

Abstract
It has been repeatedly claimed that neuronal death in the hippocampal CA1 sector after untreated global ischemia occurs via apoptosis. This is based largely on DNA laddering, nick end labeling, and light microscopy. Delineation of apoptosis requires fine structural examination to detect morphological events of cell death. We studied the light and ultrastructural characteristics of CA1 injury after 5 min of untreated global ischemia in gerbils. To increase the likelihood of apoptosis, some ischemic gerbils were subjected to delayed postischemic hypothermia, a treatment that mitigates injury and delays the death of some neurons. In these gerbils, 2 d of mild hypothermia was initiated 1, 6, or 12 hr after ischemia, and gerbils were killed 4, 14, or 60 d later. Ischemia without subsequent cooling killed 96% of CA1 neurons by day 4, whereas all hypothermia-treated groups had significantly reduced injury at all survival times (2–67% loss). Electron microscopy of ischemic neurons with or without postischemic hypothermia revealed features of necrotic, not apoptotic, neuronal death even in cells that died 2 months after ischemia. Dilated organelles and intranuclear vacuoles preceded necrosis. Unique to the hypothermia-treated ischemic groups, some salvaged neurons were persistently abnormal and showed accumulation of unusual, morphologically complex secondary lysosomes. These indicate selective mitochondrial injury, because they were closely associated with normal and degenerate mitochondria, and transitional forms between mitochondria and lysosomes occurred. The results show that untreated global ischemic injury has necrotic, not apoptotic, morphology but do not rule out programmed biochemical events of the apoptotic pathway occurring before neuronal necrosis.
Keywords: ischemia, hypothermia, gerbil, CA1, necrosis, apoptosis, electron microscopy, mitochondria, lysosome
The mechanisms by which postischemic hippocampal CA1 neurons die remain controversial. Although initial reports suggested that ischemic cell death was by necrosis, recent papers propose an apoptotic death. Brief global cerebral ischemia in rodents (Kirino, 1982; Pulsinelli et al., 1982) and humans (Petito et al., 1987; Horn and Schlote, 1992) results in a selective loss of hippocampal CA1 neurons that typically succumb over a 24–72 hr period, a time course that could be consistent with apoptosis. However, ultrastructural reports describe an early proliferation of endoplasmic reticulum (Kirino, 1982; Kirino and Sano, 1984; Yamamoto et al., 1990), disaggregation of polyribosomes (Kirino and Sano, 1984; Deshpande et al., 1992), and selective dendritic swelling (Johansen et al., 1984;Yamamoto et al., 1990), findings that are not indicative of apoptosis as a mechanism.
Nevertheless, it has been repeatedly suggested, largely on the basis of biochemical criteria, that untreated neuronal death in the CA1 zone is attributable to apoptosis (MacManus et al., 1993, 1995; Okamoto et al., 1993; Kihara et al., 1994; Iwai et al., 1995; Nitatori et al., 1995;Honkaniemi et al., 1996; MacManus and Linnik, 1997; Ni et al., 1998). In this study we examined the ultrastructural events associated with delayed CA1 neuronal death in global cerebral ischemia in gerbils. Because there is evidence that milder insults are more likely to result in apoptosis than necrosis (Kerr, 1971; Portera-Cailliau et al., 1977;Wyllie et al., 1980; Harmon et al., 1990; Fukuda et al., 1993; Bonfoco et al., 1995; Choi, 1996; Du et al., 1996; Endres et al., 1998), we used prolonged postischemic hypothermia, which results in some CA1 neurons dying over weeks to months (Colbourne and Corbett, 1994, 1995), to mitigate the insult and create experimental conditions possibly more favorable for apoptosis over necrosis.
MATERIALS AND METHODS
Animals. This experiment conformed to the guidelines of the Canadian Council on Animal Care and was approved by a local animal care committee of the University of Calgary. Female Mongolian gerbils were obtained from High Oak Ranch (Baden, Ontario, Canada) and used at 16 weeks of age (∼56 gm). Gerbils were subjected to either normothermic ischemia (ISCH, HYPO-1, HYPO-6, and HYPO-12) or sham operation (SHAM, SHAM+HYPO). Cooling was instituted 1 (HYPO-1,n = 12), 6 (HYPO-6, n = 13; SHAM+HYPO,n = 3), or 12 hr (HYPO-12, n = 12) after the end of ischemia/sham occlusion. The SHAM (n = 7) and ISCH (n = 15) groups were not subjected to postischemic hypothermia. One animal died during ischemia, and two were excluded because of poor reflow, established by direct visual inspection of the carotid artery. One HYPO-12 gerbil died approximately 2 months after ischemia of unknown cause. These animals were not included in the data analysis.
Brain temperature. Similar to previous work (Colbourne et al., 1996), brain temperature (dorsal striatum) was measured by wireless AM probes (model XM-FH, Mini-Mitter Co., Sunriver, OR) that were secured inside a guide cannula. A 5.0 mm cannula was implanted at the dural surface 4 d before ischemia/sham occlusion. Two days after cannula placement, probes were inserted, and brain temperature was collected up to 4 d after the time of ischemia/sham surgery. Temperature was collected (DataQuest IV, DataSciences, St. Paul, MN) while animals were individually housed in cages that rested on telemetry receivers. Animals had free access to food and water and were housed under diurnal lighting conditions.
Ischemia. Surgical techniques were similar to those reported in previous work (Colbourne and Corbett, 1995). Briefly, gerbils were anesthetized with 2.0% halothane (70% N2O, 30% O2) followed by maintenance at 1.5%. They were then wrapped in a homeothermic body blanket (Harvard Apparatus, South Natick, MA). After a ventral neck incision, the carotid arteries were isolated and occluded for 5 min under 0.5–1% halothane (except SHAM and SHAM+HYPO gerbils). The neck wound was sutured, and a large portion of the back and abdomen was shaved to facilitate cooling. The duration of surgery was ∼20 min. At the end of surgery, animals were individually housed in cages for up to 4 d of postischemic temperature measurement/control.
Temperature control. Core temperature was held near 37.0°C during ischemia, and brain temperature was maintained close to a desired 36.4°C. The latter was regulated by feedback control of an infrared lamp (250 W) placed ∼50 cm above the gerbil (Colbourne et al., 1996). All groups except SHAM, which freely thermoregulated, were subjected to postischemic temperature control. In an effort to minimize variability, the untreated ischemia groups (ISCH) were regulated for 24 hr to mimic a mild hyperthermic pattern taken from an average of 37 untreated ischemic gerbils who spontaneously developed this hyperthermia (Colbourne and Corbett, 1994, 1995). After this, hypothermia (<35.5°C) was prevented for an additional 24 hr. The cooled groups were similarly maintained until the initiation of hypothermia at 1, 6, or 12 hr after ischemia. At that time, groups were slowly (1.0°C/30 min) cooled to 32.0°C and held at that temperature for 24 hr. They were then gradually rewarmed to 34.0°C where they were kept for 24 hr. Finally, they were warmed to 35.0°C and kept within the 35.0–36.0°C range for 12 hr, after which they were monitored until 96 hr after ischemia. The slow rates of cooling/warming were chosen to mimic the clinical situation. Postischemic temperature control was achieved in the awake, freely moving animal by a thermoregulator program that controlled a lamp (100 W incandescent or 175 W infrared lamp), fan, and water-misting system for each animal (Colbourne et al., 1996). Temperature was typically held to within 0.25°C of the desired value except for one SHAM+HYPO gerbil in which induction of hypothermia was ∼1 hr slower than desired for no apparent reason.
Light microscopy. Gerbils were killed at 4, 14, or 60 d after ischemia by an overdose of sodium pentobarbital. They were then perfused with ∼15 ml of heparinized saline followed by ∼60 ml of Karnovsky’s fixative (3% glutaraldehyde, 4% formaldehyde). Brains were refrigerated (∼4°C) in situ overnight before extraction and blocked into anterior and posterior sections (∼2 mm posterior to bregma). The anterior block, initially fixed with Karnovsky’s solution, was soaked in 10% formalin for several days before being embedded in paraffin, sectioned at 6 μm, and stained with hematoxylin and eosin. Glutaraldehyde was replaced with formaldehyde to improve paraffin embedding and tissue-sectioning quality. The number of viable CA1 neurons (Colbourne and Corbett, 1995) in each of medial (adjacent to subiculum), middle (apex of CA1), and lateral sectors (next to CA2) was then examined at ∼1.7 mm posterior to bregma (Loskoto et al., 1975). CA1 counts were summated over the six sectors (three per hemisphere).
The ISCH and HYPO groups were killed at 4, 14, and 60 d, whereas the SHAM gerbils were killed at 60 d only and the SHAM+HYPO gerbils were killed at 14 d. There were four or five gerbils per survival time for each of the HYPO groups. The ISCH groups had five gerbils at each survival time. These numbers were deemed sufficient because untreated gerbils subjected to 5 min of normothermic ischemia consistently have near-total CA1 loss (Colbourne and Corbett, 1994,1995).
Electron microscopy. Immediately after the removal of the brain from the skull, the posterior hippocampi at ∼2.0–2.3 mm posterior to bregma were dissected free, and small slices of CA1 were taken and placed in 2.5% glutaraldehyde solution. This level of CA1 has a similar degree of neuronal injury as that taken for cell counts (Colbourne and Corbett, 1994, 1995; Nurse and Corbett, 1994). Care was taken to discard adjacent subiculum and CA2/CA3 regions. Usually, four to eight semithin (1 μm) sections were stained with toluidine blue, whereas trimmed ultrathin (600 Å) sections were stained with uranyl acetate and lead citrate and examined under a transmission electron microscope (Hitachi M600). Approximately 40,000 neurons or cell remnants were examined in this study, and 638 photographic plates were prepared.
As a positive control for apoptosis, three naive postnatal day 6 (PD6) gerbils were similarly processed, and semithin and ultrathin sections of the striatum and midbrain colliculi were examined for signs of developmental apoptosis.
Statistics.The CA1 cell counts and the temperature data were analyzed with ANOVA and specific contrasts as performed previously (Colbourne and Corbett, 1994, 1995). When heterogeneity of variance was detected by a significant Levene’s test, we used separate ttests instead of the usual pooled t tests (BMDP statistical package). Electron microscopy (EM) findings were not statistically analyzed because these data were qualitative.
RESULTS
Temperature and weight data
Brain temperature (Table 1) during occlusion or in the first hour thereafter was maintained close to the baseline temperature of 36.4°C, with no notable differences between ischemic groups (ISCH, HYPO-1, HYPO-6, HYPO-12). Brain temperature after ischemia (Fig. 1) was regulated to the desired profiles outlined in Materials and Methods. There were no notable differences between subgroups (i.e., different survival times).
Table 1.
Mean (±SD) brain temperature (°C) during the day before ischemia (baseline), during ischemia/sham occlusion, and in the first hour
| Group | Baseline | Occlusion | First hour |
|---|---|---|---|
| SHAM | 36.3 ± 0.2 | 36.5 ± 0.1 | 37.1 ± 0.4 |
| SHAM+HYPO | 36.2 ± 0.3 | 36.5 ± 0.0 | 36.8 ± 0.3 |
| ISCH | 36.4 ± 0.3 | 36.0 ± 0.4 | 37.2 ± 0.4 |
| HYPO-1 | 36.3 ± 0.2 | 36.2 ± 0.3 | 37.2 ± 0.2 |
| HYPO-6 | 36.5 ± 0.3 | 36.2 ± 0.2 | 37.2 ± 0.2 |
| HYPO-12 | 36.3 ± 0.3 | 36.3 ± 0.2 | 37.0 ± 0.4 |
Note that all ischemic groups had similar temperatures. Differences between survival subgroups were negligible.
Fig. 1.

Brain temperature averaged every hour starting after ischemia/sham occlusion to 4 d later. Data were collected every 20 or 30 sec but plotted as averaged 1 hr intervals. All ischemic groups and the SHAM+HYPO group were regulated for 2 d after ischemia (see Materials and Methods), whereas SHAM animals were not manipulated. Servo-control was achieved in awake, freely moving gerbils by independent use of fan, lamp, and a water-misting system. See Table 1 for baseline, occlusion, and first hour mean temperatures.
All gerbils weighed ∼56 gm on the day of ischemia/sham occlusion. ISCH gerbils lost 2 gm on average by day 4, whereas the hypothermic animals lost ∼5 gm, similar to previous findings (Colbourne and Corbett, 1995). Animals eventually regained this weight but the HYPO-12 group was slower to normalize than the HYPO-1 and HYPO-6 groups.
Quantitative CA1 cell counts (hematoxylin and eosin-stained tissue)
CA1 cell counts 1.7 mm posterior to bregma are presented in Figure2. There was no damage (i.e., eosinophilia) in the SHAM and SHAM+HYPO gerbils, and thus they were combined for subsequent statistical analyses. Damage in the untreated ISCH group was near total (∼96% loss) by 4 d, and because cell counts were the same at 4, 14, and 60 d these groups were combined for statistical comparisons. Hypothermia significantly reduced CA1 injury in all groups at all survival times. The near-total protection in the HYPO-1 groups was similar (t < 1) at the 4, 14, and 60 d survival times. There was a significant decline in cell counts from 4 to 14 d in the HYPO-6 groups (t7 = 3.55, p = 0.0094), whereas the other comparisons were not statistically significant (p ≥ 0.1236). Although the decline in cell counts with longer survival times in the HYPO-12 groups indicated a continued loss of CA1 neurons, this was not significant (p ≥ 0.2309).
Fig. 2.

CA1 cell counts expressed as a mean percentage of normal (SHAM and SHAM+HYPO groups) ±SD. Cell counts in SHAM and SHAM+HYPO groups were the same and thus were combined. Likewise, cell counts in the three ISCH sub groups were very similar (differed by only a few cells) and were combined into one group for statistical comparisons. Hypothermia significantly blunted injury in all groups (*p < 0.05, **p < 0.01, ***p < 0.001), but the greatest benefit occurred with the 1 hr intervention delay where CA1 injury was almost totally abolished. There was no overlap between CA1 counts in any HYPO group and the ISCH groups.
Light microscopy of semithin sections (toluidine blue staining)
A few dark neurons were noted in toluidine blue-stained semithin sections from the SHAM group (Fig.3A). Dark neuronal profiles are known to occur independently of brain regions of ischemia (Garcia et al., 1995) and even to occasionally appear in aldehyde-fixed tissue of normal CNS tissue (Cohen and Pappas, 1969).
Fig. 3.

Light microscopic CA1 neuronal findings. Dark neurons were rare but were found in all groups, as is characteristic of aldehyde-fixed tissue. The one illustrated (A) is from a SHAM animal. Hypothermia-treated ischemic animals had robust CA1 neuroprotection (B), but occasional neurons (arrow) had granular inclusions (also see Figs. 7, 8). Untreated ischemia produced near-total CA1 loss by 4 d after ischemia (C), and neuronal cytolysis and karyorrhectic debris (arrow) predominate in the field. Necrotic neurons from HYPO groups had similar light microscopic morphology as from ISCH groups. Large clumps of karyorrhectic debris might be confused with apoptotic bodies under lower magnification. However, no apoptotic body was found in any ISCH or HYPO group. For control, an apoptotic body (compacted chromatin enclosed within a membrane) is shown (D) from the tectum of a neonatal gerbil. The same apoptotic body in D is shown with briefer exposure to illustrate the chromatin mass within the dense body (inset). Tissue was embedded in Epon, and sections were stained with toluidine blue. Scale bar (shown in Dapplies to A–D): 20 μm.
Almost all neurons in the CA1 sector of ISCH animals had died by 4 d, as indicated by the quantitative cell counts (Fig. 2). Although many of these neurons were in an advanced state of degeneration, clumping chromatin in aggregates of various sizes was still quite evident by light microscopy (LM) (Fig. 3C). Concomitant with extensive degeneration was a marked gliosis at 4 and 14 d survival.
Hypothermia treatment significantly reduced the number of injured or killed cells, but some protected neurons showed cytoplasmic granules (Fig. 3B). Regardless, LM signs of degeneration in HYPO gerbils (e.g., clumped chromatin), which occurred at all survival times, were similar to those seen in ISCH animals. Apoptotic bodies (Fig. 3D) seen in our positive control of developmental cell death were not found in any adult ischemic animal. However, under low magnification the coarse, punctate chromatin and globular karyorrhectic aggregates of necrotic cells could be misconstrued as apoptotic bodies.
Electron microscopy
Although there were no damaged neurons in the SHAM and SHAM+HYPO groups (Fig. 4A), an occasional dark neuron was found (Fig. 4B). These were shrunken, but there were no signs of degeneration. Occasionally, small intranuclear vacuoles (Fig. 4A) were noted in the SHAM group, but these were never as extensive as that found in ISCH and HYPO groups. ISCH gerbils at day 4 revealed near-total CA1 pyramidal cell disintegration of nuclear and cell membranes and amorphous cytoplasm (Fig. 4C) with phagocytic microglia and macrophages. At later survival times, such neurons consisted only of debris, whereas some other cells, which presumably had recently died, had earlier ultrastructural signs of necrosis. The qualitative ultrastructural characteristics of ischemic CA1 injury were generally identical in ISCH and HYPO groups at all survival times, but quantitatively, there were fewer injured cells in the hypothermia groups.
Fig. 4.
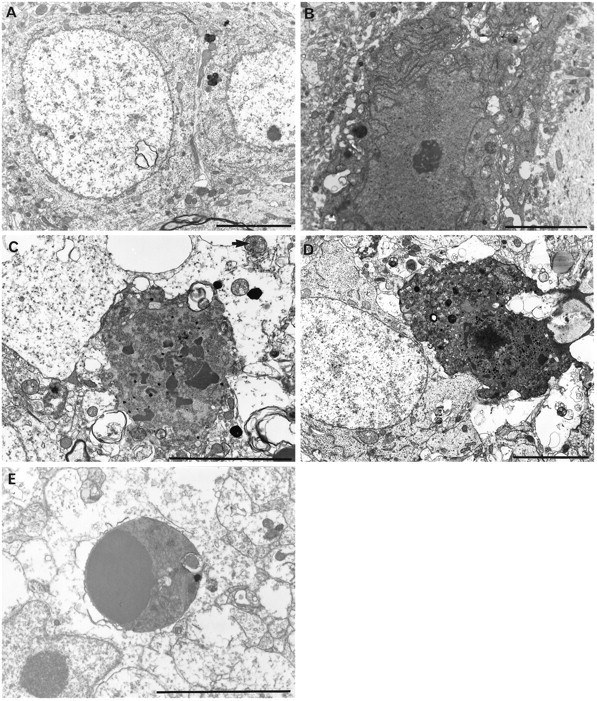
CA1 neurons from control animals (SHAM) showing small lysosomes and some intranuclear vacuoles (A) and a dark but undamaged neuron (B). CA1 cell necrosis in untreated ischemia (ISCH) showed nuclear and plasma membrane breaks, clumped tigroid chromatin, and amorphous organelles with mitochondrial flocculent densities after 4 d survival (C). Although less prevalent after hypothermic-treated ischemia (D, HYPO-12, 4 d survival), neuronal necrosis has an identical ultrastructural appearance. Necrotic neurons had variable electron density. Apoptotic bodies, like those found in neonatal gerbil striatum (E), were never found in any adult gerbil examined by light and electron microscopy. Scale bars, 5 μm.
Lethally injured neurons had clumped tigroid chromatin, discontinuous nuclear and plasma membranes, and amorphous organelles (Fig.4C,D). Mitochondrial flocculent densities, which are another sign of cell death, were also observed (Fig. 4C). Surrounding tissue was highly vacuolated with numerous membranous whorls within neurons and in the neuropil. Necrotic neurons that had an electron-dense and often microvacuolated appearance were easily distinguished from dark, uninjured neurons (e.g., from SHAM) (Fig.4B) because the latter did not have organelle or membrane damage. Morphological signs of apoptosis (i.e., apoptotic bodies containing chromatin) were never found in any ischemic or hypothermia-treated ischemic gerbil. Conversely, apoptotic bodies were easily found by LM (Fig. 3D) and EM (Fig.4E) in the neonatal gerbil brain.
Most of the few remaining CA1 neurons in the ISCH groups were injured, whereas a much smaller percentage of the remaining CA1 neurons in HYPO groups were damaged. Prelethal signs of injury in ISCH and HYPO groups, which were similar at 4, 14, and 60 d, included dilated organelles [i.e., Golgi apparatus, rough endoplasmic reticulum (RER), and mitochondria] (Fig. 5A,B), massive intranuclear vacuoles, and membranous whorls. The occurrence of intranuclear vacuoles was much more common in ischemic than in SHAM animals, where it was rare. Large lysosomal aggregates (Fig.5C) were occasionally found in viable neuronal processes in the ISCH and HYPO groups. These aggregates contained both primary and secondary lysosomes. Rarely, surviving neurons showed a proliferation and lamellar appearance of RER (Fig. 5D). Extensively dilated axons, dendrites, and astrocytes were also a common finding.
Fig. 5.

In contrast to the terminal necrotic changes shown in Fig. 4, some of the remaining neurons showed sublethal alterations that were either unique or present far in excess of their incidence in controls. Dilated organelles, including RER, Golgi apparatus, and mitochondria as well as extensive intranuclear vacuoles (arrows) (A, ISCH 4 d survival; B, HYPO-6, 14 d survival) and large clusters of primary and secondary lysosomes (C, HYPO-12, 2 month survival) were seen in untreated and hypothermia-treated ischemic groups. Rarely, neurons contained stacks of proliferated RER (D, HYPO-12, 4 d survival). Scale bars, 5 μm.
Densely stained inclusions were frequently found in dendrites (Fig.6A) and axons (Fig.6B) at 4 d and especially 14 and 60 d survival times. These inclusions were admixed among dense, pyknotic, or degenerate mitochondria. These structures were deemed to be mitochondria from their double-membrane structure and visible internal cristae. These pyknotic mitochondria and other small inclusions were found in both ISCH and HYPO gerbils as well as in neonatal gerbils (Fig. 6C).
Fig. 6.
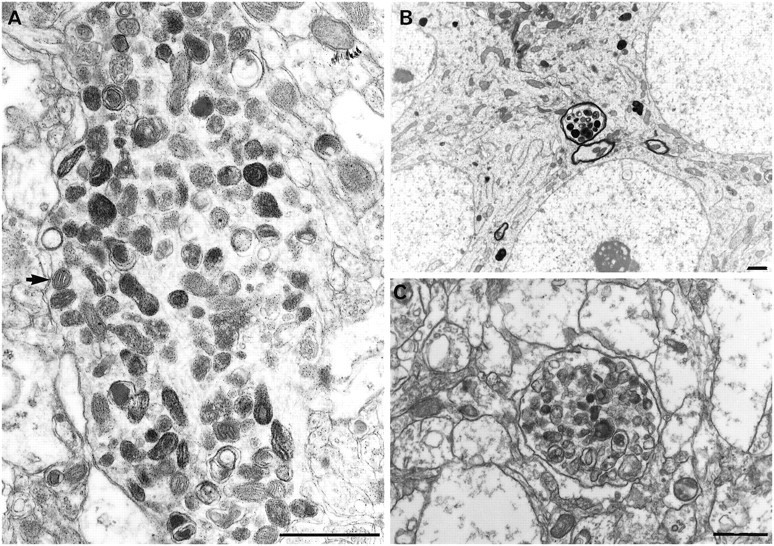
Dendritic (A, HYPO-12, 14 d survival) and axonal (B, HYPO-1, 2 month survival) pyknotic inclusions as found in HYPO and ISCH groups. Similar clusters were found in PD6 gerbils (C, colliculus). Because normal (A, arrow) and degenerate mitochondria can be seen with these clusters, these dense inclusions are likely degenerating mitochondria. Scale bars, 1 μm.
The granules seen with LM in hypothermia-treated ischemic gerbils proved to be lysosomal bodies on EM (Fig.7, 8). The few CA1 sector neurons containing these complexes were otherwise healthy looking. Adjacent neurons were often normal (Fig. 7). These inclusions were sometimes membrane bound and frequently tubulovesicular in appearance (Fig.8A). Transitional forms between mitochondria and lysosomes suggested a progression between the two. These differed from, or were a more advanced form of, the more typical mitochondrial autolysosome (Fig.9B,D) yet were distinct from the large lysosomes found in some cells (Fig. 5C) and also from the pyknotic mitochondrial clusters (Fig. 6) found in axons and dendrites.
Fig. 7.
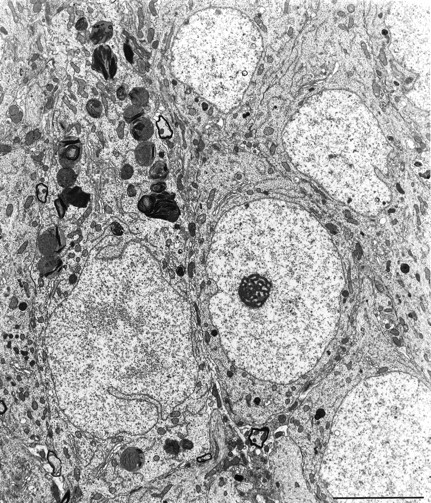
Some CA1 neurons of all HYPO groups at all survival times showed large numbers of accumulating bodies, probably derived from mitochondria (see Fig. 9). The neuron containing this large number of autolysosomes was otherwise healthy looking (HYPO-1, 14 d survival). Adjacent neurons look normal. Scale bar, 5 μm.
Fig. 8.

View of early (A) and late (B) mitochondrial evolution into lysosomes. On the left are seen a group of mitochondria with variably disorganized cristae and a tubulovesicular internal structure. The appearance of homogenous electron dense streaks suggests lipid concentration within them. Other groups of cytoplasmic bodies (B) appear more electron dense throughout most of their internal structure, apart from the small, round body left of center, which has a double membrane (arrows) indicating a mitochondrial origin. Scale bars, 1 μm.
Fig. 9.
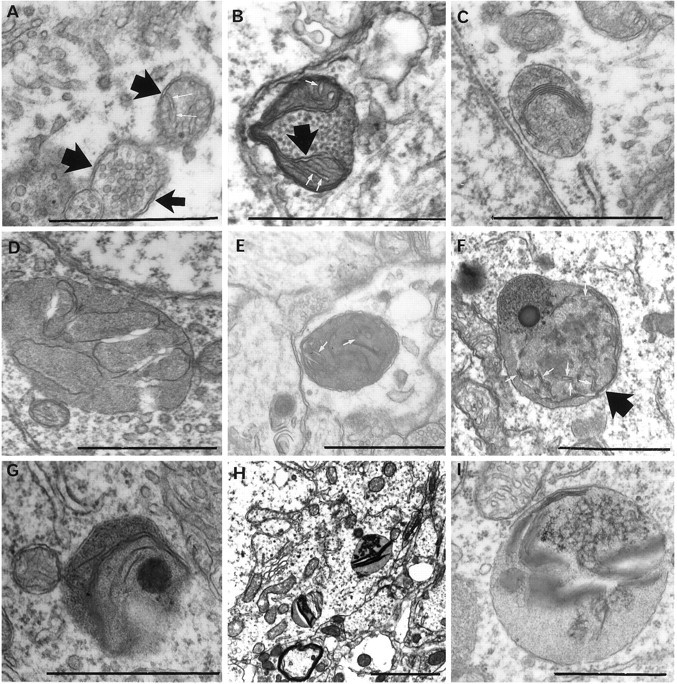
Progression of mitochondrial injury to autolysosomes. All of these forms were found only in hypothermia-treated ischemic animals at all survival periods. Double membranes (broad arrows, A, B, F) as well as remnant cristae (white arrows, A, B, E, F) attest to the mitochondrial origin of these structures. Collapsing internal membranes are seen (C, D, G). Increased internal electron density (F–I) suggests eventual accumulation of lipid or lipofuscin-like material. Scale bars, 1 μm.
Glial apoptosis was looked for (Petito et al., 1998), but no evidence for this was found here.
DISCUSSION
It has been repeatedly suggested that global cerebral ischemia can produce apoptosis of CA1 sector cells (MacManus et al., 1993, 1995; Okamoto et al., 1993; Kihara et al., 1994; Iwai et al., 1995; Nitatori et al., 1995; Honkaniemi et al., 1996; MacManus and Linnik, 1997; Ni et al., 1998). This mode of cell death is thought to be more prevalent after milder insults (Kerr, 1971; Wyllie et al., 1980; Harmon et al., 1990; Fukuda et al., 1993; Bonfoco et al., 1995;Choi, 1996; Du et al., 1996; Endres et al., 1998). However, the present examination of an estimated 40,000 neurons failed to find any morphological evidence of apoptosis. Attenuation of the massive CA1 ischemic neuronal death using hypothermia led to mitigated neuronal injury and the discovery of new inclusion bodies in neurons, but still no evidence of apoptosis, even in neurons that died more slowly.
Apoptosis and necrosis have distinct morphology, and EM is necessary to distinguish these. Necrosis is seen as a primarily cytoplasmic event. Thus, the present morphological findings of early organelle (mitochondria, RER, Golgi apparatus) swelling, disaggregation of polyribosomes, and subsequent lethal findings of mitochondrial flocculent densities, and cell and nuclear membrane breaks are clearly indicative of neuronal necrosis, not apoptosis. Other ultrastructural studies in ischemia either have found features that do not argue for either apoptosis or necrosis, such as proliferation of rough endoplasmic reticulum (Kalimo et al., 1977; Kirino and Sano, 1984;Yamamoto et al., 1990; Tomimoto et al., 1993) or have found early features of excitotoxic necrosis, such as dendritic swelling (Johansen et al., 1984; Yamamoto et al., 1990). Indeed, most ultrastructural studies of global ischemic CA1 neuronal death have found features of necrosis, not apoptosis (Kirino and Sano, 1984; Yamamoto et al., 1990;Deshpande et al., 1992; Tomimoto et al., 1993). Only a single (Nitatori et al., 1995) electron microscopic study has claimed apoptotic morphology after transient forebrain ischemia, but the cell remnant labeled as apoptotic resembles our Figure 4C,D and is necrotic debris, because it lacks a cell membrane.
We show here that the features of necrosis were constant, regardless of the time of cell death or the application of postischemic hypothermia. The novel cytoplasmic inclusions in CA1 neurons were limited to the hypothermia-treated ischemic gerbils. Although CA1 neurons containing these bodies were otherwise normal in appearance, the existence of these massive numbers of secondary lysosomes, and transitional forms from mitochondria, indicates that they received a borderline or sublethal insult perhaps affecting mitochondria selectively. The selective involvement of mitochondria per se does not argue against apoptosis or necrosis, because mitochondria are involved in both modes of cell death (Zamzami et al., 1996, 1997). Although these inclusions were distinct from the more classic autolysosomes, the latter may eventually form the larger structures (Ghadially, 1988). Although primary mitochondrial pathology has been proposed to contribute to ischemic cell death (Abe et al., 1995), mitochondrial autolysosomes were only found if ischemia was followed by hypothermia. Perhaps this is because hypothermia allowed ischemic neurons to survive long enough for turnover and accumulation of degenerate mitochondria. The duration of hypothermia or the species (rat vs gerbil) might also be a factor, because autolysosomes were not reported in rats subjected to global ischemia with and without treatment with 3 hr of immediate postischemic hypothermia (Dietrich et al., 1993).
Mitochondrial injury was also commonly observed in axons and dendrites of ischemic and hypothermic groups in variably sized clusters of small inclusions. Similar figures have been illustrated previously, but not identified, within the hippocampus (Bonnekoh et al., 1990) and cortex (Nishikawa et al., 1989) of ischemic animals. These small inclusions intermixed with dense mitochondria appear to be nonspecific signs of mitochondrial cellular injury, because they resemble the pyknotic mitochondria in renal necrosis induced with d-serine (Wachstein and Besen, 1964), in cortical neurons treated with glycine antagonist (Auer, 1997), in Wallerian degeneration (Symmens, 1986), and in neonatal gerbils (present study). Thus, these small whorled membranous bodies found intermingled with dense mitochondria probably derive from mitochondria undergoing involution (Ghadially, 1988) after cell death or serious cellular injury.
Biochemical, genetic (MacManus and Linnik, 1997), and pharmacological (Goto et al., 1990; Chen et al., 1998; Himi et al., 1998) studies have been interpreted to show that CA1 sector death after global ischemia occurs by an apoptotic pathway. Caspase activation has been shown to occur in CA1 cells in ischemia (Chen et al., 1998; Ni et al., 1998), but whether such activation is causal or merely a concomitant of neuronal death remains to be shown. Some of these results may merely reflect DNA breaks occurring with necrosis (Petito et al., 1997). Apoptotic-like DNA fragmentation can occur in necrotic cells (Collins et al., 1992; Bicknell and Cohen, 1995; Dong et al., 1997). The widespread reliance on only terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining to label apoptotic cells is fraught with hazard, because TUNEL also tags necrotic neurons (Gold et al., 1994; Grasl-Kraupp et al., 1995; de Torres et al., 1997).
In summary, only our positive control for apoptosis (i.e., PD6 gerbil brain) showed membrane-delimited apoptotic bodies. Demonstration of membrane delimitation requires EM. We found no neuron subjected to ischemia or inadequately protected by delayed hypothermia that showed features other than prelethal or lethal signs of necrosis. However, a novel feature in hypothermically salvaged neurons was the cytoplasmic aggregation of lysosomal-like bodies in persistently surviving CA1 neurons. Given that these large inclusions appear to originate from mitochondria, a prolonged mitochondrial pathology may contribute to the very slow cell death that can occur after postischemic hypothermia.
Footnotes
We gratefully acknowledge research support by the Medical Research Council of Canada (MT-9935) to R.N.A. and by the Heart and Stroke Foundation of Canada to G.R.S. F.C. was the recipient of a fellowship from the Heart and Stroke Foundation. We are also grateful to the Foothills Hospital electron microscopy support staff and to Dr. D. Rakić for histology processing and Drs. D. Corbett and K. A. Sharkey for their helpful comments on this manuscript.
Correspondence should be addressed to Dr. Roland N. Auer, Department of Pathology, Faculty of Medicine, University of Calgary, 3330 Hospital Drive N.W., Calgary, Alberta, Canada, T2N 4N1.
REFERENCES
- 1.Abe K, Aoki M, Kawagoe J, Yoshida T, Hattori A, Kogure K, Itoyama Y. Ischemic delayed neuronal death. A mitochondrial hypothesis. Stroke. 1995;26:1478–1489. doi: 10.1161/01.str.26.8.1478. [DOI] [PubMed] [Google Scholar]
- 2.Auer RN. Structural neurotoxicologic investigation of the glycine antagonist 5-nitro-6,7-dichloroquinoxalinedione (ACEA1021). Neurotoxicology. 1997;18:53–62. [PubMed] [Google Scholar]
- 3.Bicknell GR, Cohen GM. Cleavage of DNA to large kilobase pair fragments occurs in some forms of necrosis as well as apoptosis. Biochem Biophys Res Commun. 1995;207:40–47. doi: 10.1006/bbrc.1995.1150. [DOI] [PubMed] [Google Scholar]
- 4.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonnekoh P, Barbier A, Oschlies U, Hossmann KA. Selective vulnerability in the gerbil hippocampus: morphological changes after 5-min ischemia and long survival times. Acta Neuropathol (Berl) 1990;80:18–25. doi: 10.1007/BF00294217. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Nagayama T, Jin K, Stetler RA, Zhu R, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi DW. Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol. 1996;6:667–672. doi: 10.1016/s0959-4388(96)80101-2. [DOI] [PubMed] [Google Scholar]
- 8.Cohen EB, Pappas GD. Dark profiles in the apparently normal central nervous system: a problem in the electron microscopic identification of early anterograde axonal degeneration. J Comp Neurol. 1969;136:375–396. doi: 10.1002/cne.901360403. [DOI] [PubMed] [Google Scholar]
- 9.Colbourne F, Corbett D. Delayed and prolonged post-ischemic hypothermia is neuroprotective in the gerbil. Brain Res. 1994;654:265–272. doi: 10.1016/0006-8993(94)90488-x. [DOI] [PubMed] [Google Scholar]
- 10.Colbourne F, Corbett D. Delayed postischemic hypothermia: a six month survival study using behavioral and histologic assessments of neuroprotection. J Neurosci. 1995;15:7250–7260. doi: 10.1523/JNEUROSCI.15-11-07250.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colbourne F, Sutherland GR, Auer RN. An automated system for regulating brain temperature in awake and freely moving rodents. J Neurosci Methods. 1996;67:185–190. [PubMed] [Google Scholar]
- 12.Collins RJ, Harmon BV, Gobe GC, Kerr JFR. Internucleosomal DNA cleavage should not be the sole criterion for identifying apoptosis. Int J Radiat Biol. 1992;61:451–453. doi: 10.1080/09553009214551201. [DOI] [PubMed] [Google Scholar]
- 13.de Torres C, Munell F, Ferrer I, Reventos J, Macaya A. Identification of necrotic cell death by the TUNEL assay in the hypoxic-ischemic neonatal rat brain. Neurosci Lett. 1997;230:1–4. doi: 10.1016/s0304-3940(97)00445-x. [DOI] [PubMed] [Google Scholar]
- 14.Deshpande J, Bergstedt K, Lindèn T, Kalimo H, Wieloch T. Ultrastructural changes in the hippocampal CA1 region following transient cerebral ischemia: evidence against programmed cell death. Exp Brain Res. 1992;88:91–105. doi: 10.1007/BF02259131. [DOI] [PubMed] [Google Scholar]
- 15.Dietrich WD, Busto R, Alonso O, Globus MY-T, Ginsberg MD. Intraischemic but not postischemic brain hypothermia protects chronically following global forebrain ischemia in rats. J Cereb Blood Flow Metab. 1993;13:541–549. doi: 10.1038/jcbfm.1993.71. [DOI] [PubMed] [Google Scholar]
- 16.Dong Z, Saikumar P, Weinberg JM, Venkatachalam MA. Internucleosomal DNA cleavage triggered by plasma membrane damage during necrotic cell death. Am J Pathol. 1997;151:1205–1213. [PMC free article] [PubMed] [Google Scholar]
- 17.Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after mild focal cerebral ischemia: a role for apoptosis? J Cereb Blood Flow Metab. 1996;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gómez-Isla T, Hyman BT, Moskowitz MA. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J Cereb Blood Flow Metab. 1998;18:238–247. doi: 10.1097/00004647-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 19.Fukuda K, Kojiro M, Chiu J-F. Demonstration of extensive chromatin cleavage in transplanted morris hepatoma 7777 tissue: apoptosis or necrosis? Am J Pathol. 1993;142:935–946. [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia JH, Liu K-F, Ho K-L. Neuronal necrosis after middle cerebral artery occlusion in Wistar rats progresses at different time intervals in the caudoputamen and the cortex. Stroke. 1995;26:636–643. doi: 10.1161/01.str.26.4.636. [DOI] [PubMed] [Google Scholar]
- 21.Ghadially FN. Ultrastructural pathology of the cell and matrix, Vol 1, Ed 3. Butterworths; London: 1988. [Google Scholar]
- 22.Gold R, Schmied M, Giegerich G, Breitschopf H, Hartung HP, Toyka KV, Lassmann H. Differentiation between cellular apoptosis and necrosis by the combined use of in situ tailing and nick translation techniques. Lab Invest. 1994;71:219–225. [PubMed] [Google Scholar]
- 23.Goto K, Ishige A, Sekiguchi K, Iizuka S, Sugimoto A, Yuzurihara M, Aburada M, Hosoya E, Kogure K. Effects of cycloheximide on delayed neuronal death in rat hippocampus. Brain Res. 1990;534:299–302. doi: 10.1016/0006-8993(90)90144-z. [DOI] [PubMed] [Google Scholar]
- 24.Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995;21:1465–1468. doi: 10.1002/hep.1840210534. [DOI] [PubMed] [Google Scholar]
- 25.Harmon BV, Corder AM, Collins RJ, Gobe GC, Allen J, Allan DJ, Kerr JFR. Cell death induced in a murine mastocytoma by 42–47°C heating in vitro: evidence that the form of death changes from apoptosis to necrosis above a critical heat load. Int J Radiat Biol. 1990;58:845–858. doi: 10.1080/09553009014552221. [DOI] [PubMed] [Google Scholar]
- 26.Himi T, Ishizaki Y, Murota S-i. A caspase inhibitor blocks ischaemia-induced delayed neuronal death in the gerbil. Eur J Neurosci. 1998;10:777–781. doi: 10.1046/j.1460-9568.1998.00073.x. [DOI] [PubMed] [Google Scholar]
- 27.Honkaniemi J, Massa SM, Breckinridge M, Sharp FR. Global ischemia induces apoptosis-associated genes in hippocampus. Mol Brain Res. 1996;42:79–88. doi: 10.1016/s0169-328x(96)00121-0. [DOI] [PubMed] [Google Scholar]
- 28.Horn M, Schlote W. Delayed neuronal death and delayed neuronal recovery in the human brain following global ischemia. Acta Neuropathol (Berl) 1992;85:79–87. doi: 10.1007/BF00304636. [DOI] [PubMed] [Google Scholar]
- 29.Iwai T, Hara A, Niwa M, Nozaki M, Uematsu T, Sakai N, Yamada H. Temporal profile of nuclear DNA fragmentation in situ in gerbil hippocampus following transient forebrain ischemia. Brain Res. 1995;671:305–308. doi: 10.1016/0006-8993(94)01363-m. [DOI] [PubMed] [Google Scholar]
- 30.Johansen FF, Jørgensen MB, von Lubitz DKJE, Diemer NH. Selective dendrite damage in hippocampal CA1 stratum radiatum with unchanged axon ultrastructure and glutamate uptake after transient cerebral ischemia in the rat. Brain Res. 1984;291:373–377. doi: 10.1016/0006-8993(84)91272-1. [DOI] [PubMed] [Google Scholar]
- 31.Kalimo H, Garcia JH, Kamijyo Y, Tanaka J, Trump BF. The ultrastructure of brain death. II. Electron microscopy of feline cortex after complete ischemia. Virchows Archiv B Cell Pathol. 1977;25:207–220. [PubMed] [Google Scholar]
- 32.Kerr JFR. Shrinkage necrosis: a distinct mode of cellular death. J Pathol. 1971;105:13–20. doi: 10.1002/path.1711050103. [DOI] [PubMed] [Google Scholar]
- 33.Kihara S, Shiraishi T, Nakagawa S, Toda K, Tabuchi K. Visualization of DNA double strand breaks in the gerbil hippocampal CA1 following transient ischemia. Neurosci Lett. 1994;175:133–136. doi: 10.1016/0304-3940(94)91097-9. [DOI] [PubMed] [Google Scholar]
- 34.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 35.Kirino T, Sano K. Fine structural nature of delayed neuronal death following ischemia in the gerbil hippocampus. Acta Neuropathol (Berl) 1984;62:209–218. doi: 10.1007/BF00691854. [DOI] [PubMed] [Google Scholar]
- 36.Loskoto WJ, Lomax P, Verity MA. A stereotaxic atlas of the Mongolian gerbil brain (Meriones unguiculatus). Ann Arbor Science; Ann Arbor, MI: 1975. [Google Scholar]
- 37.MacManus JP, Linnik MD. Gene expression induced by cerebral ischemia: an apoptotic perspective. J Cereb Blood Flow Metab. 1997;17:815–832. doi: 10.1038/aj.jcbfm.9590266. [DOI] [PubMed] [Google Scholar]
- 38.MacManus JP, Buchan AM, Hill IE, Rasquinha I, Preston E. Global ischemia can cause DNA fragmentation indicative of apoptosis in rat brain. Neurosci Lett. 1993;164:89–92. doi: 10.1016/0304-3940(93)90864-h. [DOI] [PubMed] [Google Scholar]
- 39.MacManus JP, Hill IE, Preston E, Rasquinha I, Walker T, Buchan AM. Differences in DNA fragmentation following transient cerebral or decapitation ischemia in rats. J Cereb Blood Flow Metab. 1995;15:728–737. doi: 10.1038/jcbfm.1995.93. [DOI] [PubMed] [Google Scholar]
- 40.Ni B, Wu X, Su Y, Stephenson D, Smalstig EB, Clemens J, Paul SM. Transient global forebrain ischemia induces a prolonged expression of the caspase-3 mRNA in rat hippocampal CA1 pyramidal neurons. J Cereb Blood Flow Metab. 1998;18:248–256. doi: 10.1097/00004647-199803000-00003. [DOI] [PubMed] [Google Scholar]
- 41.Nishikawa Y, Takahashi T, Shimoda A. Morphological studies on cerebral cortical lesions induced by transient ischemia in Mongolian gerbil-diffuse and peripheral pallor of the neuronal perikarya. Acta Neuropathol (Berl) 1989;78:1–8. doi: 10.1007/BF00687395. [DOI] [PubMed] [Google Scholar]
- 42.Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nurse S, Corbett D. Direct measurement of brain temperature during and after intraischemic hypothermia: correlation with behavioral, physiological, and histological endpoints. J Neurosci. 1994;14:7726–7734. doi: 10.1523/JNEUROSCI.14-12-07726.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okamoto M, Matsumoto M, Ohtsuki T, Taguchi A, Mikoshiba K, Yanagihara T, Kamada T. Internucleosomal DNA cleavage involved in ischemia-induced neuronal death. Biochem Biophys Res Commun. 1993;196:1356–1362. doi: 10.1006/bbrc.1993.2402. [DOI] [PubMed] [Google Scholar]
- 45.Petito CK, Feldmann E, Pulsinelli WA, Plum F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology. 1987;37:1281–1286. doi: 10.1212/wnl.37.8.1281. [DOI] [PubMed] [Google Scholar]
- 46.Petito CK, Torres-Munoz J, Roberts B, Olarte JP, Nowak TS, Jr, Pulsinelli WA. DNA fragmentation follows delayed neuronal death in CA1 neurons exposed to transient global ischemia in the rat. J Cereb Blood Flow Metab. 1997;17:967–976. doi: 10.1097/00004647-199709000-00006. [DOI] [PubMed] [Google Scholar]
- 47.Petito CK, Olarte JP, Roberts B, Nowak TS, Jr, Pulsinelli WA. Selective glial vulnerability following transient global ischemia in rat brain. J Neuropathol Exp Neurol. 1998;57:231–238. doi: 10.1097/00005072-199803000-00004. [DOI] [PubMed] [Google Scholar]
- 48.Portera-Cailliau C, Price DL, Martin LJ. Non-NMDA and NMDA receptor-mediated excitotoxic neuronal deaths in adult brain are morphologically distinct: further evidence for an apoptosis-necrosis continuum. J Comp Neurol. 1997;378:88–104. [PubMed] [Google Scholar]
- 49.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 50.Symmens WSC. Symmens systemic pathology, Vol 4. Churchill Livingstone; New York: 1986. [Google Scholar]
- 51.Tomimoto H, Yamamoto K, Homburger HA, Yanagihara T. Immunoelectron microscopic investigation of creatine kinase BB-isoenzyme after cerebral ischemia in gerbils. Acta Neuropathol (Berl) 1993;86:447–455. doi: 10.1007/BF00228579. [DOI] [PubMed] [Google Scholar]
- 52.Wachstein M, Besen M. Electron microscopy of renal coagulative necrosis due to dl-serine, with special reference to mitochondrial pyknosis. Am J Pathol. 1964;44:383–400. [PMC free article] [PubMed] [Google Scholar]
- 53.Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 54.Yamamoto K, Hayakawa T, Mogami H, Akai F, Yanagihara T. Ultrastructural investigation of the CA1 region of the hippocampus after transient cerebral ischemia in gerbils. Acta Neuropathol (Berl) 1990;80:487–492. doi: 10.1007/BF00294608. [DOI] [PubMed] [Google Scholar]
- 55.Zamzami N, Susin SA, Marchetti P, Hirsch T, Gomez-Monterrey I, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zamzami N, Hirsch T, Dallaporta B, Petit PX, Kroemer G. Mitochondrial implication in accidental and programmed cell death: apoptosis and necrosis. J Bioenerg Biomembr. 1997;19:185–193. doi: 10.1023/a:1022694131572. [DOI] [PubMed] [Google Scholar]