

Abstract
Although the metabolic half-life of muscle endplate acetylcholine receptor (AChR) changes during development and after denervation in the adult, little is known about the molecular mechanisms that influence receptor stability. We have investigated the effect on AChR turnover of its interaction with rapsyn, a 43 kDa peripheral membrane protein that is closely associated with the AChR in muscle cells and is required for its clustering at endplates. Both in transfected COS cells and in cultured myotubes from rapsyn-negative and rapsyn-positive mice, we have found that the presence of rapsyn slows the turnover of AChRs by as much as twofold. The effect was similar for both embryonic (α2βδγ) and adult (α2βδε) AChRs and for AChRs whose β subunit lacked a putative tyrosine phosphorylation site. Neither colchicine nor cytochalasin D altered AChR turnover or prevented the rapsyn effect. Mutant rapsyn proteins whose N-terminal myristoylation signal was eliminated, or whose C terminus or zinc-finger domains were deleted, failed to change the rate of receptor turnover. Each of these mutations affects the association of the AChR with rapsyn, suggesting that AChR stability is altered by interaction between the two proteins. Our results suggest that, in addition to its role in AChR clustering, rapsyn also functions to metabolically stabilize the AChR.
Keywords: nicotinic receptors, rapsyn, 43 kDa protein, receptor turnover, acetylcholine, neuromuscular junction, endplate, myotubes
In developing and adult muscle, acetylcholine receptors (AChRs) at the neuromuscular junction undergo dramatic changes in their metabolic stability that correspond to the innervation state of muscle fibers. Thus, in uninnervated myotubes, at newly formed synapses, and in denervated muscle fibers, AChRs turn over at a relative rapid rate (t1/2 ≈ 1 d), whereas at mature adult synapses the AChRs turn over much more slowly (t1/2 ≈ 10 d) (Berg and Hall, 1974; Chang and Huang, 1975; Fambrough, 1979; Steinbach et al., 1979; Salpeter, 1987). Recent investigation of AChRs at denervated endplates has revealed other populations of AChRs with intermediate turnover times (Andreose et al., 1993; O’Malley et al., 1993). Although the physiological modulation of AChR stability has been long-known, relatively little is understood about the molecular interactions that influence AChR stability. Interactions of receptor subunits with cytoskeletal components have been postulated to be important (Fambrough, 1979; Steinbach et al., 1979; Xu and Salpeter, 1995, 1997), but little evidence is available about the significance of interactions with specific proteins.
One protein that is known to interact with the AChR is rapsyn, a peripheral membrane protein found at synapses in Torpedo electric organs and vertebrate muscle cells in 1∶1 stoichiometry with the AChR (for review, see Froehner, 1991). Rapsyn, which associates with the inner face of the membrane through an amino-terminal myristoyl group (Phillips et al., 1991b), has recently been shown to play a key role in AChR clustering. Thus, rapsyn can induce AChR clustering in heterologous cells (Froehner et al., 1990; Phillips et al., 1991a), and rapsyn-deficient mice fail to form AChR clusters at synapses in embryonic myotubes (Gautam et al., 1995). At developing neuromuscular junctions, rapsyn appears concurrently with AChR clusters at the very beginning of synaptogenesis (Burden, 1985; Noakes et al., 1993). Chemical cross-linking studies and structural analysis using electron microscopy and x-ray diffraction suggest that rapsyn may interact directly with the cytoplasmic domains of the AChR (Sealock et al., 1984; Mitra et al., 1989), either through a specific interaction with the β subunit (Burden et al., 1983) or through interactions with each of the subunits (Maimone and Merlie, 1993; Yu and Hall, 1994). Rapsyn is also thought to interact with other postsynaptic proteins such as those in the utrophin-associated complex of proteins at the neuromuscular junction (Phillips et al., 1993; Apel and Merlie, 1995;Apel et al., 1995). Because of its multiple interactions, rapsyn is postulated to play an adaptor role, mediating the interaction of the AChR with cytoskeletal elements at the synapse (Walker et al., 1984;Froehner, 1991; Apel and Merlie, 1995; Glass and Yancopoulos, 1997).
Because of its close association with the AChR, we have investigated whether rapsyn influences the metabolic stability of the receptor. We find, both in heterologous cells and in muscle cells, that association with rapsyn slows the degradation of the AChR and thus increases its metabolic stability.
A preliminary account of these results has been published previously (Wang and Hall, 1996).
MATERIALS AND METHODS
cDNAs and expression vectors. Full-length cDNA clones coding for the α (Isenberg et al., 1986), β (Buonanno et al., 1986), γ (Yu et al., 1986), and δ subunits (Lapolla et al., 1984) of mouse muscle nicotinic AChR were gifts from Drs. J. P. Merlie (Washington University, St. Louis, MO) and N. Davidson (California Institute of Technology, Pasadena, CA). The full-length cDNA for the mouse ε subunit was obtained from Dr. P. D. Gardner [University of Texas Health Science Center, San Antonio, TX; see Gardner (1990)]. A cDNA construct encoding the extracellular ligand binding domain of the α7 neuronal AChR and the transmembrane domains and cytoplasmic loop of the 5-HT3 serotonergic receptor (α7-5HT3 V201 chimera) was a gift from Dr. J. W. Patrick [Baylor College of Medicine, Houston, TX; see Dineley and Patrick (1998); Eisele et al., 1993]. A full-length cDNA clone encoding mouse rapsyn was kindly provided by Dr. S. Froehner [University of North Carolina, Chapel Hill, NC; see Froehner (1989)]. Each of the cDNAs was subcloned into an SV40-based expression vector,pSM (Brodsky et al., 1990). Anti-rapsyn antibodies mAb 1579A and mAb1234A were generous gifts from Dr. S. Froehner (Scotland et al., 1993).
Transfection of cells. COS cells grown in DMEM-H21 supplemented with 10% fetal bovine serum were transfected using an adenovirus-aided, DEAE-dextran transfection procedure (Forsayeth and Garcia, 1994, Wang et al., 1996a,b). Cells at ∼70% confluence in a 60 mm dish were incubated for 2 hr at 37°C with a transfection mixture containing plasmid DNAs in DMEM-H21 (2.5 ml), an E1 defective adenovirus lysate (0.5 ml), and DEAE-dextran (120 μg). The amounts of plasmid DNA used in the transfection mixture for α, β, γ or ε, and δ subunits were (in μg): 1.32, 0.66, 1.0, and 0.26, respectively. They were empirically determined to produce maximal cell surface AChR expression. For coexpression of rapsyn in the cells, 2 μg of plasmid DNA encoding rapsyn was added per 60 mm dish, except where indicated otherwise. After 2 hr of incubation, the transfection mixture was removed, and the cells were rinsed with PBS twice before being returned to the incubator in 4 ml of growth medium. After 24 hr at 37°C, the cells were trypsinized and plated into three wells of 24-well plates. Surface AChR binding was measured after an additional 24 hr in culture.
Culture of rapsyn-deficient mouse myotubes. The rapsyn−/− muscle cell line was derived using rapsyn−/− mutants bearing an immortalizing transgene consisting of a temperature-sensitive SV40 large T antigen (tsA58) under the control of a γ-interferon (γ-IFN)-inducible mouseH-2kb Class I MHC promoter (Jat et al., 1991). Mice bearing the immortalizing transgene were obtained from matings between rapsyn+/− mice and rapsyn heterozygotes that were also positive for the SV40 T antigen transgene (M. Gautam, unpublished data). Cells were placed on gelatin-coated plastic dishes and maintained at 33°C in DMEM containing 20% fetal bovine serum, 3% chick embryo extract, and 4 U/ml mouse γ-IFN. After 2–3 d, cells were switched to 37°C in DMEM with 2% horse serum and no γ-IFN to promote myotube formation. Surface 125I-α-bungarotoxin binding and receptor turnover assays were performed 24–48 h after the myotubes were switched to the fusion medium.
Surface AChR assay and receptor turnover. Surface AChR expression was determined by incubating cells for 90 min at 37°C in fresh growth medium containing 4 nm125I-α-bungarotoxin (α-BuTx, 220 Ci/mmol; Amersham, Arlington Heights, IL). Nonspecific binding was measured by including excess unlabeled α-BuTx (1 μm) in the assay medium or by assaying sham-transfected cells. At the end of the incubation period, unbound 125I-α-BuTx was removed by washing the cells three times with PBS. Cells were solubilized in 0.1N NaOH, and radioactivity was counted with a Wallac gamma counter.
The degradation rate of surface AChR was measured by labeling the receptor with 125I-α-BuTx for 90 min. Unbound125I-α-BuTx was removed by washing the cells three times with PBS. The cells in growth medium were then returned to the 37°C CO2 incubator. At various intervals thereafter, duplicate aliquots of culture medium were collected and replaced with fresh medium. At the end of the experiments, cells were solubilized in 0.1N NaOH. Radioactivity in both the aliquots and cells was measured with a Wallac gamma counter. The total radioactivity on the cell surface at the beginning of the experiment was calculated by addition of that found in the medium and solubilized cells. The data were used to calculate the turnover rates, and degradation curves were fitted by linear regression and normalized to 100% at time 0 after surface labeling.
Mutagenesis of rapsyn. A full-length mouse cDNA encoding wild-type rapsyn was cloned into the mammalian expression vectorpSM under the control of an SV40 early promoter. This clone was used to create rapsyn mutants by site-directed mutagenesis as described (Phillips et al., 1993). To make a myristoylation-defective rapsyn (RapsynA2), we mutated the Gly2 residue in the wild-type rapsyn to Ala2 and left the remainder of the polypeptide sequence unchanged. RapsynΔ16–254, a leucine zipper deletion mutant, was created by deleting the cDNA sequences encoding amino acids 16–254 with an in-frame fusion of Gln15 to Ile255. The C-terminal truncation mutant rapsynΔ255–412 was made by replacing amino acid 255 with a stop codon to cause early termination of the polypeptide at amino acid 254. To disrupt the two tandem zinc finger domains, we constructed the mutant rapsynH→Q by changing amino acids 384 and 387 from histidine to glutamine. To eliminate the putative serine phosphorylation site in the C terminus of rapsyn, we made mutant rapsynS→A by replacing serine 405 and 406 with alanine (Scotland et al., 1993).
Gel electrophoresis and immunoblotting. Transfected COS cells were extracted with a Triton X-100-containing buffer as described (Wang et al., 1996a). Cell lysates were denatured by boiling for 5 min in SDS gel-loading buffer containing β-mecaptoethanol, electrophoresed on a 12.5% SDS-polyacrylamide gel (Laemmli, 1970), and then transferred to nitrocellulose membranes. After preincubation in PBS/0.3% Tween 20 containing 3% bovine serum albumin to block nonspecific binding, the membranes were incubated with primary antibodies against rapsyn in the same buffer and washed with PBS/Tween 20. Antibodies bound to the membrane were detected with horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescent reagents (ECL, Amersham).
Immunofluorescent staining. Transfected COS cells grown on chamber slides were incubated with rhodamine-conjugated α-BuTx for 90 min, fixed for 10 min in 2% paraformaldehyde, and washed in cold PBS. After a short incubation in PBS containing 10% bovine serum and 0.05% saponin at 4°C for 20 min to permeabilize the membrane, cells were incubated at 4°C for 1 h with a monoclonal rat antibody to the α subunit of the mouse AChR. After a brief rinse in cold PBS, cells were incubated with an FITC-conjugated goat anti-rat secondary antibody. The slides were examined under a fluorescence microscope equipped with the appropriate filters.
RESULTS
Rapsyn slows turnover of AChRs in transfected cells
We first examined the effect of rapsyn on the metabolic half-life of the mouse muscle nicotinic AChR expressed on the surface of transfected COS cells. COS cells were transfected with cDNAs for all four subunits of either the embryonic (α2βδγ) or adult (α2βδε) forms of the AChR, either with or without the inclusion of rapsyn cDNA. Forty-eight hours after transfection, the surface AChRs were labeled with125I-α-bungarotoxin, and AChR degradation was followed by the subsequent appearance of radioactivity in the medium, as described by Gu et al. (1990). In both cases, the inclusion of rapsyn cDNA significantly increased the half-life of AChRs on the cell surface. For adult AChRs, the half-life of the receptor in the absence of rapsyn was 11 h; in the presence of 1 μg of rapsyn cDNA per 60 mm dish, the half-life was increased to 17 h. The half-life of the embryonic form of the AChR was similarly increased by coexpression of rapsyn (Fig. 1A). We observed no significant difference between the turnover times of the adult and embryonic forms of the receptor, either with or without rapsyn (see Discussion).
Fig. 1.
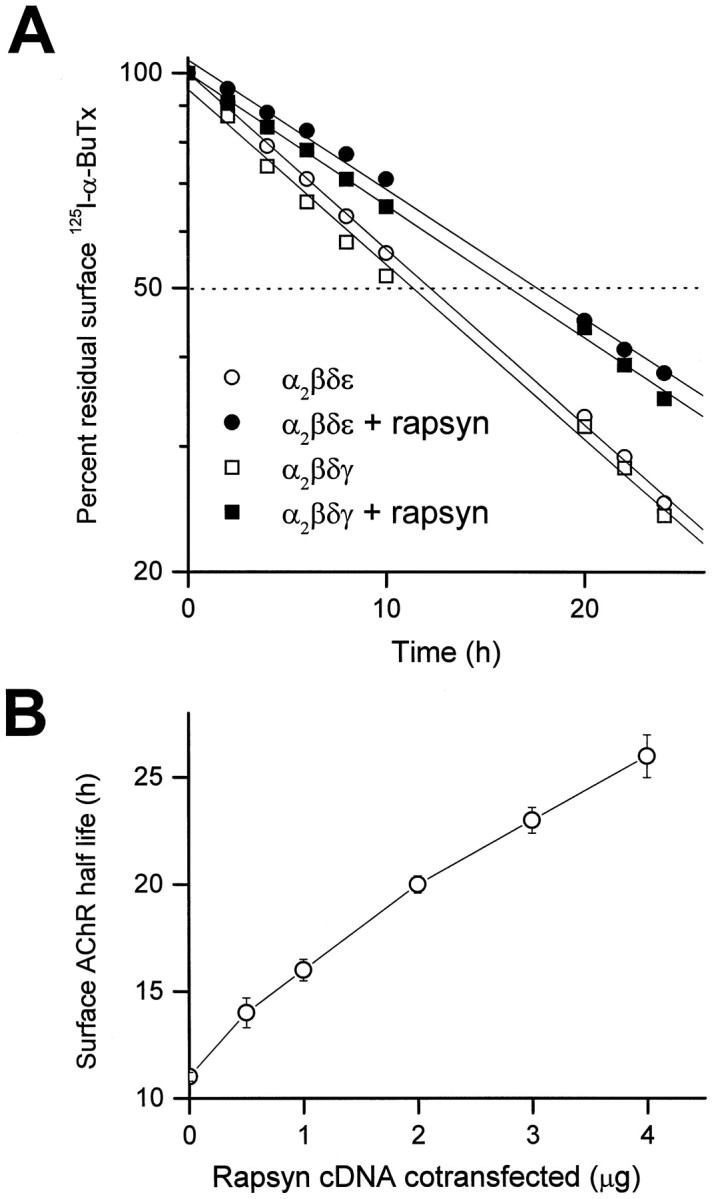
Metabolic stabilization of surface AChR by coexpression of rapsyn in COS cells. A, Cotransfection of mouse rapsyn cDNA (1 μg/60 mm dish) significantly reduced the degradation rate of surface AChRs labeled by125I-α-bungarotoxin. Transfected COS cells in culture were labeled with 4 nm125I-α-BuTx for 90 min, washed, and incubated in growth medium. The radioactivity released into the media was measured at the times indicated, and the data were used to calculate the turnover rates. Degradation curves were fitted by linear regression and normalized to 100% at time 0 after surface labeling. Metabolic stabilization of surface AChR by rapsyn was seen in cells transfected with subunit cDNAs encoding either embryonic-type (α2βδγ) or adult-type (α2βδε) AChRs. B, The effect on AChR (α2βδε) turnover was more prominent with increased amounts of rapsyn cDNA cotransfected per 60 mm dish. Each data point represents the mean ± SEM of four separate experiments.
The increase in half-life of the AChR depended monotonically on the amount of rapsyn cDNA that was added, up to a value of 28 h, seen with 4 μg of rapsyn cDNA per 60 mm dish, the maximum amount that could be used without reducing surface expression of the AChR (Fig.1B). This continued increase suggests that with larger amounts of rapsyn cDNA, either a larger fraction of the AChR was present in a form that turned over more slowly or that all molecules turned over at progressively slower rates with increased expression of rapsyn. Our data do not distinguish between these two possibilities.
The effect of rapsyn is specific
Other proteins were coexpressed with the adult form of the AChR to determine whether they would have the same effect as rapsyn. The proteins chosen were CD8, an integral membrane protein normally found on the surface of lymphocytes (Littman, 1987), and two peripheral membrane proteins that are present at the neuromuscular junction, syntrophin and α-dystroglycan. Syntrophin is a 58 kDa protein that, like rapsyn, is associated with the cytoplasmic side of the membrane (Adams et al., 1993; Froehner et al., 1997); α-dystroglycan is a heavily glycosylated protein that is attached to the extracellular matrix on the outside of the membrane (Ibraghimov-Beskrovnaya et al., 1992). Both α-dystroglycan and syntrophin are part of the dystrophin-associated complex of proteins that link the cytoskeleton and the extracellular matrix; both have also been implicated in the molecular mechanisms underlying the formation and maintenance of AChR clusters (Fallon and Hall, 1994; Apel and Merlie, 1995; Froehner et al., 1997). When coexpressed with the subunits of the adult form of the AChR, none of the three proteins altered the AChR turnover time (Fig.2).
Fig. 2.
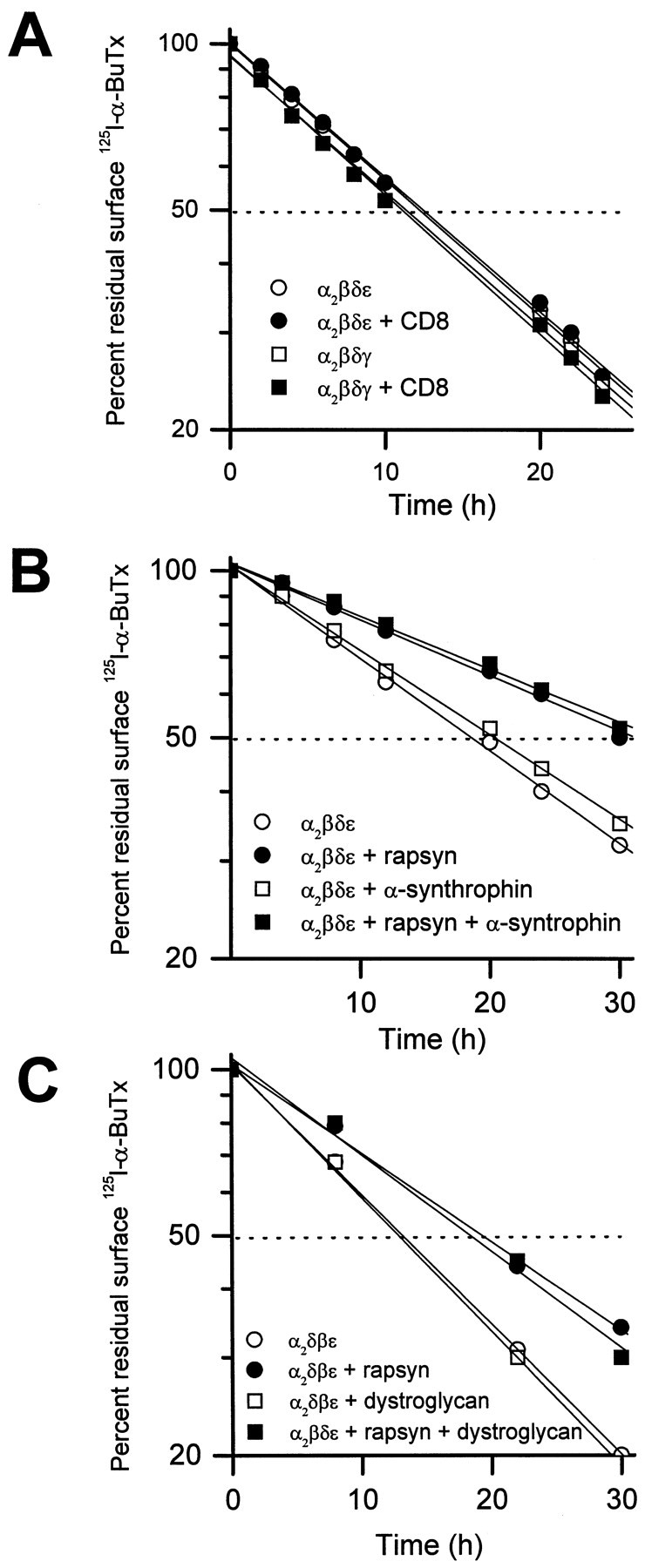
The effect of rapsyn on metabolic stability of the AChR is specific because other membrane proteins fail to change the rate of receptor degradation. When coexpressed in COS cells, lymphocyte surface molecule CD8 was incapable of stabilizing the AChR (A). Coexpression of postsynaptic muscle proteins, α1-syntrophin (B), and dystroglycan (C) neither affected the turnover rate of surface AChR (α2βδε) nor changed the stabilizing effect of rapsyn on the receptor. For each 60 mm culture dish, 4 μg of each cDNA encoding CD8, α-syntrophin, and dystroglycan were used for transfection, respectively. The amount of rapsyn cDNA cotransfected was 4 μg in B and 2 μg in C. Each data point represents the mean of three determinations.
To determine whether the metabolic stabilization induced by rapsyn is specific for muscle-type AChRs, we have sought to examine the effect of rapsyn on turnover of neuronal α7 AChR. When α7 subunit is expressed in COS cells, the surface receptor level is below the detection limit of the 125I-α-BuTx binding assay, although toxin binding sites and pentameric receptor can be found in whole cell lysates (Cooper and Millar, 1997; Dineley and Patrick, 1998). To circumvent this problem, we have used a previously characterized chimeric construct α7-5HT3 (V201). This construct encodes the extracellular ligand binding domain of the α7 nicotinic receptor fused at valine 201 to the transmembrane domains and the cytoplasmic loop of the 5HT3 receptor (Eisele et al., 1993). When expressed in COS cells, the chimeric receptor is transported to the cell surface, where it can be detected by125I-α-BuTx binding. In addition, the chimeric protein migrates as a pentamer in a sucrose gradient (Dineley and Patrick, 1998). Using the degradation of bound, radioactive toxin as an assay, we tested whether coexpressed rapsyn would alter the turnover time of this protein. We found that it did not. Both when expressed with or without rapsyn, the α7-5HT3 (V201) receptor was degraded with a half-life of ∼8 h (Fig.3A). These results suggest that the stabilization effect of rapsyn does not extend to other membrane-associated receptor proteins. Immunocytochemical experiments showed that coexpression with rapsyn also did not alter the surface distribution of the α7-5HT3 protein, suggesting that the two are not associated (data not shown).
Fig. 3.
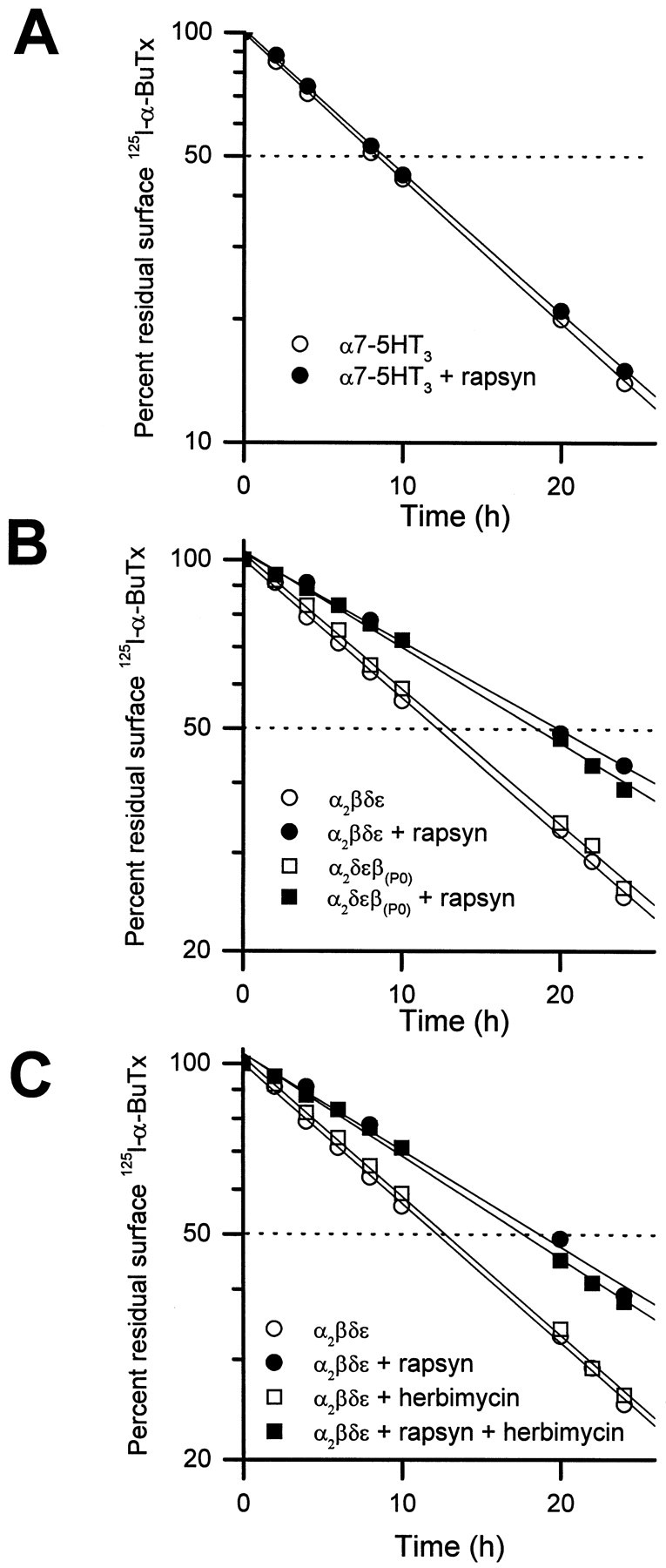
The effect of rapsyn is specific for the muscle-type AChRs and is independent of tyrosine phosphorylation of AChR subunits. A, Degradation of an α7-5HT3chimeric receptor on COS cell surface was unaffected by coexpression of rapsyn (2 μg cDNA/60 mm dish). The α7-5HT3 chimera was made by fusing the NH2-terminal extracellular domain of the neuronal nicotinic α7 subunit at valine 201 to the complementary C-terminal domain of the serotonergic 5-HT3 receptor subunit (see Results; also see Eisele et al., 1993; Dineley and Patrick, 1998). B, Rapsyn was effective in slowing the degradation of surface AChRs whose β subunit lacked the putative tyrosine phosphorylation site in the cytoplasmic loop [β(p0)]. The seven amino acids GTDEYFI containing the site of tyrosine phosphorylation on the β cytoplasmic loop between the third and fourth transmembrane domains were mutated to those of the α subunit PSRDKQE (containing no tyosine residue) (Yu and Hall, 1994).C, Herbimycin A, a potent inhibitor of tyrosine kinases, did not alter the decreased turnover rate of surface AChRs induced by rapsyn. The drug (1 μm) was added to the culture medium 6 h before 125I-α-BuTx labeling and was continuously present in the labeling and growth media thereafter. Control cells were treated similarly with the solvent DMSO without the drug. Each data point represents the averaged results of three determinations.
Tyrosine phosphorylation of AChR β subunit is not required for the effect of rapsyn
Rapsyn can be cross-linked in situ to the β subunit of the AChR (Burden et al., 1983), suggesting that the two polypeptides are in close association, presumably through interactions of rapsyn with the long cytoplasmic domain connecting transmembrane regions M3 and M4 of the β subunit. The loop contains a putative site of tyrosine phosphorylation (Huganir and Miles, 1989; Wagner et al., 1991). When this site was eliminated by replacing the amino acids GTDEYFI with the corresponding amino acids from the α subunit, which has no site of tyrosine phosphorylation [PSRDKQE;Yu and Hall (1994)], rapsyn-induced stabilization of surface AChR metabolism was unaffected (Fig. 3B). Moreover, treatment of transfected COS cells with a potent tyrosine kinase inhibitor, herbimycin A (1 μm), did not alter the effect of rapsyn on surface AChR turnover (Fig. 3C). Because herbimycin A has been shown to effectively block the clustering of muscle AChR and prevent the phosphorylation of the β subunit (Ferns et al., 1996), we conclude that neither AChR clustering nor tyrosine phosphorylation of the receptor is essential for metabolic stabilization of AChRs by rapsyn in COS cells.
Metabolic stabilization is not inhibited by agents that disrupt the cytoskeleton
The cytoskeleton of muscle cells may play an important role in maintaining rapsyn-induced AChR clusters by serving as an anchor for the rapsyn-receptor complex (for review, see Froehner, 1991; Apel and Merlie, 1995). It remains unclear whether rapsyn binds to the cytoskeleton directly or through links provided by other postsynaptic proteins such as utrophin or the dystrophin–glycoprotein complex (Phillips et al., 1993; Apel et al., 1995). Previous studies, however, have shown that receptor clusters are stabilized by actin-containing filaments and that the movement of receptors in the plane of the membrane requires microtubules (Bloch, 1983; Connolly, 1984;Walker et al., 1984). To examine whether rapsyn-induced metabolic stabilization of the AChR in COS cells involves interaction with the cytoskeleton, we tested the effects of microfilament- and microtubule-disrupting drugs on receptor turnover. Neither cytochalasin D (2 μg/ml), which disrupts actin filaments, nor colchicine (10 μm), which depolymerizes microtubules, affected the turnover rate of the AChR. In addition, neither of these drugs interfered with the metabolic stabilization effect of rapsyn on surface receptors (Fig. 4).
Fig. 4.
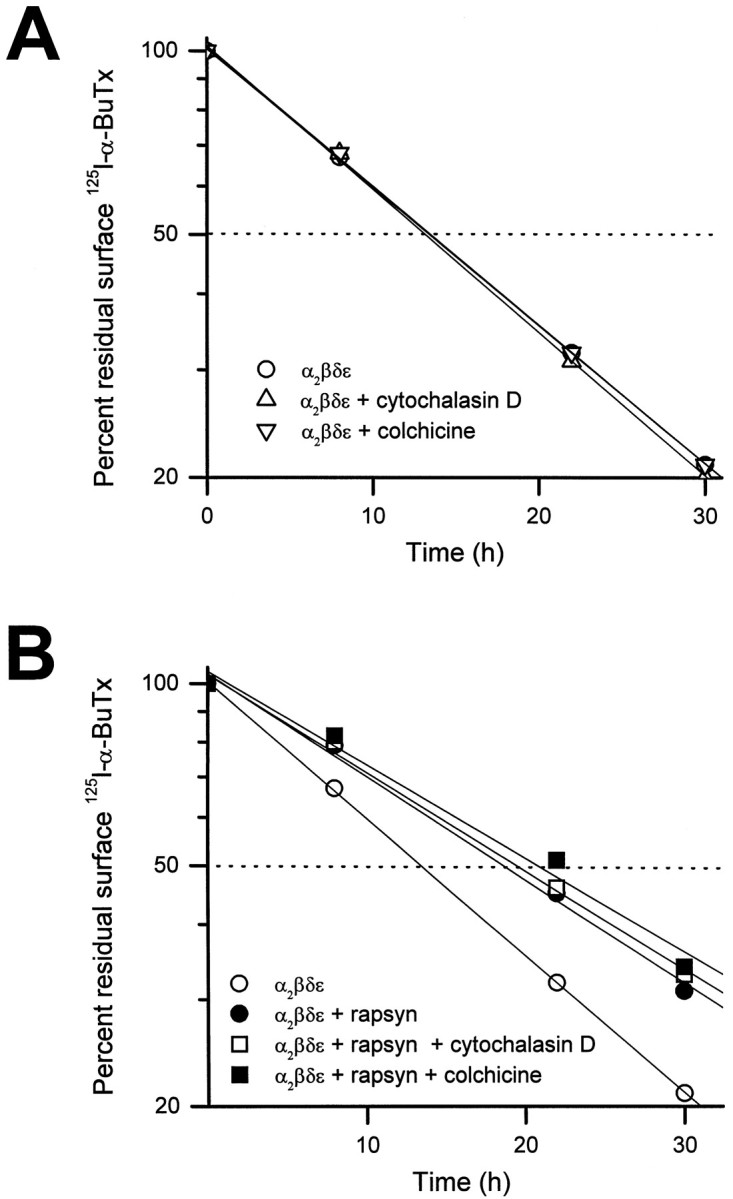
Cytochalasin D (2 μg/ml), which disrupts actin filaments, and colchicine (10 μm), which depolymerizes microtubules, failed to change the degradation rate of surface AChR (A) and the stabilizing effect of rapsyn on the receptor (B). The drugs were applied to the culture medium immediately after the cells were labeled with125I-α-BuTx for determination of surface AChR degradation rate. Two micrograms of rapsyn cDNA were used for cotransfection experiments in B. Each data point represents the averaged results of three measurements.
The rapsyn domains involved in AChR clustering are also required for metabolic stabilization of the receptor
Three domains of rapsyn have been identified as being essential for its ability to stimulate AChR clustering (Phillips et al., 1991b;Scotland et al., 1993). At its N terminus, rapsyn is normally modified by the addition of an N-linked myristate moiety to glycine2 (Fig.5A). When the signal for myristoylation is removed by mutation of the glycine to alanine, the mutated protein associates poorly with the membrane and is inactive in clustering the AChR. The carboxyl region of rapsyn contains a zinc-finger domain that has also been shown to be necessary for clustering, and the central portion of the protein contains a leucine zipper motif, which is necessary for AChR interaction (Fig.5A,B). We examined mutations in each of these regions for their effect on the metabolic stability of the AChR.
Fig. 5.

Schematic diagram of wild-type and mutant forms of rapsyn protein. A, G represents glycine residue, the site for myristoylation. The zig-zag line before Gindicates the myristate moiety. A indicates the substitution of alanine to prevent myristoylation. The potential leucine zipper (striated bar), zinc fingers (black bar), and serine phosphorylation site (S) were also illustrated. B, The two histidine residues at positions 384 and 387 were mutated to glutamine (Q) to disrupt the tandem zinc-finger domains of rapsyn. Serines 405 and 406 were substituted by alanines to mutate the putative phosphorylation site in the C terminus of rapsyn.
When oligonucleotide-directed mutagenesis was used to change the N-terminal Gly2 of rapsyn to Ala2, AChR clustering was drastically reduced as reported (Fig. 6C) (Phillips et al., 1991b), and the mutated rapsyn no longer affected the turnover rate of the AChR (Fig. 6, bottom left). Mutant rapsynΔ255–412, which lacked 158 residues on the C terminus of the protein, impaired the clustering activity of rapsyn (Fig. 6E). Likewise, mutation of the two histidine residues at positions 384 and 387, which disrupts the tandem zinc-finger domains in the C terminus of rapsyn, efficiently prevented metabolic stabilization of the AChR and disrupted clustering (Fig.6F). Thus the myristoylation signal, the C-terminal domain, and the zinc-finger motif are all required for the effect of rapsyn on AChR turnover. In contrast, mutant rapsynΔ16–254, which lacked the leucine zipper domain, still retained some receptor clustering function as shown by immunofluorescent staining of the transfected COS cells (Fig.6D) and had a partial effect on AChR turnover (Fig.6, bottom left). Thus, the leucine zipper motif is not an absolute requirement for metabolic stabilization by rapsyn.
Fig. 6.

Mutagenesis of mouse rapsyn protein delineates domains involved in metabolic stabilization of surface AChRs.A–G, Cells were transfected with cDNAs encoding α, β, δ, and ε subunits of the AChR alone (A), or in combination with each of the cDNAs for wild-type rapsyn (B), mutant rapsynA2(D), rapsynΔ16–254(D), rapsynΔ255–412(E), rapsynH→Q(F), or rapsynS→A(G). Two days after transfection, cells were fixed and stained with a monoclonal rat antibody to the α subunit of the AChR (mAb210), followed by FITC-conjugated secondary antibody.Bottom, Effects of rapsyn mutants on the degradation rate of surface AChRs in COS cells. The cells were transfected with cDNAs encoding the adult-type AChR (α2βδε subunits) alone or in combination with each of the mutant rapsyn cDNAs as indicated (4 μg/60 mm dish). Each data point represents the averaged results of four determinations.
The C-terminal region of rapsyn contains a putative serine phosphorylation site by PKA or PKC (Froehner, 1991; Scotland et al., 1993). To examine its role in receptor clustering and stabilization, we mutated the site by changing serines 405 and 406 to alanines (Fig.5B). When coexpressed with the AChR in COS cells, this mutant protein induced clustering and also slowed the degradation of surface AChR (Fig. 6G and bottom right). We conclude that the serine phosphorylation sites are not essential for metabolic stabilization of the AChR by rapsyn.
Surface AChRs in myotubes of rapsyn-deficient mice are metabolically unstable
Recent genetic studies have shown that the AChR fails to become clustered at the postsynaptic membrane of neuromuscular junctions in mice with a targeted disruption of the rapsyn gene (Gautam et al., 1995). To examine whether the deficiency of rapsyn also alters the rate of receptor turnover, we compared AChR degradation rates in cultured muscle cells of homozygous (−/−) and heterozygous (+/−) rapsyn knockout mice. The myoblasts obtained from these mice carry a temperature-sensitive, SV40 large T antigen under control of the class I MHC promoter (Jat et al., 1991). In the presence of γ-interferon, the myoblasts can be maintained in an undifferentiated state at the permissive temperature (33°C). When switched to fusion medium and grown at 37°C, they differentiate into multinucleated myotubes (M. Gautam, unpublished data). In rapsyn+/−myotubes, spontaneous AChR clusters were present but very few in number. When soluble, recombinant agrin was added to the culture medium, the number of receptor clusters increased dramatically. In contrast, rapsyn−/− myotubes had neither spontaneous nor agrin-induced receptor clusters (data not shown). Thus, when the metabolic stability of AChRs was examined, the surface AChRs labeled with 125I-α-bungarotoxin in rapsyn+/− cells were found to have a half-life of ∼37 h, comparable to those of noninnervated, embryonic muscle and cultured C2 myotubes. In contrast, the AChR half-life in rapsyn−/− muscle cells was reduced to ∼19 h (Fig. 7). Thus, in muscle cells, as well as in heterologous cells, the presence of rapsyn alters the metabolic stability of AChRs.
Fig. 7.

The surface AChRs on cultured myotubes of rapsyn-deficient (rapsyn−/−) mice degrade significantly faster than those of control (rapsyn+/−) mice. The myotubes in culture were labeled with 4 nm125I-α-BuTx for 90 min, washed, and incubated in growth medium. The radioactivity released into the media was measured at the times indicated, and the data were used to calculate the turnover rates. Degradation curves were fitted by linear regression and normalized to 100% at time 0 after surface labeling. Each data point represents the mean ± SEM of four separate determinations.
DISCUSSION
The primary finding of this paper is that rapsyn modifies the metabolic turnover rate of cell surface AChR. This effect is seen both in heterologous COS cells, when rapsyn and the AChR are coexpressed, and in cultured muscle cells, when wild-type myotubes are compared with cells derived from genetically altered mice that lack rapsyn. The effect appears to be specific, because coexpression of other synaptic muscle proteins in COS cells, including α-dystroglycan and syntrophin, as well as the unrelated lymphocyte membrane protein CD8, did not change the rate of degradation of the AChR.
In COS cells, as in Xenopus oocytes and quail fibroblasts, rapsyn also changes the distribution of AChRs at the cell surface (Froehner et al., 1990; Phillips et al., 1991a; Yu and Hall, 1994). When expressed alone, rapsyn occurs at or near the cell surface in aggregates; in contrast, AChRs are diffusely distributed when expressed alone, but occur in clusters that are coextensive with the rapsyn aggregates when the two proteins are expressed together. Experiments with mutated forms of rapsyn indicate that alterations that affect the ability of rapsyn to cluster AChRs, such as removal of the myristoylation site or deletion of the zinc-finger or leucine zipper domains (Phillips et al., 1991b; Scotland et al., 1993), also affect the ability of rapsyn to alter AChR turnover (Figs. 5, 6). One interpretation of these findings is that the alteration in AChR metabolic stability caused by rapsyn is the result of clustering, as suggested by a recent research report (Phillips et al., 1997). The two effects appear to be separate, however, because rapsyn has the same effect in muscle cells in which most AChRs are not clustered (Fig. 7). In addition, it has been long known that AChRs in clusters formed at sites of nerve contact in cultured myotubes have the same turnover rate as diffusely distributed surface AChRs (Salpeter et al., 1982;O’Malley et al., 1993). Furthermore, in contrast to recent results ofPhillips et al. (1997), we observed that the addition of neural agrin to C2 myotubes in culture only slightly enhanced the metabolic stability of surface AChRs (t1/2 = 13.4 ± 0.2 hr in control vs 14.3 ± 0.5 hr in agrin-treated group;p > 0.05), although neural agrin caused the formation of numerous AChR clusters on cell membrane (Z. Wang and Z. Hall, unpublished observations). Thus, the effect of rapsyn on turnover time is not secondary to cluster formation, but is a distinct effect, independent of clustering.
The results with the mutated forms of rapsyn suggest that similar domains in the protein mediate the effects of rapsyn on AChR turnover and AChR clustering. In both cases, direct interaction between rapsyn and the AChR is likely, suggesting that these domains are necessary for association of the two proteins. Recently, Ramarao and Cohen (1998) have identified residues 298–331 of rapsyn protein as a potential coiled-coil domain and established that mutants disrupting this region prevent AChR clustering. Although we have not tested whether the sequence 298–331 is required for metabolic stabilization by rapsyn, our result with rapsynΔ255–412, which lacked this putative domain, is consistent with the data of this recent report. Drugs that disrupt actin filaments or microtubules did not affect turnover, suggesting that the changes in AChR degradation are not attributable to an overall change in cytoskeletal organization.
Although direct evidence is lacking, several observations suggest that AChRs that are unclustered, as well as those in clusters, are associated with rapsyn (Noakes et al., 1993; Peng and Froehner, 1985). The 1∶1 correspondence of AChRs and rapsyn (LaRochelle and Froehner, 1986, 1987) and the coprecipitation of rapsyn and the AChR in C2 myotubes in culture (C. Fuhrer and Z. Hall, unpublished observations) are both consistent with this idea. The ability of rapsyn to affect the turnover rate of AChRs in uninnervated myotubes in which clusters are rare (Fig. 7) provides further support for the idea that rapsyn is associated with both clustered and unclustered AChRs.
The association of one protein with another is known to affect metabolic stability in a number of cases. For instance, association of the α and δ subunits of AChR in the endoplasmic reticulum during receptor assembly stabilizes the α subunit by decreasing its degradation rate (Blount et al., 1990). Such change may be brought about by a conformational change of the protein after its association, which renders it resistant to protease degradation. A conformational change of the AChR after association with rapsyn is unlikely, however, because the interactions do not change the channel properties of the receptor and its ligand-binding activity (Brennan et al., 1992). A second alternative is that rapsyn obscures a sequence on the AChR that serves as a degradation signal for the receptor. Such signals are present in other membrane receptor proteins, including members of the EGF receptor, where they are thought to play a critical role in mediating receptor endocytosis, thus affecting the metabolic stability of membrane proteins (Kornilova et al., 1996; Kurten et al., 1996). Alternatively, the change in turnover time may occur because the two proteins, AChR and rapsyn, when associated, are metabolized together. This possibility is unlikely, however, because the half-life of rapsyn was found to be short (∼3 h), and the presence of AChR does not affect the degradation rate of rapsyn in muscle cells (Frail et al., 1989).
Although rapsyn is thought to bind directly to the AChR, little is known about the receptor domains involved in mediating the interaction. Rapsyn in situ can be chemically cross-linked to the β subunit of the AChR (Burden et al., 1983), and studies on AChR clustering suggest that each of the subunits of the AChR can interact with rapsyn, presumably via the long cytoplasmic loop between transmembrane regions III and IV (Maimone and Merlie, 1993; Yu and Hall, 1994). Specific sequences that mediate the interaction have not been detected. Our finding that when the N-terminal domain of the nicotinic α7 subunit was expressed on the surface as an α7-5HT3 chimeric receptor, rapsyn coexpression had no effect on either its surface distribution or its degradation rate (Fig.3A) is consistent with the idea that rapsyn interacts with an intracellular domain of the AChR. Mutation of a presumptive phosphorylation sequence on the β subunit did not alter the effect of rapsyn on either AChR distribution (Yu and Hall, 1994) or AChR turnover (Fig. 3B). These results, plus the observation that herbimycin A was without effect, suggest that tyrosine phosphorylation is not required for the interaction of rapsyn with the AChR. Recently it has been possible to coimmunoprecipitate rapsyn and the AChR from extracts of C2 myotubes, which will allow further investigation of the interaction (C. Fuhrer and Z. Hall, unpublished observations).
Rapsyn was equally effective in slowing degradation of both adult (α2βεδ) and embryonic (α2βδγ) forms of the AChR, indicating that its interaction is not dependent on the presence of either ε or γ subunit. We also found in the present study that surface AChRs containing either the γ or ε subunits have indistinguishable half-lives (∼11 h) when expressed in COS cells. This result coincides with those from two other laboratories, using Xenopusoocytes or CHO cells (Kopta and Steinbach, 1994; Liu et al., 1994), respectively, but differs from an earlier report from our own laboratory in which we looked at AChRs expressed in COS cells (Gu et al., 1990) and a recent report on AChRs in muscle cells (Sala et al., 1997). Our own interpretation of these conflicting results is that there is a small intrinsic difference between the turnover times of the two AChR types that is variably detected because of differences in cell-type and experimental conditions.
During development, the embryonic form of the AChR at endplates, which undergoes rapid turnover (half-time ∼1 d) equivalent to that seen in unclustered AChRs, is replaced by the adult form of the AChR, with a concomitant change in turnover to an approximately 10-fold lower rate (half-time ∼10 d). After denervation of the endplate in the adult, the ε-AChR changes its half-time of turnover to ∼3 d and is then replaced by γ-AChRs with a turnover time of 1 d (Burden, 1977; for review, see Fambrough, 1979; Salpeter, 1987). These experiments indicate that AChR turnover time at the endplate depends on subunit composition and on other factors controlled by innervation. Although rapsyn is presumably an integral part of the complex of proteins that regulate the metabolic stability of AChRs at the endplate, variations in rapsyn concentration are unlikely to be an active regulatory element. Rapsyn is present at AChR clusters at the very beginning of synaptogenesis (Burden, 1985; Noakes et al., 1993), and message levels for both proteins are coordinately regulated during development (Froehner, 1989; LaRochelle et al., 1990). In the absence of rapsyn, AChR clusters cannot form, and AChRs do not become stabilized. Our results showing the effects of association with rapsyn on AChR stability may have broader relevance to understanding the effects of other receptor or ion channel-associated cytoskeletal elements such as PSD 95, GRIP, gephrin, and ankyrin on the metabolism of membrane receptors and ion channels (Sheng and Kim, 1996; Colledge and Froehner, 1998; Gee et al., 1998).
Footnotes
This work was supported by grants from the Muscular Dystrophy Association, the National Institute of Neurological Disorders and Stroke, and the intramural research program of the National Institute of Mental Health. We thank Drs. N. Davidson, S. Froehner, P. Gardner, J. Lindstrom, J. Merlie, and K. Campbell for antibodies and cDNAs. We also thank Dr. Joshua Sanes for helpful discussions, and members of the Hall laboratory for comments and advice.
Correspondence should be addressed to Dr. Zuo-Zhong Wang at his present address: Department of Neurobiology, University of Pittsburgh School of Medicine, 3500 Terrace Street, E1440 BST, Pittsburgh, PA 15261.
Dr. Hall’s present address: Department of Physiology, University of California School of Medicine, 513 Parnassus Avenue, San Francisco, CA 94143-0410.
REFERENCES
- 1.Adams ME, Butler MH, Dwyer TM, Peters MF, Murnane AA, Froehner SC. Two forms of mouse syntrophin, a 58 kd dystrophin-associated protein, differ in primary structure and tissue distribution. Neuron. 1993;11:531–540. doi: 10.1016/0896-6273(93)90157-m. [DOI] [PubMed] [Google Scholar]
- 2.Andreose JS, Xu R, Lomo T, Salpeter MM, Fumagalli G. Degradation of two AChR populations at rat neuromuscular junctions: regulation in vivo by electrical stimulation. J Neurosci. 1993;13:3433–3438. doi: 10.1523/JNEUROSCI.13-08-03433.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Apel ED, Merlie JP. Assembly of the postsynaptic apparatus. Curr Opin Neurobiol. 1995;5:62–67. doi: 10.1016/0959-4388(95)80088-3. [DOI] [PubMed] [Google Scholar]
- 4.Apel ED, Roberds SL, Campbell KP, Merlie JP. Rapsyn may function as a link between the acetylcholine receptor and the agrin-binding dystrophin-associated glycoprotein complex. Neuron. 1995;15:115–126. doi: 10.1016/0896-6273(95)90069-1. [DOI] [PubMed] [Google Scholar]
- 5.Berg DK, Hall ZW. Fate of α-bungarotoxin bound to acetylcholine receptor in normal and denervated muscle. Science. 1974;184:473–475. doi: 10.1126/science.184.4135.473. [DOI] [PubMed] [Google Scholar]
- 6.Bloch RJ. Acetylcholine receptor clustering in rat myotubes: requirement for Ca2+ and effects of drugs which depolymerize microtubules. J Neurosci. 1983;3:2670–2680. doi: 10.1523/JNEUROSCI.03-12-02670.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blount P, Smith MM, Merlie JP. Assembly intermediates of the mouse muscle nicotinic acetylcholine receptor in stably transfected fibroblasts. J Cell Biol. 1990;111:2601–2611. doi: 10.1083/jcb.111.6.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brennan C, Scotland PB, Froehner SC, Henderson LP. Functional properties of acetylcholine receptor coexpressed with the 43K protein in heterologous cell systems. Dev Biol. 1992;149:100–111. doi: 10.1016/0012-1606(92)90267-k. [DOI] [PubMed] [Google Scholar]
- 9.Brodsky MH, Warton M, Myers RM, Littman DR. Analysis of the site in CD4 that binds to the HIV envelope glycoprotein. J Immunol. 1990;144:3078–3086. [PubMed] [Google Scholar]
- 10.Buonanno A, Mudd J, Shah V, Merlie JP. A universal oligonucleotide probe for acetylcholine receptor genes: selection and sequencing of cDNA clones for the mouse β subunit. J Biol Chem. 1986;261:16451–16458. [PubMed] [Google Scholar]
- 11.Burden S. Acetylcholine receptors at the neuromuscular junction: developmental change in receptor turnover. Dev Biol. 1977;61:79–85. doi: 10.1016/0012-1606(77)90343-8. [DOI] [PubMed] [Google Scholar]
- 12.Burden SJ. The subsynaptic 43-kDa protein is concentrated at developing nerve-muscle synapses in vitro. Proc Natl Acad Sci USA. 1985;82:8270–8273. doi: 10.1073/pnas.82.23.8270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burden SJ, DePalma RL, Gottesman GS. Crosslinking of proteins in acetylcholine receptor-rich membranes: association between the beta-subunit and the 43 KD subsynaptic protein. Cell. 1983;35:687–692. doi: 10.1016/0092-8674(83)90101-0. [DOI] [PubMed] [Google Scholar]
- 14.Chang CC, Huang MC. Turnover of junctional and extrajunctional acetylcholine receptors of the rat diaphragm. Nature. 1975;253:643–644. doi: 10.1038/253643a0. [DOI] [PubMed] [Google Scholar]
- 15.Colledge M, Froehner SC. To muster a cluster: anchoring neurotransmitter receptors at synapses. Proc Natl Acad Sci USA. 1998;95:3341–3343. doi: 10.1073/pnas.95.7.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Connolly JA. Role of the cytoskeleton in the formation, stabilization, and removal of acetylcholine receptor clusters in cultured muscle cells. J Cell Biol. 1984;99:148–154. doi: 10.1083/jcb.99.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooper ST, Millar NS. Host cell-specific folding and assembly of the neuronal nicotinic acetylcholine receptor alpha7 subunit. J Neurochem. 1997;68:2140–2151. doi: 10.1046/j.1471-4159.1997.68052140.x. [DOI] [PubMed] [Google Scholar]
- 18.Dineley KT, Patrick JW. Surface receptor expression determinants: mechanism. Soc Neurosci Abstr. 1998;24:838. [Google Scholar]
- 19.Eisele J-L, Bertrand S, Galzi J-L, Devillers-Thiery A, Changeux J-P, Bertrand D. Chimaeric nicotinic-serotonergic receptor combines distinct ligand binding and channel specificities. Nature. 1993;366:479–483. doi: 10.1038/366479a0. [DOI] [PubMed] [Google Scholar]
- 20.Fallon JR, Hall ZW. Building synapses: agrin and dystroglycan stick together. Trends Neurosci. 1994;17:469–473. doi: 10.1016/0166-2236(94)90135-x. [DOI] [PubMed] [Google Scholar]
- 21.Fambrough DM. Control of acetylcholine receptors in skeletal muscle. Physiol Rev. 1979;59:165–227. doi: 10.1152/physrev.1979.59.1.165. [DOI] [PubMed] [Google Scholar]
- 22.Ferns M, Deiner M, Hall ZW. Agrin-induced acetylcholine receptor clustering in mammalian muscle requires tyrosine phosphorylation. J Cell Biol. 1996;132:937–944. doi: 10.1083/jcb.132.5.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forsayeth JR, Garcia PD. Adenovirus-mediated transfection of cultured cells. Biotechniques. 1994;17:354–358. [PubMed] [Google Scholar]
- 24.Frail DE, Musil LS, Buonanno A, Merlie JP. Expression of RAPsyn (43K protein) and nicotinic acetylcholine receptor genes is not coordinately regulated in mouse muscle. Neuron. 1989;2:1077–1086. doi: 10.1016/0896-6273(89)90232-8. [DOI] [PubMed] [Google Scholar]
- 25.Froehner SC. Expression of RNA transcript for the postsynaptic 43 kDa protein in innervated and denervated rat skeletal muscle. FEBS Lett. 1989;249:229–233. doi: 10.1016/0014-5793(89)80629-5. [DOI] [PubMed] [Google Scholar]
- 26.Froehner SC. The submembrane machinery for nicotinic acetylcholine receptor clustering. J Cell Biol. 1991;114:1–7. doi: 10.1083/jcb.114.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Froehner SC, Luetje CW, Scotland PB, Patrick J. The postsynaptic 43K protein clusters muscle nicotinic acetylcholine receptors in Xenopus oocytes. Neuron. 1990;5:403–410. doi: 10.1016/0896-6273(90)90079-u. [DOI] [PubMed] [Google Scholar]
- 28.Froehner SC, Adams ME, Peters MF, Gee SH. Syntrophins: modular adapter proteins at the neuromuscular junction and the sarcolemma. Soc Gen Physiol Ser. 1997;52:197–207. [PubMed] [Google Scholar]
- 29.Gardner PD. Nucleotide sequence of the epsilon-subunit of the mouse muscle nicotinic acetylcholine receptor. Nucleic Acids Res. 1990;18:6714. doi: 10.1093/nar/18.22.6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gautam M, Noakes PG, Mudd J, Nichol M, Chu GC, Sanes JR, Merlie JP. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature. 1995;377:232–236. doi: 10.1038/377232a0. [DOI] [PubMed] [Google Scholar]
- 31.Gee SH, Madhavan R, Levinson SR, Caldwell JH, Sealock R, Froehner SC. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J Neurosci. 1998;18:128–137. doi: 10.1523/JNEUROSCI.18-01-00128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glass DJ, Yancopoulos GD. Sequential roles of agrin, MuSK and rapsyn during neuromuscular junction formation. Curr Opin Neurobiol. 1997;7:379–384. doi: 10.1016/s0959-4388(97)80066-9. [DOI] [PubMed] [Google Scholar]
- 33.Gu Y, Franco A, Gardner PD, Lansman JB, Hall ZW. Properties of embryonic and adult muscle acetylcholine receptors transiently expressed in COS cells. Neuron. 1990;5:147–157. doi: 10.1016/0896-6273(90)90305-y. [DOI] [PubMed] [Google Scholar]
- 34.Huganir RL, Miles K. Protein phosphorylation of nicotinic acetylcholine receptors. Crit Rev Biochem Mol Biol. 1989;24:183–215. doi: 10.3109/10409238909082553. [DOI] [PubMed] [Google Scholar]
- 35.Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 36.Isenberg KE, Mudd J, Shah V, Merlie JP. Nucleotide sequence of the mouse muscle acetylcholine receptor α subunit. Nucleic Acids Res. 1986;14:5111. doi: 10.1093/nar/14.12.5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jat PS, Noble MD, Ataliotis P, Tanaka Y, Yannoutsos N, Larsen L, Kioussis D. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci USA. 1991;88:5096–5100. doi: 10.1073/pnas.88.12.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kopta C, Steinbach JH. Comparison of mammalian adult and fetal nicotinic acetylcholine receptors stably expressed in fibroblasts. J Neurosci. 1994;14:3922–3933. doi: 10.1523/JNEUROSCI.14-06-03922.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kornilova E, Sorkina T, Beguinot L, Sorkin A. Lysosomal targeting of epidermal growth factor receptors via a kinase-dependent pathway is mediated by the receptor carboxyl-terminal residues 1022–1123. J Biol Chem. 1996;271:30340–30346. doi: 10.1074/jbc.271.48.30340. [DOI] [PubMed] [Google Scholar]
- 40.Krikorian JG, Bloch RJ. Treatments that extract the 43K protein from acetylcholine receptor clusters modify the conformation of cytoplasmic domains of all subunits of the receptor. J Biol Chem. 1992;267:9118–9128. [PubMed] [Google Scholar]
- 41.Kurten RC, Cadena DL, Gill GN. Enhanced degradation of EGF receptors by a sorting nexin, SNX1. Science. 1996;272:1008–1010. doi: 10.1126/science.272.5264.1008. [DOI] [PubMed] [Google Scholar]
- 42.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 43.Lapolla RJ, Mayne KM, Davidson N. Isolation and characterization of a cDNA clone for the complete protein coding region of the δ subunit of the mouse acetylcholine receptor. Proc Natl Acad Sci USA. 1984;81:7970–7974. doi: 10.1073/pnas.81.24.7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.LaRochelle WJ, Froehner SC. Determination of the tissue distributions and relative concentrations of the postsynaptic 43-kDa protein and the acetylcholine receptor in Torpedo. J Biol Chem. 1986;261:5270–5274. [PubMed] [Google Scholar]
- 45.LaRochelle WJ, Froehner SC. Comparison of the postsynaptic 43-kDa protein from muscle cells that differ in acetylcholine receptor clustering activity. J Biol Chem. 1987;262:8190–8195. [PubMed] [Google Scholar]
- 46.LaRochelle WJ, Witzemann V, Fiedler W, Froehner SC. Developmental expression of the 43K and 58K postsynaptic membrane proteins and nicotinic acetylcholine receptors in Torpedo electrocytes. J Neurosci. 1990;10:3460–3467. doi: 10.1523/JNEUROSCI.10-10-03460.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Littman DR. The structure of the CD4 and CD8 genes. Annu Rev Immunol. 1987;5:561–584. doi: 10.1146/annurev.iy.05.040187.003021. [DOI] [PubMed] [Google Scholar]
- 48.Liu E, Hamill OP, Salpeter MM. Mouse muscle ε- and γ-containing acetylcholine receptors expressed in Xenopus laevis oocytes do not differ in their degradation half-lives. Neurosci Lett. 1994;174:77–80. doi: 10.1016/0304-3940(94)90123-6. [DOI] [PubMed] [Google Scholar]
- 49.Maimone MM, Merlie JP. Interaction of the 43 kd postsynaptic protein with all subunits of the muscle nicotinic acetylcholine receptor. Neuron. 1993;11:53–66. doi: 10.1016/0896-6273(93)90270-2. [DOI] [PubMed] [Google Scholar]
- 50.Mitra AAK, McCarthy MP, Stroud RM. Three dimensional structure of the nicotinic acetylcholine receptor, and location of the major associated 43-kD cytoskeletal protein, determined at 22 Å by low dose electron microscopy and x-ray diffraction to 12.5 Å. J Cell Biol. 1989;109:755–774. doi: 10.1083/jcb.109.2.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Noakes PG, Phillips WD, Hanley TA, Sanes JR, Merlie JP. 43K protein and acetylcholine receptors colocalize during the initial stages of neuromuscular synapse formation in vivo. Dev Biol. 1993;155:275–280. doi: 10.1006/dbio.1993.1025. [DOI] [PubMed] [Google Scholar]
- 52.O’Malley JP, Rubin LL, Salpeter MM. Two populations of AChR in rat myotubes have different degradation rates and responses to cAMP. Exp Cell Res. 1993;208:44–47. doi: 10.1006/excr.1993.1220. [DOI] [PubMed] [Google Scholar]
- 53.Peng HB, Froehner SC. Association of the postsynaptic 43K protein with newly formed acetylcholine receptor clusters in cultured muscle cells. J Cell Biol. 1985;100:1698–1705. doi: 10.1083/jcb.100.5.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Phillips WD, Kopta C, Blount P, Gardner PD, Steinbach JH, Merlie JP. ACh receptor-rich domains organized in fibroblasts by recombinant 43-kilodalton protein. Science. 1991a;251:568–570. doi: 10.1126/science.1703661. [DOI] [PubMed] [Google Scholar]
- 55.Phillips WD, Maimone MM, Merlie JP. Mutagenesis of the 43-kD postsynaptic protein defines domains involved in plasma membrane targeting and AChR clustering. J Cell Biol. 1991b;115:1713–1723. doi: 10.1083/jcb.115.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Phillips WD, Noakes PG, Roberds SL, Campbell KP, Merlie JP. Clustering and immobilization of acetylcholine receptors by the 43-kD protein: a possible role for dystrophin-related protein. J Cell Biol. 1993;123:729–740. doi: 10.1083/jcb.123.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Phillips WD, Vladeta D, Han H, Noakes PG. Rapsyn and agrin slow the metabolic degradation of the acetylcholine receptor. Mol Cell Neurosci. 1997;10:16–26. doi: 10.1006/mcne.1997.0634. [DOI] [PubMed] [Google Scholar]
- 58.Ramarao MK, Cohen JB. Mechanism of nicotinic acetylcholine receptor cluster formation by rapsyn. Proc Natl Acad Sci USA. 1998;95:4007–4012. doi: 10.1073/pnas.95.7.4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salpeter MM. Development and neural control of the neuromuscular junction and of the junctional acetylcholine receptor. In: Salpeter MM, editor. The vertebrate neuromuscular junction. Alan R. Liss; New York: 1987. pp. 55–115. [Google Scholar]
- 60.Salpeter MM, Spanton S, Holly K, Podleski TR. Brain extract causes acetylcholine receptor redistribution which mimics some early events at developing neuromuscular junctions. J Cell Biol. 1982;93:417–425. doi: 10.1083/jcb.93.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sala C, O’Malley J, Xu R, Fumagalli G, Salpeter MM. Epsilon subunit-containing acetylcholine receptors in myotubes belong to the slowly degrading population. J Neurosci. 1997;17:8937–8944. doi: 10.1523/JNEUROSCI.17-23-08937.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scotland PB, Colledge M, Melnikova I, Dai Z, Froehner SC. Clustering of the acetylcholine receptor by the 43-kD protein: involvement of the zinc finger domain. J Cell Biol. 1993;123:719–728. doi: 10.1083/jcb.123.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sealock R, Wray BE, Froehner SC. Ultrastructural localization of the Mr 43,000 protein and acetylcholine receptor in Torpedo postsynaptic membranes using monoclonal antibodies. J Cell Biol. 1984;98:2239–2244. doi: 10.1083/jcb.98.6.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sheng M, Kim E. Ion channel associated proteins. Curr Opin Neurobiol. 1996;6:602–608. doi: 10.1016/s0959-4388(96)80091-2. [DOI] [PubMed] [Google Scholar]
- 65.Steinbach JH, Merlie J, Heinemann S, Bloch R. Degradation of junctional and extrajunctional acetylcholine receptors by developing rat skeletal muscle. Proc Natl Acad Sci USA. 1979;76:3547–3551. doi: 10.1073/pnas.76.7.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wagner K, Edson K, Heginbotham L, Post M, Huganir RL, Czernik AJ. Determination of the tyrosine phosphorylation sites of the nicotinic acetylcholine receptor. J Biol Chem. 1991;266:23784–23789. [PubMed] [Google Scholar]
- 67.Walker JH, Boustead CM, Witzemann V. The 43-K protein, v1, associated with acetylcholine receptor containing membrane fragments is an actin-binding protein. EMBO J. 1984;3:2287–2290. doi: 10.1002/j.1460-2075.1984.tb02127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang ZZ, Hall ZW. Coexpression of rapsyn (43 kDa protein) slows the turnover of the nicotinic acetylcholine receptor in COS cells. Soc Neurosci Abstr. 1996;22:1034. [Google Scholar]
- 69.Wang ZZ, Hardy SF, Hall ZW. Assembly of the nicotinic acetylcholine receptor: the first transmembrane domains of truncated α and δ subunits are required for heterodimer formation in vivo. J Biol Chem. 1996a;271:27575–27584. doi: 10.1074/jbc.271.44.27575. [DOI] [PubMed] [Google Scholar]
- 70.Wang ZZ, Hardy SF, Hall ZW. Membrane tethering enables an extracellular domain of the acetylcholine receptor α subunit to form a heterodimeric ligand-binding site. J Cell Biol. 1996b;135:809–817. doi: 10.1083/jcb.135.3.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu R, Salpeter MM. Protein kinase A regulates the degradation rate of Rs acetylcholine receptors. J Cell Physiol. 1995;165:30–39. doi: 10.1002/jcp.1041650105. [DOI] [PubMed] [Google Scholar]
- 72.Xu R, Salpeter MM. Acetylcholine receptors in innervated muscles of dystrophic mdx mice degrade as after denervation. J Neurosci. 1997;17:8194–8200. doi: 10.1523/JNEUROSCI.17-21-08194.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yu L, LaPolla RJ, Davidson N. Mouse muscle nicotinic acetylcholine receptor γ subunit: cDNA sequence and gene expression. Nucleic Acids Res. 1986;14:3539–3555. doi: 10.1093/nar/14.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu X-M, Hall ZW. The role of the cytoplasmic domains of individual subunits of the acetylcholine receptor in 43 kDa protein-induced clustering in COS cells. J Neurosci. 1994;14:785–795. doi: 10.1523/JNEUROSCI.14-02-00785.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]