

Abstract
Aberrant reorganization of dentate granule cell axons, the mossy fibers, occurs in human temporal lobe epilepsy and rodent epilepsy models. Whether this plasticity results from the remodeling of preexisting mossy fibers or instead reflects an abnormality of developing dentate granule cells is unknown. Because these neurons continue to be generated in the adult rodent and their production increases after seizures, mossy fibers that arise from either developing or mature granule cells are potential substrates for this network plasticity. Therefore, to determine whether seizure-induced, mossy fiber synaptic reorganization arises from either developing or mature granule cell populations, we used low-dose, whole-brain x-irradiation to eliminate proliferating dentate granule cell progenitors in adult rats. A single dose of 5 Gy irradiation blocked cell proliferation and eliminated putative progenitor cells in the dentate subgranular proliferative zone. Irradiation 1 d before pilocarpine-induced status epilepticus significantly attenuated dentate granule cell neurogenesis after seizures. Two irradiations, 1 d before and 4 d after status epilepticus, essentially abolished dentate granule cell neurogenesis but failed to prevent mossy fiber reorganization in the dentate molecular layer. These results indicate that dentate granule cell neurogenesis in the mature hippocampal formation is vulnerable to the effects of low-dose ionizing irradiation. Furthermore, the development of aberrant mossy fiber remodeling in the absence of neurogenesis suggests that mature dentate granule cells contribute substantially to seizure-induced network reorganization.
Keywords: hippocampus, dentate gyrus, dentate granule cells, neurogenesis, pilocarpine, seizures, epilepsy, mossy fiber sprouting, network reorganization, synaptic plasticity, brain irradiation
Axons of the fascia dentata granule cells, the mossy fibers, undergo network reorganization in both human temporal lobe epilepsy (TLE) and experimental epilepsy models in rodents (Tauck and Nadler, 1985; Cronin and Dudek, 1988; de Lanerolle et al., 1989; Sutula et al., 1989; Houser et al., 1990; Babb et al., 1991; Mello et al., 1993). After seizure-induced hippocampal injury in adult rats, mossy fibers project aberrantly to the supragranular inner molecular layer of the dentate gyrus, where they form connections with novel targets (Isokawa et al., 1993; Franck et al., 1995; Okazaki et al., 1995). Although the anatomical features and physiological consequences of this axonal remodeling have been studied in considerable detail (Cronin et al., 1992; Sloviter, 1992; Isokawa et al., 1993; Franck et al., 1995; Okazaki et al., 1995; Wuarin and Dudek, 1996), the cellular and molecular mechanisms that underlie mossy fiber synaptic reorganization remain poorly understood (for review, seeParent and Lowenstein, 1997). Indeed, even the proposed source of this network plasticity, i.e., newly “sprouted” axon collaterals that arise from mature dentate granule cells (DGCs), is unproved. Because new DGCs continue to be generated during postnatal and adult life in rodents and at least several other mammalian species including humans (Altman and Das, 1965, 1967; Kaplan and Hinds, 1977; Gueneau et al., 1982; Cameron et al., 1993; Kuhn et al., 1996; Eriksson et al., 1998;Gould et al., 1998), developing axons arising from newly born DGCs are another potential source of abnormally targeted mossy fibers after hippocampal injury.
Neural progenitor cells persist in the dentate gyrus of adult rats (Kaplan and Hinds, 1977; Kaplan and Bell, 1984; Cameron et al., 1993; Kuhn et al., 1996). These precursors proliferate in the dentate subgranular proliferative zone (SGZ) at the border of the granule cell layer and hilus, and their progeny migrate into the DGC layer where they differentiate into mature granule neurons (Cameron et al., 1993;Seki and Arai, 1993; Kuhn et al., 1996; Parent et al., 1997). A smaller number of progeny probably also differentiates into radial glia-like cells in the granule cell layer (Cameron et al., 1993), although pluripotentiality of individual SGZ progenitor cells in vivohas not been established. Newly differentiating DGCs appear to integrate normally into existing hippocampal networks, as demonstrated by the projection of developing mossy fibers to appropriate distant targets in area CA3 of the hippocampus proper (Stanfield and Trice, 1988; Seki and Arai, 1993; Parent et al., 1997). The physiological role of continued granule neuron birth in the mature brain is unknown. The rate of DGC neurogenesis during early postnatal and adult life appears to be influenced, at least in part, by factors such as aging, environmental stimulation, glucocorticoid hormone levels, and glutamatergic input to the DGC layer (for review, see Gage et al., 1998). Furthermore, recent investigations using a variety of experimentally induced pathologies, including seizures (Bengzon et al., 1997; Parent et al., 1997, 1998; Gray and Sundstrom, 1998; Scott et al., 1998), cerebral ischemia (Liu et al., 1998), and focal toxin injection (Gould and Tanapat, 1997), have identified significant alterations of DGC neurogenesis after brain injury.
Several recent studies have shown that DGC neurogenesis in adult rats increases in various chemoconvulsant and electrical stimulation models of TLE (Bengzon et al., 1997; Parent et al., 1997, 1998; Gray and Sundstrom, 1998; Scott et al., 1998). One of these epilepsy models, using pilocarpine to induce an episode of status epilepticus, is associated with robust, aberrant mossy fiber reorganization that begins during the second week after status epilepticus (SE) and peaks after ∼2 months (Cavalheiro et al., 1991; Mello et al., 1993). We demonstrated recently that cell proliferation in the dentate SGZ increases within 3 d after pilocarpine-induced SE and persists for at least 2 weeks (Parent et al., 1997). Thus, the birth and subsequent differentiation of increased numbers of developing DGCs induced by prolonged seizure activity parallel the time course of mossy fiber remodeling. In addition, at least a portion of these newly generated DGCs participates in the aberrant axonal reorganization that occurs in both area CA3 and the dentate inner molecular layer after pilocarpine-induced SE (Parent et al., 1997). These findings raise the questions of whether seizure-induced mossy fiber remodeling arises principally from the developing axons of newly generated DGCs and whether mature granule cells can actually give rise to new axonal projections. To examine these questions, we used low-dose, whole-brain x-irradiation to inhibit DGC neurogenesis in adult rats. We then investigated whether the elimination of DGC progenitors altered seizure-induced mossy fiber reorganization in the pilocarpine TLE model. The results indicate that DGC progenitors are selectively vulnerable to low-dose radiation treatment and that seizure-induced mossy fiber reorganization develops and persists despite prolonged inhibition of neurogenesis. These results provide the first evidence that newly sprouted axon collaterals from preexisting, mature DGCs can serve as a substrate for mossy fiber remodeling after seizure-induced injury.
MATERIALS AND METHODS
Brain irradiation. Young adult (170–250 gm), male Sprague Dawley rats were used in all studies, and the protocols were approved by the Committee on Animal Research at the University of California, San Francisco. Irradiation was done using a Phillips orthovoltage x-ray system operated at 150 kVp and 20 mA. Rats were anesthetized with a mixture of ketamine, xylazine, and acepromazine, placed in sternal recumbency on a treatment table, and then irradiated individually. The head was centered in a 5 cm × 6 cm treatment field, and the neck and body were protected by lead shielding. Dosimetry was done using a Keithly electrometer–ionization chamber calibrated with lithium fluoride thermal luminescent dosimeters. The corrected dose rate was ∼175 cGy/min at a source-to-skin distance of 21 cm. Sham-irradiation controls for all experiments received anesthesia only. The radiation dose–response effects on the hippocampal formation were determined by irradiating animals with single doses of 1, 3, or 5 Gy (n = 3 per group), with an additional sham-irradiation group (n = 3). These doses were based on a previous study of radiation-induced cell death of subventricular zone progenitor cells in the adult rat (Shinohara et al., 1997). 5-Bromo-2′-deoxyuridine (BrdU; Boehringer Mannheim, Indianapolis, IN) was administered at 24 and 30 hr after irradiation (see below), and animals were killed 1–2 hr after the last BrdU injection. To examine cell death in the hippocampal formation after radiation treatment, we irradiated animals with a dose of 5 Gy (n = 3) or sham irradiation (n = 3) 6 hr before perfusion. Brains were processed for terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick-end labeling (TUNEL) stains and morphological analysis of DNA fragmentation (see below). A dose of 5 Gy was chosen for all subsequent radiation studies.
Seizure induction and BrdU labeling. SE was induced with pilocarpine hydrochloride (340 mg/kg, i.p.; Sigma, St. Louis, MO) 20 min after pretreatment with atropine methylbromide (5 mg/kg, i.p.; Sigma). Seizure activity was monitored behaviorally, and after 2 hr of convulsive SE, seizures were terminated with diazepam (10 mg/kg, i.p.; Elkins-Sinn, Cherry Hill, NJ). Only rats that displayed continuous, convulsive seizure activity after pilocarpine treatment were used in these experiments. Control rats received the same injections of atropine and diazepam, but saline was given instead of pilocarpine. To label S phase cells, BrdU (50 mg/kg, i.p., in PBS) was administered in all cases (Miller and Nowakowski, 1988).
The goal of the combined seizure and radiation experiments was to eliminate proliferating dentate gyrus progenitor cells selectively without preventing the reorganizational response to seizure-induced injury. Therefore, we first sought to determine whether the progeny of DGC precursors that were constitutively proliferating before seizure activity contributed significantly to increased neurogenesis after SE. To label constitutively proliferating cells before SE, we injected rats with BrdU twice, 6 hr apart, on the day before pilocarpine or saline treatment. Animals were killed 2, 4, 7, 10, or 14 d (n = 3–4 per time point) after seizure induction and were examined for BrdU labeling and TOAD-64 (turned-on-after-division protein, 64 kDa) immunostaining (see below). Subsequently, we irradiated rats 1 d before pilocarpine treatment (n = 3), injected BrdU twice, 6 hr apart, on the fourth day after SE, and perfused the animals after a 24 hr survival period. Control groups received sham irradiation and pilocarpine (n = 4) or sham irradiation and saline (n = 3) and underwent identical BrdU labeling and survival duration. Additional groups of animals experienced the same treatments except that BrdU was given the day before pilocarpine and the animals were killed on day 7 after SE. Because cell proliferation in the dentate SGZ was not entirely obliterated at 5 d after a single 5 Gy dose of radiation, animals in the last set of radiation and seizure experiments (n = 6) received 5 Gy doses 1 d before and 4 d after pilocarpine-induced SE. Three control groups (n = 4 per group) were used: (1) sham irradiation and pilocarpine treatment, 2) irradiation and saline treatment, and 3) sham irradiation and saline treatment. Treatment and control groups received two daily injections of BrdU, 6 hr apart, on days 3–5 and were killed 28 d after pilocarpine or saline administration.
Tissue preparation, TUNEL staining, and histochemistry. For cell death studies, rats received an anesthetic overdose and were transcardially perfused with 350 ml of a 10% buffered formalin solution (Sigma). Brains were post-fixed in 10% buffered formalin for 3 d, and then 5-mm-thick coronal sections containing the hippocampal formation were cut and embedded in paraffin. Five-micrometer-thick coronal sections from rostral, middle, and caudal hippocampal levels were cut with a microtome and placed on polylysine-coated glass slides. Sections were deparaffinized and processed for TUNEL staining and morphological analysis (see below). For all other studies, animals were given an anesthetic overdose and transcardially perfused at a rate of 30 ml/min as follows: normal saline (3 min); 2% paraformaldehyde in 0.1 m sodium acetate, pH 6.5 (3 min); and 2% paraformaldehyde and 0.1% glutaraldehyde in 0.1 m borate, pH 8.5 (12 min). Rats surviving for 28 d were also perfused with 0.37% sulfide solution, pH 7.2 (4 min), before aldehydes, for subsequent Timm staining. After post-fixation in situ overnight, brains were removed, washed once in PBS, and cryoprotected with 30% sucrose in PBS. Forty-micrometer-thick coronal frozen sections through the entire rostrocaudal extent of the hippocampus were cut with a cryostat and used for histochemistry or immunohistochemical stains.
Dying cells in 5 μm coronal brain sections were identified by a distinct morphological pattern of nuclear fragmentation and by using the TUNEL method to label specifically the 3′-hydroxyl termini of DNA strand breaks (Gavrieli et al., 1992; Thomas et al., 1995). For the TUNEL procedure, all reagents were part of a kit (ApopTag; Oncor, Gaithersburg, MD). Procedures were performed according to the manufacturer’s instructions, except that the TdT enzyme to reaction buffer ratio was changed to 1:40 and specimens were incubated for 20 min in 0.2% H2O2 (Sigma) in deionized distilled water containing 0.1 m sodium azide (Sigma) to quench endogenous peroxidase activity. Gill’s hematoxylin was used as a counterstain. For Timm and Nissl staining, 40 μm coronal sections from rostral, middle, and caudal hippocampal levels were placed on coated slides (Super-Frost Plus; Fisher Scientific, Houston, TX) and air-dried overnight. To assess patterns of mossy fiber growth, Timm staining was performed on brain sections from rats in the 28 d after-pilocarpine or saline survival groups (8–10 sections per animal) and was done according to the modified Timm’s procedure described bySloviter (1982). Control and treatment groups were batch processed, and incubation times were the same for all groups (75 min). Some sections were counterstained with 0.1% cresyl violet, and the remainder of the sections were used for densitometric analysis of Timm staining (see below). Nissl staining (1% cresyl violet) was performed on adjacent sections after dehydration and rehydration in graded ethanols and xylenes, and Timm- and Nissl-stained sections were dehydrated and coverslipped with Permount (Fisher Scientific).
Immunohistochemistry. Immunohistochemistry was performed on 40-μm-thick coronal free-floating sections obtained throughout the septotemporal extent of the hippocampus. For BrdU immunostaining, DNA was first denatured by incubating tissue sections in 2N HCl for 30 min at 37°C followed by a 10 min wash in 0.1 m borate solution, pH 8.5. Diaminobenzidine peroxidase immunohistochemistry for BrdU and TOAD-64 and double-label immunofluorescence to detect polysialylated-neural cell adhesion molecule (PSA-NCAM) and glial fibrillary acidic protein (GFAP) were done as described previously (Parent et al., 1997). Primary antibody dilutions used were 1:1000 for BrdU (mouse monoclonal; Boehringer Mannheim), 1:10,000 for TOAD-64 (rabbit polyclonal; a gift of S. Hockfield, Yale University, New Haven, CT), 1:200 for GFAP (rat monoclonal; a gift of V. Lee, University of Pennsylvania), and 1:1000 for PSA-NCAM (mouse IgM monoclonal; a gift of G. Rougon, Université de Luminy, Marseille, France). All incubations in primary antibody were for 36 hr at 4°C, except for that in BrdU antibody, for which an overnight incubation at room temperature was used. Secondary antibodies for immunofluorescence were goat anti-rat IgG conjugated to fluorescein isothiocyanate and goat anti-mouse IgM conjugated to Texas Red (both from Jackson ImmunoResearch, West Grove, PA) at 1:200 dilutions. Immunofluorescence images were obtained using a Bio-Rad MRC 1024 confocal laser microscope (Hercules, CA), visualized with NIH Image version 1.61 software, and transferred to Adobe Photoshop software (Adobe Systems, Mountain View, CA).
Quantification and statistical analyses. For cell death studies, the number of cells in the dentate gyrus exhibiting TUNEL-positive nuclei and showing nuclear DNA fragmentation was counted separately in six hippocampal regions per animal. Cell counts were performed at a magnification of 400× in an area encompassing the entire DGC layer (superior and inferior blades) and extending approximately two cell body widths deep into the hilus (SGZ). Statistical comparisons of TUNEL- and nuclear DNA fragmentation–positive cells from irradiated and control groups were made using a paired Student’s t test. BrdU immunostaining was quantified densitometrically via a digital image analysis system and NIH Image version 1.61 software as described previously (Parent et al., 1997). Six to seven sections per animal (12–14 hippocampal regions) divided between the rostral, middle, and caudal one-thirds of the hippocampus were analyzed using the same area of the dentate gyrus as described above for cell counts. The mean percentage of area immunostained was determined for each group, and ANOVA with Fisher’s protected least significant difference (PLSD) post hoc test was performed using StatView software (Abacus Concepts, Berkeley, CA). Densitometric analysis of Timm staining was done with the same system on images captured at 200× magnification and imported into NIH Image software. Six circular regions, each with an area of 215 pixels, were placed over the inner molecular layer, and the mean density was calculated for each region. To account for differences in background staining intensity between sections, the mean density from four identical circular regions placed in the outer molecular layer of each hippocampal area was determined and used to calculate a corrected inner molecular layer Timm-staining density value. Values from eight DGC layer regions per animal in irradiated and SE and in sham-irradiated and SE groups were averaged, and statistical comparisons were made using a two-tailed Student’s t test. Timm-staining density in irradiated and saline and in sham-irradiated and saline control groups was not quantified because of the absence of inner molecular layer Timm staining in these animals.
RESULTS
DGC progenitors are selectively vulnerable to low-dose x-irradiation
To examine whether DGC progenitors are vulnerable to whole-brain ionizing irradiation, we first compared the effects of escalating radiation doses on cell proliferation in the mature dentate SGZ and granule cell layer. Adult rats received single doses of 1, 3, or 5 Gy or sham irradiation, and cell proliferation was measured by systemic administration of BrdU. At 31 hr after radiation treatment, BrdU labeling declined significantly in the dentate gyrus in a dose-responsive manner (Fig.1A,B,E). Although multiple clusters of BrdU-immunoreactive (-IR) nuclei were seen in sham-irradiated animals (Fig. 1A), animals treated with 3 or 5 Gy radiation doses exhibited only a few scattered and isolated BrdU-IR nuclei or no immunostaining in the dentate SGZ (Fig. 1B). Unlike in controls, some BrdU-labeled nuclei were apparent in the hilus after 3 Gy and, to a greater extent, 5 Gy irradiation (Fig. 1B), suggesting that a slight glial proliferative response was induced by the radiation treatment. To determine whether the decrease in cell proliferation after brain irradiation corresponded with a decline in cells exhibiting an immature DGC phenotype, we performed immunohistochemistry for TOAD-64, a member of the collapsin response mediator protein family that is transiently expressed in postmitotic, immature neurons (Minturn et al., 1995; Wang and Strittmatter, 1996). Similar to the decrease in BrdU labeling, a qualitative dose-dependent decline in TOAD-64–IR occurred in the DGC layer after brain irradiation (Fig. 1C,D) (data not shown).
Fig. 1.
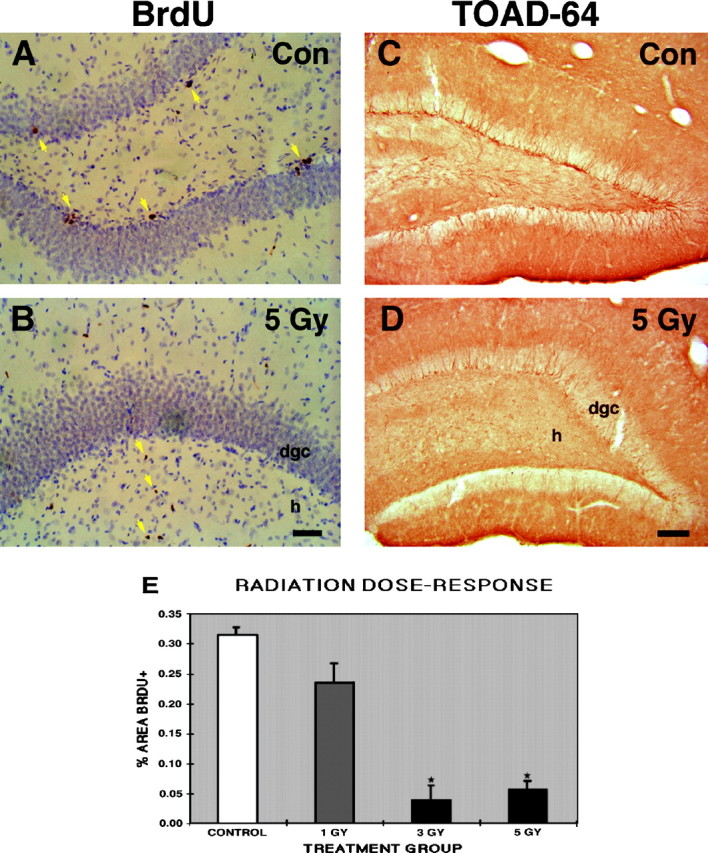
Low-dose x-irradiation inhibits cell proliferation in the mature dentate gyrus. A, B, BrdU immunostaining in the dentate gyrus shows a typical pattern of clustered BrdU-labeled nuclei in the dentate SGZ and interior DGC layer in a sham-irradiated control rat (yellow arrowsin A), whereas no BrdU labeling is seen in the same regions 31 hr after a single 5 Gy x-ray dose (B). Hilar BrdU-positive cells are found after x-ray treatment (yellow arrow in B) but not in controls (A). These cells probably represent reactive glial proliferation. BrdU was given 24 and 30 hr after treatment. C, D, TOAD-64 immunostaining of the dentate gyrus from animals in the same treatment conditions described in A and B is shown. Note the paucity of TOAD-64–immunostained, immature neurons in the hilar aspect of the DGC layer 31 hr after irradiation (D) compared with that in the control adult rat (C).E, A dose-dependent decrease in BrdU labeling occurred after a single x-ray dose, with significantly less BrdU labeling in the 3 and 5 Gy groups versus controls (mean ± SEM;asterisks denote p < 0.005, ANOVA with Fisher’s PLSD). Scale bars: A, B, 50 μm; C, D, 100 μm.Con, Control; dgc, dentate granule cell layer; h, hilus.
We next sought to establish whether the decrease in both BrdU labeling and cells expressing an immature neuronal phenotype resulted from the selective death of progenitor cells. TUNEL staining and a morphological assessment of nuclear fragmentation at the light microscopic level were performed in rats 6 hr after a single 5 Gy radiation dose or sham irradiation. In agreement with previous findings in the adult rat subventricular zone (Bellinzona et al., 1996; Shinohara et al., 1997), radiation treatment produced cell death in the hippocampal formation selectively in regions where cell proliferation is known to be ongoing—the dentate SGZ and the hilar aspect of the DGC layer (Fig.2). Dying cells appeared singly or in groups resembling the clusters of mitotically active SGZ cells seen with BrdU labeling (see and compare Figs. 2B with 1A, 4A). The numbers of TUNEL-positive cells and cells with nuclear fragmentation were dramatically higher in irradiated rats (Fig. 2) compared with the negligible amounts seen in sham-irradiated controls (mean ± SEM, TUNEL-stained, 53.25 ± 3.08 irradiated group vs 0.75 ± 0.22 control group; p < 0.0001; mean ± SEM, nuclear fragmentation, 31.83 ± 6.20 irradiated group vs 0.33 ± 0.19 control group; p < 0.0009). Furthermore, the number of dying cells was much greater than that expected after a pulse label of BrdU, suggesting that cells in proliferative states other than S phase, and perhaps immature postmitotic cells in this location, are vulnerable to this dose of ionizing irradiation. No TUNEL-positive cells were seen in the hippocampus proper. Taken together, these findings indicate that DGC progenitors in the mature hippocampal formation are selectively vulnerable to the effects of low-dose x-rays.
Fig. 2.

Low-dose x-irradiation selectively kills cells in the dentate subgranular proliferative zone of adult rats.A, B, TUNEL stain of the dentate gyrus from an irradiated adult rat 6 hr after a single dose of 5 Gy shows dying cells (brown areas) almost exclusively located in the dentate SGZ and hilar aspect of the DGC layer. Higher magnification of the DGC layer reveals nuclear fragmentation (yellow arrows in B) as well as TUNEL labeling. Five-micrometer-thick sections are counterstained with Gill’s hematoxylin. Scale bars: A, 50 μm; B, 25 μm. dgc, Dentate granule cell layer;h, hilus.
Prolonged seizures activate constitutively proliferating DGC progenitors
The main aim of this study was to determine the contributions of developing and mature DGC populations to network reorganization after SE. Because our strategy involved inhibiting DGC neurogenesis with brain irradiation, we wanted to use a protocol that would effectively eliminate neurogenesis while minimizing the impact of x-irradiation on other responses to seizure-induced injury. We especially sought to avoid irradiating in the immediate period after SE, because this is the time when there is maximal proliferation and activation of glial and other non-neuronal cell types. This led us to explore first whether the constitutively proliferating precursor population (i.e., DGC progenitors normally undergoing mitosis in the mature dentate gyrus) contributes to the increased neurogenesis that occurs after SE. If this is the case, then it would be possible to irradiate rats before pilocarpine treatment and minimize the postseizure effects of irradiation. Therefore, we performed BrdU labeling in adult rats 1 d before the induction of SE and then examined dentate gyrus BrdU labeling after several different survival periods. In saline-treated controls surviving 3–15 d after BrdU administration (Fig.3A,C), a mixture of strongly and weakly BrdU-IR nuclei was found predominantly in the inner DGC layer adjacent to the hilus. The number of BrdU-labeled cells qualitatively increased only slightly between 2 and 7 d after saline (3–8 d after BrdU administration) and then remained stable at the 10 and 14 d time points. In pilocarpine-treated animals, BrdU-IR cells similarly appeared in the hilar aspect of the DGC layer. However, an increased number of lightly BrdU-IR cells was present within 4–10 d after seizures (5–11 d after BrdU injection) compared with that in controls (Fig.3A–D). These cells were most often seen in large clusters but also were occasionally diffusely separated throughout the cell layer. The number of weakly BrdU-labeled nuclei markedly expanded 10–14 d after SE and constituted a nearly continuous band within the interior DGC layer (Fig. 3D). The progressive increase in lightly BrdU-IR cells in the DGC layer with increasing time after seizures suggested a dilution of BrdU content in each nucleus because of multiple cell divisions. Therefore, each constitutively proliferating progenitor cell appeared to divide more often after SE than in the normal state. The corresponding increase in TOAD-64–IR, immature neurons in this same region 7–14 d after SE in these (Fig.3E,F) and similarly treated rats (Parent et al., 1997) indicates that many of the progeny of this constitutively dividing population differentiate into neurons. Immunofluorescence double-labeling of the cells lightly BrdU-immunostained using the immunoperoxidase method was not possible given the reduced sensitivity of immunofluorescence for detecting low levels of BrdU.
Fig. 3.
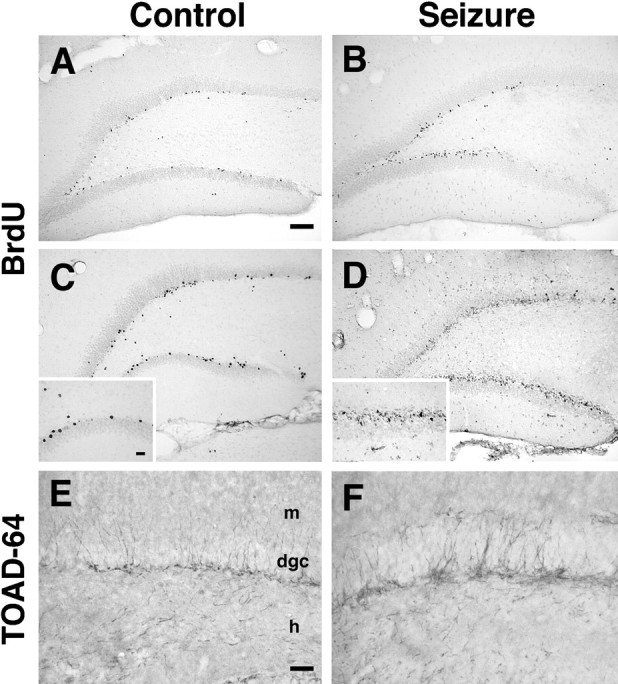
Prolonged seizures activate constitutively proliferating progenitor cells. A–D, Adult rats were given BrdU (2 doses; 6 hr apart) to label proliferating (S phase) cells 1 d before pilocarpine (B, D) or saline (A, C) treatment. BrdU-IR in the dentate gyrus of controls increased only slightly between 4 d (A) and 10 d (C) after saline, with intensely immunostained cells scattered in the interior DGC layer (inset in C). The number of BrdU-IR cells first appeared to increase by 4 d after pilocarpine-induced SE (B). Ten days after pilocarpine (D), many more cells were found, again predominantly in the hilar aspect of the DGC layer, and most with lightly BrdU-immunostained nuclei (inset inD) consistent with a dilution of nuclear BrdU content by multiple cell divisions. E, F, TOAD-64 immunostaining in the same animals analyzed in C andD shows an increase in TOAD-64–IR in the interior DGC layer after pilocarpine treatment (F) compared with that in controls (E). Note that the location of the TOAD-64–positive, immature neurons (putative DGCs) corresponds to that of the BrdU labeling. Scale bars:A–D, 100 μm; insets inC, D, 25 μm; E,F, 50 μm. dgc, Dentate granule cell layer; h, hilus; m, molecular layer.
Despite the fact that both astroglia and DGCs proliferate after seizure-induced injury (Niquet et al., 1994; Parent et al., 1997), BrdU labeling of the progeny of constitutively proliferating cells in both pilocarpine-treated and control rats was remarkably confined to the SGZ and inner DGC layer (Fig. 3). Because astroglial proliferation after prolonged seizures predominates in the hilus and molecular layers of the dentate gyrus (Niquet et al., 1994; Khurgel and Ivy, 1996) (see Fig. 6E), the progeny of the constitutively proliferating progenitors do not appear to be involved in the glial response to SE. In fact, the pattern of cell proliferation identified when BrdU is given 1 d after pilocarpine-induced SE differs markedly, with intense BrdU labeling in the regions of active glial proliferation in the dentate hilus and molecular layers (data not shown). These findings suggest that most astrocyte proliferation after seizure-induced injury arises from a different population of nonconstitutively proliferating precursor cells—either mature glia that reenter the cell cycle or a quiescent progenitor pool. Alternatively, it is possible that detection of more slowly proliferating cells was limited by the timing of BrdU injections on the day before SE or that more rapid proliferation of these cells after seizures diluted the nuclear BrdU content beyond detection. The latter explanation is unlikely, however, because BrdU labeling in glial proliferative zones was not found even as early as 2 d after SE (data not shown).
Fig. 6.
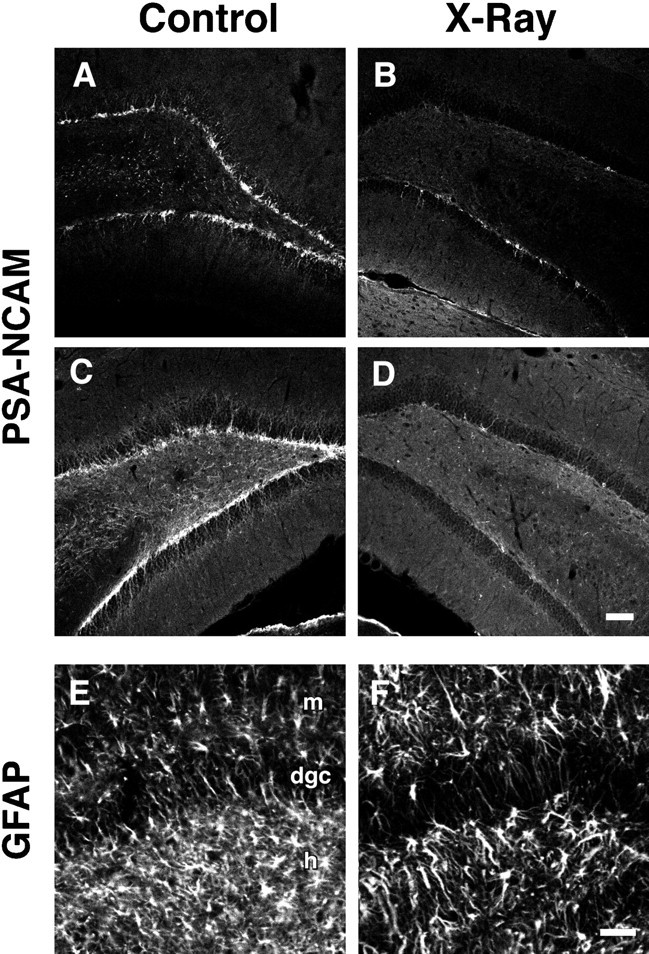
Brain irradiation alters neuronal and glial proliferation after SE. A–D, PSA-NCAM immunofluorescence of the dentate gyrus from irradiated (B, D) versus sham-irradiated (A, C) rats treated with pilocarpine (C, D) or saline (A,B). In saline-treated controls, two radiation treatments (B) eliminated the normal pattern of neurogenesis in the DGC layer (A). Four weeks after SE, the amount of neurogenesis increased (C), but this was also eliminated by double-dose irradiation (D). E, F, GFAP immunofluorescence in the dentate gyrus of the same animals analyzed inC and D, respectively. Radiation treatment (F) caused a much smaller decrease in the typical degree of glial proliferation seen after SE (E) relative to changes in neurogenesis. Scale bars: A–D, 100 μm; E,F, 50 μm. dgc, Dentate granule cell layer; h, hilus; m, molecular layer.
Single-dose irradiation attenuates neurogenesis after seizures
We next determined whether irradiation 1 d before pilocarpine treatment would inhibit the accelerated neurogenesis known to occur after seizures. When constitutively dividing cells were labeled with BrdU 1 d before SE (i.e., on the day of radiation treatment), BrdU labeling was absent or minimal in irradiated animals surviving 7 d after pilocarpine-induced SE (data not shown) compared with the typical patterns in nonirradiated saline- or pilocarpine-treated rats (Fig. 3). However, irradiated rats that received BrdU 4 d after SE and were killed 24 hr later exhibited more BrdU labeling than did sham-irradiated, saline-treated controls (Fig.4A,B). Both of these groups had significantly less BrdU labeling than did sham-irradiated, pilocarpine-treated animals (Fig.4A–D). In irradiated compared with sham-irradiated animals, BrdU-IR cells in the SGZ were fewer in number and more often seen as individual immunostained nuclei or in smaller clusters. In addition, TOAD-64 immunostaining revealed an even larger decrease in immature neurons in irradiated rats compared with controls at the same time point after seizures (Fig.4E,F). TOAD-IR cells were localized to the hilar portion of the DGC layer in irradiated and control groups, but irradiated animals exhibited fewer and less clustered TOAD-IR cell bodies. The radiation treatment had no observable effects on the episode of status epilepticus itself, because both irradiated and sham-irradiated groups had similar times to seizure onset, behavioral seizures, and mortality after pilocarpine treatment.
Fig. 4.

Preseizure x-ray treatment attenuates cell proliferation and neurogenesis after status epilepticus.A, BrdU labeling and immunohistochemistry in a control adult rat 5 d after saline administration and 6 d after sham irradiation revealed clusters of BrdU-labeled cells in the dentate SGZ.B, C, BrdU immunostaining 5 d after pilocarpine treatment in adult rats that received a single dose of 5 Gy x-irradiation (B) or sham irradiation (C) 1 d before SE is shown. In the sham-irradiated controls, pilocarpine treatment produced a marked increase in dentate SGZ BrdU labeling (C) compared with that in saline treatment (A). This increase was significantly attenuated by a single radiation dose 1 d before SE (B). D, Quantification of BrdU labeling in animals treated as described inA–C showed significantly decreased BrdU labeling in the sham x-rays and saline controls and in the x-rays and SE group compared with the sham x-rays and SE group (mean ± SEM;single asterisk denotes p = 0.002;double asterisks denote p = 0.006). BrdU was given twice, 6 hr apart, on day 4 after pilocarpine or saline treatment for all groups. E, F, TOAD-64 immunostaining 5 d after SE in the same animals analyzed inB and C was similarly decreased by radiation treatment (E) compared with sham irradiation (F). Scale bars:A–C, 100 μm; E,F, 50 μm. dgc, Dentate granule cell layer; h, hilus; m, molecular layer.
Although a single 5 Gy x-ray dose markedly attenuated neurogenesis at 5 d after seizures, some BrdU labeling in the SGZ and DGC layer persisted (Fig. 4B). This cell proliferation most likely represented a slow return of mitotic activity in progenitor cells resistant to radiation treatment (perhaps quiescent precursor cells), because BrdU labeling in this region increased 2 d later (i.e., 7 d after seizures; data not shown). Interestingly, TOAD-64–IR was also more prevalent 7 d after SE (8 d after irradiation) with immunostained cells seen in small clusters similar to the clustering of BrdU-IR nuclei in this region (data not shown). These findings indicate that low-dose x-irradiation before pilocarpine treatment significantly, although incompletely, reduced the accelerated cell proliferation and neurogenesis induced by SE.
Seizure-induced mossy fiber sprouting develops despite elimination of neurogenesis by double-dose irradiation
To examine whether seizure-induced, aberrant mossy fiber reorganization is altered by inhibiting neurogenesis, we irradiated adult rats both before and after pilocarpine-induced SE. On the basis of the partial preservation of cell proliferation in the dentate gyrus 5 d after a single dose of 5 Gy irradiation described above, we chose to irradiate rats with two doses of 5 Gy radiation given 1 d before and 4 d after SE. The patterns of neurogenesis and mossy fiber projections were then assessed 4 weeks after seizures by BrdU labeling, immunohistochemical detection of immature neuronal markers, and Timm staining. Although many BrdU-IR cells were found in the DGC layer of sham-irradiated controls (Fig.5A), BrdU labeling was absent in the SGZ and DGC layer in irradiated rats 4 weeks after saline (data not shown) or pilocarpine (Fig. 5B) treatment. Because the BrdU was administered only during the first week after seizures (see Materials and Methods), absent BrdU-IR at 4 weeks does not preclude a rebound in neurogenesis during weeks 2–4 after SE. Therefore, we next examined the effects of radiation on neurogenesis by performing immunohistochemistry for TOAD-64 and PSA-NCAM. These markers are expressed specifically in immature neurons in the mature dentate gyrus (Seki and Arai, 1993; Parent et al., 1997), and the pattern of immunostaining gives a more cumulative sampling of changes in the rate of neurogenesis after seizures than does BrdU labeling (Parent et al., 1997). TOAD-64– and PSA-NCAM–IR increased after seizures as compared with that in saline-treated controls in the two sham-irradiation groups (Figs. 5C,6A,C), and this immunostaining was entirely or nearly abolished in pilocarpine- and saline-treated, irradiated rats (Figs. 5D,6B,D). Although a modest reduction in glial proliferation as identified by the degree of GFAP-IR (Latov et al., 1979) also occurred after radiation treatment (Fig.6E,F), this decrease was much less than the corresponding decline in neurogenesis. Also, a slight increase in GFAP immunostaining in the hilus and dentate molecular layers was seen in irradiated, saline-treated rats compared with the corresponding sham-irradiated controls (data not shown). This finding, together with the minimal hilar BrdU labeling induced by a single dose of 5 Gy radiation (Fig. 1B), suggests that brain irradiation produced a mild glial proliferative response. No colocalization of PSA-NCAM– and GFAP-IR was observed in the dentate gyrus of sham-irradiated controls that received either pilocarpine or saline (data not shown).
Fig. 5.

Double-dose irradiation inhibits DGC neurogenesis after prolonged seizures. Two doses of 5 Gy x-rays, 1 d before and 4 d after pilocarpine treatment, nearly abolished cell proliferation and DGC neurogenesis in the dentate SGZ and granule cell layer as evidenced by BrdU (A, B) and TOAD-64 (C, D) immunostaining. Irradiated rats (B, D) exhibited a marked reduction of BrdU- (B) and TOAD-64–IR (D) cells in the dentate SGZ and interior granule cell layer compared with that in sham-irradiated (A,C) seizure controls. The amount of TOAD-IR shown inD represents the maximal seen in the irradiated group; most sections had no TOAD-IR cells. BrdU was given on days 3–5 after SE, and animals were killed 28 d after SE. Scale bar, 100 μm.
We next performed Timm staining to assess the patterns of mossy fiber projections in the hippocampal formation of irradiated, pilocarpine-treated rats and controls. As expected, the dentate gyrus of saline-treated controls that received two doses of 5 Gy irradiation or sham irradiation showed a normal pattern of Timm staining (Fig.7A,B). In contrast, aberrant supragranular mossy fiber reorganization in the dentate inner molecular layer occurred in Timm-stained sections of all pilocarpine-treated rats, both in the double-dose irradiation and sham-irradiation groups (Fig. 7C,D). The band of inner molecular layer Timm staining appeared qualitatively more intense and thicker in irradiated rats compared with sham-irradiated animals at 4 weeks after SE; however, quantification of Timm stain intensity with densitometric analysis showed no statistically significant difference between the two groups (p= 0.10, Student’s t test). Nissl staining revealed the typical pattern of pyramidal and hilar cell loss in pilocarpine-treated rats from both radiation and sham-radiation groups, and no cell loss was evident in hippocampi from irradiated or sham-irradiated controls (data not shown). Although we did not specifically assess recurrent seizures in the two pilocarpine-treated groups, no obvious differences were observed between irradiated and sham-irradiated groups in the episode of behavioral SE or the incidence of typical handling-induced seizures 4 weeks later. These results indicate that robust, seizure-induced mossy fiber reorganization occurs in the mature dentate gyrus even in the absence of neurogenesis.
Fig. 7.
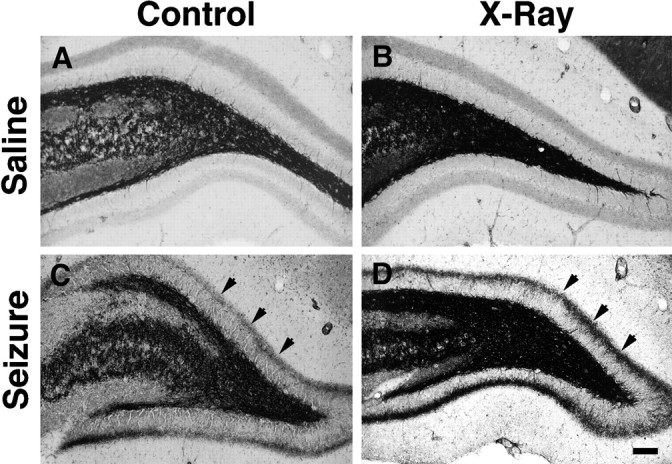
Mossy fiber reorganization persists after double-dose x-irradiation. Timm-stained sections through the dentate gyrus of rats from the same groups shown in Figure6A–D. The saline-treated, sham-irradiated control (A) displayed a normal pattern of mossy fiber Timm staining (black), and two radiation treatments alone had no effect on this pattern (B). Pilocarpine-induced SE produced a new band of Timm-stained fibers in the inner molecular layer 4 weeks after seizures (arrows in C), and this aberrant band also appeared in a rat treated with 5 Gy x-rays 1 d before and 4 d after SE (arrows in D). Note that the inner molecular layer Timm staining is darker in the irradiated (D) than in the sham-irradiated (C) rat. Scale bar, 100 μm.
DISCUSSION
The central finding of this study is that sprouting of axon collaterals from the mature DGC population gives rise to seizure-induced, aberrant mossy fiber remodeling. This mechanism has been assumed to underlie mossy fiber synaptic plasticity in the mature dentate gyrus since the initial description of DGC axonal reorganization after chemoconvulsant-induced status epilepticus over a decade ago (Tauck and Nadler, 1985). Neuroanatomic studies of hippocampi from humans with mesial TLE and rodent epilepsy models confirmed that aberrantly targeted axons in the dentate inner molecular layer arise from DGCs (Isokawa et al., 1993; Franck et al., 1995;Okazaki et al., 1995). However, these investigations did not take into account the presence of both mature and developing DGC populations in the adult mammalian dentate gyrus. The recent findings that DGC neurogenesis markedly increases before the development of seizure-induced mossy fiber remodeling (Parent et al., 1997; Gray and Sundstrom, 1998) suggest that newly developing DGCs are a potential source of aberrantly targeted mossy fibers. However, our data demonstrate that mossy fiber reorganization after pilocarpine-induced SE occurs even in the absence of DGC neurogenesis. This provides the first evidence that sprouting of axon collaterals from mature DGCs contributes to this network remodeling.
Several findings indicate that brain irradiation successfully inhibits neurogenesis at the doses used in our study. A single dose of 5 Gy markedly decreased the numbers of proliferating cells and immature neurons in the DGC layer identified by BrdU labeling and TOAD-64–IR, respectively, and this effect persisted for at least 5 d after irradiation (and 4 d after seizures). Furthermore, the same 5 Gy dose resulted in cell death localized to the dentate SGZ. After two x-ray doses, no BrdU was incorporated into proliferating cells during the first week after SE when BrdU was administered. Importantly, animals that were irradiated twice and survived for 4 weeks after seizures also had few or no immature neurons in the dentate gyrus detectable by TOAD-64 or PSA-NCAM immunostaining. TOAD-64–IR appears to accumulate in the DGC layer between 1 and 4 weeks after pilocarpine-induced SE, because increased TOAD-64 immunostaining is first evident within 1 week and peaks at ∼4 weeks after seizures (Parent et al., 1997). By this time, cell proliferation has returned to the lower basal rate. PSA-NCAM appears to be expressed in the same immature neuronal populations, both in normal and pilocarpine-treated adult rats (Figs. 5, 6) (Seki and Arai, 1993). Although we cannot entirely exclude that some neurogenesis occurred between 1 and 4 weeks after seizures, the amount is unlikely to be significant because TOAD-64–IR and PSA-NCAM–IR was still well below the normal basal level of expression at the 4 week time point. The much longer inhibition of neurogenesis after two radiation treatments compared with that after a single dose most likely reflects the greater degree of cell death produced by higher cumulative x-ray doses (Bellinzona et al., 1996; Shinohara et al., 1997).
Ionizing irradiation has numerous direct and indirect effects on cell function (Maity et al., 1994). Although we show that x-irradiation alone does not produce aberrant mossy fiber reorganization (Fig.7B), it is conceivable that x-rays may promote axonal sprouting in the setting of seizure-induced injury. We observed an increase in Timm staining in the molecular layer of irradiated rats compared with that of sham-irradiated controls 4 weeks after pilocarpine-induced SE. Although this was not a statistically significant difference, the relatively small sample sizes and limitations in quantification of Timm stain density may have masked a true increase in aberrant mossy fiber sprouting after irradiation and SE. This potential increase in sprouting may relate directly to the effects of brain irradiation. Alternatively, the presence of developing DGCs may normally inhibit new axon collateral outgrowth from mature granule neurons, and the elimination of neurogenesis could have removed such an effect. Also, it is unclear whether the apparent qualitative increase in seizure-induced mossy fiber remodeling after irradiation reflects a change in the rate of axonal sprouting, because only one time point after SE was used to assess Timm staining.
Although the present study indicates that newly generated DGCs are not necessary for aberrant axonal reorganization in the pilocarpine model of TLE, it does not preclude their involvement in the formation of abnormal networks after seizure-induced injury. It is possible, although unlikely, that mossy fiber remodeling under normal conditions arises exclusively from developing granule cells and that only in their absence do preexisting DGCs sprout. Even if this is not the case, it is clear that newly born DGCs are found in atypical locations in the dentate gyrus after prolonged seizures and that these differentiating neurons appear to integrate both normally and abnormally into the existing hippocampal circuitry (Parent et al., 1997). Mossy fiber network reorganization has been implicated in the development of hippocampal hyperexcitability associated with epileptogenesis, based on both anatomical and electrophysiological evidence that abnormally projecting DGC axon collaterals form recurrent excitatory synapses onto other DGCs (Tauck and Nadler, 1985; Cronin et al., 1992; Isokawa et al., 1993; Franck et al., 1995; Okazaki et al., 1995; Wuarin and Dudek, 1996). However, other studies have suggested that mossy fiber remodeling primarily enhances inhibition within the dentate gyrus (Sloviter, 1992). How the different populations of DGCs together contribute to seizure-induced plasticity and how these structural changes effect hippocampal function remain to be determined.
The normal function of persistent DGC neurogenesis in the adult mammalian brain and its regulation by both physiological and pathological processes are poorly understood. Because seizure activity induces a marked and consistent increase in dentate gyrus progenitor cell proliferation, seizure models offer potentially useful systems to investigate the mechanisms underlying neural development in the mature hippocampal formation. Our results indicate that a population of constitutively proliferating precursor cells in the dentate gyrus is activated by pilocarpine-induced SE and undergoes accelerated proliferation. After an apparent latent period of several days, the constitutively dividing cells in pilocarpine-treated rats, labeled by BrdU before SE, proliferate more rapidly than do those in controls. This results in an increased number of progeny, identified by markedly diluted nuclear BrdU content, that likely accounts for a significant proportion of the increase in newly generated DGCs after a prolonged seizure episode. Whether a population of quiescent progenitor cells is also activated by seizures remains to be determined. Furthermore, we found that the progeny of the constitutively proliferating cells in both pilocarpine-treated and control animals were remarkably localized to the SGZ and hilar aspect of the DGC layer. Because proliferating cells in these regions give rise mainly to granule neurons and occasional radial glia-like cells (Cameron et al., 1993), it is unlikely that astroglial proliferation after SE arises from the constitutively dividing precursor cell population in the dentate gyrus. However, further studies are needed to identify more precisely the phenotypes of proliferating cells in regions of astrogliosis and thus determine the contributions to the glial response of activated progenitor cells or mature astrocytes that reenter the cell cycle after seizure-induced hippocampal injury.
Our data also show that proliferating DGC progenitors are exquisitely vulnerable to low-dose x-irradiation. This is not unexpected given the sensitivity of proliferating cells to the effects of irradiation (Núñez et al., 1996), as well as previous reports demonstrating radiation-induced apoptosis of neural progenitors in the adult rat subventricular zone (Bellinzona et al., 1996; Shinohara et al., 1997). Ionizing irradiation is commonly used, with cumulative doses much higher than those given in this study, to treat malignant brain tumors in humans. One of the complications of this therapy is cognitive dysfunction, often involving difficulties with learning and memory (Crossen et al., 1994; Roman and Sperduto, 1995). Given the putative role of the hippocampal formation in learning and memory (Milner et al., 1998) and the recent evidence of continued DGC neurogenesis in the adult human brain (Eriksson et al., 1998), radiation-induced death of DGC progenitors may play a role in some of the untoward effects of radiation therapy. Together, these findings argue for the importance of further study of both neurogenesis in the human dentate gyrus and the specific effects of radiation on the human hippocampal formation.
Footnotes
This project was sponsored by a grant from the Epilepsy Foundation of America and National Institutes of Health (NIH) Grant NS02006 to J.M.P., by NIH Grant CA13525 to J.R.F., and by NIH Grants NS32062 and NS35628 and a grant from the March of Dimes Birth Defects Foundation to D.H.L. We thank Drs. S. Hockfield for providing TOAD-64 antibody, V. Lee for providing GFAP antibody, and G. Rougon for providing PSA-NCAM (MenB) antibody. We also thank Dr. R. Sloviter for critical review of this manuscript.
Correspondence should be addressed to Dr. Daniel H. Lowenstein, Department of Neurology, Box 0114, 513 Parnassus Avenue, University of California, San Francisco, San Francisco, California 94143-0114.
REFERENCES
- 1.Altman J, Das GD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol. 1965;124:319–335. doi: 10.1002/cne.901240303. [DOI] [PubMed] [Google Scholar]
- 2.Altman J, Das GD. Postnatal neurogenesis in the guinea pig. Nature. 1967;214:1098–1101. doi: 10.1038/2141098a0. [DOI] [PubMed] [Google Scholar]
- 3.Babb TL, Kupfer WR, Pretorious JK, Crandall PH, Levesque MF. Synaptic reorganization by mossy fibers in human epileptic fascia dentata. Neuroscience. 1991;42:351–363. doi: 10.1016/0306-4522(91)90380-7. [DOI] [PubMed] [Google Scholar]
- 4.Bellinzona M, Gobbel GT, Shinohara C, Fike JR. Apoptosis is induced in the subependyma of young adult rats by ionizing irradiation. Neurosci Lett. 1996;208:163–166. doi: 10.1016/0304-3940(96)12572-6. [DOI] [PubMed] [Google Scholar]
- 5.Bengzon J, Kokaia Z, Elmer E, Nanobashvili A, Kokaia M, Lindvall O. Apoptosis and proliferation of dentate gyrus neurons after single and intermittent limbic seizures. Proc Natl Acad Sci USA. 1997;94:10432–10437. doi: 10.1073/pnas.94.19.10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cameron HA, Woolley CS, McEwen BS, Gould E. Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience. 1993;56:337–344. doi: 10.1016/0306-4522(93)90335-d. [DOI] [PubMed] [Google Scholar]
- 7.Cavalheiro EA, Leite JP, Bortolotto ZA, Turski WA, Ikonomidou C, Turski L. Long-term effects of pilocarpine in rats: structural damage of the brain triggers kindling and spontaneous recurrent seizures. Epilepsia. 1991;32:778–782. doi: 10.1111/j.1528-1157.1991.tb05533.x. [DOI] [PubMed] [Google Scholar]
- 8.Cronin J, Dudek FE. Chronic seizures and collateral sprouting of dentate mossy fibers after kainic acid treatment in rats. Brain Res. 1988;474:181–184. doi: 10.1016/0006-8993(88)90681-6. [DOI] [PubMed] [Google Scholar]
- 9.Cronin J, Obenaus A, Houser CR, Dudek FE. Electrophysiology of dentate granule cells after kainate-induced synaptic reorganization of the mossy fibers. Brain Res. 1992;573:305–310. doi: 10.1016/0006-8993(92)90777-7. [DOI] [PubMed] [Google Scholar]
- 10.Crossen JR, Garwood D, Glatstein E, Neuwelt EA. Neurobehavioral sequelae of cranial irradiation in adults: a review of radiation-induced encephalopathy. J Clin Oncol. 1994;12:627–642. doi: 10.1200/JCO.1994.12.3.627. [DOI] [PubMed] [Google Scholar]
- 11.de Lanerolle NC, Kim JH, Robbins RJ, Spencer DD. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989;495:387–395. doi: 10.1016/0006-8993(89)90234-5. [DOI] [PubMed] [Google Scholar]
- 12.Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn A, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- 13.Franck JE, Pokorny J, Kunkel DD, Schwartzkroin PA. Physiologic and morphologic characteristics of granule cell circuitry in human epileptic hippocampus. Epilepsia. 1995;36:543–558. doi: 10.1111/j.1528-1157.1995.tb02566.x. [DOI] [PubMed] [Google Scholar]
- 14.Gage FH, Kemperman G, Palmer TD, Peterson DA, Ray J. Multipotent progenitor cells in the adult dentate gyrus. J Neurobiol. 1998;36:249–266. doi: 10.1002/(sici)1097-4695(199808)36:2<249::aid-neu11>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 15.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gould E, Tanapat P. Lesion-induced proliferation of neuronal progenitors in the dentate gyrus of the adult rat. Neuroscience. 1997;80:427–436. doi: 10.1016/s0306-4522(97)00127-9. [DOI] [PubMed] [Google Scholar]
- 17.Gould E, Tanapat P, McEwen BS, Flügge G, Fuchs E. Proliferation of granule cell precursors in the dentate gyrus of adult monkeys is diminished by stress. Proc Natl Acad Sci USA. 1998;95:3168–3171. doi: 10.1073/pnas.95.6.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gray WP, Sundstrom LE. Kainic acid increases the proliferation of granule cell progenitors in the dentate gyrus of the adult rat. Brain Res. 1998;790:52–59. doi: 10.1016/s0006-8993(98)00030-4. [DOI] [PubMed] [Google Scholar]
- 19.Gueneau G, Privat A, Drouet J, Court L. Subgranular zone of the dentate gyrus of young rabbits as a secondary matrix: a high-resolution autoradiographic study. Dev Neurosci. 1982;5:345–358. doi: 10.1159/000112694. [DOI] [PubMed] [Google Scholar]
- 20.Houser CR, Miyashiro JE, Swartz BE, Walsh GO, Rich JR, Delgado-Escueta AV. Altered patterns of dynorphin immunoreactivity suggest mossy fiber reorganization in human hippocampal epilepsy. J Neurosci. 1990;10:267–282. doi: 10.1523/JNEUROSCI.10-01-00267.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Isokawa M, Levesque MF, Babb TL, Engel J., Jr Single mossy fiber axonal systems of human dentate granule cells studied in hippocampal slices from patients with temporal lobe epilepsy. J Neurosci. 1993;13:1511–1522. doi: 10.1523/JNEUROSCI.13-04-01511.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaplan MS, Bell DH. Mitotic neuroblasts in the 9-day-old and 11-month-old rodent hippocampus. J Neurosci. 1984;4:1429–1441. doi: 10.1523/JNEUROSCI.04-06-01429.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaplan MS, Hinds JW. Neurogenesis in adult rat: electron microscopic analysis of light radioautographs. Science. 1977;197:1092–1094. doi: 10.1126/science.887941. [DOI] [PubMed] [Google Scholar]
- 24.Khurgel M, Ivy GO. Astrocytes in kindling: relevance to epileptogenesis. Epilepsy Res. 1996;26:163–175. doi: 10.1016/s0920-1211(96)00051-4. [DOI] [PubMed] [Google Scholar]
- 25.Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16:2027–2033. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Latov N, Nilaver G, Zimmerman EA, Johnson WG, Silverman AJ, Defendini R, Cote L. Fibrillary astrocytes proliferate in response to brain injury: a study combining immunoperoxidase technique for glial fibrillary acidic protein and radioautography of tritiated thymidine. Dev Biol. 1979;72:381–384. doi: 10.1016/0012-1606(79)90127-1. [DOI] [PubMed] [Google Scholar]
- 27.Liu J, Solway K, Messing RO, Sharp FR. Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J Neurosci. 1998;18:7768–7778. doi: 10.1523/JNEUROSCI.18-19-07768.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maity A, McKenna G, Muschel RJ. The molecular basis for cell cycle delays following ionizing radiation: a review. Radiother Oncol. 1994;31:1–13. doi: 10.1016/0167-8140(94)90408-1. [DOI] [PubMed] [Google Scholar]
- 29.Mello LE, Cavalheiro EA, Tan AM, Kupfer WR, Pretorius JK, Babb TL, Finch DM. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: cell loss and mossy fiber sprouting. Epilepsia. 1993;34:985–995. doi: 10.1111/j.1528-1157.1993.tb02123.x. [DOI] [PubMed] [Google Scholar]
- 30.Miller MW, Nowakowski RS. Use of bromodeoxyuridine-immunohistochemistry to examine the proliferation, migration and time of origin of cells in the central nervous system. Brain Res. 1988;457:44–52. doi: 10.1016/0006-8993(88)90055-8. [DOI] [PubMed] [Google Scholar]
- 31.Milner B, Squire LR, Kandel ER. Cognitive neuroscience and the study of memory. Neuron. 1998;20:445–468. doi: 10.1016/s0896-6273(00)80987-3. [DOI] [PubMed] [Google Scholar]
- 32.Minturn JE, Geschwind DH, Fryer HJL, Hockfield S. Early postmitotic neurons transiently express TOAD-64, a neural specific protein. J Comp Neurol. 1995;355:369–379. doi: 10.1002/cne.903550304. [DOI] [PubMed] [Google Scholar]
- 33.Niquet J, Ben-Ari Y, Represa A. Glial reaction after seizure induced hippocampal lesion: immunohistochemical characterization of proliferating glial cells. J Neurocytol. 1994;23:641–656. doi: 10.1007/BF01191558. [DOI] [PubMed] [Google Scholar]
- 34.Núñez MI, McMillan TJ, Valenzuela MT, Ruiz de Almodóvar JM, Pedraza V. Relationship between DNA damage, rejoining and cell killing by radiation in mammalian cells. Radiother Oncol. 1996;39:155–165. doi: 10.1016/0167-8140(96)01732-x. [DOI] [PubMed] [Google Scholar]
- 35.Okazaki MM, Evenson DA, Nadler JV. Hippocampal mossy fiber sprouting and synapse formation after status epilepticus in rats: visualization after retrograde transport of biocytin. J Comp Neurol. 1995;352:515–534. doi: 10.1002/cne.903520404. [DOI] [PubMed] [Google Scholar]
- 36.Parent JM, Lowenstein DH. Mossy fiber reorganization in the epileptic hippocampus. Curr Opin Neurol. 1997;10:103–109. doi: 10.1097/00019052-199704000-00006. [DOI] [PubMed] [Google Scholar]
- 37.Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parent JM, Janumpalli S, McNamara JO, Lowenstein DH. Increased dentate granule cell neurogenesis following amygdala kindling in the adult rat. Neurosci Lett. 1998;247:9–12. doi: 10.1016/s0304-3940(98)00269-9. [DOI] [PubMed] [Google Scholar]
- 39.Roman DD, Sperduto PW. Neuropsychological effects of cranial radiation: current knowledge and future directions. Int J Radiat Oncol Biol Phys. 1995;31:983–998. doi: 10.1016/0360-3016(94)00550-8. [DOI] [PubMed] [Google Scholar]
- 40.Scott BW, Wang S, Burnham WM, De Boni U, Wojtowicz JM. Kindling-induced neurogenesis in the dentate gyrus of the rat. Neurosci Lett. 1998;248:73–76. doi: 10.1016/s0304-3940(98)00355-3. [DOI] [PubMed] [Google Scholar]
- 41.Seki T, Arai Y. Highly polysialylated neural cell adhesion molecule (NCAM-H) is expressed by newly generated granule cells in the dentate gyrus of the adult rat. J Neurosci. 1993;13:2351–2358. doi: 10.1523/JNEUROSCI.13-06-02351.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shinohara C, Gobbel GT, Lamborn KR, Tada E, Fike JR. Apoptosis in the subependyma of young adult rats after single and fractionated doses of X-rays. Cancer Res. 1997;57:2694–2702. [PubMed] [Google Scholar]
- 43.Sloviter RS. A simplified Timm stain procedure compatible with formaldehyde fixation and routine embedding of rat brain. Brain Res Bull. 1982;8:771–774. doi: 10.1016/0361-9230(82)90104-6. [DOI] [PubMed] [Google Scholar]
- 44.Sloviter RS. Possible functional consequences of synaptic reorganization in the dentate gyrus of kainate-treated rats. Neurosci Lett. 1992;137:91–96. doi: 10.1016/0304-3940(92)90306-r. [DOI] [PubMed] [Google Scholar]
- 45.Stanfield BB, Trice JE. Evidence that granule cells generated in the dentate gyrus of adult rats extend axonal projections. Exp Brain Res. 1988;72:399–406. doi: 10.1007/BF00250261. [DOI] [PubMed] [Google Scholar]
- 46.Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- 47.Tauck D, Nadler J. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas LB, Gates DJ, Richfield EK, O’Brien JB, Schweitzer JB, Steindler DA. DNA end labeling (TUNEL) in Huntington’s disease and other neuropathological conditions. Exp Neurol. 1995;133:265–272. doi: 10.1006/exnr.1995.1029. [DOI] [PubMed] [Google Scholar]
- 49.Wang L, Strittmatter SM. A family of rat CRMP genes is differentially expressed in the nervous system. J Neurosci. 1996;16:6197–6207. doi: 10.1523/JNEUROSCI.16-19-06197.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wuarin J, Dudek FE. Electrographic seizures and new recurrent excitatory circuits in the dentate gyrus of hippocampal slices from kainate-treated epileptic rats. J Neurosci. 1996;16:4438–4448. doi: 10.1523/JNEUROSCI.16-14-04438.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]