

Abstract
Brain dopamine (DA) systems are involved in the modulation of the sensorimotor gating phenomenon known as prepulse inhibition (PPI). The class of D2-like receptors, including the D2, D3, and D4 receptor subtypes, have all been implicated in the control of PPI via studies of DA agonists and antagonists in rats. Nevertheless, the functional relevance of each receptor subtype remains unclear because these ligands are not specific. To determine the relevance of each receptor subtype, we used genetically altered strains of “knock-out” mice lacking the DA D2, D3, or D4 receptors. We tested the effects of each knock-out on both the phenotypic expression of PPI and the disruption of PPI produced by the indirect DA agonist d-amphetamine (AMPH). No phenotypic differences in PPI were observed at baseline. AMPH significantly disrupted PPI in the D2 (+/+) mice but had no effect in the D2 (−/−) mice. After AMPH treatment, both DA D3 and D4 receptor (+/+) and (−/−) mice had significant disruptions in PPI. These findings indicate that the AMPH-induced disruption of PPI is mediated via the DA D2 receptor and not the D3 or D4 receptor subtypes. Uncovering the neural mechanisms involved in PPI will further our understanding of the substrates of sensorimotor gating and could lead to better therapeutics to treat gating disorders, such as schizophrenia.
Keywords: prepulse inhibition, startle, mice, dopamine receptors, genetics, amphetamine
Prepulse inhibition (PPI) is a cross-species measure of a sensorimotor gating phenomenon in which startle magnitude is reduced when the startling stimulus is preceded by a low-intensity prepulse. Disruptions in PPI have been reported in certain psychiatric disorders associated with dopamine (DA) dysregulation, including schizophrenia (Braff et al., 1992). Previous animal studies have demonstrated a contribution of DA and its receptor subtypes to the modulation of PPI. Consistent with the DA hypothesis of schizophrenia, indirect DA agonists, such as amphetamine (AMPH), and direct DA agonists, such as apomorphine, disrupt PPI in rats (Mansbach et al., 1988). These apomorphine-induced disruptions of PPI are prevented by typical and atypical antipsychotics (Mansbach et al., 1988; Swerdlow et al., 1991; Swerdlow and Geyer, 1993).
Many studies have been conducted in rats using selective DA D2 family antagonists and agonists to elucidate the involvement of the DA D2, D3, and/or D4 receptor (D2R, D3R, and D4R, respectively) subtypes in the modulation of PPI. The D2 receptor was implicated because apomorphine disrupted PPI, and haloperidol and the D2-like receptor antagonist raclopride could reverse the apomorphine-induced disruption of PPI (Mansbach et al., 1988; Swerdlow et al., 1991). Furthermore, the preferential D2-like receptor agonist quinpirole was shown to disrupt PPI (Peng et al., 1990). Reports that the D3 agonists 7-OH-DPAT, PD128907, and quinelorane also disrupted PPI indicated that the D3 receptor might be involved in the modulation of PPI (Caine et al., 1995; Bristow et al., 1996; Varty and Higgins, 1998), although the preferential D3 antagonist UH232 failed to restore an apomorphine-induced disruption of PPI (Caine et al., 1995). Support for a role of the D4 receptor in the modulation of PPI came from reports that D4 antagonists, such as CP-293,019, U-101,387, and PD167021, restored an apomorphine-induced disruption of PPI (Corbin and Heffner, 1997; Mansbach et al., 1998), although conflicting reports of the efficacy of other selective antagonists followed. L-745,870 was reported as reversing an apomorphine-induced disruption of PPI (Mansbach et al., 1998), whereas others found it ineffective (Bristow et al., 1997a; Corbin and Heffner, 1997). Although it is clear that the D2 family of receptors is involved in the modulation of PPI, the specific role of each receptor subtype has remained unclear because of limitations in the availability of selective ligands.
Recent reports have shown that mice display robust PPI and that DA agonists disrupt PPI in mice (Dulawa and Geyer, 1996; Paylor and Crawley, 1997; Bullock et al., 1998). Thus, to overcome the problems of the lack of selective DA receptor compounds, we used genetically mutated mice to assess the involvement of the DA D2 family of receptors in the modulation of PPI. Specifically, we used D2R, D3R, and D4R mutant mice to determine whether the removal of a specific receptor subtype would change the normal expression of either the acoustic startle response (ASR), PPI, or the ability of AMPH to disrupt PPI.
MATERIALS AND METHODS
Subjects. D2R and D4R mice were generated at Oregon Health Sciences University (Portland, OR). The D2R mice (Kelly et al., 1997, 1998) were incipient congenic, having been backcrossed for five generations (N5) to the inbred C57BL/6J (The Jackson Laboratory, Bar Harbor, ME) strain of mice. The F2 hybrid D4R mice (Rubinstein et al., 1997) were a combination of C57BL/6J and 129/Ola genetic backgrounds. The D3R mutant mice were generated at Duke University Medical Center (Durham, NC). Initial characterization has indicated no genotypic or phenotypic differences between this line and the other two previously reported D3R knock-out (KO) lines (Accili et al., 1996; Xu et al., 1997). Full characterization of this line will be published elsewhere (Y.-M. Wang, R. R. Gainetdinov, and M. G. Caron, unpublished observations). The F2 D3R mice were a crossbreed between the C57BL/6J and 129 SvJ strains (The Jackson Laboratory). All mice were allowed to acclimate to the vivarium at University of California at San Diego (San Diego, CA) for at least 1 week before behavioral testing in an American Association for Accreditation of Laboratory Animal Care-approved animal facility. This facility meets all federal and state requirements for animal care, and federal and state guidelines for the care and treatment of laboratory animals are followed. All genotyping of the mice was performed by the home institutions. The mice were group housed (segregated by sex when applicable) in a climate-controlled animal colony with a reversed 12 hr light/dark cycle (lights on at 7:00 P.M., lights off at 7:00 A.M.). All behavioral testing occurred between 9:00 A.M. and 5:00 P.M. Food (Harlan Teklab, Madison, WI) and water were available throughout the experiments, except during behavioral testing.
Drugs. d-Amphetamine sulfate (AMPH) was obtained from Sigma (St. Louis, MO) and was dissolved in 0.9% saline. Free base drug weights were used in all drug calculations. Injections of 10.0 mg/kg AMPH or saline were given intraperitoneally immediately before behavioral testing at a volume of 5 ml/kg body weight. Data from pilot studies were used to determine that 10 mg/kg was the optimal dose of AMPH necessary to disrupt PPI in mice (data not shown).
Apparatus. Startle reactivity was measured using four startle chambers (SR-LAB; San Diego Instruments, San Diego, CA). Each chamber consisted of a clear nonrestrictive Plexiglas cylinder resting on a platform inside a ventilated box. A high-frequency loudspeaker inside the chamber produced both a continuous background noise of 65 dB and the various acoustic stimuli. Vibrations of the Plexiglas cylinder caused by the whole-body startle response of the animal were transduced into analog signals by a piezoelectric unit attached to the platform. These signals were then digitized and stored on a computer. Sixty-five readings were taken at 1 msec intervals, starting at stimulus onset, and the average amplitude was used to determine the ASR. Sound levels in dB(A) sound pressure level were measured as described previously (Dulawa et al., 1997), and the SR-LAB calibration unit was used routinely to ensure consistent stabilimeter sensitivity between test chambers and over time (Geyer and Swerdlow, 1998).
Test sessions. All PPI test sessions consisted of startle trials (PULSE-ALONE), prepulse trials (PREPULSE+PULSE), and no-stimulus trials (NOSTIM). The PULSE-ALONE trial consisted of a 40 msec, 120 dB pulse. The PREPULSE+PULSE trials consisted of a 20 msec prepulse, 100 msec delay and then a 40 msec, 120 dB startle pulse (120 msec onset-to-onset interval). Prepulse intensities were 4, 8, and 16 dB above the 65 dB background noise. The NOSTIM trial consisted of background noise only. Each test session began and ended with five presentations of the PULSE-ALONE trial; in between, each trial type was presented 10 times in a pseudorandom order. There was an average of 15 sec (range, 12–30 sec) between trials. After the mice were placed in the startle chambers, a 65 dB background noise level was presented for a 10 min acclimation period and continued throughout the test session. Each animal was always tested in the same startle chamber.
Data analyses. The amount of PPI was calculated as a percentage score for each prepulse trial type: % PPI = 100 − {[(startle response for PREPULSE+PULSE)/(startle response for PULSE-ALONE)] × 100}. Startle magnitude was calculated as the average response to all of the PULSE-ALONE trials, excluding the first and last blocks of five PULSE-ALONE trials. Data from the first and last blocks of PULSE-ALONE trials were analyzed but are not included because they are similar to the startle reactivity data presented. ANOVAs were used to compare means, and where applicable, Tukey’s tests were used for post hoc analysis (see each experiment description). For brevity, main effects of prepulse intensity (which were always significant) will not be discussed, andF values will be reported only when significant. All data were also analyzed using difference scores, where PPI difference = (PULSE-ALONE − PREPULSE+PULSE) for each prepulse trial type. Using these difference scores, the same ANOVAs were performed, and the same effect of AMPH on PPI was found in the D2R, D3R, and D4R mice (data not shown). Data from the NOSTIM trials are not included in Results because the effects of AMPH were inconsistent and inconclusive. Response values on the NOSTIM trials were trivial relative to values on trials containing startle stimuli.
Experiment 1. Nine wild-type (WT) (+/+), 25 heterozygote (+/−), and 12 knock-out (−/−) male and 9 wild-type (+/+), 12 heterozygote (+/−), and 11 knock-out (−/−) female D2R mice were characterized in a baseline test session to determine the ASR and PPI levels. Three days later, mice from each genotype were assigned to receive either 10.0 mg/kg AMPH or vehicle (balanced for baseline ASR, PPI, startle chamber assignment, and treatment) and were tested in the PPI session. The mice were placed into the startle chambers immediately after each injection. The mice were tested again 1 week later, counterbalanced for drug treatment, to complete a within-subjects design. The baseline PPI data were analyzed using a three-way ANOVA, with gender and genotype as between-subjects factors and prepulse intensity as a within-subjects factor. Startle magnitude was analyzed using a two-way ANOVA, with gender and genotype as between-subjects factors. After the administration of AMPH, PPI data were analyzed using a four-way ANOVA, with gender and genotype as between-subjects factors and drug treatment and prepulse intensity as within-subjects factors. In post hoc ANOVAs, the α level was adjusted top < 0.025 to accommodate the removal of genotype as a factor. Startle magnitude was analyzed using a three-way ANOVA, with drug treatment as a within-subjects factor and gender and genotype as between-subjects factors.
Experiment 2. Ten wild-type (+/+) and nine knock-out (−/−) male D3R mice were characterized initially to establish their baseline ASR and PPI levels. Three days later, mice from each genotype were assigned to either 10.0 mg/kg AMPH or vehicle treatment groups (balanced for baseline ASR, PPI, startle chamber assignment, and treatment) and tested in a PPI session. The mice were tested again 1 week later, counterbalanced for drug treatment, to complete a within-subjects design. The same ANOVA structures used in experiment 1 were used to analyze the D3R PPI and startle magnitude data.
Experiment 3. Twenty-eight wild-type (+/+) and 29 knock-out (−/−) male and 28 wild-type (+/+) and 29 knock-out (−/−) female D4R mice were characterized initially to ascertain their baseline ASR and PPI levels. Three days later, mice from each genotype were assigned to test groups (male: WT vehicle, 9; drug, 7; KO vehicle, 10; drug, 9; female: WT vehicle, 10; drug, 9; KO vehicle, 9; drug, 8) balanced for baseline startle magnitude, PPI, and startle chamber assignment. Each animal received an injection of either vehicle or 10 mg/kg AMPH before being tested. The same ANOVA structures used in experiment 1 were used to analyze the D4R data, except drug treatment was a between-subjects factor.
RESULTS
D2 receptor mice
There were no significant differences between D2R genotypes in the initial characterization of PPI (Fig.1A) or the ASR (Table1), although there was a gender effect on startle reactivity in which female D2R mice had lower ASR than male D2R mice (F(1,85) = 7.7; p < 0.01). Because there was no effect of or interaction with gender in the AMPH experiment, male and female PPI data were combined. There were main effects of drug treatment on PPI (F(1,154) = 32.8; p < 0.01) and a drug treatment by genotype interaction (F(2,154) = 6.4; p< 0.05). Data were segregated by genotype, and two-way ANOVAs were completed with drug treatment and prepulse intensity as a within-subjects factors. As expected from previous work (Mansbach et al., 1988; Dulawa and Geyer, 1996), AMPH reduced PPI significantly in D2R wild-type (+/+) mice (F(1,17) = 34.7;p < 0.01) (Fig.2A), and there was a trend toward a disruption in the heterozygous (+/−) mice (F(1,36) = 4.9; p = 0.03) (Fig.2B). In contrast, AMPH had no significant effect on PPI in the knock-out (−/−) mice (Fig. 2C). There was a significant main effect of gender on startle reactivity after AMPH treatment (F(1,74) = 4.6; p < 0.05). Further analyses revealed that there were significant main effects of AMPH on startle reactivity in both male (F(1,45) = 23; p < 0.05) and female (F(1,29) = 19.3; p < 0.01) D2R mice (Table 2). AMPH significantly reduced startle reactivity in all genotypes in both male and female D2R mice.
Fig. 1.
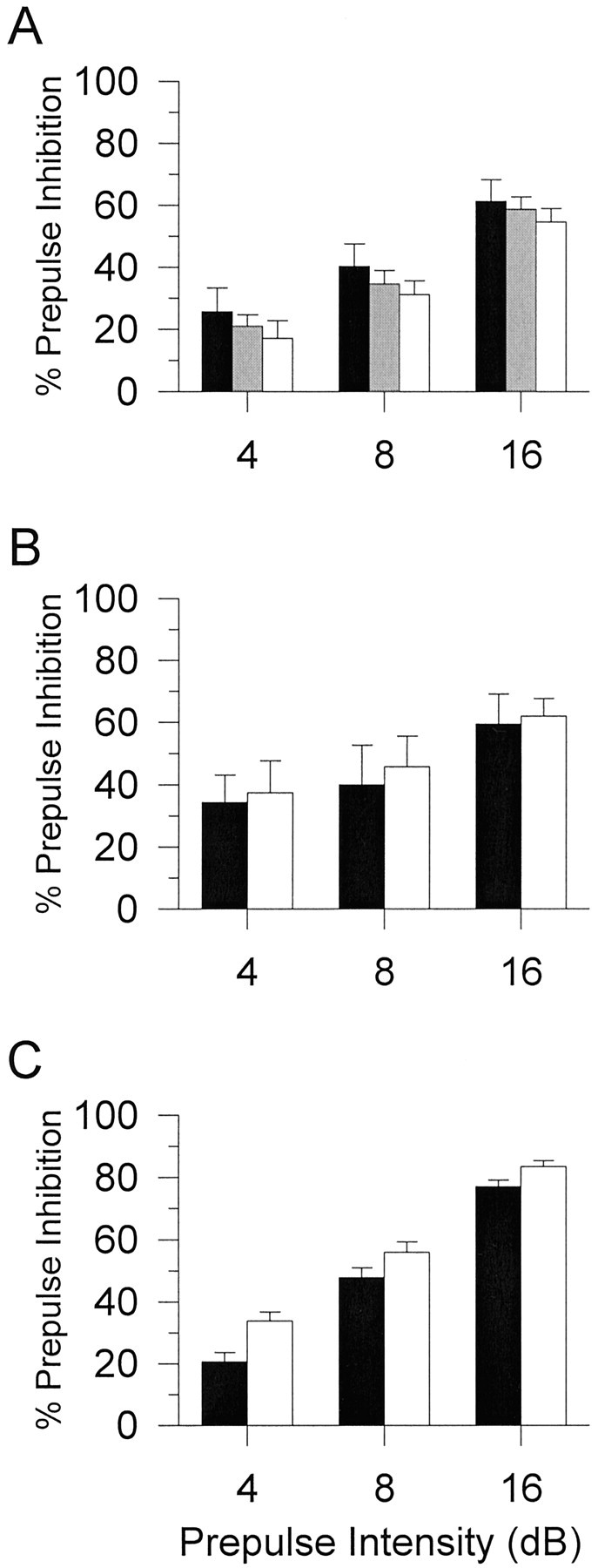
Baseline PPI for D2R (A), D3R (B), and D4R (C) mutant mice. There were comparable levels of PPI between wild-type (+/+) (black bars), heterozygous (+/−) (gray bars), and knock-out (−/−) (white bars) within each mutant line of mice. Values represent mean ± SEM percentage of PPI for each prepulse intensity.
Table 1.
Baseline acoustic startle response for D2R, D3R, and D4R mutant mice
| Wild-type (+/+) | Heterozygote (+/−) | Knock-out (−/−) | |
|---|---|---|---|
| D2R Male | 236.6 ± 34.8 | 347.4 ± 30.2 | 289.4 ± 41.4 |
| D2R Female | 202.2 ± 35.6 | 223.0 ± 22.6 | 204.4 ± 22.2 |
| D3R Male | 174.7 ± 56.4 | — | 212.7 ± 80.0 |
| D4R Male | 308.6 ± 29.2 | — | 235.0 ± 31.6 |
| D4R Female | 231.2 ± 26.6 | — | 204.2 ± 23.6 |
There were no significant differences in the startle response between genotypes for each mutant strain tested (p > 0.05). Values (arbitrary units) represent mean ± SEM startle magnitude.
Fig. 2.
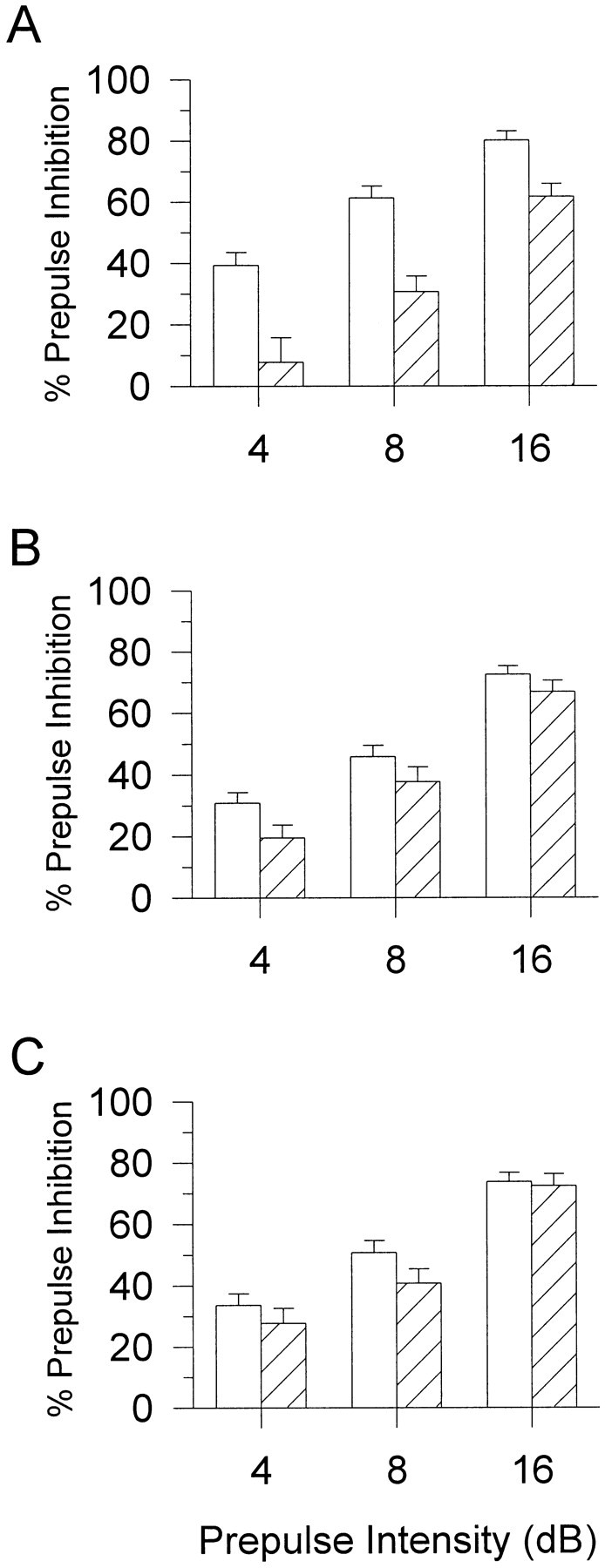
PPI for D2R mutant mice with vehicle control (0.9% saline; open bars) and AMPH (10 mg/kg;hatched bars). Post hoc ANOVAs revealed that AMPH significantly reduced PPI at the 4, 8, and 16 dB prepulse intensities (p < 0.01) in D2R wild-type (+/+) mice (A) but had no significant effect in the heterozygote (+/−) (B) or knock-out (−/−) (C) mice. Values represent mean ± SEM percentage of PPI for each prepulse intensity.
Table 2.
Effects of amphetamine on the acoustic startle response for D2R, D3R, and D4R mutant mice
| Wild-type (+/+) | Heterozygote (+/−) | Knock-out (−/−) | |
|---|---|---|---|
| D2R Male | |||
| Vehicle | 211.4 ± 34.0 | 273.4 ± 30.8 | 252.4 ± 47.2 |
| AMPH | 102.2 ± 23.6* | 217.6 ± 26.4* | 176.6 ± 26.4* |
| D2R Female | |||
| Vehicle | 191.6 ± 45.6 | 202.8 ± 40.0 | 159.2 ± 27.8 |
| AMPH | 90.8 ± 20.4** | 152.6 ± 27.8** | 102.0 ± 21.0** |
| D3R Male | |||
| Vehicle | 138.2 ± 62.6 | — | 244.5 ± 78.2 |
| AMPH | 59.1 ± 21.9** | — | 67.9 ± 20.8** |
| D4R Male | |||
| Vehicle | 349.2 ± 47.2 | — | 234.6 ± 55.6 |
| AMPH | 75.4 ± 15.2** | — | 69.0 ± 9.6** |
| D4R Female | |||
| Vehicle | 321.0 ± 56.6 | — | 256.4 ± 38.6 |
| AMPH | 91.6 ± 22.2** | — | 108.6 ± 30.6** |
Amphetamine (10.0 mg/kg, 5 ml/kg volume) significantly reduced the acoustic startle response for D2R, D3R, and D4R mutant mice. Values (arbitrary units) represent mean ± SEM startle magnitude.
*p < 0.05; **p < 0.01 versus vehicle control (0.9% saline).
D3 receptor mice
There were no differences in either PPI (Fig.1B) or the ASR (Table 1) between the D3R wild-type (+/+) and knock-out (−/−) mice at baseline testing. After AMPH treatment, there were main effects of drug treatment on both PPI (F(1,17) = 19.3; p < 0.01) and startle reactivity (F(1,17) = 9.9;p < 0.01), but there were no interactions. As in the baseline test, there were no significant main effects of genotype on PPI or startle reactivity. AMPH significantly reduced PPI (Fig.3) and the ASR (Table 2) in both the D3R wild-type (+/+) and knock-out (−/−) mice.
Fig. 3.
PPI for D3R mutant mice with vehicle control (0.9% saline; open bars) and AMPH (10 mg/kg;hatched bars). After AMPH treatment, both wild-type(+/+) (A) and knock-out (−/−) (B) mice had significantly disrupted PPI (p < 0.01). Values represent mean ± SEM percentage of PPI for each prepulse intensity.
D4 receptor mice
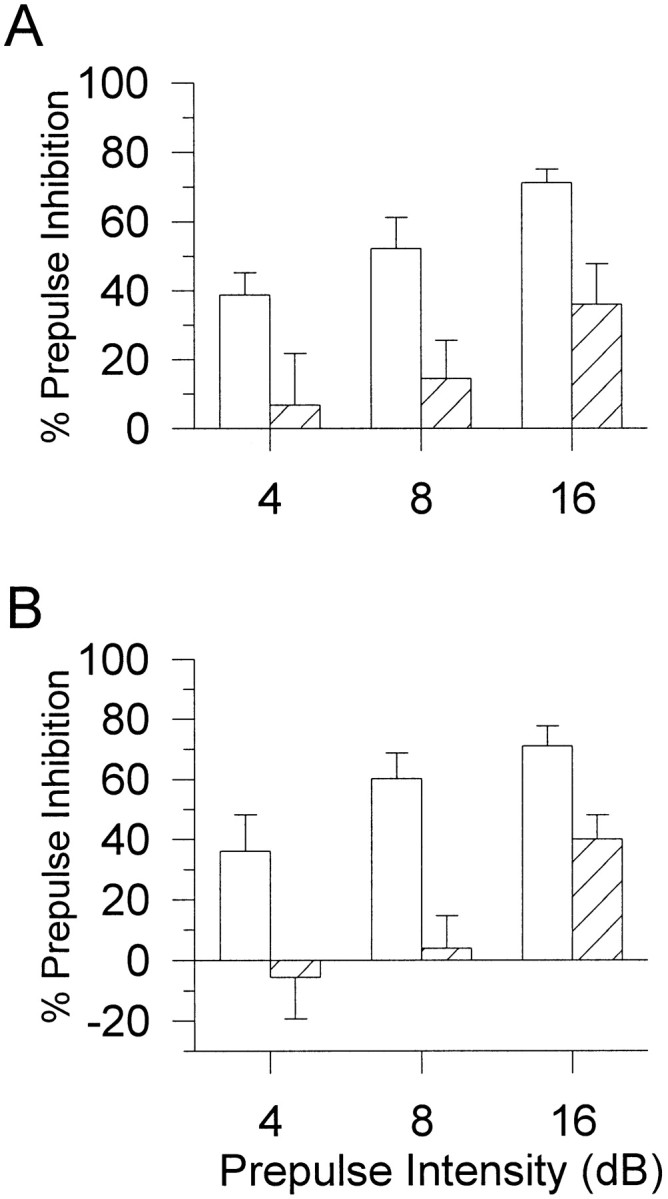
In the initial characterization of PPI, there was no main effect of or interactions with gender; therefore, male and female were considered together. There was a significant main effect of genotype (F(1,113) = 7.6; p < 0.01) in which D4R knock-out (−/−) mice had higher PPI than wild-type (+/+) mice (Fig. 1C). There were no significant differences in the ASR between the D4R wild-type (+/+) and knock-out (−/−) mice at baseline testing (Table 2). In the AMPH experiment, female and male D4R data were considered separately because there was a gender by treatment interaction on PPI (F(1,63) = 4.6;p < 0.05). In contrast to the baseline study, there were no significant main effects of genotype on PPI or startle reactivity in the AMPH experiment in the female D4R mice. There were, however, significant main effects of AMPH treatment on PPI (F(1,32) = 26.3; p < 0.01) and startle reactivity (F(1,32) = 23.5;p < 0.01), but there were no interactions between genotype and treatment. AMPH significantly reduced PPI (Fig.4A,B) and startle magnitude (Table 2) in both the D4R wild-type (+/+) and knock-out (−/−) female mice. As with the female mice, there were no main effects of genotype on PPI or startle reactivity in the AMPH experiment with male D4R mice. There was a trend toward a main effect of treatment on PPI (F(1,31) = 3.5;p = 0.07), but there were no main effects or interactions with genotype (Fig. 4C,D). There was a main effect of AMPH on startle magnitude (F(1,29) = 32.6; p < 0.01).Post hoc analyses confirmed that AMPH reduced startle in both D4R wild-type (+/+) and knock-out (−/−) mice (Table 2).
Fig. 4.

PPI for male and female D4R mutant mice with vehicle control (0.9% saline; open bars) and AMPH (10 mg/kg; hatched bars). After AMPH treatment, both female wild-type (+/+) (A) and knock-out (−/−) (B) mice had significantly disrupted PPI (p < 0.01). In the male D4R mice, however, there was only a trend (p = 0.07) for AMPH treatment to disrupt PPI in wild-type (+/+) (C) and knock-out (−/−) (D) mice, with no significant interaction with genotype. Values represent mean ± SEM percentage of PPI for each prepulse intensity.
DISCUSSION
We characterized the startle behavior of DA D2R, D3R, and D4R mutant mice and found that all strains displayed robust PPI and ASR, regardless of genotype. After AMPH treatment, all groups of mice exhibited disrupted PPI, except for the mice lacking the DA D2 receptor subtype. The D2R, D3R, and D4R mice all had significant reductions in startle reactivity after treatment with AMPH, with the various missing receptor subtypes having no effect on the ability of AMPH to reduce ASR in mice. Thus, the D2R subtype of the D2 family of receptors appears to be critical to the PPI-disruptive effects of an indirect DA agonist, but the role of the D2R in the effects of the indirect agonist on startle reactivity remains unclear. Although the lack of AMPH effect in the D2R knock-out (−/−) mice might be attributed to the addition of C57BL/6J genetic material through backcrossing, AMPH was effective in disrupting PPI in the D2R wild-type (+/+) mice, which have the closest genetic make-up to the D2R mutant mouse. AMPH also disrupted PPI in dose response studies of the 129SvEv, 129SvJ, and C57BL/6J strains of mice (R. J. Ralph and M. A. Geyer, unpublished observations). It will be necessary, however, to conduct further studies using fully congenic lines of each of the mutant mice described here (once they are created) to further verify our results.
In the experiments using the D2R and D4R mice, some gender and genotype effects were detected on both PPI and ASR. Gender influenced startle magnitude in the D2R mice both at baseline and after AMPH, but despite these main effects of gender, the disruptive effect of AMPH on PPI and the AMPH-induced reduction in startle reactivity were consistent across genotypes. These findings indicate that the gender effect seen in the D2R mice is unrelated to the effects of AMPH on PPI or ASR. The D2R heterozygote (+/−) mice reportedly have ∼50% of the D2R (Kelly et al., 1997) and have demonstrated phenotypic responses that were intermediate to those of the wild-type (+/+) and knock-out (−/−) mice when given haloperidol (Kelly et al., 1998). Interestingly, there was a trend toward an effect of AMPH on PPI in the D2R heterozygote, suggesting a gene dosage effect. As only one dose of AMPH was tested here, an AMPH dose response might reveal further phenotypic distinctions in the D2R mice. In the D4R mice, the small but significant main effect of genotype on PPI at baseline testing was not accompanied by an effect of genotype on startle reactivity. Thus, the increase in PPI seen in the D4R knock-out mice was not attributable to a difference in ASR between genotypes. This effect was not consistent, however, because there was no significant effect of genotype in the AMPH experiments in either male or female D4R mice. Similarly, this phenotypic difference in PPI has not been observed consistently in other cohorts of D4R mice (S. C. Dulawa and M. A. Geyer, unpublished observations). The gender by treatment interaction found in the D4R AMPH experiment is consistent with the observation of a significant AMPH-induced disruption of PPI in the female D4R, with only a strong trend toward a disruption in PPI in the male D4R mice. Although it is not clear why AMPH appears to be less effective in the D4R wild-type mice, there were robust effects of AMPH in both the female D4R wild-type mice and in the male D4R knock-out mice. Although various effects of gender and genotype were found, ultimately the effects of AMPH were consistent on PPI and ASR in both the D2R and D4R mice.
Previous reports demonstrate that the nonselective DA agonists AMPH and apomorphine (Mansbach et al., 1988; Geyer et al., 1990), as well as the more selective D2 agonist quinpirole (Peng et al., 1990), disrupt PPI in rats, supporting the role of the DA D2R in the modulation of PPI. Furthermore, the D2R selective antagonists haloperidol and raclopride are effective in restoring an apomorphine-induced disruption in PPI (Mansbach et al., 1988; Swerdlow et al., 1991). The present study using genetically altered mice confirms that indeed the D2R is a necessary part of the network of receptors that mediate the effects of AMPH on PPI, because AMPH had no effect on PPI in mice that lacked the D2R subtype. In both the D3R and D4R mice, PPI was disrupted by AMPH, regardless of genotype, suggesting that neither the D3R nor the D4R is mandatory for an indirect DA agonist to disrupt PPI. This result seems to contradict findings that D3R agonists, including 7-OH-DPAT, PD128907, and quinelorane, disrupt PPI in rats (Caine et al., 1995;Bristow et al., 1996; Varty and Higgins, 1998), and selective D4R antagonists restore apomorphine-induced disruptions in PPI (Corbin and Heffner, 1997; Mansbach et al., 1998). There may be several reasons for these discrepancies. One possibility is that a difference in the dopaminergic systems between the rat and the mouse accounts for the relative receptor contributions to the modulation of PPI. A more plausible reason might be that these compounds are not sufficiently selective to identify subtype-specific effects. All of the “D3R agonists” have some affinity for the D2 receptor (Seeman and Van Tol, 1994; Bowery et al., 1996). As described by Seeman and Van Tol (1994), however, it is difficult to obtain accurate comparisons of agonist selectivities in vitro and in vivo because of limitations in the affinity states of receptors in cultured cells. There are also discrepancies in the reported relative affinities of the D4R antagonists for the D4R versus the D2R (Bristow et al., 1997b;Mansbach et al., 1998). A related explanation for the inconsistent findings using “selective” D4R antagonists may be that D4R antagonism is only effective in restoring an apomorphine-induced disruption of PPI at the highest doses tested (Corbin and Heffner, 1997; Mansbach et al., 1998), which may have been acting at the D2R (Bristow et al., 1997a).
Regardless of genotype, D2R, D3R, and D4R mutant mice displayed reduced startle reactivity after administration of AMPH, a finding that is consistent with other reports (Dulawa and Geyer, 1996). Thus, the ability of AMPH to reduce startle reactivity may not depend on a DA D2 family receptor subtype. Of note is the fact that the D2R (−/−) mice had normal PPI, but a reduced ASR, after receiving AMPH. This finding lends further support for the dissociation between the effects of dopaminergic manipulations on PPI versus the ASR indicated by previous studies in rats. For example, 6-OHDA lesions of the nucleus accumbens (an area rich in D2R) reduced the ability of AMPH to disrupt PPI but did not affect the ability of AMPH to affect the ASR (Swerdlow et al., 1990). Similarly, some groups find that AMPH increases startle reactivity in rats (Davis et al., 1975; Mansbach et al., 1988; Swerdlow et al., 1990), whereas others find no change in the ASR after AMPH treatment (Bakshi et al., 1995; Johannson et al., 1995). It remains unclear why AMPH would have differential effects on startle reactivity in rats and mice, but the effects of another DA agonist, apomorphine, on the ASR seem to depend on the strain of rat being tested (Mansbach et al., 1988; Davis et al., 1990; Rigdon, 1990; Varty and Higgins, 1994).
Measures of deficient PPI in animals have been used widely in studies addressing the neurobiology of impaired sensorimotor gating in patients with schizophrenia (Geyer et al., 1990; Swerdlow et al., 1994). Symptomatic patients with schizophrenia or nonmedicated schizotypal patients exhibit deficits in PPI (Braff et al., 1992; Cadenhead et al., 1993) that appear similar to those produced by DA agonists in rodents. As predicted by the DA hypothesis of schizophrenia, activation of the DA system disrupts sensorimotor gating in rodents (Mansbach et al., 1988; Dulawa and Geyer, 1996). It is widely accepted that antagonists of the D2-like family of receptors are efficacious in the treatment of schizophrenia (Creese et al., 1976). Indeed, the ability of an antipsychotic drug to restore an apomorphine-induced disruption of PPI in rats correlates with its affinity for D2 receptors (Swerdlow et al., 1994). The present findings that the D2R plays a crucial role in the AMPH-induced disruption of sensorimotor gating in mice provides further support for this relationship and specifically identifies the D2R subtype of the D2 family of receptors as being the most relevant to this effect of an indirect DA agonist. Conversely, these findings suggest that neither the D3R nor the D4R provide important contributions to sensorimotor gating deficits induced by dopaminergic activation. Antagonists at either the D3R or D4R subtypes have been proposed as candidates for antipsychotic treatments (Sokoloff et al., 1990; Seeman et al., 1997). To the extent that the disruption of PPI in rodents produced by dopaminergic activation provides a predictive measure of the therapeutic actions of antipsychotic drugs (Geyer et al., 1990; Swerdlow et al., 1994), the present observations support the hypothesis that the D2R subtype, but not the D3R or D4R subtypes, of DA receptors is most relevant to antipsychotic drug action.
Footnotes
This work was supported by grants from the National Alliance for Research on Schizophrenia and Depression and the National Institute on Drug Abuse. R.J.R. was supported by predoctoral National Research Service Award Grant F31-MH12086 from the National Institute of Mental Health (NIMH). M.G.C. is an Investigator, and M.R. is an International Research Scholar of the Howard Hughes Medical Institute. M.A.G. has an equity interest in San Diego Instruments, Inc. and was supported by Research Scientist Award K05-MH01228 from NIMH. We thank Virginia Lehmann-Masten, Darlene Giracello, and Elizabeth Lutz for their technical assistance.
Correspondence should be addressed to Dr. Mark A. Geyer, Department of Psychiatry, University of California at San Diego, La Jolla, CA 92093-0804.
REFERENCES
- 1.Accili D, Fishburn CS, Drago J, Steiner H, Lachowicz JE, Park BH, Gauda EB, Lee EJ, Cool MH, Sibley DR, Gerfen CR, Westphal H, Fuchs S. A targeted mutation of the D3 dopamine receptor gene is associated with hyperactivity in mice. Proc Natl Acad Sci USA. 1996;93:1945–1949. doi: 10.1073/pnas.93.5.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bakshi VP, Geyer MA, Taaid N, Swerdlow NR. A comparison of the effects of amphetamine, strychnine and caffeine on prepulse inhibition and latent inhibition. Behav Pharmacol. 1995;6:801–809. [PubMed] [Google Scholar]
- 3.Bowery BJ, Razzaque F, Emms F, Patel S, Freedman S, Bristow L, Kulagowski J, Seabrook GR. Antagonism of the effects of (+)-PD 128907 on midbrain dopamine neurones in rat brain slices by a selective D2 receptor antagonist L-741,626. J Pharmacol. 1996;199:1491–1497. doi: 10.1111/j.1476-5381.1996.tb16063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braff DL, Grillon C, Geyer MA. Gating and habituation of the startle reflex in schizophrenic patients. Arch Gen Psychiatry. 1992;49:206–215. doi: 10.1001/archpsyc.1992.01820030038005. [DOI] [PubMed] [Google Scholar]
- 5.Bristow LJ, Cook GP, Gay JC, Kulagowski JJ, Landon L, Murray F, Saywell KL, Young L, Hutson PH. The behavioural and neurochemical profile of the putative dopamine D3 agonist, (+)-PD 128907, in the rat. Neuropharmacology. 1996;35:285–294. doi: 10.1016/0028-3908(96)00179-7. [DOI] [PubMed] [Google Scholar]
- 6.Bristow LJ, Collinson N, Cook GP, Curtis N, Freedman SB, Kulagowski JJ, Leeson PD, Patel S, Ragan CI, Ridgill M, Saywell KL, Tricklebank MD. L-745,870, a subtype selective dopamine D4 receptor antagonist, does not exhibit a neuroleptic-like profile in rodent behavioral tests. J Pharmacol Exp Ther. 1997a;283:1256–1263. [PubMed] [Google Scholar]
- 7.Bristow LJ, Kramer MS, Kulagowski J, Patel S, Ragan CI, Seabrook GR. Schizophrenia and L-745,870, a novel dopamine D4 receptor antagonist. Trends Pharmacol Sci. 1997b;18:186–188. doi: 10.1016/s0165-6147(97)01066-3. [DOI] [PubMed] [Google Scholar]
- 8.Bullock AE, Slobe BS, Vazquez V, Collins AC. Inbred mouse strains differ in the regulation of startle and prepulse inhibition of the startle response. Behav Neurosci. 1998;111:1353–1360. doi: 10.1037//0735-7044.111.6.1353. [DOI] [PubMed] [Google Scholar]
- 9.Cadenhead KS, Geyer MA, Braff DL. Impaired startle prepulse inhibition and habituation in schizotypal patients. Am J Psychiatry. 1993;150:1862–1867. doi: 10.1176/ajp.150.12.1862. [DOI] [PubMed] [Google Scholar]
- 10.Caine SB, Geyer MA, Swerdlow NR. Effects of D3/D2 dopamine receptor agonists and antagonists on prepulse inhibition of acoustic startle in the rat. Neuropsychopharmacology. 1995;12:139–145. doi: 10.1016/0893-133X(94)00071-7. [DOI] [PubMed] [Google Scholar]
- 11.Corbin AE, Heffner TG. Effects of dopamine (DA) D4 antagonists on prepulse inhibition of acoustic startle and amphetamine-stimulated locomotion in rats. Soc Neurosci Abstr. 1997;23:2417. [Google Scholar]
- 12.Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Nature. 1976;192:481–483. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- 13.Davis M, Svensson TH, Aghajanian GK. Effects of d- and l-amphetamine on habituation and sensitization of the acoustic startle response in rats. Psychopharmacologia. 1975;43:1–11. doi: 10.1007/BF00437607. [DOI] [PubMed] [Google Scholar]
- 14.Davis M, Mansbach RS, Swerdlow NR, Campeau S, Braff DL, Geyer MA. Apomorphine disrupts the inhibition of acoustic startle induced by weak prepulses in rats. Psychopharmacology (Berl) 1990;102:1–4. doi: 10.1007/BF02245735. [DOI] [PubMed] [Google Scholar]
- 15.Dulawa SC, Geyer MA. Psychopharmacology of prepulse inhibition in mice. Chin J Physiol. 1996;39:139–146. [PubMed] [Google Scholar]
- 16.Dulawa SC, Hen R, Scearce-Levie K, Geyer MA. Serotonin-1B receptor modulation of startle reactivity, habituation, and prepulse inhibition in wild-type and serotonin-1B knockout mice. Psychopharmacology (Berl) 1997;132:125–134. doi: 10.1007/s002130050328. [DOI] [PubMed] [Google Scholar]
- 17.Geyer MA, Swerdlow NR. Measurement of startle response, prepulse inhibition, and habituation. In: Crawley JN, Skolnick P, editors. Current protocols in neuroscience. Wiley; New York: 1998. pp. 8.7.1–8.7.15. [DOI] [PubMed] [Google Scholar]
- 18.Geyer MA, Swerdlow NR, Mansbach RS, Braff DL. Startle response models of sensorimotor gating and habituation deficits in schizophrenia. Brain Res Bull. 1990;25:485–498. doi: 10.1016/0361-9230(90)90241-q. [DOI] [PubMed] [Google Scholar]
- 19.Johannson C, Jackson DM, Zhang J, Svensson L. Prepulse inhibition of acoustic startle, a measure of sensorimotor gating: effects of antipsychotics and other agents in rats. Pharmacol Biochem Behav. 1995;52:649–654. doi: 10.1016/0091-3057(95)00160-x. [DOI] [PubMed] [Google Scholar]
- 20.Kelly MA, Rubinstein M, Asa SL, Zhang G, Saez C, Bunzow JR, Allen RG, Hnasko R, Ben-Jonathon N, Grandy DK, Low MJ. Pituitary lactotroph hyperplasia and chronic hyperprolactinemia in dopamine D2 receptor-deficient mice. Neuron. 1997;19:103–113. doi: 10.1016/s0896-6273(00)80351-7. [DOI] [PubMed] [Google Scholar]
- 21.Kelly MA, Rubinstein M, Phillips TJ, Lessov CN, Burkhart-Kasch S, Zhang G, Bunzow JR, Fang Y, Gerhardt GA, Grandy DK, Low MJ. Locomotor activity in D2 dopamine receptor-deficient mice is determined by gene dosage, genetic background, and developmental adaptations. J Neurosci. 1998;18:3470–3479. doi: 10.1523/JNEUROSCI.18-09-03470.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mansbach RS, Geyer MA, Braff DL. Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacology (Berl) 1988;94:507–514. doi: 10.1007/BF00212846. [DOI] [PubMed] [Google Scholar]
- 23.Mansbach RS, Brooks EW, Sanner MA, Zorn SH. Selective dopamine D4 receptor antagonists reverse apomorphine-induced blockade of prepulse inhibition. Psychopharmacology (Berl) 1998;135:194–200. doi: 10.1007/s002130050501. [DOI] [PubMed] [Google Scholar]
- 24.Paylor R, Crawley JN. Inbred strain differences in prepulse inhibition of the mouse startle response. Psychopharmacology (Berl) 1997;132:107–124. doi: 10.1007/s002130050333. [DOI] [PubMed] [Google Scholar]
- 25.Peng RY, Mansbach RS, Braff DL, Geyer MA. A D2 dopamine receptor agonist disrupts sensorimotor gating in rats. Neuropsychopharmacology. 1990;3:211–218. [PubMed] [Google Scholar]
- 26.Rigdon GC. Differential effects of apomorphine on prepulse inhibition of acoustic startle reflex in two rat strains. Psychopharmacology (Berl) 1990;102:419–421. doi: 10.1007/BF02244115. [DOI] [PubMed] [Google Scholar]
- 27.Rubinstein M, Phillips TJ, Bunzow JR, Falzone TL, Dziewczapolski G, Zhang G, Fang Y, Larson JL, McDougall JA, Chester JA, Saez C, Pugsley TA, Gershanik O, Low MJ, Grandy DK. Mice lacking dopamine D4 receptors are supersensitive to ethanol, cocaine, and methamphetamine. Cell. 1997;90:991–1001. doi: 10.1016/s0092-8674(00)80365-7. [DOI] [PubMed] [Google Scholar]
- 28.Seeman P, Van Tol HHM. Dopamine receptor pharmacology. Trends Pharmacol Sci. 1994;15:264–270. doi: 10.1016/0165-6147(94)90323-9. [DOI] [PubMed] [Google Scholar]
- 29.Seeman P, Corbett R, Van Tol HHM. Atypical neuroleptics have low affinity for dopamine D2 receptors or are selective for D4 receptors. Neuropsychopharmacology. 1997;16:93–110. doi: 10.1016/S0893-133X(96)00187-X. [DOI] [PubMed] [Google Scholar]
- 30.Sokoloff P, Giros B, Martres M-P, Bouthenet ML, Schwartz J-C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature. 1990;347:146–151. doi: 10.1038/347146a0. [DOI] [PubMed] [Google Scholar]
- 31.Swerdlow NR, Geyer MA. Clozapine and haloperidol in an ani-mal model of sensorimotor gating deficits in schizophrenia. Pharmacol Biochem Behav. 1993;44:741–744. doi: 10.1016/0091-3057(93)90193-w. [DOI] [PubMed] [Google Scholar]
- 32.Swerdlow NR, Mansbach RS, Geyer MA, Pulvirenti L, Koob GF, Braff DL. Amphetamine disruption of prepulse inhibition of acoustic startle is reversed by depletion of mesolimbic dopamine. Psychopharmacology (Berl) 1990;100:413–416. doi: 10.1007/BF02244616. [DOI] [PubMed] [Google Scholar]
- 33.Swerdlow NR, Keith VA, Braff DL, Geyer MA. Effects of spiperone, raclopride, SCH 23390 and clozapine on apomorphine inhibition of sensorimotor gating of the startle response in the rat. J Pharmacol Exp Ther. 1991;256:530–536. [PubMed] [Google Scholar]
- 34.Swerdlow NR, Braff DL, Taaid N, Geyer MA. Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenic patients. Arch Gen Psychiatry. 1994;51:139–154. doi: 10.1001/archpsyc.1994.03950020063007. [DOI] [PubMed] [Google Scholar]
- 35.Varty GB, Higgins GA. Differences between three rat strains in sensitivity to prepulse inhibition of an acoustic startle response: influence of apomorphine and phencyclidine pretreatment. J Psychopharmacol. 1994;8:148–156. doi: 10.1177/026988119400800302. [DOI] [PubMed] [Google Scholar]
- 36.Varty GB, Higgins GA. Dopamine agonist-induced hypothermia and disruption of prepulse inhibition: evidence for a role of D3 receptors? Behav Pharmacol. 1998;9:445–455. doi: 10.1097/00008877-199809000-00008. [DOI] [PubMed] [Google Scholar]
- 37.Xu M, Koeltzow TE, Santiago GT, Moratalla R, Cooper DC, Hu XT, White NM, Graybiel AM, White FJ, Tonegawa S. Dopamine D3 receptor mutant mice exhibit increased behavioral sensitivity to concurrent stimulation of D1 and D2 receptors. Neuron. 1997;19:837–848. doi: 10.1016/s0896-6273(00)80965-4. [DOI] [PubMed] [Google Scholar]