

Abstract
Interleukin-1 (IL-1) is a pleotrophic cytokine implicated in a variety of central activities, including fever, sleep, ischemic injury, and neuromodulatory responses, such as neuroimmune, and neuroendocrine interactions. Although accumulating evidence is available regarding the expression pattern of this cytokine, its receptors in the CNS, and its mechanistic profile under pathological levels, it is unclear whether this substance modulates central neurons under physiological concentrations. Further, in light of the functional and spatial overlap between the adenosine and IL-1 systems, it is not known whether these two systems are coupled. We report here that, in rat brain slices, brief application of sub-femtomolar IL-1β causes a profound decrease of glutamate transmission, but not GABAergic inhibition, in hippocampal CA1 pyramidal neurons. This decrease by IL-1β is prevented by pharmacological blockade of adenosine A1 receptors. In addition, we show that IL-1β failed to suppress glutamate transmission at room temperature. Because the production and release of adenosine in the CNS is thought to be metabolically dependent, this observation suggests that one of the functions of IL-1β is to increase the endogenous production of adenosine. Together, these data suggest for the first time that sub-femtomolar levels of IL-1 can effectively modulate glutamate excitation in hippocampal neurons via an adenosine-dependent mechanism.
Keywords: adenosine, brain slices, cytokine, electrophysiology, femtomolar, glutamatergic transmission, hippocampus, interleukin-1β
Numerous pathological conditions, such as central and peripheral manifestations of inflammation, and ischemic episodes are associated with increased interleukin-1 (IL-1) protein and gene expression. The pathophysiology accompanying these states, such as neuronal cell death, fever, reductions in food gathering, and sexual behavior, and increases in sleep and lethargy can be attenuated by treatment with IL-1 antibodies and IL-1 receptor antagonist (for review, see Dinarello et al., 1990; Dinarello, 1991,1994; Schobitz et al., 1994; Rothwell and Hopkins, 1995). Because of this, IL-1 has been traditionally considered as a mediator arising in pathological situations. More recently, however, many studies have been converging on the hypothesis that IL-1 is involved in normal physiological processes. Immunocytochemistry studies show IL-1 bioactivity in the normal healthy rat CNS (Quan et al., 1996). In addition, IL-1 has been implicated in the mediation of physiological sleep (for review, see Krueger et al., 1995; Takahashi et al., 1996), synaptic plasticity, neuroimmune, and neuroendocrine interactions (Nguyen et al., 1998; Schneider et al., 1998) in normal healthy animals.
When applied directly onto central neurons in vitro, IL-1 has been shown to produce several electrophysiological changes, such as alteration of firing patterns (Nakashima et al., 1989; Kuriyama et al., 1990; Li et al., 1992; Yamashita et al., 1995; Mo et al., 1996), inhibition of voltage-gated calcium currents (Plata-Salaman and Ffrench-Mullen 1992, 1994), and modulation of excitatory (Katsuki et al., 1990; Bellinger et al., 1993; Yu and Shinnick-Gallagher, 1994; Cunningham et al., 1996; Coogan and O’Connor 1997; D’Arcangelo et al., 1997) and/or inhibitory (Miller et al., 1991; Zeise et al., 1992; Yu and Shinnick-Gallagher, 1994; Pringle et al., 1996) synaptic responses. Although these studies demonstrate that IL-1 can induce neurophysiological changes, they are all limited to some extent by the high picomolar to nanomolar concentrations of IL-1 used, which are characteristic of pathological conditions (Symons et al., 1987; Jacobs and Tabor, 1990; Cacabelos et al., 1991). When considering the effects of IL-1 on central neurons, it is becoming apparent that it is important to make a distinction between the effects of pathological versus physiological levels of IL-1. Scheider et al. (1998) highlight this in a recent study, which demonstrates that physiological levels of IL-1 are necessary for the maintenance of long-term potentiation. Contrary to this, others have shown that high levels of IL-1 (nanomolar) inhibit long-term potentiation (Katsuki et al., 1990; Bellinger et al., 1993; Cunningham et al., 1996). Becausein vivo animal model and cell culture data show that femtomolar IL-1 (fm IL-1) is able to cause physical and cellular changes (for review, see Sundar et al., 1989; Dinarello, 1994; Rosoff et al., 1988), one of our objectives was to determine whether physiological levels of IL-1 (femtomolar to low picomolar) could alter synaptic activity of central neurons in the brain slice model.
One of the characteristic properties of the IL-1 system is its ability to induce slow-wave sleep (for review, see Krueger et al., 1995). Much evidence indicates that this role is played out in normal physiological conditions, in addition to situations of infection and inflammation. IL-1β injected peripherally or centrally has the effect of inducing slow-wave sleep; IL-1 receptor antagonists have the ability to attenuate normal spontaneous sleep (Takahashi et al., 1996), and animals with IL-1 type I receptor knock-outs are deficient in sleep (Fang et al., 1998). Consistent with these findings, IL-1 activity is elevated in cat CSF with entry into sleep (Lue et al., 1988), and its mRNA is upregulated during sleep deprivation (Mackiewicz et al., 1996) and varies diurnally with the sleep–wake cycle, declining in correlation to the increasing amounts of previous sleep (Taishi et al., 1997).
A neurotransmitter that shares striking functional and spatial similarities to IL-1 is adenosine. Microdialysis measurements in freely behaving cats demonstrate that extracellular concentrations of adenosine in the brain progressively increased during wakefulness and declined slowly during recovery sleep. Furthermore, increases in the amounts of slow-wave sleep seen after prolonged wakefulness are mimicked by central administration of adenosine transport inhibitor [S-(4-nitrobenzyl)-6-thioinosine], which raises extracellular adenosine (Porkka-Heiskanen et al., 1997). Immunocytochemistry and autoradiography studies have revealed that adenosine A1 and IL-1 receptors are coexpressed in discreet regions in the CNS (for review, see Goodman and Synder, 1982; Fastbom et al., 1987a,b; Cunningham et al., 1992, 1993; Schöbitz et al., 1994). In light of these previous reports, we decided to investigate whether these two systems were coupled in any respect.
MATERIALS AND METHODS
To determine whether low levels of IL-1 modulate central synaptic activity and whether there is any coupling between the adenosine and IL-1 systems, standard electrophysiological recording techniques were applied to the hippocampal CA1 region of rat brain slices. The hippocampus was ideal for testing our hypothesis because its local networks are well characterized; moreover, it represents a brain region with the highest densities of both adenosine A1 and IL-1 receptors (see introductory remarks).
Brain slice preparation and electrophysiological recordings were performed as described previously (Zhang et al., 1991). Briefly, male Wistar rats (25- to 50-d-old) were anesthetized by halothane and decapitated. The brain was quickly dissected out and sliced transversely to 400 μm sections in an ice-cold artificial CSF (ACSF). Brain slices were then maintained in oxygenated (5% CO2–95% O2) ACSF at room temperature (22–23°C) for at least 1 hr before recording. The composition of the ACSF was (in mm): NaCl 125, KCl 2.5, NaH2PO4 1.25, CaCl2 2, MgSO4 1.8, NaHCO3 26, and glucose 10.
Electrophysiological recordings were done in a fully submerged chamber at 32–33°C except when indicated. The slice perfusion rate was 4–5 ml/min. Field synaptic potentials were recorded extracellularly using a NaCl-filled glass pipette in stratum radiatum of the CA1 region. Schaffer collateral–CA1 afferents were electrically stimulated by placing a bipolar tungsten electrode in the stratum radiatum at the CA1–CA2 border. Constant current pulses of 0.1 msec duration was generated via a Grass stimulator (S88; Grass Instruments, Quincy, MA) and delivered via an isolation unit every 15 sec.
For whole-cell patch recordings, the patch pipette was filled with a solution of 150 mm potassium methylsulfate, 2 mm HEPES, and 0.1 mm K-EGTA, pH 7.25 (osmolarity of 280 ± 10 mOsm) (Zhang et al., 1994). The tip resistance of the filled patch pipette was ∼4 MΩ, and the series resistance after membrane breakthrough was <15 MΩ. Signals were recorded via an Axopatch amplifier (200B; Axon Instruments, Foster City, CA). The low-pass filter was set at 5 kHz, and the series resistance compensation was ∼80%. Data were acquired, digitized, and stored using pClamp software (version 6.3) and a 12-bit analog-to-digital interface (Digidata 1200; Axon Instruments).
Recombinant rat IL-1β, recombinant mouse IL-1 receptor antagonist (IL-1ra), and anti-rat IL-1β polyclonal antibody were obtained from R & D Systems (Minneapolis, MN). The specific activity for rat IL-1β was determined by R & D Systems using a mouse D10.G4.1 helper T cell line proliferation assay, and the ED50 was between 1–3 ng/ml. IL-1β was initially dissolved in sterile PBS that contained 0.1% albumin as the carrier protein. The stock solution was stored at −80°C and then appropriately diluted to the ACSF at desired concentrations just before each experiment. Before the application of IL-1β, the slices were perfused with the control ACSF that contained the same amount of PBS–albumin until stable responses were reached. To minimize nonspecific binding of IL-1β, all apparatus that came into contact with IL-1 was thoroughly and routinely siliconated with Aquasil (Pierce, Rockford, IL). We found that it was critical to thoroughly and routinely siliconize all apparatus in contact with IL-1 to obtain measurable results. Adenosine A1 antagonists 8-(p-sulfophenyl)theophyilline (8-PST) and 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), as well as the adenosine A1 receptor agonist adenosine amine congener (ADAC), were obtained from Research Biochemicals (Natick, MA).
Adenosine in superfusates from the slices was determined essentially as described previously (Hoehn and White, 1990; Semba and White, 1997). Briefly, samples were deproteinated with 0.3 mZnSO4 and 0.3 m Ba(OH)2, and the supernatants were placed in a boiling water bath with 5.4% chloroacetaldehyde to form ethenoadenosine. Samples were concentrated under N2, and ethenoadenosine detected by HPLC with fluorescence detection. Adenosine release was expressed as picomoles per milliliter of supernatant.
RESULTS
Robust inhibition of field EPSPs by IL-1β
After electrical stimulation of Schaffer collateral afferents (the major glutamatergic input to the CA1 region) at maximal strength, field EPSPs (fEPSP) were recorded extracellularly in the hippocampal CA1 area. These fEPSP were fully blocked by 20 μm6-cyano-7-nitroquinozaline-2,3-dione (CNQX), confirming their mediation by AMPA glutamate receptors (n = 5) (Shinno et al., 1997).
Perfusion of slices with recombinant rat IL-1β, at concentrations as low as 10−17m, caused decreases in the amplitude of fEPSPs compared with baseline control (p < 0.01; paired t test) (Fig.1A). The decrease in fEPSPs started 1 min after beginning IL-1β perfusion, achieved a plateau within the 4 min application period, and fully recovered after washing (Fig. 1B). This decrease exhibited a linear concentration dependency between 10−19 and 10−15m IL-1β. Saturation was observed at concentrations ≥10−15mIL-1β, where fEPSPs were inhibited to ∼38% of peak baseline amplitude (Fig. 1C). The decrease in fEPSPs by IL-1β at different concentrations was not accompanied by any substantial changes in the presynaptic volleys (Fig. 1A). In a set of slices in which the presynaptic volley was clearly visualized (n = 14), their amplitude was unchanged after application of IL-1β (0.65 ± 0.11 and 0.64 ± 0.11 mV measured before and after IL-1β application, respectively).
Fig. 1.
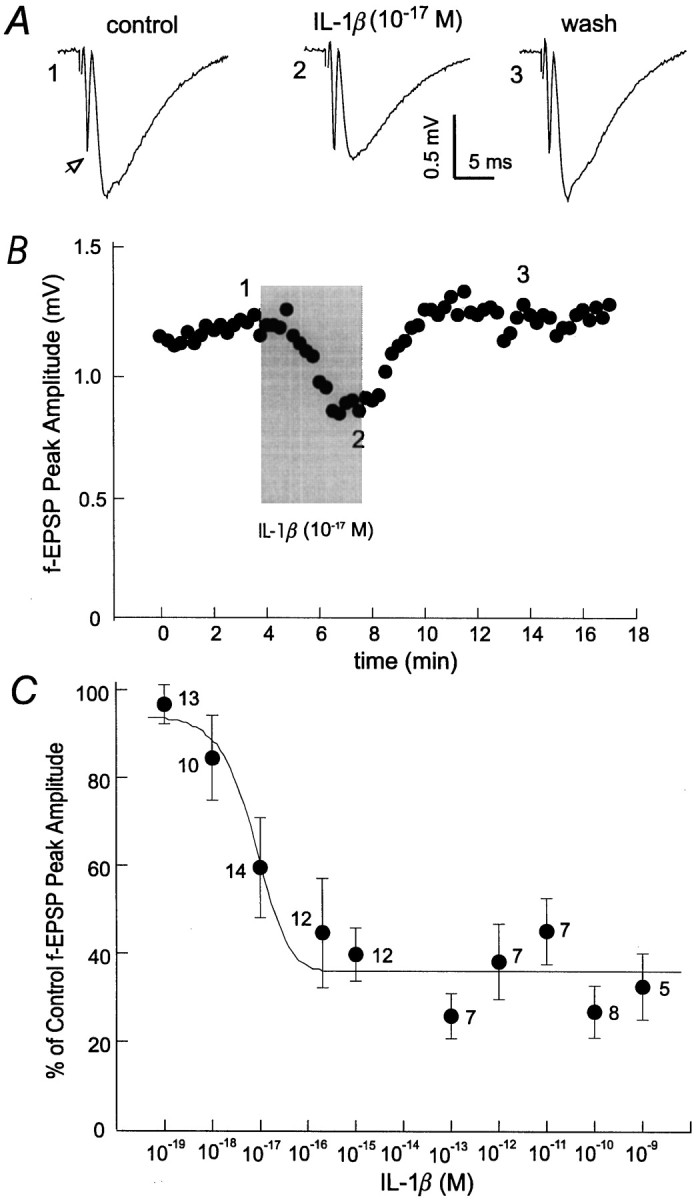
Suppression of fEPSPs by sub-fmIL-1β. A, fEPSPs were recorded from the hippocampal CA1 region of a brain slice after constant afferent stimulation every 15 sec. Each trace was averaged from three consecutive measurements and collected before, during, or after washing out IL-1β (10−17m). The open arrow indicates the presynaptic volley. B, Amplitudes of fEPSPs were plotted versus time; numbered data points correspond to the traces illustrated inA. The shaded column indicates the time period of IL-1β application. Recordings were taken after stabilized fEPSPs were achieved. C, Concentration–response relationship for IL-1β suppression of fEPSPs. Changes in fEPSPs amplitudes were normalized as percentages of baseline control; this was plotted versus the concentrations of IL-1β used. The line through the data points is a computed Lorentzian fit using the following equation:y = a + b/(1 + ((x −c)/d)2), wherea = 36.1, b = 321.3,c = −1.4 × 10−17, andd = 6.6 × 10−18;r2 = 0.92. The number of slices examined for each data point is indicated.
To test whether the effect of IL-1β on glutamate transmission was related to the strength of afferent stimulation, CA1 fEPSPs were evoked by afferent stimulation at near-threshold, half-maximal, and maximal strength, and changes were then monitored after the perfusion of 10−16m IL-1β (n = 6). All resultant fEPSPs were reduced by ∼60% compared with baseline (p < 0.001; paired t test) (Fig.2A,B), indicating a general suppression by sub-fm IL-1β on stimulated hippocampal glutamatergic synapses.
Fig. 2.
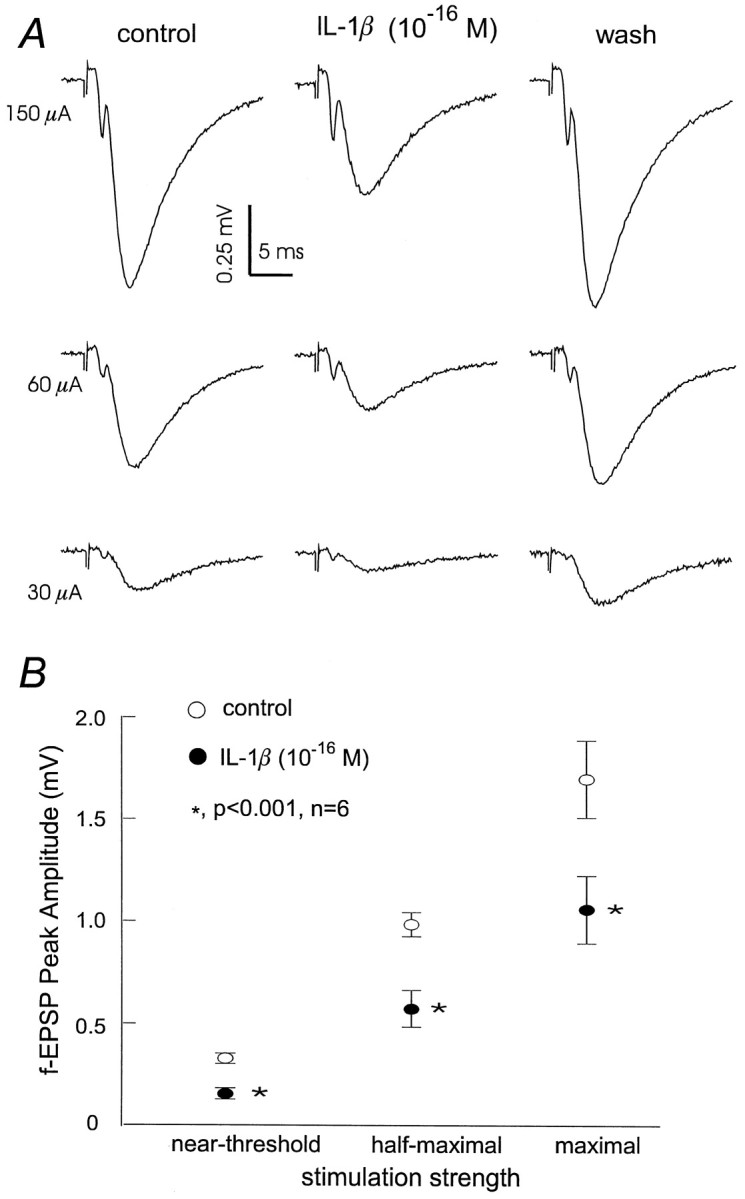
Sub-fm IL-1β exerts a generalized depression on CA1 fEPSPs. A, Representativetraces of fEPSPs recorded from a slice after afferent stimulation at near-threshold, half-maximal, or maximal strength. Eachtrace was averaged from three consecutive measurements before, during, and after 10−16mIL-1β application. B, Peak amplitude of fEPSPs plotted versus afferent stimulation at near-threshold, half-maximal, or maximal strength, respectively. Data were collected from a set of six slices.Open and filled circles represent measurements obtained before or after IL-1β application (10−16m, for 4 min), respectively. Mean ± SEM are indicated.
IL-1 receptor-mediated inhibition of fEPSPs
To determine that our observations were mediated via activated IL-1 receptors and were IL-1β-specific, IL-1ra was continuously perfused in the hippocampal isolate before IL-1β application (n = 7) (Fig.3Aii). Control perfusion of 1.5 × 10−12m IL-1ra alone for 10 min caused no significant changes in fEPSPs (5.6 ± 8.0% from baseline of 1.88 ± 0.04 mV). Application of 10−13m IL-1β (the level at which the suppression of fEPSPs was saturated and most robust) (Figs.1C, 3Ai) in the presence of IL-1ra attenuated its ability to inhibit fEPSPs: 29.1 ± 12.2% compared with 74.1 ± 5.1% by IL-1β alone (p < 0.001; one-way ANOVA) (Fig. 3B). In a separate experiment, IL-1β was neutralized with a polyclonal antibody raised against rat IL-1β (10−13m IL-1β in 3.3 × 10−12m anti-rat IL-1β antibody) (Fig. 3Aiii). The application of antibody-neutralized IL-1β produced a 12.0 ± 6.7% decrease in fEPSPs, again greatly attenuated compared with that induced by nontreated IL-1β at the same concentration (p < 0.001; one-way ANOVA) (Fig.3B).
Fig. 3.
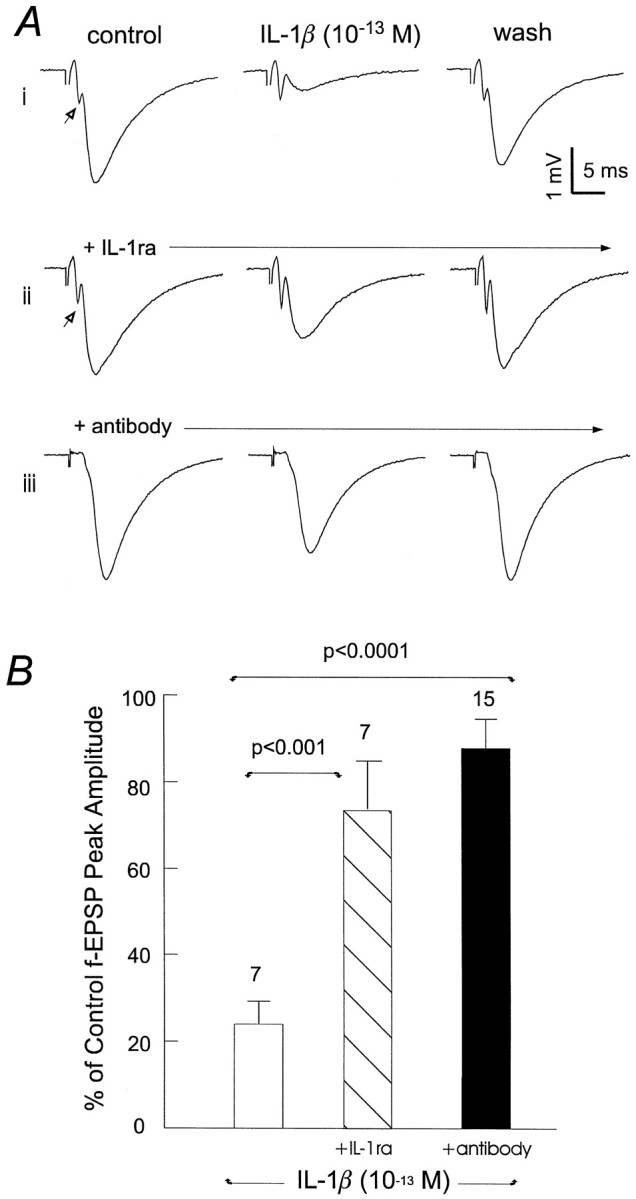
IL-1 receptor-mediated fEPSP depression.A, Records were collected from three separate slices, and each individual trace was averaged from three measurements. i, IL-1β alone. ii, IL-1β plus IL-1ra. iii, Antibody-neutralized IL-1β. To control for any nonspecific effects, the antibody and receptor antagonist were maintained at a constant concentration throughout the recording period. Open arrows indicate the presynaptic volley. B, Percent of baseline control fEPSP peak amplitude measured in the presence of 10−13m IL-1β alone, 10−13mIL-1β plus 1.5 × 10−12mIL-1ra, and 10−13m IL-1β neutralized by 3.3 × 10−12m anti-IL-1β antibody. Mean ± SEM and the number of slices examined in each group are indicated. Statistical significance was calculated via one-way ANOVA.
Selective decrease of EPSCs but not IPSCs by IL-1β
To assess whether a similar pattern of decreased synaptic transmission by IL-1β occurred at the single cell level, synaptic currents evoked by afferent stimulation were recorded from individual CA1 pyramidal neurons in the whole-cell voltage-clamp mode. At a holding potential of approximately −60 mV, CA1 neurons displayed transient, inward currents, referred to as EPSCs (Fig.4A, top). We have shown previously that these EPSCs are blocked by CNQX (Shinno et al., 1997; Ouanounou et al., 1999), indicating their mediation by AMPA glutamate receptors. Perfusion of slices with 10−15m IL-1β for 4–5 min reversibly suppressed the EPSCs by 70% (n = 8) (Fig.4A,B), paralleling the results obtained by extracellular recordings.
Fig. 4.
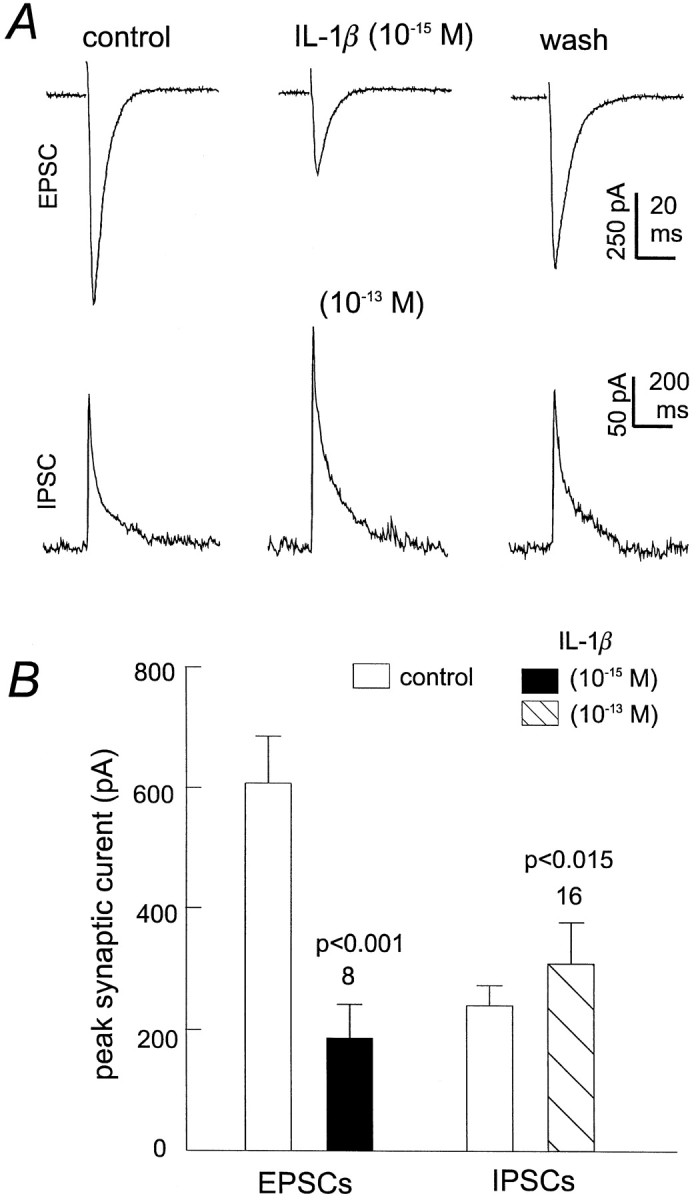
Sub-fm IL-1β inhibits EPSCs and slightly potentates IPSCs. A, Representative EPSCs (top) and IPSCs (bottom) recorded from two CA1 pyramidal neurons at the holding potential of −60 or −50 mV, respectively. Each trace was averaged from three consecutive measurements and was recorded before, at the end of IL-1β application (10−15 or 10−13m, 5–7 min), and after washing. IPSCs were isolated pharmacologically by perfusing slices with 20 μm CNQX throughout the recording period. B, Peak amplitude of EPSCs or IPSCs measured before and after application of IL-1β. Mean ± SEM and the number of CA1 pyramidal neurons examined in each group are indicated, and statistical significance was calculated via a paired t test.
IPSCs were evoked in the presence of 20 μm CNQX. Subsequent application of IL-1β at 10−13m caused a slight but significant increase in the amplitude of IPSC from 265.4 ± 44.7 to 319.5 ± 59.6 pA (n = 16; p < 0.015; pairedt test) (Fig. 4A, bottom,B). Our laboratory has shown previously that these pharmacologically isolated IPSCs are mediated by Cl−-dependent GABAA-mediated currents (Zhang et al., 1991, 1993, 1998). Therefore, IL-1β may weakly potentiate GABAergic transmission in the rat hippocampus.
Decrease in fEPSPs by IL-1β was prevented by adenosine A1 receptor antagonists
Stimulation of adenosine A1 receptors in the hippocampal CA1 region has been shown to inhibit glutamatergic transmission without suppressing GABAergic transmission (Lambert and Teyler, 1991; Yoon and Rothman, 1991; Thompson et al., 1992; Khazipov et al., 1995). This similarity shared by adenosine A1receptor stimulation and IL-1β application suggested that these two systems might indeed be coupled. To test this idea, slices were first perfused thoroughly with 20 μm DCPCX, an adenosine A1 receptor antagonist. After application of DCPCX for >10 min, CA1 fEPSPs stabilized at 2.05 ± 0.09 mV, no significant difference from baseline at 1.97 ± 0.09 mV (n = 15). In the presence of DCPCX, subsequent applications of 10−13m IL-1β for 5 min caused no substantial decrease in fEPSPs (Δ, 4.2 ± 1.6%;n = 15). We also examined the effect of 8-PST, a water soluble adenosine A1 receptor antagonist, on IL-1β induced synaptic inhibition, using a similar protocol to that of DPCPX application. In the presence of 5–10 μm 8-PST (concentrations that are preferential for adenosine A1receptors; Bruns et al., 1980; Rainnie et al., 1994), subsequent applications of 10−13mIL-1β for 5 min caused no substantial decrease in fEPSPs (Δ, 8.8 ± 6.4%; n = 10) (Fig.5A,C). These observations were in sharp contrast to those observed in control slices in which the similar application of IL-1β alone greatly inhibited CA1 fEPSPs by 61.0 ± 5.9% (p < 0.0001; paired t test; n = 17) (Fig.5A,C).
Fig. 5.
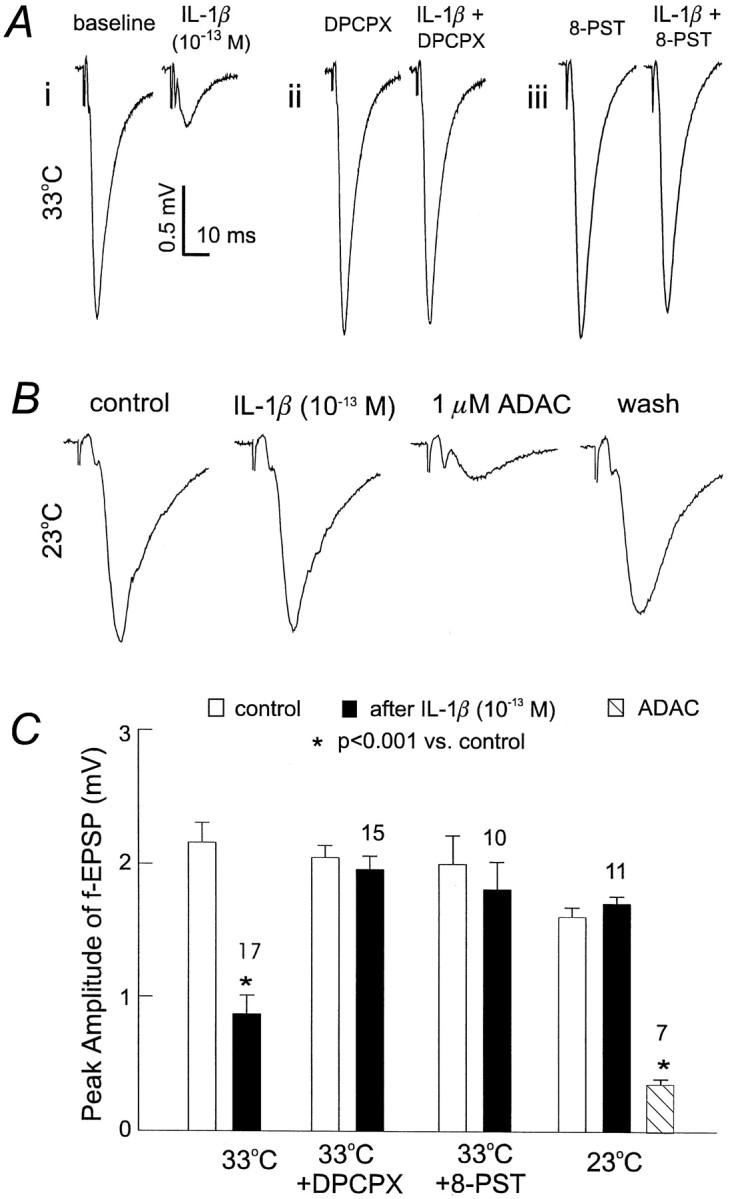
Adenosine-dependent suppression of fEPSPs by IL-1β. A, fEPSPs were recorded from three slices. Eachtrace was averaged from three consecutive measurements and collected before or after the application of IL-1β at 33°C.i, IL-1β alone. ii, In the presence of 20 μm DCPCX, a adenosine A1 receptor antagonist. iii, In the presence of 10 μm8-PST, a water soluble adenosine A1 receptor antagonist.B, Representative fEPSPs were recorded from a single slice at 23°C; each trace was averaged from three consecutive measurements. Left two traces were collected before and after IL-1β application. Right two traceswere recorded in the presence of ADAC, a stable adenosine A1 receptor agonist, and after wash. C, Changes in fEPSPs by IL-1β examined under four conditions (33°C, 33°C plus DPCPX, 33°C plus 8-PST, and 23°C). Open columns represent baseline control, filled columns represent measurement in the presence of IL-1β with one of the four conditions, and the hatched columnrepresents measurement in the presence of ADAC. Mean ± SEM and number of slices examined are indicated. Statistically significant decreases from the baseline control, *p < 0.0001; paired t test.
Promotion of endogenous adenosine release by IL-1B?
To provide additional evidence for the possibility that adenosine mediates IL-1β-induced glutamatergic suppression, we examined the effects of IL-1β at room temperature (22–23°C). Because the synthesis and transmembrane flux of adenosine is directly related to energy metabolism (for review, see Brundege and Dunwiddie, 1997), we hypothesized that the suppression of fEPSPs by IL-1β would be attenuated at room temperature. As expected, CA1 fEPSPs recorded at room temperature showed no substantial inhibition after exposure to 10−13m IL-1β for 5–6 min (Fig.5B). However, they were strongly suppressed by subsequent application of 1 μm ADAC, a stable adenosine A1 receptor agonist, indicating that the adenosine A1 receptor cascade remained functional at room temperature (Fig. 5B).
To measure the possible elevation of extracellular adenosine by IL-1β, perfused ACSF was collected before and at the end of IL-1β application (10−13m for ∼4 min) when the fEPSPs were decreased by 78 ± 6.8% from baseline (n = 8). The collected ACSF was frozen immediately to −70°C and analyzed via HPLC and fluorescence assays (Hoehn and White, 1990; Semba and White, 1997). The decreased fEPSPs were not associated with any significant observable changes in extracellular adenosine (−5 ± 8%), from a basal level of 6–8 pmol/ml collected before the IL-1β application.
DISCUSSION
Two major findings emerge from the present experiments: (1) brief application of sub-fm IL-1β selectively decreases glutamate AMPA receptor-mediated transmission in the rat hippocampus; and (2) the decrease by IL-1β is mediated through an adenosine-dependent pathway. These data suggest for the first time that there is a coupling between the adenosine and IL-1 systems. Further, our data reinforce the idea that the IL-1 system plays a regulatory role at physiological levels found in the normal mammalian CNS. However, it should be noted that the in vivo action of IL-1 is much more complex than observed in an isolated brain slice. Our results do not in any way prove that the slow-wave sleep-promoting aspects of IL-1 are mediated via adenosine; rather, our data provide clues for further investigation into these issues.
The concentration–response curve shown in Figure 1 highlights two important aspects. First, during infection and inflammation, IL-1β is found in the CNS at picomolar to nanomolar levels; it is at this concentration that IL-1 assumes the role of a primary immune mediator. We observed that, at this level, IL-1β suppression of glutamatergic fEPSP was saturated at 70% of baseline control, whereas the linear portion of the concentration–response curve was observed at the sub-femtomolar to femtomolar range. These data demonstrate that the activation profile of IL-1β is characteristic of a neuromodulatory substance, in that IL-1β alters, but does not abolish, glutamatergic excitation. Second, these data illustrate that sub-fmIL-1β can effectively regulate glutamate transmission and thus suggest that subtle modifications in the basal levels of IL-1βin vivo could greatly effect normal brain functioning.
Because application of IL-1 receptor antagonist and neutralized IL-1β via its specific polyclonal antibody attenuated the ability of IL-1β to suppress fEPSPs, we concluded that the inhibition by IL-1β of glutamatergic AMPA receptor-mediated EPSP–EPSCs is conveyed via IL-1 receptors and is IL-1β-specific. Our findings are in apparent contradiction to previously published data reporting that IL-1β inhibits NMDA, but not AMPA, receptor-mediated fEPSP in the dentate gyrus region of brain slices (Coogan and O’Connor, 1997). However, the length and dose of application used in that study is well above the level we used in the present experiments.
The decrease in glutamatergic transmission by IL-1β manifests itself in both field and single cell recordings and was not associated with substantial changes in the afferent axonal potentials. These observations suggest that IL-1β suppression of the glutamate response results from decreased glutamate release (McGahon and Lynch, 1995;Murray et al., 1997) and not the attenuation of presynaptic excitability.
Adenosine is an important modulatory neurotransmitter implicated in a variety of brain activities, particularly those related to sleep and ischemic–hypoxic episodes (for review, see Phillis and Wu, 1981;Snyder, 1985; Brundege and Dunwiddie, 1997; Porkka-Heiskanen et al., 1997). Of the multiple neurophysiological actions, inhibition of glutamate transmission by adenosine has been noted for some time in several brain regions (Dunwiddie, 1985; Greene and Haas, 1991) and is likely a result of the inhibition of presynaptic calcium influx (Wu and Saggau, 1994). In the hippocampal CA1 region, adenosine-induced decreases in glutamate transmission are mediated via A1 subtype receptors, and this suppression occurs without directly affecting GABAergic transmission (Yoon and Rothman 1991;Capogna et al., 1993). We believe that the decrease of EPSPs by IL-1β presented here is conveyed through endogenous adenosine acting on A1 receptors based on the following observations. First, the decrease by IL-1β is only seen in glutamate EPSPs–EPSCs but not in GABAergic IPSCs. Second, IL-1β-induced fEPSP suppression is fully blocked by the adenosine A1 receptor antagonists DPCPX and 8-PST. Third, when examined at room temperature, the evoked fEPSPs were insensitive to IL-1β but were greatly suppressed by application of the adenosine A1 receptor agonist ADAC (Fig.5B,C). Our interpretation of these three observations is that the adenosine A1 receptor signal cascade can be turned on at room temperature after direct agonist stimulation and that the failure by IL-1β to suppress fEPSPs at room temperature reflects insufficient stimulation of adenosine A1 receptors. Therefore, in light of metabolic–temperature dependence of endogenous adenosine release and the subsequent modulation of glutamate transmission (Masino and Dunwiddie, 1999), we propose that IL-1 acts to suppress glutamate transmission via promoting endogenous adenosine release rather than by sensitizing the adenosine A1 receptor-mediated signal cascade.
However, an increase by IL-1β in extracellular adenosine was not detectable by HPLC measurement in the present experiments. It is possible that IL-1β promotes a local release of adenosine at sites near the activated glutamatergic synapses, at levels that are sufficient to suppress the glutamate transmission but too low to be measured in the perfusate collected from the entire slice. Previous studies have noted that enhanced adenosine release is characteristically found only in the proximity of activated synapses (Manzoni et al., 1994; Brundege and Dunwiddie, 1996). The stimulation afforded by our experimental settings only activates a small amount of the total glutamate synapses in the hippocampal slice at any one time. Therefore, the lack of a detectable increase in extracellular adenosine by IL-1β in the hippocampal slice is likely caused by the nature of adenosine release only at activated synapses. Although further experiments are needed to resolve the full nature of this mechanism, our observations with adenosine A1 receptor antagonists and the temperature dependency of the actions of IL-1β are consistent with the above assumption.
In summary, the present data suggest that IL-1β can effectively modulate glutamate transmission in the hippocampus at sub-femtomolar concentrations, likely via an adenosine-dependent pathway. The strong coexpression of both IL-1 receptors and adenosine A1receptors in the mammalian CNS suggests that IL-1 may act via adenosine-mediated mechanisms in other regions of the brain. However, this does not preclude IL-1 actions via other pathways, because we have observed the enhancement of IL-1β of GABAergic IPSCs, which do not appear to be adenosine-dependent (Yoon and Rothman, 1991; Capogna et al., 1993). What remains to be seen is whether such regulation of synaptic activity by IL-1β occurs in vivo and its relevance to the induction of slow-wave sleep and other physiological activities.
Footnotes
This work was supported by Canadian Medical Research Council Grant MT-12943 to L.Z. and by a Toronto Psychiatric Research Foundation grant to W.P.L. L.Z. is a Research Scholar of the Heart and Stroke Foundation of Canada.
W. P. Luk and Y. Zhang contributed equally to this work.
Correspondence should be addressed to Dr. Liang Zhang, Toronto Hospital (Western Division), 399 Bathurst Street, McLaughlin Pavillion, Room 13-411, Toronto, Ontario, Canada M5T 2S8.
REFERENCES
- 1.Bellinger FP, Madamba S, Siggins GR. Interleukin 1β inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993;628:227–234. doi: 10.1016/0006-8993(93)90959-q. [DOI] [PubMed] [Google Scholar]
- 2.Brundege JM, Dunwiddie TV. Modulation of excitatory synaptic transmission by adenosine released from single hippocampal pyramidal neurons. J Neurosci. 1996;16:5603–5612. doi: 10.1523/JNEUROSCI.16-18-05603.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brundege JM, Dunwiddie TV. Role of adenosine as a modulator of synaptic activity in the central nervous system. Adv Pharmacol. 1997;39:353–391. doi: 10.1016/s1054-3589(08)60076-9. [DOI] [PubMed] [Google Scholar]
- 4.Bruns RF, Daly JW, Snyder SH. Adenosine receptors in brain membranes: binding of N6-cyclohexyl[3H]adenosine and 1,3-diethyl-8-[3H]phenylxanthine. Proc Natl Acd Sci USA. 1980;77:5547–5551. doi: 10.1073/pnas.77.9.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cacabelos R, Barquero M, Garcia P, Alvarez XA, Deseijas EV. Cerebrospinal fluid interleukin-1β (IL-1β) in Alzheimer’s disease and neurological disorders. Methods Find Exp Clin Pharmacol. 1991;13:455–458. [PubMed] [Google Scholar]
- 6.Capogna M, Gahwiler BH, Thompson SM. Mechanisms of μ-opiod receptor-mediated presynaptic inhibition in rat hippocampus in vitro. J Physiol (Lond) 1993;470:539–558. doi: 10.1113/jphysiol.1993.sp019874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coogan A, O’Connor JJ. Inhibition of NMDA receptor-mediated synaptic transmission in the rat dentate gyrus in vitro by IL-1β. NeuroReport. 1997;8:2107–2110. doi: 10.1097/00001756-199707070-00004. [DOI] [PubMed] [Google Scholar]
- 8.Cunningham AJ, Murray CA, O’Neill LA, Lynch MA, O’Connor JJ. Interleukin-1β (IL-1β) and tumor necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett. 1996;203:17–20. doi: 10.1016/0304-3940(95)12252-4. [DOI] [PubMed] [Google Scholar]
- 9.Cunningham ET, Wada E, Carter DB, Tracey DE, Battey JE, DeSouza EB. In situ histochemical localization of type I interleukin-1 receptor messenger RNA in the central nervous system, pituitary, and adrenal gland of the mouse. J Neurosci. 1992;12:1101–1114. doi: 10.1523/JNEUROSCI.12-03-01101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cunningham ET, Jr, De Souza EB. Interleukin 1 receptors in the brain and endocrine tissues. Immunol Today. 1993;14:171. doi: 10.1016/0167-5699(93)90281-o. [DOI] [PubMed] [Google Scholar]
- 11.D’Arcangelo G, Dodt HU, Zieglgansberger W. Reduction of excitation by interleukin-1β in rat neocortical slices visualized using infrared-darkfield videomicroscopy. NeuroReport. 1997;8:2079–2083. doi: 10.1097/00001756-199705260-00053. [DOI] [PubMed] [Google Scholar]
- 12.Dinarello CA. Interleukin-1 and interleukin-1 antagonism. Blood. 1991;77:1627–1652. [PubMed] [Google Scholar]
- 13.Dinarello CA. The interleukin-1 family: 10 years of discovery. FASEB J. 1994;8:1314–1325. [PubMed] [Google Scholar]
- 14.Dinarello CA, Clark BD, Ikejima T, Puren AJ, Savage N, Rosoff PM. Interleukin-1 receptors and biological responses. Yale J Biol Med. 1990;63:87–93. [PMC free article] [PubMed] [Google Scholar]
- 15.Dunwiddie TV. The physiological role of adenosine in the central nervous system. Int Rev Neurobiol. 1985;27:63–139. doi: 10.1016/s0074-7742(08)60556-5. [DOI] [PubMed] [Google Scholar]
- 16.Fang JD, Wang Y, Krueger JM. Effects of interleukin-1β on sleep are mediated by the type I receptor. Am J Physiol. 1998;43:R655–R660. doi: 10.1152/ajpregu.1998.274.3.R655. [DOI] [PubMed] [Google Scholar]
- 17.Fastbom J, Pazos A, Probst A, Palacios JM. Adenosine A1 receptors in the human brain: a quantitative autoradiographic study. Neuroscience. 1987a;22:827–839. doi: 10.1016/0306-4522(87)92962-9. [DOI] [PubMed] [Google Scholar]
- 18.Fastbom J, Pazos A, Palacios JM. The distribution of adenosine A1 receptors and 5′-nucleotidase in the brain of some commonly used experimental animals. Neuroscience. 1987b;22:813–826. doi: 10.1016/0306-4522(87)92961-7. [DOI] [PubMed] [Google Scholar]
- 19.Goodman RR, Synder SH. Autoradiographic localization of adenosine receptors in rat brain using [3H]cyclohexyladenosine. J Neurosci. 1982;2:1230–1241. doi: 10.1523/JNEUROSCI.02-09-01230.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greene RW, Haas HL. The electrophysiology of adenosine in the mammalian central nervous system. Prog Neurobiol. 1991;36:329–341. doi: 10.1016/0301-0082(91)90005-l. [DOI] [PubMed] [Google Scholar]
- 21.Hoehn K, White TD. Glutamate-evoked release of endogenous adenosine from cortical synaptosomes is mediated by glutamate uptake and not by receptors. J Neurochem. 1990;54:1716–1724. doi: 10.1111/j.1471-4159.1990.tb01226.x. [DOI] [PubMed] [Google Scholar]
- 22.Jacobs RF, Tabor DR. The immunology of sepsis and meningitis–cytokine biology. Scand J Infect Dis Suppl. 1990;73:7–15. [PubMed] [Google Scholar]
- 23.Katsuki H, Nakai S, Hirai Y, Akaji K, Kiso Y, Satoh M. Interleukin-1β inhibits long term potentiation in the CA3 region of mouse hippocampal slices. Eur J Pharmacol. 1990;181:323–326. doi: 10.1016/0014-2999(90)90099-r. [DOI] [PubMed] [Google Scholar]
- 24.Khazipov R, Congar P, Ben-Ari Y. Hippocampal CA1 lacunosum-moleculare interneurons: comparison of effects of anoxia on excitatory and inhibitory postsynaptic currents. J Neurophysiol. 1995;74:2138–2149. doi: 10.1152/jn.1995.74.5.2138. [DOI] [PubMed] [Google Scholar]
- 25.Krueger JM, Takahashi S, Kapas L, Bredow S, Roky R, Fang J, Floyd R, Renegar KB, Guha-Thakurta N, Novitsky S. Cytokines in sleep regulation. Adv Neuroimmunol. 1995;5:171–188. doi: 10.1016/0960-5428(95)00007-o. [DOI] [PubMed] [Google Scholar]
- 26.Kuriyama K, Hori T, Mori T, Nakashima T. Actions of interferon α and interleukin-1β on the glucose-responsive neurons in the ventromedial hypothalamus. Brain Res Bull. 1990;24:803–810. doi: 10.1016/0361-9230(90)90143-n. [DOI] [PubMed] [Google Scholar]
- 27.Lambert NA, Teyler TJ. Adenosine depresses excitatory but not fast inhibitory synaptic transmission in area CA1 of the rat hippocampus. Neurosci Lett. 1991;122:50–52. doi: 10.1016/0304-3940(91)90190-5. [DOI] [PubMed] [Google Scholar]
- 28.Li Z, Inenaga K, Kawano S, Kannan H, Yamashita H. Interleukin-1β directly excites hypothalamic supraoptic neurons in rats in vitro. NeuroReport. 1992;3:91–93. doi: 10.1097/00001756-199201000-00024. [DOI] [PubMed] [Google Scholar]
- 29.Lue FA, Bail M, Jephthah-Ochola J, Carayanniotis K, Gorczynski R, Moldofsky H. Sleep and cerebrospinal fluid interleukin-1 like activity in the cat. Int J Neurosci. 1988;42:179–183. doi: 10.3109/00207458808991595. [DOI] [PubMed] [Google Scholar]
- 30.Mackiewicz M, Sollars PJ, Ogilvie MD, Pack AI. Modulation of IL-1β gene expression in the rat CNS during sleep deprivation. NeuroReport. 1996;7:529–533. doi: 10.1097/00001756-199601310-00037. [DOI] [PubMed] [Google Scholar]
- 31.Manzoni OJ, Manabe T, Nicoll RA. Release of adenosine by activation of NMDA receptors in the hippocampus. Science. 1994;265:2098–2101. doi: 10.1126/science.7916485. [DOI] [PubMed] [Google Scholar]
- 32.Masino S, Dunwiddie TV. Temperature-dependent modulation of excitatory transmission in hippocampal slices is mediated by extracellular adenosine. J Neurosci. 1999;19:1932–1939. doi: 10.1523/JNEUROSCI.19-06-01932.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGahon B, Lynch MA. A study of the synergism between metabotropic glutamate receptor activation and arachidonic acid in the rat hippocampus. NeuroReport. 1995;5:2353–2357. doi: 10.1097/00001756-199411000-00036. [DOI] [PubMed] [Google Scholar]
- 34.Miller LG, Galpern WR, Dunlap K, Dinarello CA, Turner TJ. Interleukin-1 augments γ-aminobyryric acidA receptor function in brain. Mol Pharmacol. 1991;39:105–108. [PubMed] [Google Scholar]
- 35.Mo ZL, Katafuchi T, Hori T. Effects of IL-1β on neuronal activities in the dorsal motor nucleus of the vagus in rat brain slices. Brain Res Bull. 1996;41:249–255. doi: 10.1016/s0361-9230(96)00196-7. [DOI] [PubMed] [Google Scholar]
- 36.Murray CA, McGahon B, McBennett S, Lynch MA. Interleukin-1β inhibits glutamate release in hippocampus of young, but not aged, rats. Neurobiol Aging. 1997;18:343–348. doi: 10.1016/s0197-4580(97)80317-x. [DOI] [PubMed] [Google Scholar]
- 37.Nakashima T, Hori T, Mori T, Kuriyama K, Mizuno K. Recombinant human interleukin-1β alters the activity of preoptic thermosensitive neurons in vitro. Brain Res Bull. 1989;23:209–213. doi: 10.1016/0361-9230(89)90149-4. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen KT, Deak T, Owens SM, Kohno T, Fleshner M, Watkins LR, Maier SF. Exposure to acute stress induces brain interleukin-1β protein in the rat. J Neurosci. 1998;18:2239–2246. doi: 10.1523/JNEUROSCI.18-06-02239.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ouanounou A, Zhang Y, Zhang L 1999 Changes in calcium dependence of glutamate transmission in the hippocampal CA1 region following brief hypoxia-hypoglycemia. J Neurophysiol, in press. [DOI] [PubMed]
- 40.Phillis JW, Wu P. The role of adenosine and its nucleotides in central synaptic transmission. Prog Neurobiol. 1981;16:187–239. doi: 10.1016/0301-0082(81)90014-9. [DOI] [PubMed] [Google Scholar]
- 41.Plata-Salaman CR, Ffrench-Mullen JM. Interleukin-1β depresses calcium currents in CA1 hippocampal neurons at pathophysiological concentrations. Brain Res Bull. 1992;29:221–223. doi: 10.1016/0361-9230(92)90029-w. [DOI] [PubMed] [Google Scholar]
- 42.Plata-Salaman CR, Ffrench-Mullen JM. Interleukin-1β inhibits Ca2+ channel currents in hippocampal neurons through protein kinase C. Eur J Pharmacol. 1994;266:1–10. doi: 10.1016/0922-4106(94)90202-x. [DOI] [PubMed] [Google Scholar]
- 43.Porkka-Heiskanen T, Strecker R, Thakkar M, Bjorkum A, Greene R, McCarley R. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science. 1997;276:1265–1268. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pringle AK, Gardner CR, Walker RJ. Reduction of cerebral GABAA responses by interleukin-1 (IL-1) through an indomethacin insensitive mechanism. Neuropharmacology. 1996;35:147–152. doi: 10.1016/0028-3908(95)00161-1. [DOI] [PubMed] [Google Scholar]
- 45.Quan N, Zhang ZB, Emery M, Bonsall R, Weiss JM. Detection of interleukin-1 bioactivity in various brain regions of normal healthy rats. Neuroimmunomodulation. 1996;3:47–55. doi: 10.1159/000097226. [DOI] [PubMed] [Google Scholar]
- 46.Rainnie DG, Grunze HC, McCarley RW, Greene RW. Adenosine inhibition of mesopontine cholinergic neurons: implications for EEG arousal. Science. 1994;263:689–692. doi: 10.1126/science.8303279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosoff PM, Savage N, Dinarello CA. Interleukin-1 stimulates diacylglycerol production in T lymphocytes by a novel mechanism. Cell. 1988;54:73–81. doi: 10.1016/0092-8674(88)90181-x. [DOI] [PubMed] [Google Scholar]
- 48.Rothwell NJ, Hopkins SJ. Cytokines and the nervous system. II. Actions and mechanisms of action. Trends Neurosci. 1995;18:130–136. doi: 10.1016/0166-2236(95)93890-a. [DOI] [PubMed] [Google Scholar]
- 49.Scheider H, Pitossi F, Balschun D, Wagner A, Del Rey W, Besedoveky HO. A neuromodulatory role of interleukin-1β in the hippocampus. Proc Natl Acad Sci USA. 1998;95:7778–7783. doi: 10.1073/pnas.95.13.7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schobitz B, De Kloet ER, Holsboer F. Gene expression and function of interleukin 1, interleukin 6 and tumor necrosis factor in the brain. Prog Neurobiol. 1994;44:397–432. doi: 10.1016/0301-0082(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 51.Semba K, White TD. M3 muscarinic receptor-mediated enhancement of NMDA evoked adenosine release in rat cortical slices in vitro. J Neurochem. 1997;69:1066–1072. doi: 10.1046/j.1471-4159.1997.69031066.x. [DOI] [PubMed] [Google Scholar]
- 52.Shinno K, Zhang L, Eubanks JH, Carlen PL, Wallace MC. Transient ischemia induces an early decrease of synaptic transmission in CA1 neurons of rat hippocampus: electrophysiological study in brain slices. J Cereb Blood Flow Metab. 1997;17:955–966. doi: 10.1097/00004647-199709000-00005. [DOI] [PubMed] [Google Scholar]
- 53.Snyder SH. Adenosine as a neuromodulator. Annu Rev Neurosci. 1985;8:103–124. doi: 10.1146/annurev.ne.08.030185.000535. [DOI] [PubMed] [Google Scholar]
- 54.Sundar SK, Becker KJ, Cierpial MA, Carpenter MD, Rankin LA, Fleener SL, Ritchie JC, Simson PE, Weiss JM. Intracerebroventricular infusion of interleukin 1 rapidly decreases peripheral cellular immune responses. Proc Natl Acad Sci USA. 1989;86:6398–6402. doi: 10.1073/pnas.86.16.6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Symons JA, Bundick RV, Suckling AJ, Rumsby MG. Cerebrospinal fluid interleukin-1 like activity during chronic relapsing experimental allergic encephalomyelitis. Clin Exp Immunol. 1987;68:648–654. [PMC free article] [PubMed] [Google Scholar]
- 56.Taishi P, Bredow S, Guha-Thakurta N, Obal F, Jr, Krueger JM. Diurnal variations of interleukin-1β mRNA and β-actin mRNA in rat brain. J Neuroimmunol. 1997;75:69–74. doi: 10.1016/s0165-5728(97)00002-7. [DOI] [PubMed] [Google Scholar]
- 57.Takahashi S, Kapas L, Fang J, Seyer JM, Wang Y, Krueger JM. An interleukin-1 receptor fragment inhibits spontaneous sleep and muramyl dipeptide-induced sleep in rabbits. Am J Physiol. 1996;271:R101–R108. doi: 10.1152/ajpregu.1996.271.1.R101. [DOI] [PubMed] [Google Scholar]
- 58.Thompson SM, Haas HL, Gahwiler BH. Comparison of the actions of adenosine at pre- and postsynaptic receptors in the rat hippocampus in vitro. J Physiol (Lond) 1992;451:347–363. doi: 10.1113/jphysiol.1992.sp019168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu LG, Saggau P. Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron. 1994;12:1139–1148. doi: 10.1016/0896-6273(94)90321-2. [DOI] [PubMed] [Google Scholar]
- 60.Yamashita H, Ueta Y, Inenaga K, Nagatomo T, Shibuya I, Kabashima N, Cui LN, Li Z, Yamamoto S. Cardiovascular system related peptides and hypothalamic neurons. Neurobiology. 1995;3:419–427. [PubMed] [Google Scholar]
- 61.Yoon KW, Rothman SM. Adenosine inhibits excitatory but not inhibitory synaptic transmission in the hippocampus. J Neurosci. 1991;11:1375–1380. doi: 10.1523/JNEUROSCI.11-05-01375.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu B, Shinnick-Gallagher P. Interleukin-1β inhibits synaptic transmission and induces membrane hyperpolarization in amygdala neurons. J Pharmacol Exp Ther. 1994;271:590–600. [PubMed] [Google Scholar]
- 63.Zeise ML, Madamba S, Siggins GR. Interleukin-1β increases synaptic inhibition in rat hippocampal pyramidal neurons in vitro. Regul Pept. 1992;39:1–7. doi: 10.1016/0167-0115(92)90002-c. [DOI] [PubMed] [Google Scholar]
- 64.Zhang L, Spigelman I, Carlen PL. Development of GABA-mediated, chloride-dependent inhibition in CA1 pyramidal neurones of immature rat hippocampal slices. J Physiol (Lond) 1991;44:25–49. doi: 10.1113/jphysiol.1991.sp018864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang L, Weiner JL, Carlen PL. Potentiation of GABAA receptor-mediated IPSCs by diazepam and pentobarbital in immature hippocampal CA1 neurons. J Pharmacol Exp Ther. 1993;266:1227–1235. [PubMed] [Google Scholar]
- 66.Zhang L, Weiner JL, Valiante TA, Velumian A, Walson P, Jahromi SS, Schetzer S, Pennefather P, Carlen PL. Effects of internally applied anions on the Ca2+-activated afterhyperpolarization in rat hippocampal neurons. Pflügers Arch. 1994;426:247–253. doi: 10.1007/BF00374778. [DOI] [PubMed] [Google Scholar]
- 67.Zhang L, Zhang Y, Wennberg R. Multiple actions of methohexital on hippocampal CA1 and cortical neurons of rat brain slices. J Pharmacol Exp Ther. 1998;286:177–182. [PubMed] [Google Scholar]