increases intracellular calcium toward a set point that promotes survival (Franklin and Johnson, 1992). Additionally, extensive work examining primarily NMDA receptor-mediated calcium influx has demonstrated clearly that excessive intracellular calcium results in cell death (Choi, 1995). Although our data would imply that IGF-1-induced calcium influx acts to trigger calcium-dependent survival pathways, it is not inconsistent with the concept that growth factors and/or depolarization act to stabilize the free cytosolic calcium concentration at optimal levels (Mattson, 1997). The experiments presented here exclusively address the involvement of Akt and L channels, early steps in the IGF-1 survival-promoting pathway. Downstream effect(s) may include or even be directed specifically at calcium homeostatic mechanisms.
Our data would suggest that L channel-mediated calcium influx can be optimized for survival. When we examined the ability of the L channel agonist, (−)-Bay K8644, to promote granule neuron survival in the absence of other factors, we found that the concentration dependence was a steep bell-shaped curve with a narrow range of optimal concentrations. The optimal (−)-Bay K8644 concentrations increased survival to approximately the same extent as the L channel-dependent effects of IGF-1, whereas high concentrations decreased survival below that observed in the basic survival medium alone. This is consistent with the observations that calcium influx can promote neuronal survival and differentiation (Gallo et al., 1987; Spitzer, 1994; Finkbeiner and Greenberg, 1996) and cell death (Choi, 1995). Interestingly, we also found that very brief depolarization (1 min) in high potassium is more effective in promoting survival than a 10 min depolarization. By implying that moderate influx is beneficial, but at higher levels becomes destructive, our data support the “set point” theory that calcium homeostatic mechanisms, including L channel-mediated influx, must be regulated to maintain intracellular calcium levels at or near an optimal set point (Franklin and Johnson, 1992; Mattson, 1997). The increased cell death observed at the highest (−)-Bay K8644 concentrations and with extended, 1 d, depolarization may resemble the extremely toxic conditions that occur with excessive activation of NMDA receptors, glucose deprivation and cerebral ischemia, in which calcium homeostasis is destroyed and neuronal death by both necrosis and apoptosis results (Choi, 1995; Mattson, 1997).
Recently published work demonstrates that IGF-1 induces L channel potentiation via PI 3-kinase and survival by a PI 3-kinase–Akt signaling pathway (Blair and Marshall, 1997; Dudek et al., 1997;Párrizas et al., 1997). The downstream targets of Akt are beginning to be identified and include the apoptosis signaling protein, Bad (Datta et al., 1997). We show here that the portion of IGF-1-dependent survival attributable to L channel activity proceeds through Akt, implying that L channels are another downstream target. Interestingly, studies examining the pathway regulating depolarization-dependent survival have concluded that it was independent of the IGF-1 pathway, because survival persisted in the presence of PI 3-kinase inhibitors (D’Mello et al., 1997; Soler et al., 1998). However, if calcium influx was downstream, depolarization-induced L channel activity would bypass the earlier steps of PI 3-kinase and Akt activation, raising the possibility that depolarization may activate similar or overlapping pathways as IGF-1.
In the CNS, IGF-1 is neuroprotective against excitotoxic, metabolic, and oxidative insults (Mattson, 1997). In animal models, intraventricular injection of IGF-1 after ischemic injury reduced neuronal loss in the cortex, striatum, and hippocampus (Tagami et al., 1997), whereas IGF-1 removal led to apoptotic cell death, as characterized by chromatin condensation, DNA fragmentation, and cytoplasmic vacuolization. Studies using cultured neurons have demonstrated that ischemia, glucose deprivation, and iron toxicity all induce excessive increases in intracellular calcium and that IGF-1 promotes survival by stabilizing calcium levels within an acceptable range (Cheng and Mattson, 1992; Cheng et al., 1993; Mattson et al., 1993; Zhang et al., 1993). Our results also suggest that calcium levels need to be maintained within limits; a failure to promote L channel activity results in low survival, and blocking L channels reduces survival even more. However, excessive increases, induced either by high concentrations of L channel agonists or by direct depolarization for extended periods, also result in cell death. Only intermediate levels of (−)-Bay K8644 or brief periods of strong depolarization promote survival. This suggests that the neuroprotective effect of IGF-1/L channel potentiation probably involves relatively modest increases in calcium influx.
Mechanistically, the data indicate that rapid IGF-1 potentiation of L channels induces a transient increase in intracellular calcium that is sufficient to trigger the calcium-dependent pathway(s) mediating neuroprotection. Although the downstream targets controlled by calcium are mainly unknown, one potential pathway might use calmodulin, which has been implicated in depolarization-dependent survival (Gallo et al., 1987; D’Mello et al., 1997), to induce phosphorylation of cAMP response element binding proteins (CREBs), a family of transcription factors activated by calcium influx and neurotrophin signaling (Tao et al., 1998; Xing et al., 1998). In addition, recent work using hippocampal neurons demonstrates that transient activation of L channels results in rapid (within 1–2 min) translocation of calmodulin to the nucleus, where it promotes CREB phosphorylation (Deisseroth et al., 1998). Activating CREB or other transcription factors then could lead to de novo synthesis of anti-apoptotic proteins, such as antioxidants or calbindin. Significantly, IGF-1 has been found to increase the expression of the anti-apoptotic proteins, Bcl-2 and Bcl-xL, in human neuroblastoma and glioblastoma cells (Singleton et al., 1996; Toms et al., 1998). Alternatively, transient calcium influx might activate calcium-dependent effectors that directly promote the activity of proteins such as Bcl-2 or Bcl-xL or that directly interfere with apoptotic signaling pathways, for instance, blocking caspase self-activation or interfering with the Bax or Bad proteins.
RTK ligands like IGF-1 previously have been considered primarily as mitogens. In postmitotic cells, however, they can act as neuromodulators. Our results imply that RTK-dependent neuromodulation can have dramatic long-term effects and specifically demonstrate that rapid Akt-mediated potentiation of L channels is an essential component of IGF-1-dependent neuronal survival.
Footnotes
This research was supported by National Institutes of Health Grant R29 NS33914-02 and Council for Tobacco Research Scholar Award SA047 to J.M. We thank Drs. Michael Greenberg and Sandeep Robert Datta for helpful advice on survival assays and Drs. Justin Fallon and David Wells for the use of their digital imaging system.
L.A.C.B. and K.K.B.-H. contributed equally to this work.
Correspondence should be addressed to Dr. Leslie Blair at the above address.
REFERENCES
- 1.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 2.Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings MA. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- 3.Bean BP. Whole-cell recording of calcium channel currents. Methods Enzymol. 1992;207:181–193. doi: 10.1016/0076-6879(92)07013-e. [DOI] [PubMed] [Google Scholar]
- 4.Blair LAC, Marshall J. IGF-1 modulates N and L calcium channels in a PI 3-kinase-dependent manner. Neuron. 1997;19:421–429. doi: 10.1016/s0896-6273(00)80950-2. [DOI] [PubMed] [Google Scholar]
- 5.Blair LAC, Bence KK, Marshall J. The jellyfish green fluorescent protein: a tool for studying ion channels and second messenger signalling in neurons. Methods Enzymol. 1999;302:213–225. doi: 10.1016/s0076-6879(99)02021-2. [DOI] [PubMed] [Google Scholar]
- 6.Cheng B, Mattson MP. IGF-I and IGF-II protect cultured hippocampal and septal neurons against calcium-mediated hypoglycemic damage. J Neurosci. 1992;12:1558–1566. doi: 10.1523/JNEUROSCI.12-04-01558.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng B, McMahon DG, Mattson MP. Modulation of calcium current, intracellular calcium levels, and cell survival by glucose deprivation and growth factors in hippocampal neurons. Brain Res. 1993;607:275–285. doi: 10.1016/0006-8993(93)91517-v. [DOI] [PubMed] [Google Scholar]
- 8.Choi DW. Calcium: still center stage in hypoxic-ischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- 9.Cohen CJ, McCarthy RT. Nimodipine block of calcium channels in rat anterior pituitary cells. J Physiol (Lond) 1987;387:195–225. doi: 10.1113/jphysiol.1987.sp016570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohick WS, Clemmons DR. The insulin-like growth factors. Annu Rev Physiol. 1993;55:131–153. doi: 10.1146/annurev.ph.55.030193.001023. [DOI] [PubMed] [Google Scholar]
- 11.Courtney MJ, Akerman KEO, Coffey ET. Neurotrophins protect cultured cerebellar granule neurons against the early phase of cell death by a two-component mechanism. J Neurosci. 1997;17:4201–4211. doi: 10.1523/JNEUROSCI.17-11-04201.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and Akt are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci. 1998;18:2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Datta K, Bellacosa A, Chan TO, Tsichlis PN. Akt is a direct target of the phosphatidylinositol 3-kinase. J Biol Chem. 1996;271:30835–30839. doi: 10.1074/jbc.271.48.30835. [DOI] [PubMed] [Google Scholar]
- 14.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 15.Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CRB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- 16.Delbono O, Renganathan M, Messi ML. Regulation of mouse skeletal muscle L-type Ca2+ channel by activation of the insulin-like growth factor-1 receptor. J Neurosci. 1997;17:6918–6928. doi: 10.1523/JNEUROSCI.17-18-06918.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.D’Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D’Mello SR, Borodezt K, Soltoff SP. Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathways: possible involvement of PI 3-kinase in IGF-1 signaling. J Neurosci. 1997;17:1548–1560. doi: 10.1523/JNEUROSCI.17-05-01548.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 20.Finkbeiner S, Greenberg ME. Ca2+-dependent routes to Ras: mechanisms for neuronal survival, differentiation, and plasticity? Neuron. 1996;16:233–236. doi: 10.1016/s0896-6273(00)80040-9. [DOI] [PubMed] [Google Scholar]
- 21.Fox AP, Nowycky MC, Tsien RW. Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurons. J Physiol (Lond) 1987;394:149–172. doi: 10.1113/jphysiol.1987.sp016864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franke TF, Yang S-I, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 23.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-biphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 24.Franklin JL, Johnson EM., Jr Suppression of programmed neuronal death by sustained elevation of cytoplasmic calcium. Trends Neurosci. 1992;15:501–508. doi: 10.1016/0166-2236(92)90103-f. [DOI] [PubMed] [Google Scholar]
- 25.Galli C, Meucci O, Scorziello A, Werge TM, Calissano P, Schettini G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-1 through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci. 1995;15:1172–1179. doi: 10.1523/JNEUROSCI.15-02-01172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallo V, Kingsbury A, Balazs R, Jorgensen OS. The role of depolarization in the survival and differentiation of cerebellar granule cells in culture. J Neurosci. 1987;7:2203–2213. doi: 10.1523/JNEUROSCI.07-07-02203.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh A, Ginty DD, Bading H, Greenberg ME. Calcium regulation of gene expression in neuronal cells. J Neurobiol. 1994;25:294–303. doi: 10.1002/neu.480250309. [DOI] [PubMed] [Google Scholar]
- 28.Gwag BJ, Koh JY, De Maro JA, Ying HS, Jacquin M, Choi DW. Slowly triggered excitotoxicity occurs by necrosis in cortical cultures. Neuroscience. 1997;77:393–401. doi: 10.1016/s0306-4522(96)00473-3. [DOI] [PubMed] [Google Scholar]
- 29.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 30.Hilborn MD, Vaillancourt RR, Rane SG. Growth factor receptor tyrosine kinases acutely regulate neuronal sodium channels through the src signaling pathway. J Neurosci. 1998;18:590–600. doi: 10.1523/JNEUROSCI.18-02-00590.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hinkle PM, Nelson EJ, Haymes AA. Regulation of L-type voltage-gated calcium channels by epidermal growth factor. Endocrinology. 1993;133:271–276. doi: 10.1210/endo.133.1.7686480. [DOI] [PubMed] [Google Scholar]
- 32.Jonas EA, Knox RJ, Kaczmarek LK, Schwartz JH, Solomon DH. Insulin receptor in Aplysia neurons: characterization, molecular cloning, and modulation of ion currents. J Neurosci. 1996;16:1645–1658. doi: 10.1523/JNEUROSCI.16-05-01645.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kar S, Chabot J-G, Quirion R. Quantitative autoradiographic localization of [125I]insulin-like growth factor I, [125I]insulin-like growth factor II, and [125I]insulin receptor binding sites in developing and adult rat brain. J Comp Neurol. 1993;333:375–397. doi: 10.1002/cne.903330306. [DOI] [PubMed] [Google Scholar]
- 34.Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of a constitutively active Akt ser/thr kinase in 3T3–L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271:31372–31378. doi: 10.1074/jbc.271.49.31372. [DOI] [PubMed] [Google Scholar]
- 35.LeRoith D, Roberts CT, Werner H, Bondy C, Raizada M, Adamo ML. Insulin-like growth factors in the brain. In: Loughlin SE, Fallon JH, editors. Neurotrophic factors. Academic; San Diego: 1993. pp. 391–414. [Google Scholar]
- 36.Lovisolo D, Bonelli G, Baccino FM, Peres A, Alonzo F, Munaron L. Two currents activated by epidermal growth factor in EGFR-T17 fibroblasts. Biochim Biophys Acta. 1992;1104:73–82. doi: 10.1016/0005-2736(92)90133-7. [DOI] [PubMed] [Google Scholar]
- 37.Marchetti C, Usai C. High-affinity block by nimodipine of the internal calcium elevation in chronically depolarized rat cerebellar granule neurons. Neurosci Lett. 1996;207:77–80. doi: 10.1016/0304-3940(96)12492-7. [DOI] [PubMed] [Google Scholar]
- 38.Marshall J, Molloy R, Moss GWJ, Howe JR, Hughes TE. The jellyfish green fluorescent protein: a new tool for studying ion channel expression and function. Neuron. 1995;14:211–215. doi: 10.1016/0896-6273(95)90279-1. [DOI] [PubMed] [Google Scholar]
- 39.Mattson MP. Neuroprotective signal transduction: relevance to stroke. Neurosci Biobehav Rev. 1997;21:193–206. doi: 10.1016/s0149-7634(96)00010-3. [DOI] [PubMed] [Google Scholar]
- 40.Mattson MP, Zhang Y, Bose S. Growth factors prevent mitochondrial dysfunction, loss of calcium homeostasis, and cell injury, but not ATP depletion in hippocampal neurons deprived of glucose. Exp Neurol. 1993;121:1–13. doi: 10.1006/exnr.1993.1066. [DOI] [PubMed] [Google Scholar]
- 41.Messer A. The maintenance and identification of mouse cerebellar granule cells in monolayer culture. Brain Res. 1972;130:1–12. doi: 10.1016/0006-8993(77)90838-1. [DOI] [PubMed] [Google Scholar]
- 42.Miller SA, Dykes DD, Polesky HJ. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215–1217. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller TM, Tansey MG, Johnson EM, Jr, Creedon DJ. Inhibition of phosphatidylinositol 3-kinase activity blocks depolarization- and insulin-like growth factor-1-mediated survival of cerebellar granule cells. J Biol Chem. 1997;272:9847–9853. doi: 10.1074/jbc.272.15.9847. [DOI] [PubMed] [Google Scholar]
- 44.Misra RP, Bonni A, Miranti CK, Rivera VM, Sheng M, Greenberg ME. L-type voltage-sensitive calcium channel activation stimulates gene expression by a serum response factor-dependent pathway. J Biol Chem. 1994;269:25483–25493. [PubMed] [Google Scholar]
- 45.Murphy TH, Worley PF, Baraban JM. L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron. 1991;7:625–635. doi: 10.1016/0896-6273(91)90375-a. [DOI] [PubMed] [Google Scholar]
- 46.Naumov AP, Kuryshev YA, Mozhayeva GN. Multiple conductance levels of calcium-permeable channels activated by epidermal growth factor in A431 carcinoma cells. Biochim Biophys Acta. 1993;1145:273–278. doi: 10.1016/0005-2736(93)90299-f. [DOI] [PubMed] [Google Scholar]
- 47.Ono T, Kudo Y, Kohara K, Kawashima S, Ogura A. Activity-dependent survival of rat cerebellar granule neurons is not associated with sustained elevation of intracellular C2+. Neurosci Lett. 1997;228:123–126. doi: 10.1016/s0304-3940(97)00383-2. [DOI] [PubMed] [Google Scholar]
- 48.Párrizas M, Saltiel AR, LeRoith D. Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3′-kinase and mitogen-activated protein kinase pathway. J Biol Chem. 1997;272:154–161. doi: 10.1074/jbc.272.1.154. [DOI] [PubMed] [Google Scholar]
- 49.Peppelenbosch MP, Tertoolen LGJ, deLaat SW. Epidermal growth factor-activated calcium and potassium channels. J Biol Chem. 1991;266:19938–19944. [PubMed] [Google Scholar]
- 50.Peppelenbosch MP, Tertoolen LGJ, den Hertog J, deLaat SW. Epidermal growth factor activates calcium channels by phospholipase A2/5-lipoxygenase-mediated leukotriene C4 production. Cell. 1992;69:295–303. doi: 10.1016/0092-8674(92)90410-e. [DOI] [PubMed] [Google Scholar]
- 51.Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- 52.Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosen LB, Greenberg ME. Stimulation of growth factor receptor signal transduction by activation of voltage-sensitive calcium channels. Proc Natl Acad Sci USA. 1996;93:1113–1118. doi: 10.1073/pnas.93.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosen LB, Ginty DD, Weber MJ, Greenberg ME. Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron. 1994;12:1207–1221. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt MF, Atkinson PB, Kater SB. Transient elevations in intracellular calcium are sufficient to induce sustained responsiveness to the neurotrophic factor bFGF. J Neurobiol. 1996;31:333–344. doi: 10.1002/(SICI)1097-4695(199611)31:3<333::AID-NEU6>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 56.Selinfreund RH, Blair LAC. Insulin-like growth factor-1 induces a rapid increase in calcium currents and spontaneous membrane activity in clonal pituitary cells. Mol Pharmacol. 1994;45:1215–1220. [PubMed] [Google Scholar]
- 57.Sheng ZH, Rettig J, Cook T, Catterall WA. Calcium-dependent interaction of N-type calcium channels with the synaptic core complex. Nature. 1996;379:451–454. doi: 10.1038/379451a0. [DOI] [PubMed] [Google Scholar]
- 58.Singleton JR, Dixit VM, Feldman EL. Type 1 insulin-like growth factor receptor activation regulates apoptotic proteins. J Biol Chem. 1996;271:31791–31794. doi: 10.1074/jbc.271.50.31791. [DOI] [PubMed] [Google Scholar]
- 59.Soler RM, Egea J, Mintenig GM, Sanz-Rodriguez C, Iglesias M, Comella JX. Calmodulin is involved in membrane depolarization-mediated survival of motoneurons by phosphatidylinositol 3-kinase- and MAPK-independent pathways. J Neurosci. 1998;18:1230–1239. doi: 10.1523/JNEUROSCI.18-04-01230.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spitzer NC. Spontaneous Ca2+ spikes and waves in embryonic neurons: signaling systems for differentiation. Trends Neurosci. 1994;17:115–118. doi: 10.1016/0166-2236(94)90120-1. [DOI] [PubMed] [Google Scholar]
- 61.Tagami M, Ikeda K, Nara Y, Fujino H, Kubota A, Numano F, Yamori Y. Insulin-like growth factor-1 attenuates apoptosis in hippocampal neurons caused by cerebral ischemia and reperfusion in stroke-prone spontaneously hypertensive rats. Lab Invest. 1997;76:613–617. [PubMed] [Google Scholar]
- 62.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 63.Toledo-Aral JJ, Brehm P, Halegoua S, Mandel G. A single pulse of nerve growth factor triggers long-term neuronal excitability through sodium channel gene induction. Neuron. 1995;14:607–611. doi: 10.1016/0896-6273(95)90317-8. [DOI] [PubMed] [Google Scholar]
- 64.Tomlinson WJ, Stea A, Bourinet E, Charnet P, Nargeot J, Snutch TP. Functional properties of a neuronal class C L-type calcium channel. Neuropharmacology. 1993;32:1117–1126. doi: 10.1016/0028-3908(93)90006-o. [DOI] [PubMed] [Google Scholar]
- 65.Toms SA, Hercbergs A, Lui J, Kondo S, Haqqi T, Casey G, Iwasaki K, Barnett GH, Barna BP. Antagonist effect of insulin-like growth factor-1 on protein kinase inhibitor-mediated apoptosis in human glioblastoma cells in association with Bcl-2 and Bcl-xL. J Neurosurg. 1998;88:884–889. doi: 10.3171/jns.1998.88.5.0884. [DOI] [PubMed] [Google Scholar]
- 66.Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of an N-type calcium channel α1 subunit. Neuron. 1992;9:1099–1115. doi: 10.1016/0896-6273(92)90069-p. [DOI] [PubMed] [Google Scholar]
- 67.Xing J, Kornhauser JM, Xia Z, Thiele EA, Greenberg ME. Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol Cell Biol. 1998;18:1946–1955. doi: 10.1128/mcb.18.4.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y, Tatsuno T, Carney JM, Mattson MP. Basic FGF, NGF, and IGFs protect hippocampal and cortical neurons against iron-induced degeneration. J Cereb Blood Flow Metab. 1993;13:378–388. doi: 10.1038/jcbfm.1993.51. [DOI] [PubMed] [Google Scholar]