

Abstract
After peripheral nerve transection, axons distal to the cut site rapidly degenerate, a process termed Wallerian degeneration. In wild-type mice the compound action potential (CAP) disappears by 3 d. Previous studies have demonstrated that cold temperatures and lower extracellular calcium ion (Ca2+) concentrations can slow the rate of Wallerian degeneration. We have incubated isolated sciatic nerve segments from wild-type and C57BL/Wld mice (which carry a gene slowing Wallerian degeneration) in vitro at 25 and 37°C. At 25°C we found that the degeneration rate of wild-type axons was slowed dramatically, with the CAP preserved up to 7 d post-transection. In contrast, at 37°C the CAPs were minimal at 2 d. When the temperature of wild-type nerves was raised to 37°C after 24–72 hr at 25°C, degeneration occurred within the subsequent 24 hr. Wld nerves, too, were preserved longer at 25°C but, on return to 37°C, degenerated promptly. Cooling the nerve within 12 hr after axotomy enhanced axonal preservation. Neither wild-type nor Wld nerves showed different degeneration rates when they were incubated with 250 μm or 5 or 10 mm extracellular Ca2+ for 1–2 d, suggesting that an abrupt increase in intracellular Ca2+ occurs at the time of axonal destruction. Wallerian degeneration, thus, appears to progress through three distinct stages. Initiation occurs at the time of injury with subsequent temperature-dependent and -independent phases. Nerves appear to remain intact and are able to exclude Ca2+ from entering until an as yet unknown process finally increases axolemmal permeability.
Keywords: Wallerian degeneration, temperature, C57BL/Wld mice, axonal degeneration, calcium, in vitro
Transection of a peripheral nerve initiates the processes of Wallerian degeneration. In giant squid, crayfish, and fish the isolated axons may be preserved for months, with proteins continuing to be transported in the axoplasm and supplied by surrounding glia (Sheller and Bittner, 1992; Tanner et al., 1995a,b;Raabe et al., 1996). In contrast, injured axons in rodents degenerate within 3 d, and the nerve consequently becomes unable to conduct a compound action potential (CAP) on application of an external electrical stimulus (Luttges et al., 1976). For a CAP to be conducted, the plasma membrane and the transmembrane voltage-gated ion channels must remain intact, and the normal intracellular ion levels must be preserved. The existence of the C57BL/Wld (formerly known as the C57BL/Ola) mouse strain, carrying an autosomal dominant mutation (Wlds) slowing the rate of Wallerian degeneration (CAPs can be conducted up to 3 weeks post-transection), demonstrated for the first time that rapid degeneration in mammals is not a necessary consequence of axotomy (Lunn et al., 1989; Perry et al., 1990a; Tsao et al., 1994). It is also clear that Wallerian degeneration is an active process, with an initiating mechanism that sets into motion a cascade of events leading to the final destruction of axons. The precise sequence of events that leads to axonal destruction is not known.
Experiments on Wallerian degeneration in mammalian, lower vertebrate, and invertebrate axons have all demonstrated that cold temperatures can slow the rate of progression (Gamble et al., 1957; Gamble and Jha, 1958; Usherwood et al., 1968; Wang, 1985; Bittner, 1988; Sea et al., 1995). This effect has been attributed variously to an alteration in the activity of degradative enzymes (changing the Q10 by threefold) or the rate of axoplasmic transport (Cancalon, 1982, 1985). Additionally, researchers have demonstrated a link between the intracellular and extracellular calcium ion concentration ([Ca2+]i/o) and the time course of axonal sealing and degeneration (Schlaepfer and Bunge, 1973;Schlaepfer, 1974, 1977; Krause et al., 1994; Fern et al., 1995; George et al., 1995; Eddleman et al., 1997, 1998). Axoplasmic Ca2+ concentrations of 100 μm induce vesicle-mediated sealing, although a higher [Ca2+]i may be needed for the deleterious effects (Krause et al., 1994; Eddleman et al., 1997, 1998). The experiments of Schlaepfer and Bunge (1973), Glass et al. (1994), and George et al. (1995) suggest that degeneration proceeds only when [Ca2+]o is >300 μm.
In wild-type axons axonal inability to continue conducting CAPs occurs simultaneously with axonal destruction (McDonald, 1972). In Wld mice the first electrophysiologically detectable event in Wallerian degeneration is axonal inability to continue conducting CAPs although the morphology remains relatively preserved (Tsao et al., 1994). Via monitoring the disappearance of CAPs and axonal morphology, this study also demonstrated that axonal degeneration had similar rates in vivo and in vitro. Using an in vitro system (described in Tsao et al., 1994), we have investigated the effect of temperature and media [Ca2+] on the time course of axonal degeneration (duration of prolonged CAP and, in some experiments, the rate of decline). We asked whether axonal loss in mouse nerves could be delayed by cooling the external media temperature to 25°C and, if so, when, after injury, cooling had to be initiated to prolong axonal survival. Finally, by varying [Ca2+]o, we addressed the question of whether intra-axonal Ca2+ increases gradually after axotomy or whether there is an abrupt transition from low [Ca2+]i concentrations to high concentrations, accompanied by loss of the CAP and axonal cytoskeleton.
Parts of this paper have been published previously (Tsao and George, 1996; Tsao et al., 1997).
MATERIALS AND METHODS
Sciatic nerve preparation. C57BL/Wld mice were bred in the Johns Hopkins Hospital (Baltimore, MD) from mice originally supplied by Harlan-Olac (Bicester, UK). C57BL/6N mice (control, wild-type) were supplied by Harlan Sprague Dawley (Indianapolis, IN). Twelve-week-old mice were used in the experiments and were killed by cervical dislocation. The sciatic nerves were removed and, for the Ca2+ concentration experiments, desheathed with a microsurgical knife (Roboz Surgical Instrument, Rockville, MD) and then placed into 1 ml of serum-free Optimem I medium (Life Technologies, Gaithersburg, MD) or DME/F-12 medium (Sigma, St. Louis, MO) supplemented with CaCl2 to give a Ca2+concentration of 250 μm or 5 or 10 mm. Both media contained penicillin and streptomycin (Life Technologies), each at a concentration of 5 IU/ml. Media were gassed with 95% O2/5% CO2 to give a pH of 7.4 at 25 and 37°C in a humidified incubator and were changed every 3 d. All animals were cared for under the guidelines issued by Johns Hopkins University (Baltimore, MD).
To investigate the effect of temperature on the rate of Wallerian degeneration, we cultured one nerve from each mouse (Wld and 6N) at 25°C, with the contralateral nerve cultured at 37°C. To determine whether cooling nerves for a period of time at 25°C could slow or halt Wallerian degeneration subsequently at 37°C, we incubated wild-type nerves at 25°C for 24, 48, and 72 hr, followed by 24 hr at 37°C. Conversely, the time when degeneration could no longer be slowed by cooling was determined by incubating wild-type nerves for 3, 6, 9, 12, 14, 16, 18, and 24 hr at 37°C, followed by incubation at 25°C, for a total of 48 hr in vitro.
To examine the role of extracellular (media) Ca2+concentrations on the degeneration rate, we incubated wild-type and Wld nerves for 48 hr in 250 μm or 5 or 10 mmCa2+. Other nerves were incubated first for 24 hr in 5 or 10 mm Ca2+, followed by 24 hr in 250 μm Ca2+, or for 24 hr in 250 μm Ca2+, followed by 24 hr in 5 or 10 mm Ca2+. A further permutation was introduced by using two temperatures, 25 and 37°C.
Electrophysiology and tissue examination. Between 1 and 11 d, both A (rapidly conducting, large myelinated fiber) and C (slowly conducting, small unmyelinated fiber) waves of the CAP were measured by placing the sciatic nerves in Ringer’s solution [containing (in mm) 135 NaCl, 2.5 KCl, 10d-glucose, 1 MgCl2, 2.5 CaCl2, and 6 HEPES, pH 7.4] at room temperature. The proximal and distal ends were placed onto bipolar silver electrodes (proximal end to the stimulating electrode and distal end to the recording electrode) while the middle of the nerve remained immersed in Ringer’s solution. An inter-electrode distance of 0.5–1 cm was established so that the stimulating electrode did not interfere with the recorded signal. Adipose tissue adherent to the nerve at either the stimulating or recording end was found to decrease the CAP intensity; thus, care was taken to dissect the nerve free of such tissue.
Stimulating pulses of 200 μsec duration were delivered at 1 sec intervals via an optical isolation unit (Isoflex, A.M.P.I., Jerusalem, Israel), and the maximum fast monophasic A wave and the slower C wave were recorded. Using previously described methods (Perry et al., 1990b,1992), we analyzed the maximal peak height and peak area of both A and C waves with the Sigavg Program (Cambridge Electronic Design, Cambridge, UK) after digitization (CED 1401) at 80 kHz and found them to be equivalent (Tsao et al., 1994). Selected nerves were fixed in 2% paraformaldehyde, osmicated for 2 hr, infiltrated with glycerol, and then teased apart and examined by light microscopy.
Statistical analysis. Data were analyzed with the Statview 4.0 program (Abacus Concepts, Berkeley, CA). Significance (p < 0.05) was assessed by using either ANOVA, followed by the Student’s t test if ANOVA showed significance, or the paired t test.
RESULTS
Axonal degeneration in vivo andin vitro
We confirmed that axonal degeneration in vivo andin vitro in 6N nerves was comparable, with ∼2.5% of the CAP (relative to control) remaining at 2 d, as previously reported (Tsao et al., 1994). The act of axotomy and removing the nerves for culturing was found to reduce the CAP A wave by ∼30% within 3 hr, after which the nerve was stable for the next 21 hr (Fig.1A,B). In vivo the CAP after axotomy was found to remain constant for the first 24 hr, actually rising in the first 6 hr (Fig.2A,B). In all, 80% of the CAP was lost between 24 and 36 hr. A similar time course was foundin vitro (Fig. 2A,B).
Fig. 1.
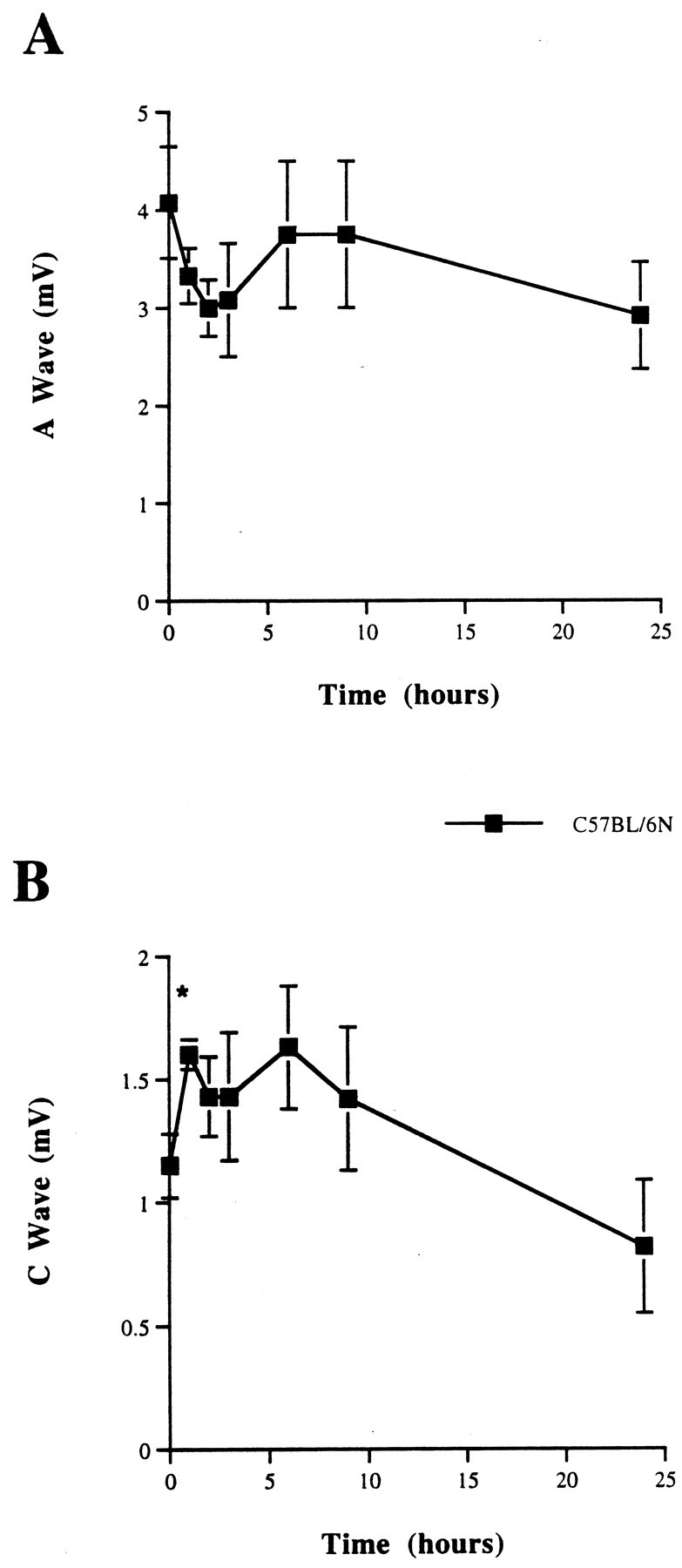
Compound action potential (CAP) from C57BL/6N sciatic nerves incubated for 0, 1, 2, 3, 6, 9, and 24 hr at 37°C in Optimem I medium. The CAP values demonstrate a decrease in CAP size in the first 3 hr after removal of the nerve to culture but stable values during the next 21 hr. A, A wave (from myelinated fibers). B, C wave (from unmyelinated fibers). *p < 0.05, relative to time 0 hr.
Fig. 2.
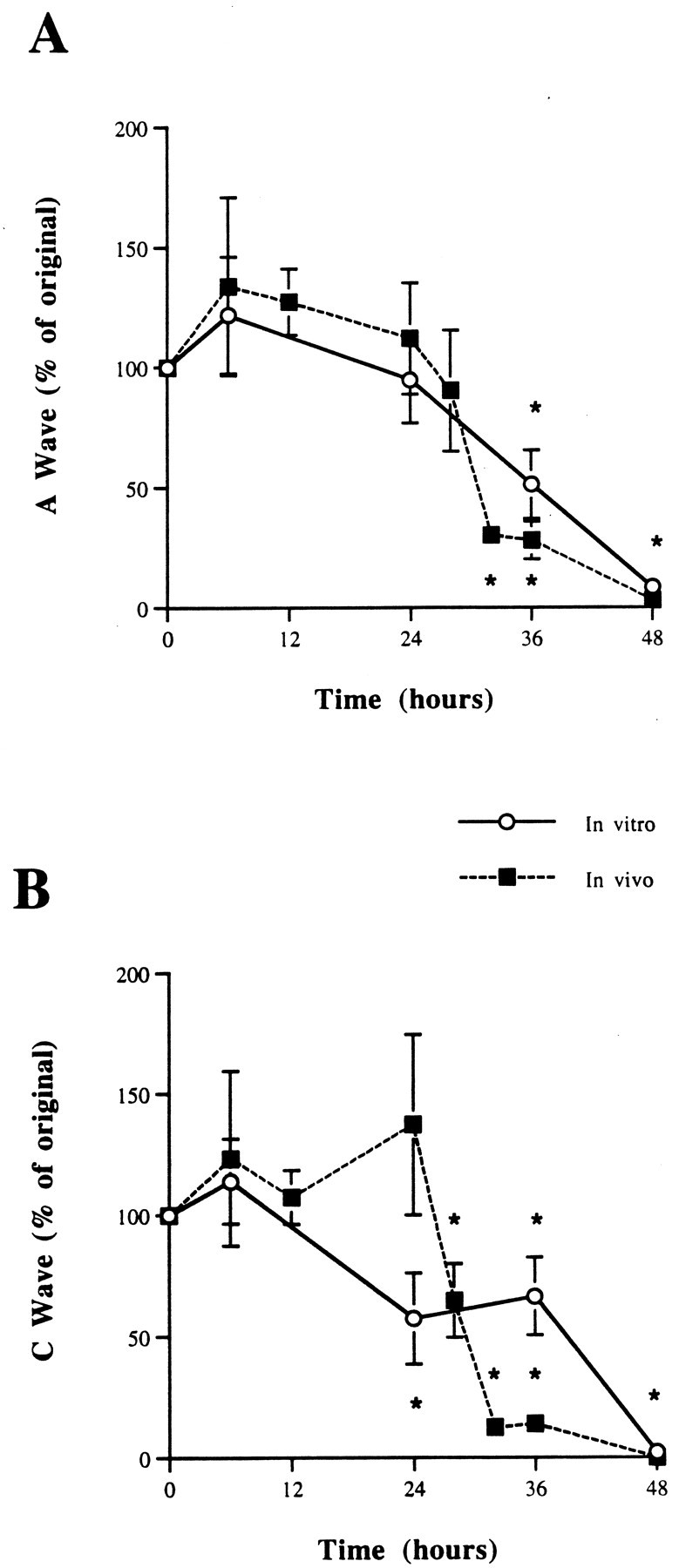
CAP from C57BL/6N sciatic nerves axotomized 0–48 hr in vivo and in vitro.A, A wave. B, C wave. In the first 6 hr after axotomy the CAP actually is increased. This is unlikely to be attributable to decreased temporal dispersion because the area of the waveform is increased. In both A and C waves, 80% of the CAP is lost between 24 and 36 hr. A similar time course for loss of the CAP is seen in vivo and in vitro. *p < 0.05.
The effect of temperature changes on the rate of axonal degeneration
Nerves from 6N mice maintained at 25°C showed a prolonged presence of CAPs (A and C waves) (Fig.3A,B). Light microscopic examination of teased fibers confirmed that myelinated axons remained intact (data not shown). The incubation of nerves at 25°C for 1–3 d, followed by 37°C for 1 d, demonstrated that previous cooling could not prevent the subsequent decrease in the CAP (Fig.4A,B), because at the end of this additional 24 hr only small CAPs could be detected. The CAP at intermediate time points after the return to 37°C was not significantly different from that seen in nerves incubated continuously at 37°C.
Fig. 3.
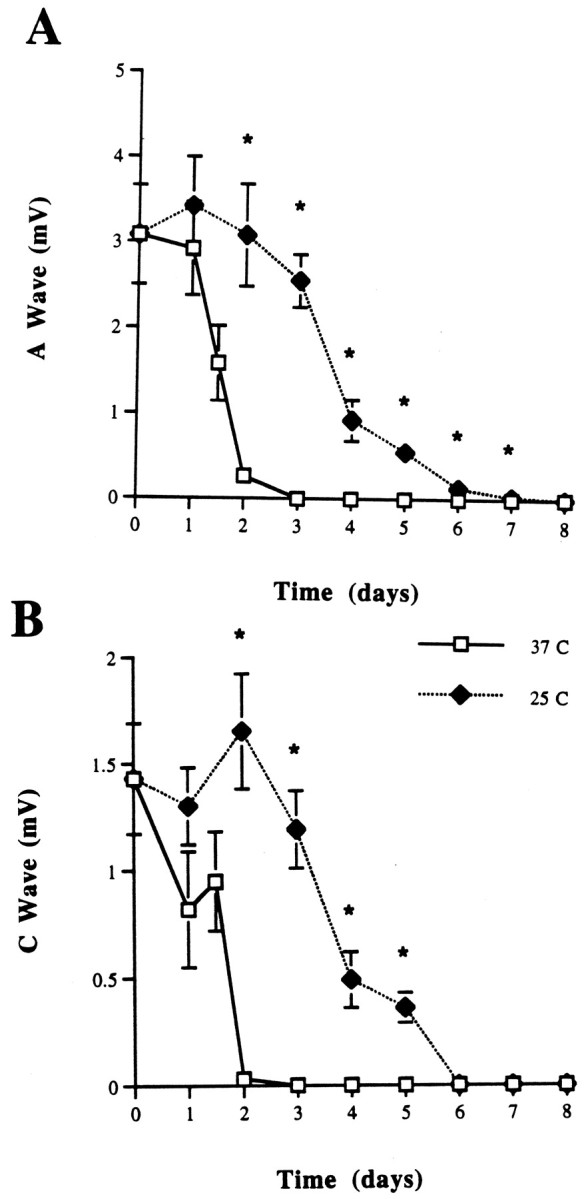
CAP from C57BL/6N sciatic nerves maintained at 25 and 37°C in Optimem I medium for 1–8 d. A, A wave.B, C wave. Note that the CAP disappears within 3 d at 37°C, whereas it is preserved to 7 d at 25°C. *p < 0.05.
Fig. 4.

CAP from C57BL/6N sciatic nerves maintained at 25°C in Optimem I medium for a period from 6 hr to 3 d, followed by 1 d at 37°C. A, A wave. B, C wave. The CAP for nerves maintained for the duration of the experiment at both 25 and 37°C (from Fig. 1) is shown for comparison. Note that the CAP disappears within the subsequent 24 hr rather than the customary 48 hr seen with newly axotomized nerves.
If they were cooled to 25°C within 12 hr, 6N nerves that were incubated initially at 37°C had complete and prolonged preservation of the CAP A and C waves (mimicking the rate seen with continuous incubation at 25°C) (Fig.5A,B). If the nerves were cooled after this time had elapsed, the CAP amplitude was found to be diminished at 48 hr.
Fig. 5.
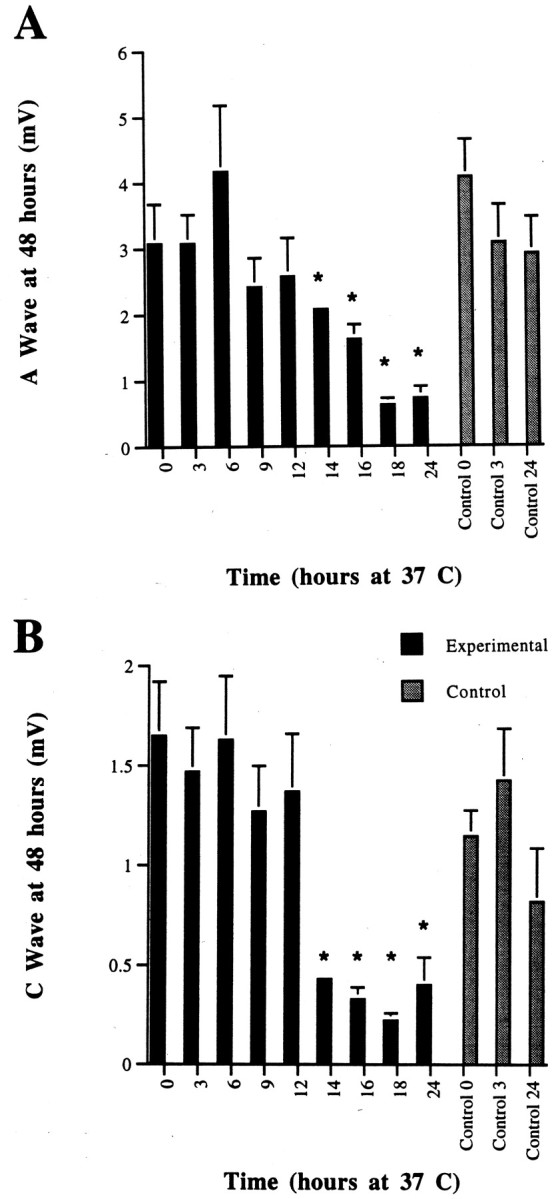
CAP from C57BL/6N sciatic nerves at 48 hr incubation in Optimem I medium. Nerves were incubated for 3–24 hr at 37°C before being cooled to 25°C. The CAP values at time 0, 3, and 24 hr at 37°C are shown for comparison to demonstratethe decrease in CAP size in the first 3 hr after removal of the nerve to culture. A, A wave. B, C wave. Complete preservation of both A and C waves is seen only if the cooling occurs by 12 hr after axotomy. *p < 0.05.
Wld nerves also showed a delay in the degeneration rate on cooling to 25°C (Fig. 6A,B). On returning the nerves to 37°C after 3–4 d at 25°C, the CAP also returned to values not significantly different from those nerves incubated for the same length of time at 37°C (data not shown), similar to the effect seen in wild-type nerves. At 37°C in vitro the nerve survival was prolonged to 9 d, further extending previous data (Perry et al., 1990b; Tsao et al., 1994).
Fig. 6.
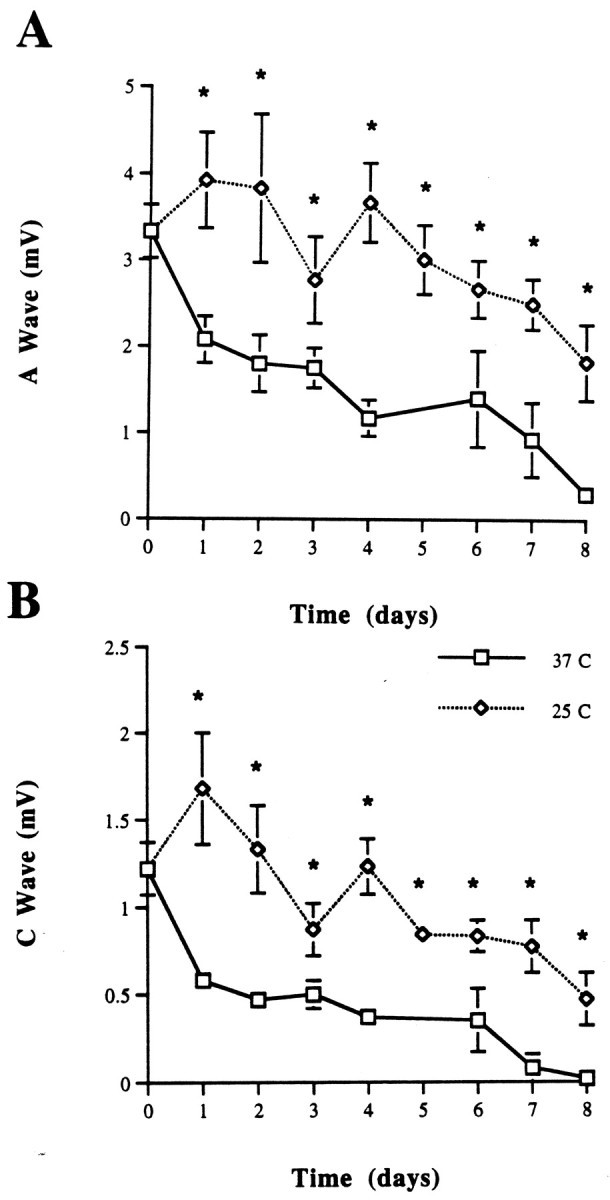
CAP from C57BL/Wld sciatic nerves maintained at 25 and 37°C in Optimem I for 1–11 d. A, A wave.B, C wave. Both waves are preserved with cooler temperatures. *p < 0.05.
The effect of extracellular calcium ion concentration on the rate of Wallerian degeneration
Incubation of wild-type nerves in the presence of 250 μm or 5 or 10 mm external Ca2+ did not affect the degeneration rate (Fig.7). Altering Ca2+concentrations after 24 hr, with or without temperature adjustment, also failed to influence the rate of degeneration (data not shown). Similarly, the rate of axonal degeneration in Wld nerves was unaffected by the three Ca2+ concentrations (data not shown).
Fig. 7.
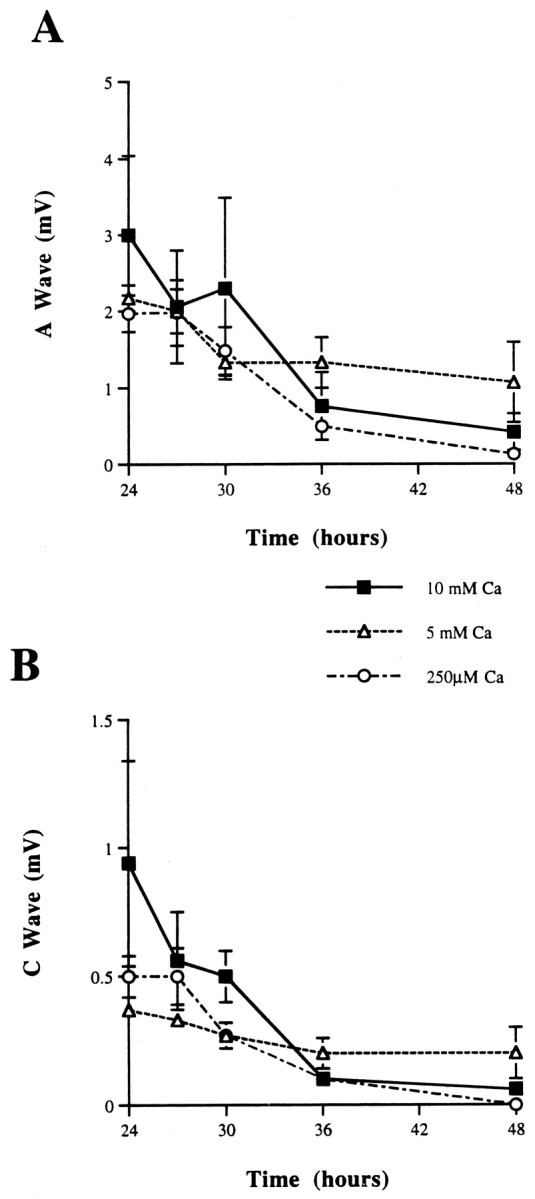
CAP from C57BL/6N sciatic nerves (desheathed) incubated with either 250 μm or 5 or 10 mmCaCl2 for 48 hr at 37°C. A, A wave.B, C wave. *p < 0.05.
DISCUSSION
The effect of cooling on the rate of Wallerian degeneration
The effect of cool external temperatures slowing Wallerian degeneration in vivo is well known (Gamble et al., 1957;Gamble and Jha, 1958; Usherwood et al., 1968; Wang, 1985; Sea et al., 1995). In rats, Sea and colleagues (1995) showed that the time course for myelinated axons to degenerate after axotomy was 3 d at 32°C and 6 d at 23°C. The current results confirm that lowering the temperature in vitro can delay the progression of Wallerian degeneration (see Fig. 3A,B). The onset of degeneration appears to be halted in wild-type nerves in the first 1–4 d by maintaining an external media temperature of 25°C. On raising the temperature to 37°C, however, the time to complete degeneration was not the customary 48 hr seen with a constant temperature of 37°C. Instead, loss of CAP occurred within 24 hr, with a rate similar to that seen in nerves continuously incubated at 37°C between 24 and 48 hr (see Fig. 4A,B). This suggests that cooling delays a “final” stage of Wallerian degeneration, but when that stage commences, it progresses rapidly to completion.
Knowing that a media temperature of 25°C would allow for the preservation of the axons and, thus, the CAP up to 7 d in vitro, we asked when after axotomy the cooling had to be initiated to delay Wallerian degeneration. We found that 100% of the CAP was preserved if the axotomized nerve was cooled from 37 to 25°C within the first 12 hr (all measurements were made at 48 hr for both A and C waves) (see Fig. 5A,B). These results suggest that there is a critical period, early after the initial injury and before the effects of Wallerian degeneration are manifest, during which activation of the sequence of events leading to axonal destruction may have begun but is not evident until later. One study supporting this hypothesis is that of Majno and Karnovsky (1958), which showed changes in nerve lipid composition within the first 6–12 hr after injury, a time when the CAP appears to be unaffected (see Fig. 2A,B). A change in temperature to 25°C would result in an alteration of both degradative enzymes and axonal transport along the Q10’s (Cancalon, 1982, 1985), either of which might affect the subsequent degeneration rate. Alternatively, Wallerian degeneration might begin only after an initial period of time, after which the necessary degradative enzymes, such as calpains, become active.
Of note also are experiments on neural damage in head trauma that have revealed that axotomy from shear injury generally does not occur. Instead, subtle changes—an increase in axolemmal permeability, neurofilament compaction, and impairment of axoplasmic transport—appear within 1–6 hr, a time when axons still appear morphologically intact (Povlishock, 1992; Povlishock and Christman, 1995; Povlishock and Pettus, 1996; Povlishock et al., 1997). The injured axons finally begin separating from their cell bodies only after 6–12 hr. The current data are consistent with the hypothesis that changes in the first 12 hr are responsible for triggering the subsequent breakdown of the axolemma and cytoskeleton. Additionally, for the first time on the basis of these results, one could theorize that intervention during this early time might allow Wallerian degeneration to be reversed or halted completely. After 12 hr have elapsed, it appears that a second stage of Wallerian degeneration has begun, which either is temperature-independent or requires even lower temperatures to stop (see Fig. 5A,B).
Interestingly, Wld nerves also showed a temperature dependence in the degeneration rate (see Fig. 6A,B), suggesting that the Wlds mutation actively acts to slow the progression of degeneration. A comparison of Figures 3 and 6 suggests that the early stages of Wallerian degeneration are prolonged similarly by cooling in both the 6N and Wld nerves. In contrast, the final stage in Wld nerves is slow and relatively unaffected by cooling, whereas the final stage of Wallerian degeneration in 6N nerves is rapid at 37°C but slowed by cooling. The data indicate that the signal initiating Wallerian degeneration is transduced in Wld axons but does not appear to propagate as rapidly as in wild-type axons.
The effect of extracellular calcium ion concentration on the rate of Wallerian degeneration
Intra- and extracellular Ca2+ concentrations are important in determining the rate of Wallerian degeneration and activation of degradative enzymes (Schlaepfer, 1974, 1977; George et al., 1995). Calcium ions are required both to help seal the damaged axon and to induce degeneration later. Axotomy is accompanied by the entry of calcium ions into the axoplasm, allowing the formation of membrane vesicles that help to seal the injured axon; this process requires a minimum axoplasmic Ca2+ concentration of 100 μm (Krause et al., 1994; Eddleman et al., 1997,1998). An external level of <250 μmCa2+ has been shown to retard degeneration of axons after axotomy (Schlaepfer and Bunge, 1973; George et al., 1995). However, it is not known whether Ca2+ enters axons at a constant rate after injury or whether the ion exclusion is maintained until a time when large amounts of Ca2+suddenly enter, reaching a concentration necessary for degradative enzyme activation. A [Ca2+]o of 5 or 10 mm provides a larger ionic driving force for Ca2+ entry than a concentration of 250 μm does (physiological [Ca2+]o is 1–2 mm). Our experiments examined whether or not maintenance of intact, desheathed sciatic nerves in low and high external Ca2+concentrations could affect the rate of Wallerian degeneration and loss of the CAP. Incubation of nerves in both 5 and 10 mmexternal Ca2+ did not speed the time course for axonal degeneration in either wild-type or Wld nerves. Conversely, incubation of matched contralateral nerves in 250 μmCa2+ also did not delay Wallerian degeneration (see Fig. 7). We were unable to lower media Ca2+ below 250 μm, because this caused the CAP to disappear within 6 hr (our unpublished observations). In both high and low Ca2+ concentrations, the size of CAPs was similar at all of the time points that were studied. Because the rate of axonal degeneration is independent of the driving force for Ca2+ entry, either the transporter for Ca2+ influx is saturated easily even at 250 μm Ca2+, or Ca2+influx is a briefer event for which the duration does not impact on the overall rate of axonal degeneration. In the latter case plasmalemmal integrity may be maintained until a point, late in axonal degeneration, when there is rapid entry of extracellular Ca2+ and a culminating final catastrophic moment of axonal destruction, rather than a gradual creeping upward of [Ca2+]i leading to a crescendoing degeneration rate.
Further supporting this hypothesis is an x-ray microprobe elemental analysis of rat sciatic nerves post-transection, which demonstrated a decrease in intracellular potassium (K+) and chloride (Cl−) ions and an increase in phosphorus (P) at 8 hr after injury in myelinated fibers (LoPachin et al., 1990). At 16 hr post-transection, elemental values remained the same as at 8 hr, with the exception of a newly elevated intracellular sodium ion (Na+) concentration. The Ca2+content of the axoplasm was unaltered at 8 and 16 hr, but its level in mitochondria was elevated significantly at 16 hr. Only after 48 hr, when all fibers had degenerated, was increased axoplasmic Ca2+ detected, with mitochondria at that time showing a >80-fold elevation in Ca2+. This disruption of elemental homeostasis appears to be concurrent with a neuronal energy deficit seen 24–48 hr after peripheral nerve axotomy, which is accompanied by decreased activity of the membrane Na+-K+-ATPase (Stewart et al., 1965; Bachelard and Silva, 1966). The elemental changes noted byLoPachin et al. (1990) are consistent with Na+-K+-ATPase inhibition; with an intact plasma membrane Na+ would be excluded, whereas K+ still would continue to leak through open ion channels, with Cl− following passively to maintain electroneutrality. With the K+ gradient dissipated, Na+ then could enter the axon, leading to the entry of Ca2+ via Na+/Ca2+ exchange or the failure of pumping mechanisms for Ca2+ extrusion or sequestration.
Our data are not consistent with the hypothesis that the act of axotomy leads to a transient, massive increase in [Ca2+]i along the entire length of the axon, which immediately activates the degradative proteases and lipases involved in Wallerian degeneration. A rise in intra-axonal Ca2+ concentration to >15 μm (a level certainly sufficient enough to activate most degradative enzymes) has been noted in Aplysia axons transected in vitro(Ziv and Spira, 1993). The authors of this study estimated that axonal Ca2+ concentrations quickly returned to basal levels of 0.05–0.1 μm after the 7–10 min that followed the sealing of the cut end within 0.5–2 min. They also determined that the elevation in Ca2+ was attributable to the influx of extracellular Ca2+ through the cut end of the axon and through the opening of voltage-gated Ca2+channels along the entire length of the axon. If proteases and lipases are activated at the initial time of axonal injury, the delayed destruction of the axon that is seen (CAP remains nearly 100% at 24 hr after axotomy and then declines by 80% over the next 12 hr) occurs once sufficient plasmalemma and cytoskeleton have been degraded. In this case the effects of cooling would be similar at all phases of axonal degeneration, which our data do not support.
An alternative hypothesis is that the transient rise in Ca2+ might activate axonal kinases and phosphatases. The subsequent action of axonal transport on these modulators of protein phosphorylation might lead to the activation of Ca2+ channels or Ca2+ porters located at the nodes of Ranvier or along the length of the axolemma. The activation of mediators of Ca2+ influx later during the process of Wallerian degeneration then allows a catastrophic rise in axonal Ca2+ concentrations, leading to sudden, rapid destruction of the axon. This hypothesis gives rise to at least two phases of axonal degeneration and so is consistent with both the observed temperature effect and the insensitivity to external Ca2+ levels.
In the latter scenario, individual axons degenerate very rapidly at the end of the multiphasic Wallerian degeneration. The distribution of axonal calibers, concentration and rate of transport of the “factor” responsible for Ca2+ channel activation, and the ratio of the number of fibers of the sensory and motor lineages all affect the percentage of fibers that remain at any given time. The gradual decline in the measured CAP between 24 and 48 hr is, thus, a measure of a declining population of axons able to conduct CAPs.
Conclusions
The data lead us to propose a three-stage model of Wallerian degeneration (Fig. 8). The first stage occurs during the first 12 hr after axotomy at 37°C and is prolonged by low temperatures. Events during the first stage appear to initiate the process leading to Wallerian degeneration or “light the fuse.” The second stage occurs during the period 12–24 hr after axotomy at 37°C. During this stage “the fuse is burning,” because axonal degeneration is not yet evident (the morphology is normal, and the CAP remains close to 100% until 24 hr) but can no longer be delayed by cooling. It may be that this stage is independent of stage 1, because a delayed stage 2 is not seen when cooled nerves are warmed. Instead, nerves cooled during stage 1 appear to enter stage 3 immediately on warming, suggesting that the events of stage 2 may proceed despite the prolongation of stage 1. Alternatively, an external temperature of 25°C may not be low enough to alter the events of stage 2.
Fig. 8.

Schematic diagram of the proposed three-stage model of Wallerian degeneration.
The third and final stage, which encompasses the rapid degeneration of axons during the period 24–48 hr (80% of the CAP is lost between 24 and 36 hr) at 37°C, occurs only after stages 1 and 2 are complete. This stage appears to correspond to the rapid enzymatic digestion of the axonal components. We speculate that catastrophic Ca2+ entry marks the transition between stages 1 and 2, which enable the Ca2+ influx, and stage 3, which results from protease and lipase activation by the Ca2+ influx.
Footnotes
This work was supported by a grant from the National Institutes of Health to J.W.G. and a National Institutes of Health National Research Service Award postdoctoral fellowship (Training Grant NS 14784) to J.W.T. We thank C. W. Tsao for assistance with the teased nerve fiber preparation.
Correspondence should be addressed to Dr. J. W. Tsao, 123 Edgewood Avenue, San Francisco, CA 94117-3712.
Dr. Tsao’s present address: Department of Neurology, University of California–San Francisco, Box 0114, 505 Parnassus Avenue, San Francisco, CA 94143.
Dr. George’s present address: Department of Neurology, Wayne State University School of Medicine, 6E University Health Center, 4201 St. Antoine, Detroit, MI 48201.
REFERENCES
- 1.Bachelard HS, Silva GD. The Na+,K+-activated adenosine triphosphatase in degenerating peripheral nerve. Arch Biochem Biophys. 1966;117:98–105. doi: 10.1016/0003-9861(66)90131-7. [DOI] [PubMed] [Google Scholar]
- 2.Bittner GD. Long-term survival of severed distal axonal stumps in vertebrates and invertebrates. Am Zool. 1988;28:1165–1179. [Google Scholar]
- 3.Cancalon P. Slow flow in axons detached from their perikarya. J Cell Biol. 1982;95:989–992. doi: 10.1083/jcb.95.3.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancalon P. Influence of temperature on various mechanisms associated with neuronal growth and nerve regeneration. Prog Neurobiol. 1985;25:27–92. doi: 10.1016/0301-0082(85)90022-x. [DOI] [PubMed] [Google Scholar]
- 5.Eddleman CS, Ballinger ML, Smyers ME, Godell CM, Fishman HM, Bittner GD. Repair of plasmalemmal lesions by vesicles. Proc Natl Acad Sci USA. 1997;94:4745–4750. doi: 10.1073/pnas.94.9.4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eddleman CS, Ballinger ML, Smyers ME, Fishman HM, Bittner GD. Endocytotic formation of vesicles and other membranous structures induced by Ca2+ and axolemmal injury. J Neurosci. 1998;18:4029–4041. doi: 10.1523/JNEUROSCI.18-11-04029.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fern R, Ransom BR, Waxman SG. Voltage-gated calcium channels in CNS white matter: role in anoxic injury. J Neurophysiol. 1995;74:369–377. doi: 10.1152/jn.1995.74.1.369. [DOI] [PubMed] [Google Scholar]
- 8.Gamble HJ, Jha BD. Some effect of temperature upon the rate and progress of Wallerian degeneration in mammalian nerve fibers. J Anat. 1958;92:171–177. [PMC free article] [PubMed] [Google Scholar]
- 9.Gamble HJ, Goldby F, Smith GMR. Effect of temperature on the degeneration of nerve fibers. Nature. 1957;179:527. doi: 10.1038/179527a0. [DOI] [PubMed] [Google Scholar]
- 10.George EB, Glass JD, Griffin JW. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci. 1995;15:6445–6452. doi: 10.1523/JNEUROSCI.15-10-06445.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glass JD, Schryer BL, Griffin JW. Calcium-mediated degeneration of the axonal cytoskeleton in the Ola mouse. J Neurochem. 1994;62:2472–2475. doi: 10.1046/j.1471-4159.1994.62062472.x. [DOI] [PubMed] [Google Scholar]
- 12.Krause TL, Fishman HM, Ballinger ML, Bittner GD. Extent and mechanism of sealing in transected giant axons of squid and earthworms. J Neurosci. 1994;14:6638–6651. doi: 10.1523/JNEUROSCI.14-11-06638.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LoPachin RM, Jr, LoPachin VR, Saubermann AJ. Effects of axotomy on distribution and concentration of elements in rat sciatic nerve. J Neurochem. 1990;54:320–332. doi: 10.1111/j.1471-4159.1990.tb13317.x. [DOI] [PubMed] [Google Scholar]
- 14.Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- 15.Luttges MW, Kelly PT, Gerren RA. Degenerative changes in mouse sciatic nerves: electrophoretic and electrophysiologic characterizations. Exp Neurol. 1976;50:706–733. doi: 10.1016/0014-4886(76)90039-x. [DOI] [PubMed] [Google Scholar]
- 16.Majno G, Karnovsky ML. A biochemical and morphological study of myelination and demyelination. II. Lipogenesis in vitro by rat nerves following transection. J Exp Med. 1958;108:197–213. doi: 10.1084/jem.108.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDonald WI. The time course of conduction failure during degeneration of a central tract. Exp Brain Res. 1972;14:550–556. doi: 10.1007/BF00236596. [DOI] [PubMed] [Google Scholar]
- 18.Perry VH, Lunn ER, Brown MC, Cahusac S, Gordon S. Evidence that the rate of Wallerian degeneration is controlled by a single autosomal dominant gene. Eur J Neurosci. 1990a;2:408–413. doi: 10.1111/j.1460-9568.1990.tb00433.x. [DOI] [PubMed] [Google Scholar]
- 19.Perry VH, Brown MC, Lunn ER, Tree P, Gordon S. Evidence that very slow Wallerian degeneration in C57BL/Ola mice is an intrinsic property of the peripheral nerve. Eur J Neurosci. 1990b;2:802–808. doi: 10.1111/j.1460-9568.1990.tb00472.x. [DOI] [PubMed] [Google Scholar]
- 20.Perry VH, Brown MC, Tsao JW. The effectiveness of the gene which slows the rate of Wallerian degeneration in C57BL/Ola mice declines with age. Eur J Neurosci. 1992;4:1000–1002. doi: 10.1111/j.1460-9568.1992.tb00126.x. [DOI] [PubMed] [Google Scholar]
- 21.Povlishock JT. Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol. 1992;2:1–12. [PubMed] [Google Scholar]
- 22.Povlishock JT, Christman CW. The pathobiology of traumatically induced axonal injury in animals and humans: a review of current thoughts. J Neurotrauma. 1995;12:555–564. doi: 10.1089/neu.1995.12.555. [DOI] [PubMed] [Google Scholar]
- 23.Povlishock JT, Pettus EH. Traumatically induced axonal damage: evidence for enduring changes in axolemmal permeability with associated cytoskeletal change. Acta Neurochir [Suppl] 1996;66:81–86. doi: 10.1007/978-3-7091-9465-2_15. [DOI] [PubMed] [Google Scholar]
- 24.Povlishock JT, Marmarou A, McIntosh T, Trojanowski JQ, Moroi J. Impact acceleration injury in the rat: evidence for focal axolemmal change and related neurofilament sidearm alteration. J Neuropathol Exp Neurol. 1997;56:347–359. [PubMed] [Google Scholar]
- 25.Raabe TD, Nguyen T, Archer C, Bittner GD. Mechanisms of the maintenance and eventual degradation of neurofilament proteins in the distal segments of severed goldfish Mauthner axons. J Neurosci. 1996;16:1605–1613. doi: 10.1523/JNEUROSCI.16-05-01605.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlaepfer WW. Calcium-induced degeneration of axoplasm in isolated segments of rat peripheral nerve. Brain Res. 1974;69:203–215. doi: 10.1016/0006-8993(74)90002-x. [DOI] [PubMed] [Google Scholar]
- 27.Schlaepfer WW. Structural alterations of peripheral nerve induced by the calcium ionophore A23187. Brain Res. 1977;136:1–9. doi: 10.1016/0006-8993(77)90126-3. [DOI] [PubMed] [Google Scholar]
- 28.Schlaepfer WW, Bunge RP. Effects of calcium ion concentration on the degeneration of amputated axons in tissue culture. J Cell Biol. 1973;59:456–470. doi: 10.1083/jcb.59.2.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sea T, Ballinger ML, Bittner GD. Cooling of peripheral myelinated axons retards Wallerian degeneration. Exp Neurol. 1995;133:85–95. doi: 10.1006/exnr.1995.1010. [DOI] [PubMed] [Google Scholar]
- 30.Sheller RA, Bittner GD. Maintenance and synthesis of proteins for an anucleate axon. Brain Res. 1992;580:68–80. doi: 10.1016/0006-8993(92)90928-3. [DOI] [PubMed] [Google Scholar]
- 31.Stewart MA, Passonneau JV, Lowry OH. Substrate changes in peripheral nerve during ischaemia and Wallerian degeneration. J Neurochem. 1965;12:719–727. doi: 10.1111/j.1471-4159.1965.tb06786.x. [DOI] [PubMed] [Google Scholar]
- 32.Tanner SL, Storm EE, Bittner GD. Maintenance and degradation of proteins in intact and severed axons: implications for the mechanisms of long-term survival of anucleate crayfish axons. J Neurosci. 1995a;15:540–548. doi: 10.1523/JNEUROSCI.15-01-00540.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanner SL, Storm EE, Bittner GD. Protein transport in intact and severed (anucleate) crayfish giant axons. J Neurochem. 1995b;64:1491–1501. doi: 10.1046/j.1471-4159.1995.64041491.x. [DOI] [PubMed] [Google Scholar]
- 34. Tsao JW, George EB. 1996. Axonal degeneration in vitro: effects of temperature and inhibitory chemical compounds. Paper presented at the XVI International Winter Meeting of the Swiss Society of Neuropathology, St. Moritz, Switzerland, March
- 35.Tsao JW, Brown MC, Carden MJ, McLean WG, Perry VH. Loss of the compound action potential: an electrophysiological, biochemical, and morphological study of early events in axonal degeneration in the C57BL/Ola mouse. Eur J Neurosci. 1994;6:516–524. doi: 10.1111/j.1460-9568.1994.tb00295.x. [DOI] [PubMed] [Google Scholar]
- 36.Tsao JW, George EB, Griffin JW. Temperature modulation shows three stages of Wallerian degeneration. Soc Neurosci Abstr. 1997;23:1404. doi: 10.1523/JNEUROSCI.19-12-04718.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Usherwood PNR, Cochrane DG, Rees D. Changes in structural, physiological, and pharmacological properties of insect excitatory nerve–muscle synapses after motor nerve section. Nature. 1968;318:589–591. doi: 10.1038/218589a0. [DOI] [PubMed] [Google Scholar]
- 38.Wang GK. The long-term excitability of myelinated nerve fibers in the transected frog sciatic nerve. J Physiol (Lond) 1985;368:309–321. doi: 10.1113/jphysiol.1985.sp015859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ziv NE, Spira ME. Spatiotemporal distribution of Ca2+ following axotomy and throughout the recovery process of cultured Aplysia neurons. Eur J Neurosci. 1993;5:657–668. doi: 10.1111/j.1460-9568.1993.tb00531.x. [DOI] [PubMed] [Google Scholar]