

Abstract
In this study we used an in situ approach to identify the oxidized nucleosides 8-hydroxydeoxyguanosine (8OHdG) and 8-hydroxyguanosine (8OHG), markers of oxidative damage to DNA and RNA, respectively, in cases of Alzheimer’s disease (AD). The goal was to determine whether nuclear and mitochondrial DNA as well as RNA is damaged in AD. Immunoreactivity with monoclonal antibodies 1F7 or 15A3 recognizing both 8OHdG and 8OHG was prominent in the cytoplasm and to a lesser extent in the nucleolus and nuclear envelope in neurons within the hippocampus, subiculum, and entorhinal cortex as well as frontal, temporal, and occipital neocortex in cases of AD, whereas similar structures were immunolabeled only faintly in controls. Relative density measurement showed that there was a significant increase (p < 0.0001) in 8OHdG and 8OHG immunoreactivity with 1F7 in cases of AD (n = 22) as compared with senile (n = 13), presenile (n = 10), or young controls (n = 4). Surprisingly, the oxidized nucleoside was associated predominantly with RNA because immunoreaction was diminished greatly by preincubation in RNase but only slightly by DNase. This is the first evidence of increased RNA oxidation restricted to vulnerable neurons in AD. The subcellular localization of damaged RNA showing cytoplasmic predominance is consistent with the hypothesis that mitochondria may be a major source of reactive oxygen species that cause oxidative damage in AD.
Keywords: Alzheimer’s disease, oxidative stress, RNA oxidation, DNA oxidation, 8-hydroxyguanosine, 8-hydroxydeoxyguanosine, mitochondrial damage
Neuron-specific oxidative stress in Alzheimer’s disease (AD) is well documented (for review, see Smith et al., 1996a; Markesbery, 1997; Smith and Perry, 1998), including oxidative damage to proteins and lipids (Smith et al., 1996b; Sayre et al., 1997) as well as the induction of specific antioxidant systems (Pappolla et al., 1992; Smith et al., 1994; Premkumar et al., 1995). One of the major findings of these studies is that oxidative damage is not limited to the pathology of AD but rather uniformly involves members of entire populations of neurons at risk of death in AD, whereas other neurons and glia remain indistinguishable from controls. This localization, together with the low diffusion of most reactive oxygen, suggests that neuronal cell bodies may be the site of reactive oxygen production that leads to oxidative stress in AD. Because mitochondria are responsible for the majority of cytoplasmic reactive oxygen production, we undertook this study to determine whether mitochondrial damage, as assessed by the oxidation of cytoplasmic DNA, shows the same restriction to vulnerable neurons.
Previous studies found higher levels of an oxidative modification of DNA, 8-hydroxy-2′-deoxyguanosine (8OHdG), in mitochondrial DNA isolated from AD brain tissue (Mecocci et al., 1993, 1994, 1997). However, these studies did not define the cell types contributing the modified DNA nor whether artifactual modifications might have been introduced during isolation (Floyd et al., 1990; Claycamp, 1992; Collins et al., 1996;Helbock et al., 1998). Addressing the relationship of mitochondrial damage to oxidative stress requires an understanding of the specific neuronal populations displaying the damage. Moreover, in AD and other neurodegenerative diseases very little is known about the extent and distribution of oxidative damage to RNA, which may be susceptible to reactive oxygen attack because of its widespread cytosolic distribution. We examined AD and control cases immunocytochemically with antibodies recognizing both oxidized nucleosides, 8-OHdG and 8-hydroxyguanosine (8OHG), which are markers of oxidative damage to DNA and RNA, respectively (Fiala et al., 1989; Rhee et al., 1995; Wamer and Wei, 1997; Wamer et al., 1997), to determine which cell types displayed the greatest differences. We found not only that the oxidized nucleosides are increased in AD but that the increase is limited to the neuronal populations showing oxidative damage to proteins and lipids. Surprisingly, instead of being restricted to mitochondrial DNA, most of the oxidized nucleoside is associated with RNA. Our findings demonstrate that oxidative damage to RNA is a prominent feature of vulnerable neurons in AD.
MATERIALS AND METHODS
Tissue. Brain tissue was obtained at autopsy from clinically and pathologically confirmed cases of AD (ages 57–93 years, average 78; n = 22), using National Institute on Aging and the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria (Khachaturian, 1985; Mirra et al., 1991), and was compared with tissue from nondemented control cases, i.e., senile controls (ages 67–86 years, average 77; n = 13), presenile controls (ages 42–64 years, average 54; n = 10), and young controls (ages 3–31 years, average 17;n = 4) with similar postmortem intervals before fixation (2–22 hr in AD cases and 4–27 hr in control cases). Hippocampal slices (∼1 cm thick and including the surrounding subiculum, entorhinal cortex, and adjacent temporal neocortex) were fixed in methacarn (methanol/chloroform/acetic acid, 6:3:1) for 16 hr at 4°C, dehydrated through graded ethanol followed by xylene, and embedded in paraffin. Sections were cut 6 μm thick and mounted on Silane-coated (Sigma, St. Louis, MO) glass slides. From the same cases, cerebellum from nine control and four AD, occipital cortex from one control and three AD, and frontal cortex from three control and three AD were examined.
Immunocytochemistry and antibodies. After deparaffinization with xylene, the sections were hydrated through graded ethanol. Endogenous peroxidase activity in the tissue was eliminated by a 30 min incubation with 3% H2O2 in methanol, and nonspecific binding sites were blocked in a 30 min incubation with 10% normal goat serum in Tris-buffered saline (150 mm Tris-HCl and 150 mm NaCl, pH 7.6). In some cases the H2O2 step was omitted as a control to assess whether H2O2-mediated oxidation during processing contributed to 8OHdG and 8OHG. Additionally, sections of three control cases were incubated with 3% H2O2 and iron [0.1 mm iron(III) citrate and 0.1 mm iron(II) chloride] in methanol for 30 min to test whether hydroxyl radicals generated by the Fenton reaction are capable of oxidizing nucleic acids as well as protein in sections. Protein oxidation was detected by increased reactive carbonyls via 2,4-dinitrophenylhydrazine labeling (Smith et al., 1998).
To detect oxidized nucleosides, we used two monoclonal antibodies developed by immunizing mice with 8OHG: 1F7 (1:30; Yin et al., 1995) (gift of Regina M. Santella, Division of Environmental Sciences, School of Public Health, Columbia University, New York, NY) and 15A3 (1:250;Park et al., 1992) (QED Bioscience, San Diego, CA). Both antibodies recognize RNA-derived 8OHG as well as DNA-derived 8OHdG (Park et al., 1992; Yin et al., 1995; Al-Abdulla and Martin, 1998). Antibody 1F7 is reported to exhibit similar binding affinities for 8OHdG and 8OHG, although affinities for deoxyguanosine and guanosine are 20,000- and 10,000-fold lower than those for 8OHdG and 8OHG, respectively (Yin et al., 1995). For 1F7 the sections were pretreated with proteinase-K (10 μg/ml in PBS, pH 7.4, for 40 min at 37°C; Boehringer Mannheim, Indianapolis, IN).
Immunostaining was developed by the peroxidase–antiperoxidase procedure (Sternberger, 1986), using 0.75 mg/ml 3,3′-diaminobenzidine cosubstrate in 0.015% H2O2 and 50 mm Tris-HCl, pH 7.6, for exactly 10 min.
Neurofibrillary tangles and senile plaques were identified by counterstaining the sections with Congo red and viewing them under plane polarized light or, alternatively, by immunostaining the adjacent section with an antiserum to tau (1:1000; Perry et al., 1991) or a mouse monoclonal antibody to phosphorylated tau, AT8 (1:500; Biosource International, Camarillo, CA). Additionally, sections of three AD cases were double-immunostained with 1F7 and the antiserum to tau.
The specificity of 1F7 and 15A3 to 8OHdG and 8OHG was confirmed by (1) comparison with adjacent sections in which the primary antibody was omitted, or (2) absorption with purified 8OHdG (Sigma), 8OHG (Cayman Chemical, Ann Arbor, MI), or guanosine (Sigma). Antibodies were incubated for 5 hr at room temperature in serial dilutions of the proteins in PBS from 0.23 mg/ml through 0.23 ng/ml and applied to the sections.
After the proteinase-K treatment, additional sections were pretreated with DNase I (10 U/μl in PBS for 1 hr at 37°C; Boehringer Mannheim), S1 DNase (10 U/μl in PBS for 1 hr at 37°C; Boehringer Mannheim), RNase (5 μg/μl in PBS for 1 hr at 37°C; Boehringer Mannheim), or a combination of all of these nucleases before incubation with 1F7 or 15A3. After enzyme treatment the DNA or RNA that was remaining was evaluated by immunostaining with a mouse monoclonal antibody to DNA, clone 16-13 (1:1000; Chemicon, Temecula, CA), or by histochemical staining with methyl green-pyronin (Bancroft and Stevens, 1996), a method that differentially stains DNA and RNA.
Relative scale of 8OHdG and 8OHG. The intensity of immunoreaction of 8OHdG and 8OHG with the 1F7 antibody was evaluated by measuring the optical density (OD). The OD in an area comprising the cytoplasm and nucleus was determined with a Quantimet 570C Image Processing and Analysis System (Leica) linked to a COHU solid state camera mounted on a Leitz Laborlux 12 ME ST microscope, according to the methods of Masliah et al. (1990). Neurons from all 27 control cases and 22 AD cases were measured in the following manner. Three adjacent fields (each field = 460 × 428 μm) of stratum pyramidale of prosubiculum adjacent to the CA1 field of hippocampus were selected. In each field of the video camera, five neurons sectioned near their equator, based on a section plane that included the nucleolus, were selected and outlined manually. The OD measurement was obtained for each of the three fields and averaged. Finally, the OD value was corrected for background by subtracting the OD of the white matter on the same section. The cross-sectional size of the selected neuron was measured also. All measurements were done under the same optical and light conditions, and an electronic shading correction was used to compensate for any unevenness that might be present in the illumination. Statistical analysis for the differences in the corrected OD value among the AD and control subgroups was performed with ANOVA, using the StatView 4.11 program (Abacus Concepts, Berkeley, CA). Fisher’s Protected Least Significant Difference was applied in the post hoc analysis.
RESULTS
When sections of hippocampus, subiculum, and entorhinal cortex as well as frontal, temporal, and occipital neocortex from cases of AD were examined, the most striking feature was the prominent immunolabeling of the oxidized nucleosides (8OHdG and 8OHG) in the neuronal cytoplasm (Fig.1A,C,E), with a similar pattern found for both antibodies, 1F7 and 15A3. Cytoplasmic immunoreaction was on granular structures with a Nissl substance-like distribution extending from the cell bodies to dendrites, but with no axonal elements displaying the immunoreaction (Fig. 1C,E). Intranuclear immunoreaction with 1F7 or 15A3 in AD cases was less intense than that found for the cytoplasmic immunoreaction. Interestingly, nuclear chromatin, demonstrated with the antibody to DNA (data not shown), was poorly detected with 1F7 or 15A3. The relative paucity of DNA-based staining can be noted clearly in the intranucleolar vacuolar region that contains chromatin (Peters et al., 1991) and is not immunolabeled with either 1F7 or 15A3 (Fig.1E). In comparison to AD the control cases, both aged and young, showed the same pattern of staining but with much reduced intensity in vulnerable neurons (Fig. 1B,D). In fact, the difference in immunoreactivity with 1F7 or 15A3 between AD and controls was most striking for cerebral pyramidal neurons, whereas the Purkinje neurons of the cerebellum showed no difference between AD and controls.
Fig. 1.
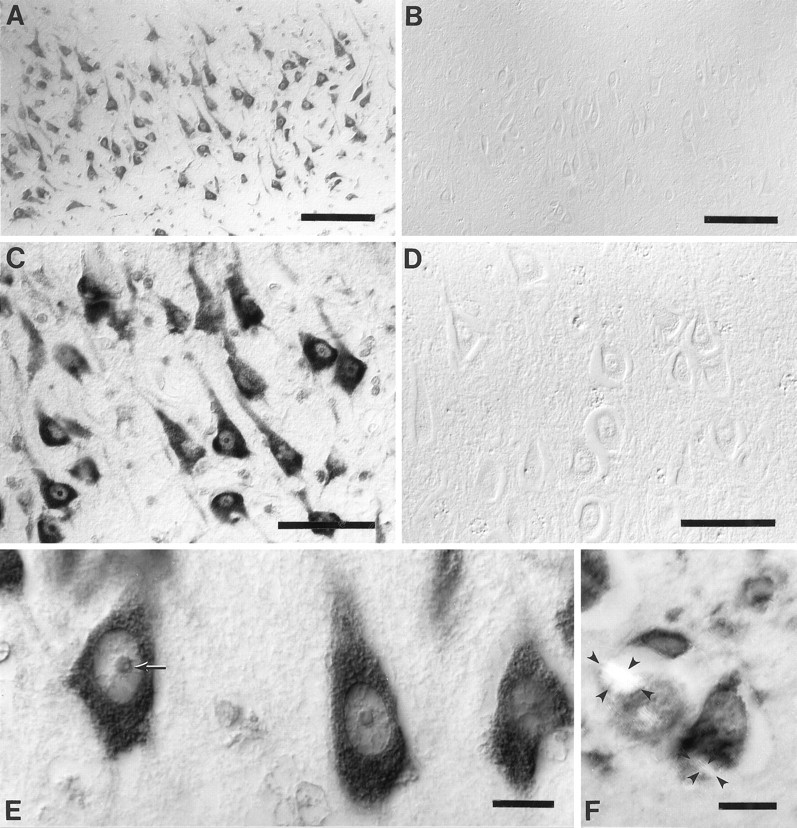
Stratum pyramidale of the hippocampal CA1 field immunostained with 1F7 antibody. Oxidized nucleosides, 8OHdG and 8OHG, are abundant in the neuronal cytoplasm for a case of Alzheimer’s disease (age, 76 years; postmortem interval, 4 hr) (A,C, E) although it is virtually undetectable in a control (age, 80 years; postmortem interval, 4 hr) (B, D). Immunolabeling of the nucleoli and nuclear envelopes is moderate and chromatin is faint as compared with the intense reaction in the cytoplasm (C, E). The relative lack of DNA staining as compared with RNA staining can be noted for the nucleolus, whereas the small vacuolar region (arrow), which is rich in DNA, remains unstained, the outer region, which contains rRNA, is stained more strongly (E). Counterstaining with Congo red and viewing under plane polarized light shows birefringent neurofibrillary tangles (outlined with small arrowheads in two neurons) and shows that the immunoreaction is not only absent from neurofibrillary tangles but also reduced in the cytoplasm excluded from the neurofibrillary tangle (F). Images are shown with differential interference contrast optics, except forF. Scale bars: A, B, 100 μm; C, D, 50 μm; E,F, 10 μm.
The presence of oxidized nucleosides in neurons was not related to the presence of neurofibrillary tangles or apposition to senile plaques, seen with tau or Congo red staining. In fact, neurons containing neurofibrillary tangles showed reduced immunoreactivity. Although in part this might be a result of the exclusion of normal cytoplasm from the region containing the neurofibrillary tangles (Fig.1F), even the remaining cytoplasm showed lower immunoreactivity with 1F7 or 15A3 than neurons lacking neurofibrillary tangles. Double immunostaining with 1F7 and antibodies to tau revealed no overlap between oxidized nucleoside and tau immunoreactivity as well as low levels of immunoreactivity with 1F7 in the cytoplasm surrounding the neurofibrillary tangles (data not shown).
Assessment of oxidized nucleoside, using a relative scale of the intensity of immunoreactivity with 1F7, demonstrated that the increase was significant (p < 0.0001) in AD when compared with each control subgroup, i.e., senile control, presenile control, or young control group (Fig. 2). These results cannot be explained by neuronal shrinkage, because the average cell profile area remained unchanged between AD cases (257–534 μm2; average of all cases, 395 μm2) and control cases (268–534 μm2; average of all cases, 380 μm2). Furthermore, we found no significant effect of postmortem intervals on the immunoreactivity with 1F7 in either control cases (r2 = 0.096;p = 0.131) or AD cases (r2 = 0.057; p = 0.285) by linear regression analysis. An agonal state before death also failed to alter the relative immunoreactivity. Indeed, similar average values for relative immunoreactivity with 1F7 was seen in controls who died from hanging (n = 1), traffic accident (n = 2), internal malignancy (n = 2), chronic cardiac failure (n = 3), or myocardial infarction (n = 3).
Fig. 2.

Relative scale of 8OHdG and 8OHG immunoreactivity with 1F7 antibody in prosubiculum neurons of 4 young controls, 10 presenile controls, and 13 senile controls, as defined in Materials and Methods, and in 22 cases of AD. Values shown are the means with SE. The difference among all controls and AD is significant by ANOVA (p < 0.0001), with post hocanalysis showing significant differences between young controls and AD (p = 0.0015), between presenile controls and AD (p = 0.0001), and between senile controls and AD (p = 0.0001).
Localization of 8OHdG and 8OHG was specific because similar results were seen with both antibodies 1F7 and 15A3, and the recognition of neurons was blocked completely by previous incubation with either 8OHdG or 8OHG at 23 ng/ml (Fig. 3), but not by over a thousand-fold greater concentration of guanosine. When antibodies were preincubated with graded 8OHdG or graded 8OHG competitively, similar blocking of immunoreaction was obtained. Therefore, 1F7 and 15A3 are equally capable of detecting oxidized DNA (8OHdG) and oxidized RNA (8OHG) by immunocytochemistry. Omission of the primary antibody yielded no immunostaining. Further, little difference in staining intensity was noted if the H2O2treatment was omitted. However, the incubation of control sections with H2O2 and iron caused a modest increase in the immunoreaction with 1F7. The increased immunoreaction of oxidized nucleosides after H2O2 and iron treatment differed from that of oxidized nucleosides created in vivo because the nucleus and cytoplasm of glial as well as neuronal cells were stained. This treatment also caused damage to protein, as noted by increased reactive carbonyls, and, as with nucleic acid, oxidative damage to protein was noted in all cell types. This contrasts with the relative limitation of the oxidized nucleoside to neuronal cytoplasm in sections from cases of AD that have not been treated with H2O2 and iron.
Fig. 3.
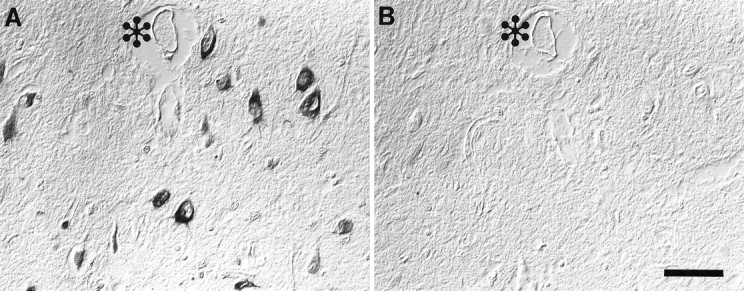
Neuronal immunolabeling with 1F7 antibody in AD (A) is abolished completely by adsorption with purified 8OHG (B). Images are shown with differential interference contrast optics. *indicates landmark blood vessel in adjacent section. Scale bar, 50 μm.
Enzymatic treatment with a mixture of both DNases and RNase essentially abolished 8OHdG and 8OHG immunoreactivity, attesting to the specificity of immunoreaction for modified nucleic acids. In separate treatments we noted a consistently greater reduction in the immunoreaction after RNase, with faint residual labeling in both nucleus and cytoplasm (Fig.4B). In contrast, DNase I treatment did not change either the intensity or the distribution of the immunoreaction significantly (Fig. 4C). The effect of DNase or RNase was confirmed by a complete absence of DNA immunostaining or RNA staining with methyl green-pyronin, respectively. Further, even after proteinase-K treatment, which does expose additional 8OHdG and 8OHG immunoreactivity, oxidized nucleosides in RNA still dominate those in DNA. Therefore, RNA is a major site of nucleic acid oxidative damage in AD.
Fig. 4.

Immunoreaction with 1F7 antibody in AD (A) was diminished greatly by the RNase treatment (B) but only slightly by DNase I treatment (C). Arrows indicate the same neurons in adjacent serial sections. Images are shown with differential interference contrast optics. Scale bar, 50 μm.
DISCUSSION
Not only do our findings confirm previous studies demonstrating that nucleic acid oxidation is increased in AD (Mecocci et al., 1993,1994, 1997), but our cytological approach allows us to determine that the increase is restricted to vulnerable neurons. This distinction is significant because it shows that oxidative damage to nucleic acid, like proteins, is limited to a defined group of neurons, the same neurons that degenerate in AD. Further, by analyzing the full spectrum of oxidized nucleic acids, we noted that oxidative damage in vulnerable neurons of AD mainly involves cytoplasmic RNA. This view is supported by the marked reduction of the immunolabeling of the oxidized nucleosides with RNase pretreatment as well as the subcellular localization of the oxidized nucleosides, with a distribution equivalent to Nissl substance. Further, the findings support cytoplasmic specificity of the damage because the nucleolus, a structure rich in RNA with an RNA/DNA ratio of 1.2 in brain (Takahashi, 1984), was immunolabeled only modestly with the antibodies to 8OHdG and 8OHG. Additionally, chromatin, a structure rich in DNA with an RNA/DNA ratio of 0.045 in brain (Takahashi, 1984), was poorly immunolabeled.
This is the first evidence of increased oxidative damage to neuronal RNA in AD. In light of the previous studies showing that cytoplasmic RNA is reduced in vulnerable neurons in AD (Mann et al., 1981; Colurso et al., 1995), we suspect that the amount of oxidized RNA as well as the extent of oxidative modification is increased in AD. It is not surprising that RNA bears substantial oxidative damage, because it is single-stranded and is not covered with protective histones like nuclear DNA. The relative paucity of DNA oxidative damage may be explained by the DNA repair mechanism, whereas the only compensation of increased RNA oxidation is higher turnover rate (Dani, 1997). Indeed, previous studies in vitro showed significant levels of RNA oxidation, without DNA oxidation, in human skin fibroblasts after exposure to ultraviolet radiation (Wamer and Wei, 1997; Wamer et al., 1997). Similarly, in vivo, RNA is more susceptible to oxidative damage than DNA in rat liver treated with a hepatocarcinogen, 2-nitropropane (Fiala et al., 1989). In consideration of the short diffusion distance of hydroxyl radicals through tissue, which is estimated in the order of several nanometers (Joenje, 1989), and the low permeability of superoxide through cell membrane (Takahashi and Asada, 1983), the predominance of oxidized nucleoside in the cytoplasm is consistent with the idea that mitochondria are a major source of damaging reactive oxygen species. Although inflammatory cells present in most senile plaques may participate in the neuronal oxidative damage by producing nitric oxide (Smith et al., 1997b), 8OHdG is not a major product of peroxynitrite-mediated oxidation of deoxyguanosine (Uppu et al., 1996). Therefore, the most likely source of oxidized nucleosides is from hydroxyl radicals formed from the reaction of highly diffusible H2O2 with redox-active metals (Schubert and Wilmer, 1991). Of note, redox-active heme and nonheme iron and copper exist within the mitochondrial and the cytosolic compartments (Prohaska, 1987), whereas iron within the nucleus is oxidized fully [i.e., iron(III)] (Smith et al., 1997a) and thus could not support hydroxyl radical formation in the nucleus.
Nucleic acid damage is not dependent on proximity to senile plaques and was reduced in neurons containing neurofibrillary tangles. Therefore, surprisingly, redox-active iron accumulation in association with AD pathology (Smith et al., 1997a) seems to make little contribution to the nucleic acid oxidation. Nucleic acid oxidation must precede lesion formation. Indeed, it is notable that neurons containing neurofibrillary tangles actually show reduced nucleic acid damage, whereas protein-related damage is increased (Good et al., 1996; Montine et al., 1996; Smith et al., 1996b, 1997b; Sayre et al., 1997). Although it could be that the reduction is attributable to the exclusion of RNA-containing structures from the neurofibrillary tangles, recent studies show that neurofibrillary tangles accumulate RNA (Ginsberg et al., 1997), and Nissl substance is abundant in the cytoplasm excluded from neurofibrillary tangles. Therefore, the lack of oxidized nucleic acid in neurofibrillary tangles and in the excluded cytoplasm suggests a real reduction in oxidative damage to RNA in those neurons. These findings may differ from protein-related damage because, although oxidatively modified proteins accumulate, RNA is turned over; thus RNA may reflect steady-state balance rather than history. Certainly, more work is required to understand these distinctions.
The cytoplasmic nucleic acid damage reported here may lie not only in RNA but also in mitochondrial DNA, which, because of the absence of histones, proximity to reactive oxygen, and poor repair mechanisms, is highly susceptible to oxidative damage (Beal, 1995). Not surprisingly, previous studies of AD showed mitochondrial DNA has greater 8OHdG levels than nuclear DNA (Mecocci et al., 1994). Mitochondrial DNA damage can be associated with increased DNA deletions or mitochondrial proliferation, both features that we have identified specifically in vulnerable neurons (K. Hirai, G. Perry, A. Nunomura, R. B. Petersen, and M. A. Smith, unpublished observations). Because the amount of mitochondrial DNA, <0.1% of total cellular DNA, is ∼3000-fold less than the amount of RNA in neurons (Davison, 1981;Giuffrida, 1983), oxidized mitochondrial DNA is inconspicuous when it is compared with oxidized RNA.
RNA seldom is analyzed for oxidative damage because such modifications are not inheritable and thus not mutagenic; further, and perhaps most importantly, analytical approaches require the isolation of specific nucleic acid fractions before analysis. Nevertheless, it has been suggested that oxidatively damaged RNA may interfere with correct base pairing and could compromise the accuracy of transcription and translation (Rhee et al., 1995). These aspects may be particularly important in the case of highly metabolic cells such as neurons. As such, it may be that oxidative damage to RNA contributes to the pathophysiology of AD.
Footnotes
This work was supported by National Institutes of Health Grants AG09287 and AG14249 and the American Health Assistance Foundation. R.W. is supported by a Ruth Salta Student Fellowship. We thank Dr. Regina M. Santella for providing the 1F7 antibody as well as the reagents for the absorption assay and Dr. Lawrence M. Sayre for critical discussions.
Correspondence should be addressed to Mark A. Smith, Ph.D., Institute of Pathology, Case Western Reserve University, 2085 Adelbert Road, Cleveland, OH 44106.
REFERENCES
- 1.Al-Abdulla NA, Martin LJ. Apoptosis of retrogradely degenerating neurons occurs in association with the accumulation of perikaryal mitochondria and oxidative damage to the nucleus. Am J Pathol. 1998;153:447–456. doi: 10.1016/S0002-9440(10)65588-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bancroft JD, Stevens A. Theory and practice of histological techniques. 1996. Fourth edition, New York: Churchill-Livingstone.
- 3.Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 4.Claycamp HG. Phenol sensitization of DNA to subsequent oxidative damage in 8-hydroxyguanine assays. Carcinogenesis. 1992;13:1289–1292. doi: 10.1093/carcin/13.7.1289. [DOI] [PubMed] [Google Scholar]
- 5.Collins AR, Dusinska M, Gedik CM, Stetina R. Oxidative damage to DNA: do we have a reliable biomarker? Environ Health Perspect. 1996;104[Suppl 3]:465–469. doi: 10.1289/ehp.96104s3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colurso GJ, Kan RK, Anthony A. Microdensitometric measures of cytoplasmic RNA and total protein in pyramidal neurons of the insular cortex and midfrontal gyrus in patients with Alzheimer’s disease. Cell Biochem Funct. 1995;13:287–292. doi: 10.1002/cbf.290130410. [DOI] [PubMed] [Google Scholar]
- 7.Dani SU. Molecular turnover and aging. In: Dani SU, Hori A, Walter GF, editors. Principles of neural aging. Elsevier; Amsterdam: 1997. pp. 83–101. [Google Scholar]
- 8.Davison AN. Biochemistry of the nervous system. In: Davison AN, Thompson RHS, editors. The molecular basis of neuropathology. Arnold; London: 1981. pp. 1–13. [Google Scholar]
- 9.Fiala ES, Conaway CC, Mathis JE. Oxidative DNA and RNA damage in the livers of Sprague Dawley rats treated with the hepatocarcinogen 2-nitropropane. Cancer Res. 1989;49:5518–5522. [PubMed] [Google Scholar]
- 10.Floyd RA, West MS, Eneff KL, Schneider JE, Wong PK, Tingey DT, Hogsett WE. Conditions influencing yield and analysis of 8-hydroxy-2′-deoxyguanosine in oxidatively damaged DNA. Anal Biochem. 1990;188:155–158. doi: 10.1016/0003-2697(90)90544-j. [DOI] [PubMed] [Google Scholar]
- 11.Ginsberg SD, Crino PB, Lee VM-Y, Eberwine JH, Trojanowski JQ. Sequestration of RNA in Alzheimer’s disease neurofibrillary tangles and senile plaques. Ann Neurol. 1997;41:200–209. doi: 10.1002/ana.410410211. [DOI] [PubMed] [Google Scholar]
- 12.Giuffrida AM. Nucleic acids in developing brain. In: Lajtha A, editor. Handbook of neurochemistry, Vol 5, Metabolic turnover in the nervous system, 2nd Ed. Plenum; New York: 1983. pp. 227–250. [Google Scholar]
- 13.Good PF, Werner P, Hsu A, Olanow CW, Perl DP. Evidence for neuronal oxidative damage in Alzheimer’s disease. Am J Pathol. 1996;149:21–28. [PMC free article] [PubMed] [Google Scholar]
- 14.Helbock HJ, Beckman KB, Shigenaga MK, Walter PB, Woodall AA, Yeo HC, Ames BN. DNA oxidation matters: the HPLC-electrochemical detection assay of 8-oxo-deoxyguanosine and 8-oxo-gaunine. Proc Natl Acad Sci USA. 1998;95:288–293. doi: 10.1073/pnas.95.1.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joenje H. Genetic toxicology of oxygen. Mutat Res. 1989;219:193–208. doi: 10.1016/0921-8734(89)90001-5. [DOI] [PubMed] [Google Scholar]
- 16.Khachaturian ZS. Diagnosis of Alzheimer’s disease. Arch Neurol. 1985;42:1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 17.Mann DMA, Neary D, Yates PO, Lincoln J, Snowden JS, Stanworth P. Alterations in protein synthetic capability of nerve cells in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1981;44:97–102. doi: 10.1136/jnnp.44.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 19.Masliah E, Terry RD, Alford M, DeTeresa R. Quantitative immunohistochemistry of synaptophysin in human neocortex: an alternative method to estimate density of presynaptic terminals in paraffin sections. J Histochem Cytochem. 1990;38:837–844. doi: 10.1177/38.6.2110586. [DOI] [PubMed] [Google Scholar]
- 20.Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann Neurol. 1993;34:609–616. doi: 10.1002/ana.410340416. [DOI] [PubMed] [Google Scholar]
- 21.Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol. 1994;36:747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- 22.Mecocci P, Beal MF, Cecchetti R, Polidori MC, Cherubini A, Chionne F, Avellini L, Romano G, Senin U. Mitochondrial membrane fluidity and oxidative damage to mitochondrial DNA in aged and AD human brain. Mol Chem Neuropathol. 1997;31:53–64. doi: 10.1007/BF02815160. [DOI] [PubMed] [Google Scholar]
- 23.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The consortium to establish a registry for Alzheimer’s disease (CERAD). II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 24.Montine TJ, Amarnath V, Martin ME, Strittmatter WJ, Graham DG. E-4-hydroxy-2-nonenal is cytotoxic and cross-links cytoskeletal proteins in P19 neuroglial cultures. Am J Pathol. 1996;148:89–93. [PMC free article] [PubMed] [Google Scholar]
- 25.Pappolla MA, Omar RA, Kim KS, Robakis NK. Immunohistochemical evidence of oxidative stress in Alzheimer’s disease. Am J Pathol. 1992;140:621–628. [PMC free article] [PubMed] [Google Scholar]
- 26.Park EM, Shigenaga MK, Degan P, Korn TS, Kitzler JW, Wehr CM, Kolachana P, Ames BN. Assay of excised oxidative DNA lesions: isolation of 8-oxoguanine and its nucleoside derivatives from biological fluids with a monoclonal antibody column. Proc Natl Acad Sci USA. 1992;89:3375–3379. doi: 10.1073/pnas.89.8.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perry G, Kawai M, Tabaton M, Onorato M, Mulvihill P, Richey P, Morandi A, Connolly JA, Gambetti P. Neuropil threads of Alzheimer’s disease show a marked alteration of the normal cytoskeleton. J Neurosci. 1991;11:1748–1755. doi: 10.1523/JNEUROSCI.11-06-01748.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peters A, Palay SL, Webster H deF. The fine structure of the nervous system, 3rd Ed. Oxford UP; New York: 1991. [Google Scholar]
- 29.Premkumar DRD, Smith MA, Richey PL, Petersen RB, Castellani R, Kutty RK, Wiggert B, Perry G, Kalaria RN. Induction of heme oxygenase-1 mRNA and protein in neocortex and cerebral vessels in Alzheimer’s disease. J Neurochem. 1995;65:1399–1402. doi: 10.1046/j.1471-4159.1995.65031399.x. [DOI] [PubMed] [Google Scholar]
- 30.Prohaska JR. Functions of trace elements in brain metabolism. Physiol Rev. 1987;67:858–901. doi: 10.1152/physrev.1987.67.3.858. [DOI] [PubMed] [Google Scholar]
- 31.Rhee Y, Valentine MR, Termini J. Oxidative base damage in RNA detected by reverse transcriptase. Nucleic Acids Res. 1995;23:3275–3282. doi: 10.1093/nar/23.16.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sayre LM, Zelasko DA, Harris PLR, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 33.Schubert J, Wilmer JW. Does hydrogen peroxide exist “free” in biological systems? Free Radic Biol Med. 1991;11:545–555. doi: 10.1016/0891-5849(91)90135-p. [DOI] [PubMed] [Google Scholar]
- 34.Smith MA, Perry G. The role of oxidative stress in the pathological sequelae of Alzheimer disease. In: Özben T, editor. Free radicals, oxidative stress, and antioxidants: pathological and physiological significance. Plenum; New York: 1998. pp. 195–204. [Google Scholar]
- 35.Smith MA, Taneda S, Richey PL, Miyata S, Yan S-D, Stern D, Sayre LM, Monnier VM, Perry G. Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc Natl Acad Sci USA. 1994;91:5710–5714. doi: 10.1073/pnas.91.12.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith MA, Sayre L, Perry G. Is Alzheimer’s a disease of oxidative stress? Alzheimer Dis Rev. 1996a;1:63–67. [Google Scholar]
- 37.Smith MA, Perry G, Richey PL, Sayre LM, Anderson VE, Beal MF, Kowall N. Oxidative damage in Alzheimer’s. Nature. 1996b;382:120–121. doi: 10.1038/382120b0. [DOI] [PubMed] [Google Scholar]
- 38.Smith MA, Harris PLR, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci USA. 1997a;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith MA, Harris PLR, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J Neurosci. 1997b;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith MA, Sayre LM, Anderson VE, Harris PLR, Beal MF, Kowall N, Perry G. Cytochemical demonstration of oxidative damage in Alzheimer disease by immunochemical enhancement of the carbonyl reaction with 2,4-dinitrophenylhydrazine. J Histochem Cytochem. 1998;46:731–735. doi: 10.1177/002215549804600605. [DOI] [PubMed] [Google Scholar]
- 41.Sternberger LA. Immunocytochemistry, 3rd Ed. Wiley; New York: 1986. [Google Scholar]
- 42.Takahashi M, Asada K. Superoxide anion permeability of phospholipid membranes and chloroplast thylakoids. Arch Biochem Biophys. 1983;226:558–566. doi: 10.1016/0003-9861(83)90325-9. [DOI] [PubMed] [Google Scholar]
- 43.Takahashi Y. Brain nuclei. In: Lajtha A, editor. Handbook of neurochemistry, Vol 7, Structural elements of the nervous system, 2nd Ed. Plenum; New York: 1984. pp. 205–230. [Google Scholar]
- 44.Uppu RM, Cueto R, Squadrito GL, Salgo MG, Pryor WA. Competitive reactions of peroxynitrite with 2′-deoxyguanosine and 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxodG): relevance to the formation of 8-oxodG in DNA exposed to peroxynitrite. Free Radic Biol Med. 1996;21:407–411. doi: 10.1016/0891-5849(96)00220-1. [DOI] [PubMed] [Google Scholar]
- 45.Wamer WG, Wei RR. In vitro photooxidation of nucleic acids by ultraviolet A radiation. Photochem Photobiol. 1997;65:560–563. doi: 10.1111/j.1751-1097.1997.tb08605.x. [DOI] [PubMed] [Google Scholar]
- 46.Wamer WG, Yin JJ, Wei RR. Oxidative damage to nucleic acids photosensitized by titanium dioxide. Free Radic Biol Med. 1997;23:851–858. doi: 10.1016/s0891-5849(97)00068-3. [DOI] [PubMed] [Google Scholar]
- 47.Yin B, Whyatt RM, Perera FP, Randall MC, Cooper TB, Santella RM. Determination of 8-hydroxydeoxyguanosine by an immunoaffinity chromatography-monoclonal antibody-based ELISA. Free Radic Biol Med. 1995;18:1023–1032. doi: 10.1016/0891-5849(95)00003-g. [DOI] [PubMed] [Google Scholar]