

Abstract
The role of enkephalin and the opioid receptors in modulating GABA release within the rat globus pallidus (GP) was investigated using whole-cell patch recordings made from visually identified neurons. Two major GP neuronal subtypes were classified on the basis of intrinsic membrane properties, action potential characteristics, the presence of the anomalous inward rectifier (Ih), and anode break depolarizations.
The μ opioid receptor agonist [d-Ala2-N-Me-Phe4-Glycol5]-enkephalin (DAMGO) (1 μm) reduced GABAAreceptor-mediated IPSCs evoked by stimulation within the striatum. DAMGO also increased paired-pulse facilitation, indicative of presynaptic μ opioid receptor modulation of striatopallidal input. In contrast, the δ opioid agonistd-Pen-[d-Pen2,5]-enkephalin (DPDPE) (1 μm) was without effect.
IPSCs evoked by stimulation within the GP were depressed by application of [methionine 5′]-enkephalin (met-enkephalin) (30 μm). Met-enkephalin also reduced the frequency, but not the amplitude, of miniature IPSCs (mIPSCs) and increased paired-pulse facilitation of evoked IPSCs, indicative of a presynaptic action. Both DAMGO and DPDPE reduced evoked IPSCs and the frequency, but not amplitude, of mIPSCs. However, spontaneous action potential-driven IPSCs were reduced in frequency by met-enkephalin and DAMGO, whereas DPDPE was without effect.
Overall, these results indicate that presynaptic μ opioid receptors are located on striatopallidal terminals and pallidopallidal terminals of spontaneously firing GP neurons, whereas presynaptic δ opioid receptors are preferentially located on terminals of quiescent GP cells. Enkephalin, acting at both of these receptor subtypes, serves to reduce GABA release in the GP and may therefore act as an adaptive mechanism, maintaining the inhibitory function of the GP in basal ganglia circuitry.
Keywords: whole–cell patch clamp, brain slices, basal ganglia, enkephalin, GABAA IPSC, opioid receptors
The external segment of the primate globus pallidus (GPe), equivalent to the rat globus pallidus (GP), occupies a critical position in basal ganglia (BG) circuitry (Alexander and Crutcher, 1990; Parent, 1990). It receives a major GABA–enkephalin projection from a discrete population of medium spiny striatal neurons (Wilson and Phelan, 1982; Gerfen and Young, 1988; Hazrati and Parent, 1992; Parent and Hazrati, 1995), in turn sending projections to the subthalamic nucleus (STN) (Smith et al., 1990; Parent and Hazrati, 1995), internal segment of the globus pallidus (entopeduncular nucleus in rodents) (Kincaid et al., 1991; Bolam and Smith, 1992), substantia nigra pars reticulata (Smith and Bolam, 1989), reticular thalamic nucleus (Cornwall et al., 1990; Hazrati and Parent, 1991; Gandia et al., 1993), and striatum (Bevan et al., 1998).
Although the prevailing model of BG function (DeLong, 1990) has recently received criticism from several sources (Parent and Hazrati, 1995; Chesselet and Delfs, 1996; Obeso et al., 1997), it remains a useful tool to explain many disorders of movement. In Parkinson’s disease, dopamine depletion is thought to lead to an increase in the activity of the indirect GABA–enkephalin striatopallidal pathway. Indeed, there is evidence for increased enkephalin gene expression in the striatum (Augood et al., 1989; Gerfen et al., 1990; Frayne et al., 1991) and increased enkephalin immunoreactivity in the GPe (Lavoie et al., 1991) in animal models of parkinsonism. In keeping with the proposal that the GABA striatopallidal pathway is overactive after dopamine depletion, injection of GABAA receptor antagonists directly into the GP has been shown to increase locomotor score in parkinsonian animals (Maneuf et al., 1994). Furthermore, direct injection of GABAA antagonists and μ and δ opioid receptor agonists into the ventral pallidum (the limbic homolog of the GP) of normal animals also promotes locomotor activity (Austin and Kalivas, 1990; Napier, 1992). The effects of opioid receptor agonists have been attributed to the presynaptic inhibition of GABA release from striatopallidal terminals (Dewar et al., 1987; Maneuf et al., 1994). Indeed, anatomical studies have provided evidence that μ opioid receptors are located on striatopallidal presynaptic terminals (Abou Khalil et al., 1984). However, more recently, ligand binding, mRNA, and immunohistochemical studies have indicated the presence of both presynaptic and postsynaptic μ and δ opioid receptors within the GP (Delfs et al., 1994; Mansour et al., 1994, 1995; Bausch et al., 1995; Peckys and Landwehrmeyer, 1999).
Two major types of GP neuron have been determined by in vivoand in vitro electrophysiological, morphological, and neurochemical experiments (DeLong, 1971; Hontanilla et al., 1994; Kita and Kitai, 1994; Nambu and Llinás, 1994, 1997). These neurons appear to display widespread intrinsic axon collaterals (Kita and Kitai, 1994; Nambu and Llinás, 1997). Therefore, the presence of presynaptic opioid receptors within the GP may reflect receptor expression on collateral terminals, as well as striatopallidal terminals.
The aim of this study was therefore to determine (1) the presence and function of μ and δ opioid receptors on pallidal and striatal terminals, (2) the presence of functional postsynaptic μ and δ opioid receptors, and (3) whether there is a correlation between GP neuronal heterogeneity and differential opioid receptor activity.
MATERIALS AND METHODS
Whole-cell patch-clamp recordings were made from single GP neurons within 300-μm-thick slices obtained from 80–120 gm male Wistar rats. Animals were first anesthetized with fluorothane and killed by cervical dislocation. The brain was quickly removed and placed in ice-cold artificial CSF (aCSF) containing (in mm): choline chloride 126, KCl 2.5, NaH2PO4 1.2, MgCl2 1.3, MgSO4 8, and glucose 10, buffered to pH 7.4 with NaHCO3 26. Slices were cut in either coronal or parasagittal plane using a DTK-1000 Microslicer (Dosaka, Japan). Slices were then transferred to a holding chamber or recording chamber at 32–34°C and perfused continuously at 2–3 ml/min with aCSF containing (in mm): NaCl 126, KCl 2.5, NaH2PO4 1.2, MgCl2 1.3, CaCl2 2.4, and glucose 10, buffered to pH 7.4 with NaHCO3 26, saturated with 95% O2–5% CO2.
Whole-cell recordings from GP neurons were made using borosilicate glass pipettes of 3–6 MΩ resistance containing (in mm): K-gluconate 125, NaCl 10, CaCl2 1, MgCl2 2, BAPTA 10, HEPES 10, GTP 0.3, Mg-ATP 2, and biocytin 5, adjusted to pH 7.25 with KOH. Individual neurons were visualized (40× water immersion objective) using a differential interference contrast infrared microscopy (BX 501; Olympus Optical, Tokyo, Japan) with CCD camera (KP-M1; Hitachi, Tokyo, Japan) and contrast enhancement system (ADV-2; Brian Reece Scientific, Berkshire, UK). Recording pipettes were advanced while under positive pressure toward individual cells in the slice. On contact, tight seals, in the order of 10–20 GΩ, were made by applying negative pressure. The membrane patch was then ruptured by suction, and membrane current and potential was monitored using an Axopatch 200B patch-clamp amplifier (Axon Instruments, Foster City, CA). Whole-cell access resistances were in the range of 7–20 MΩ before electrical compensation by 65–80%. Access resistances, after electrical compensation, were initially determined in current-clamp mode and continuously monitored in voltage clamp by measuring the size of the capacitance transient in response to a 10 mV hyperpolarizing step (Stuart et al., 1993). The access resistance was checked intermittently in current clamp, and experiments were abandoned if changes >20% were encountered. All current-clamp recordings were made in Axopatch 200B fast mode, and membrane potentials were corrected with respect to the null potential measured at the end of recording. No corrections have been made for the liquid junction potentials, estimated to be +8 mV.
Synaptic events were evoked by focal bipolar single shock stimulation (0.5 msec, 0.1–3 mA) at 15 sec intervals using a constant current stimulation unit (DS2A; Digitimer, Hertfordshire, UK). Voltage steps were generated using pCLAMP software version 6.03 (Axon Instruments), and the resulting membrane currents and those resulting from synaptic activation were filtered at 2 kHz and stored on disk and digital analog tape recorder for subsequent analysis and display on a chart recorder (Gould Easygraph; Gould Instruments, Hainault, UK). To record mIPSCs, the K-gluconate in the recording pipette was replaced with KCl. In doing this, the theoretical chloride reversal potential moved from −61 to +1 mV. mIPSCs were then recorded in tetrodotoxin (TTX) (1 μm) at a potential of −80 mV. Data were collected over 2 min periods, acquired using FETCHEX (Axon Instruments), and analyzed using Minianalysis software (Jaejin Software, Leonia, NJ).
Drugs were applied to the superfusate by exchanging the aCSF for one differing only by the addition of a known concentration of drug, with the exchange beginning after a dead time of ∼20 sec. Drugs used were as follows: 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX),dl-2-amino-5-phosphonopentanoic acid (dl-AP-5), bicuculline methiodide, picrotoxin, and ICI 174,864 (all from SEMAT); [methionine 5′]-enkephalin] (met-enkephalin), [d-Ala2-N-Me-Phe4-Glycol5]-enkephalin (DAMGO),d-Pen-[d-Pen2,5]-enkephalin (DPDPE),d-Phe-Cys-Tyr-d-Trp-Orn-Thr-Pen-Thr-NH2(CTOP), and TTX (all from Sigma, Poole, UK).
All numerical data are expressed as mean and SE unless otherwise stated. Differences in IPSC data were analyzed for statistical significance using the paired Student’s t test, and significant differences in the cumulative frequency and amplitude of spontaneous IPSCs (sIPSCs) and mIPSCs were analyzed using the Kolmogorov–Smirnov two-sample test (Jaejin Software).
RESULTS
Characterization of globus pallidus neurons
GP neurons were characterized by their electrophysiological properties and classified into two major groups. These two classes of neuron showed similar but not identical properties to the type I and II neurons described previously by Nambu and Llinás (1994). Type I neurons were spontaneously active, firing irregular action potentials at 13 ± 3.1 Hz (n = 19) at a resting membrane potential of −52.4 ± 7.8 mV (measured at I = 0). The input resistance of these cells was 349 ± 35.9 MΩ (n = 19), and action potential duration (taken as the time between the point of initiation of the maximum rate of depolarization and the equipotential point during spike repolarization) was 0.82 ± 0.1 msec (n = 19), followed by a short-lasting afterhyperpolarization (AHP) of amplitude 29.2 ± 2 mV (n = 19) (Fig.1A).
Fig. 1.

Characterization of two GP neuronal subtypes.A, i, Superimposed voltage responses from a type I neuron in response to 300 msec hyperpolarizing currents steps (25 pA increments every 5 sec) from a resting membrane potential of −50.8 mV. This neuron was spontaneously active at 10 Hz.ii, Typical action potential from a type I neuron of 0.78 msec duration and short-lasting AHP of 29 mV amplitude.B, i, Voltage traces from a type II neuron in response to 300 msec current steps (in 25 pA increments every 5 sec) from resting membrane potential of −67 mV. Hyperpolarizing steps elicited time-dependent inward rectification of membrane potential. On removal of the step, there was an anodal break depolarization accompanied by action potential firing.ii, Representative action potential from a type II GP neuron of 0.95 msec duration and a long-lasting AHP of amplitude 27 mV.
Type II neurons were identified by the presence of the time- and voltage-dependent inward rectification of membrane potential evoked by hyperpolarizing steps of 300 msec duration and anodal break rebound depolarizations. These rebound depolarizations were often accompanied by action potential firing (Fig. 1B). Type II neurons were either quiescent (22 of 38 cells) or fired regular spontaneous action potentials at a rate of 7.5 ± 1.6 Hz (16 of 38) at a resting membrane potential of −53.4 ± 2.5 mV. The input resistance of these cells was 646.1 ± 41.8 MΩ (n = 38; significantly different from type I cells,p < 0.01), and the action potential duration was 1.1 ± 0.1 msec (n = 38; significantly different from type I cells, p < 0.01), followed by a prolonged AHP of amplitude 29.3 ± 6.6 mV (n = 38).
Evoked GABA A receptor IPSCs
At a holding potential of −50 mV, single shock electrical stimulation evoked a fast inward synaptic current, followed by a slower outward current. These currents appeared to be independent of slice orientation and stimulation location. The inward current was blocked by the glutamate antagonists CNQX (10 μm) anddl-AP-5 (100 μm) (Fig.2A), whereas the slower outward current was blocked by bicuculline (10 μm;n = 4) (Fig. 2B) or picrotoxin (50 μm; n = 2) (data not shown). The outward current reversed polarity at approximately −70 mV (Fig.2C), close to the theoretical chloride equilibrium potential (ECl of −61 mV) and was therefore considered to be a GABAA receptor-mediated IPSC.
Fig. 2.

Synaptic currents evoked by local stimulation.A, At a holding potential of −50 mV, single shock bipolar electrical stimulation evoked a fast inward current that was blocked by CNQX (10 μm) and dl-AP-5 (100 μm) and a slower outward current. Eachtrace is preceded by a 10 mV hyperpolarizing step of 10 msec duration to monitor input conductance and changes in access resistance. B, Time course from a single experiment showing the reversible block of outward synaptic potential by the GABAA antagonist bicuculline (10 μm) applied to the superfusion medium for the period indicated by the bar. Each point represents the average of four traces evoked every 15 sec.C, The outward current reversed polarity at −69 mV, close to ECl.
μ Opioid receptors on striatopallidal terminals
The opioid receptor modulation of GABA release from striatopallidal terminals was studied by stimulation within the striatum (in parasagittal slices) in the presence of CNQX (10 μm) and dl-AP-5 (100 μm). Obtaining robust synaptic responses using this orientation of slice and stimulation location proved technically difficult, possibly because of the topographic nature of striatopallidal connections (Wilson and Phelan, 1982). However, application of the μ opioid receptor agonist DAMGO (1 μm) reduced the IPSCs by 53 ± 6% (n = 7) (Fig.3A), whereas the δ opioid receptor agonist DPDPE was without effect (n = 5) (Fig.3B). In each of these five cells, application of DAMGO (1 μm) significantly reduced the evoked IPSC. The action of DAMGO appeared to be independent of cell phenotype, inhibiting IPSCs in three type I and four type II cells.
Fig. 3.

Presynaptic depression of evoked striatopallidal IPSCs by μ opioid receptor activation. A, Time course showing the effect of the μ opioid receptor agonist DAMGO (1 μm) on the normalized IPSC amplitude evoked by stimulation of striatum in parasagittal slices in the presence of CNQX (10 μm) and dl-AP-5 (100 μm). DAMGO reduces the IPSC by 53 ± 6% (n = 7).B, Time course showing the effect of the δ opioid receptor agonist DPDPE (1 μm) on the normalized IPSC amplitude evoked by stimulation of the striatum in parasagittal slices in the presence of CNQX (10 μm) and dl-AP-5 (100 μm). DPDPE has no significant effect on the evoked IPSC (n = 5). However, the IPSC evoked in each of these five cells was inhibited by DAMGO (1 μm).C, Paired IPSCs recorded from the same cell in response to the same stimulation at an interval of 50 msec in control (i) and DAMGO (1 μm) (ii). iii, The pair of IPSCs recorded in the presence of DAMGO have been scaled so that the conditioning (first) responses with and without drug are of similar amplitude. The ratio of the test (second) response in DAMGO has increased, indicative of a presynaptic site of action. Paired IPSCs recorded in the presence of the μ opioid receptor antagonist CTOP (1 μm) (iv) and CTOP plus DAMGO (1 μm) (v), and ratio (vi). In the presence of CTOP, there was no depression of the conditioning IPSC by DAMGO and no change in the paired-pulse ratio. Currents resulting from a 10 mV hyperpolarizing step precede all IPSC pairs.
A paired-pulse protocol was used to show μ opioid receptor modulation of the striatal evoked IPSCs was presynaptic (Davies et al., 1990;Travagli and Williams, 1996). IPSCs were evoked by single shock electrical stimulation of equal strength and duration at an interval of 30 msec. Application of DAMGO (1 μm) reduced the amplitude of the conditioning (first) IPSC and test (second) IPSC but significantly increased the paired-pulse facilitation (21 ± 10.7%; p < 0.05; n = 4) (Fig.3C). This effect was totally abolished by preincubation (20 min) with the μ opioid receptor antagonist CTOP (1 μm;n = 3) (Fig. 3C).
Met-enkephalin reduces the IPSCs evoked by stimulation within the GP via a presynaptic mechanism
Using coronal slices, electrical stimulation within the GP, 300–900 μm away from the recording site, evoked IPSCs that were reduced reversibly by bath application of met-enkephalin (30 μm; 53.4 ± 5.1%; n = 11) (Fig.4A). To determine the locus of such modulation, the action of met-enkephalin on the paired-pulse ratio and the frequency and amplitude of action potential independent mIPSCs was studied.
Fig. 4.
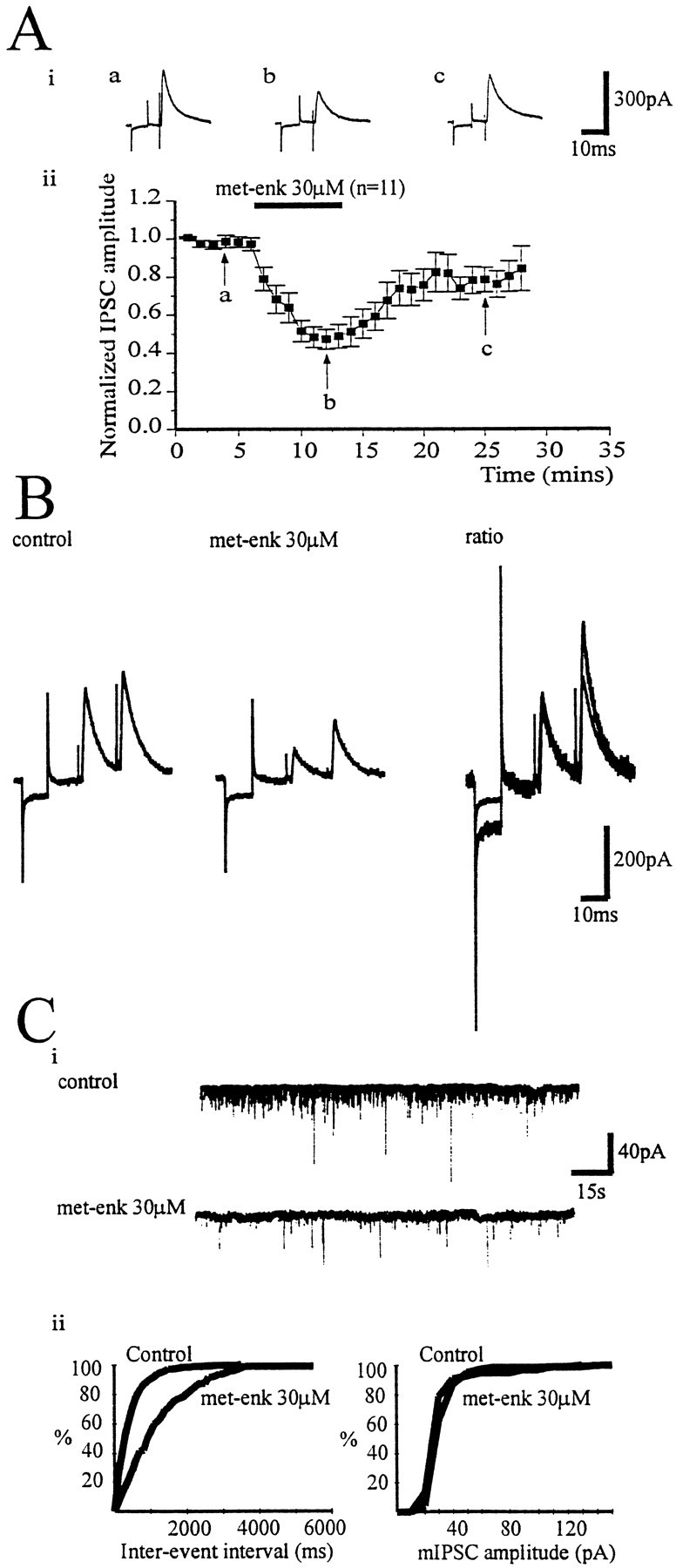
Enkephalin reduces GABAA IPSCs. IPSCs recorded at a holding potential of −50 mV in the presence of CNQX (10 μm) and dl-AP-5 (100 μm).A, i, IPSCs (mean of 4 evoked every 15 sec) before (a) and during (b) the application of met-enkephalin (30 μm) and after wash (c).A, ii, Time course showing the effect of bath application of met-enkephalin (30 μm) on normalized IPSC amplitude. B, Paired IPSCs recorded from a type II GP neuron in control (i) and met-enkephalin (10 μm) (ii). iii, The pair of IPSCs recorded in the presence of met-enkephalin has been scaled so that the conditioning (first) responses with and without drug are of similar amplitude. The increase in ratio of the test (second) response is indicative of a presynaptic site of action of met-enkephalin. C, mIPSCs were recorded from chloride loaded cells (ECl, +1 mV) in the presence of TTX (1 μm), CNQX (10 μm), and dl-AP-5 (100 μm) at a holding potential of −80 mV. mIPSCs were collected over a period of 120 sec. The same time frame was taken 3 min after drug application.i, Control mIPSCs and those recorded after application of met-enkephalin (30 μm). ii, Cumulative frequency and amplitude distributions (derived from 120 sec of data) before and after met-enkephalin. Met-enkephalin reduced the frequency of mIPSCs (p < 0.01) but not their amplitude.
Met-enkephalin (10–30 μm) reduced the amplitude of the conditioning (first) and test (second) IPSC but increased the paired-pulse ratio (20.2 ± 10.8%; p < 0.05;n = 5) (Fig. 4B).
mIPSCs were recorded in chloride loaded cells at a holding potential of −80 mV in the presence of TTX (1 μm), CNQX (10 μm), and dl-AP-5 (100 μm) (Fig.4C). IPSCs were considered “minis” after abolition by TTX (1 μm) of electrically evoked synaptic currents and action potentials during depolarizing current steps. Bath application of met-enkephalin (30 μm) significantly reduced the frequency of mIPSCs in five of seven cells (p < 0.05), whereas the amplitude of mIPSCs was unaffected by met-enkephalin in six of seven cells (Fig. 4C).
These results suggest that met-enkephalin acts via presynaptic opioid receptors leading to a reduction of GABA release within the GP. Consistent with this interpretation, changes in holding current after met-enkephalin application were rarely observed. Only 4 of 19 cells exhibited a detectable outward current, which was 27.5 ± 11.1 pA in amplitude (data from three type I and one type II neurons).
μ and δ opioid receptor agonists reduce IPSCs evoked by stimulation in the GP and mIPSCs
The δ opioid receptor agonist DPDPE (1 μm) reduced the amplitude of the IPSC evoked by stimulation in the GP (in coronal slices) by 49.9 ± 6.7% (n = 8). The depression of the evoked IPSC by the μ opioid receptor agonist DAMGO (1 μm) was more variable, ranging from 0 to 60% (mean, 29 ± 9.8%; n = 6) (Fig.5). The presynaptic action of both agonists was confirmed by analysis of mIPSCs. Bath application of DPDPE (1 μm) significantly reduced the frequency of mIPSCs (p < 0.05) in three of four cells (one type I and two type II neurons), whereas the amplitude of mIPSCs was not significantly reduced in three of four cells (Fig. 5B). Application of DAMGO (1 μm) significantly reduced the frequency of mIPSCs (p < 0.05) in five of seven cells (two I and three II type cells), although the amplitude of events was not significantly reduced in six of seven cells (Fig.5B). These results confirm the presence of both presynaptic μ and δ receptors on terminals of both type I and II GP neurons, regulating GABA release within the GP.
Fig. 5.

The δ agonist DPDPE rather than the μ opioid agonist DAMGO preferentially reduced IPSCs evoked by stimulation within the GP. A, Time course showing the effect of the δ opioid receptor agonist DPDPE (1 μm) (i) and the μ opioid receptor agonist DAMGO (1 μm) (ii) on normalized IPSC amplitude evoked by stimulation within the GP. DPDPE reduced the IPSC by 49.9 ± 6.7% (n = 8), but DAMGO only reduced the IPSC by 29 ± 9.8% (n = 6).B, Cumulative frequency (i) and amplitude (ii) distributions of mIPSCs before and after the application of DPDPE and DAMGO from two representative experiments. Both agonists reduced the frequency of the events (p < 0.01) without change in amplitude.
Spontaneous IPSCs are reduced by μ but not δ opioid receptor agonists
In the presence of CNQX (10 μm) anddl-AP-5 (100 μm), 78% of GP neurons displayed outward sIPSCs (in nonchloride loaded electrodes). These currents reversed around ECl and were abolished by TTX (1 μm; n = 3) (Fig.6A) and picrotoxin (50 μm; n = 4; data not shown), suggesting action potential-dependent activity of GABA-releasing neurons within the preparation.
Fig. 6.

Depression of sIPSCs by activation of μ opioid receptors. A, sIPSCs in a single GP neuron recorded at a holding potential of −50 mV in the presence of CNQX (10 μm) and dl-AP-5 (100 μm). Bath application of met-enkephalin (30 μm) suppressed the frequency and amplitude of sIPSCs. This effect was replicated by the μ opioid receptor agonist DAMGO (1 μm), whereas the δ opioid receptor agonist DPDPE (1 μm) was without effect. sIPSCs were blocked by TTX (1 μm). B, Cumulative frequency distributions of sIPSCs before and after the addition of met-enkephalin (p < 0.05) (i), DAMGO (p < 0.05) (ii), and DPDPE (non significant) (iii). Data were collected over a period of 120 sec. Same cell asA.
Met-enkephalin (30 μm) caused a significant reversible depression in frequency of sIPSCs in 10 of 11 cells (five I type, five II type cells; p < 0.05), an effect that was mimicked by DAMGO (1 μm; 9 of 11 cells; five I type and four II type neurons). However, DPDPE 1 μm had no effect on the frequency of sIPSCs in all nine cells tested (three type I, six type II cells) (Fig. 6).
These results suggest that presynaptic μ opioid receptors modulate GABA release from axon terminals arising from spontaneously firing neurons within the preparation and that presynaptic δ receptors are located on the terminals of axons arising from quiescent, possibly, type II GP neurons.
DISCUSSION
GP neuronal subtypes
In this study, two major subtypes of GP neuron have been characterized electrophysiologically. These neurons are similar but not identical to those reported previously (Nambu and Llinás, 1994,1997). Therefore, they were provisionally classed as type I and type II GP neurons in accordance with these previous studies.
Type I neurons (34% of total recordings) showed no evidence of inward rectification or anodal break depolarizations. These cells exhibited a low input resistance, consistent with type I neurons of Nambu and Llinás (1994). However, they differed from those reported previously because they were spontaneously active at resting membrane potentials and showed shorter action potential and AHP durations (Nambu and Llinás, 1994).
Sixty-six percent of cells recorded in the GP showed marked anomalous inward rectification on hyperpolarization (Ih) [this current was readily blocked by 2 mm cesium and 100 μm ZD 7288 (our unpublished observations)] and anodal break depolarizations often accompanied by action potential firing. These neurons also had a relatively high input resistance and were either quiescent or firing spontaneous action potentials at a tonic rate. These cells probably correspond to the type II cells described by Nambu and Llinás (1994), which exhibited rebound depolarizations and a high input resistance.
The discrepancies in neuronal characteristics between the present study and that of Nambu and Llinás (1994) may arise from differences in species (rats vs guinea pigs), slice preparation (300-μm-thick slices vs 600–1000 μm strips) or recording technique (whole-cell vs sharp microelectrode).
Presynaptic inhibition of striatopallidal GABAA IPSCs
Met-enkephalin and the μ opioid receptor agonist DAMGO depressed the GABAA receptor-mediated IPSCs evoked by stimulation within the striatum. Met-enkephalin also induced an increase in paired-pulse facilitation (an effect blocked by the selective μ opioid receptor antagonist CTOP) and reduced the frequency of mIPSCs. The δ opioid receptor agonist DPDPE had no effect on striatal evoked IPSCs. These results indicate that enkephalin, acting at presynaptic μ but not δ opioid receptors, may reduce GABA release from striatopallidal terminals. This is in agreement with binding and immunohistochemical studies, which have demonstrated the presynaptic location of μ opioid receptors on striatopallidal terminals (Abou Khalil et al., 1984; Olive et al., 1997) and suggests a possible autoinhibitory role for endogenous enkephalin released from striatopallidal terminals.
Presynaptic inhibition of pallidopallidal GABAA IPSCs in the GP
IPSCs evoked by stimulation within the GP were depressed by the activation of both μ and δ opioid receptors. The μ opioid receptor agonist DAMGO inhibited locally evoked IPSCs and reduced the frequency of mIPSCs, confirming a presynaptic locus of action. This could be caused by an action on striatopallidal terminals or intrinsic GP terminals. An indication of whether these terminals arose from striatal neurons or intrinsic GP cells was given by the study of sIPSCs, which were both TTX- and picrotoxin-sensitive, implying dependence on spontaneously firing GABA-containing neurons whose cell bodies have been preserved in the preparation. Application of either met-enkephalin or DAMGO reduced the frequency of sIPSCs. Because postsynaptic responses to opioid receptor agonists were rarely observed, this reduction in sIPSCs is most likely to be a result of the presynaptic μ opioid receptor modulation of GABA release. Because the major GABA input to the GP arises from the striatum and the majority of these neurons are quiescent, it is likely that this modulation is on presynaptic terminals of spontaneously firing GP cells. One possible candidate cell type is the type I GP neuron.
DPDPE also reduced IPSCs evoked by local GP stimulation and reduced the frequency of mIPSCs, indicative of a presynaptic location of δ receptors in the GP. The modulation of GABA release by δ opioid receptors from rat slices of GP has been reported previously (Dewar et al., 1987), although the precise location of this presynaptic modulation could not be determined. In contrast, DPDPE had no effect on sIPSCs. Therefore, the presynaptic δ receptors may well be located on the presynaptic terminals of quiescent GP neurons (possibly type II) or another nonstriatal source of GABA input to the GP.
Despite the evidence for pallidopallidal GABA terminals expressing functional presynaptic μ and δ opioid receptors, this has yet to be corroborated by anatomical studies. A number of studies have indicated scattered cells in the GP, each expressing high levels of μ opioid receptor mRNA or moderate levels of δ mRNA (Delfs et al., 1994;Mansour et al., 1994). mRNA for both μ and δ opioid receptors have also been found in scattered cells in the human GPe (Peckys and Landwehrmeyer, 1999). Therefore, opioid receptor protein transport from GP somata to presynaptic terminals remains a possibility.
Postsynaptic effects of opioid receptor activation in the GP
Immunohistochemical studies of postsynaptic opioid receptor localization are primarily in agreement with the in situhybridization data (Bausch et al., 1995; Ding et al., 1996; Olive et al., 1997). In addition, postsynaptic DOR1 (antibody to δ opioid receptors) immunostaining has been reported to be present on pallidostriatal feedback neurons (Olive et al., 1997). These neurons may be equivalent to the discrete population of GP cells, which selectively innervate subpopulations of interneurons in the striatum (Bevan et al., 1998).
We did not observe functional postsynaptic μ and δ receptors in GP neurons because changes in holding current on met-enkephalin application were rare. A small proportion of cells did show changes in mIPSC amplitude on application of DAMGO (one of seven cells) or DPDPE (one of four cells), indicative of postsynaptic receptors. However, we were unable to correlate these changes with a specific neuronal population. It may well be that there is a small population of GP neurons, as yet unidentified, which possess functional postsynaptic opioid receptors.
The lack of a measurable functional change per se does not preclude the fact that postsynaptic opioid receptors may be linked to other effector mechanisms other than those that alter membrane conductance or potential. Postsynaptic opioid receptor activation may produce long-term changes in GP cell physiology via alterations in gene expression. Indeed, systemic morphine administration has been shown to increase the expression of transcription factors, such as Fos, in the striatum and nucleus accumbens (Bontempi and Sharp, 1997).
Implications for opioid modulation of GABA transmission in the GP and relation to movement disorders
This study supports the notion that endogenous enkephalin acts at both μ presynaptic opioid receptors on striatopallidal terminals and μ and δ presynaptic opioid receptors on pallidopallidal terminals to modulate the release of GABA. Thus, enkephalin appears to regulate inhibitory synaptic activity in the GP and may guard against total repression of GP GABA output. The classical model predicts that dopamine depletion leads to this excessive striatopallidal activity, inhibition of GP neurons, and a reduction in GP GABA output. However, a number of studies have indicated that, in experimental parkinsonism, the GP shows increased excitatory responses and increases in burst firing, which may indeed enhance GABA release from GP neurons (Tremblay et al., 1989; Filion and Tremblay, 1991; Chesselet and Delfs, 1996). This apparent paradoxical increase in GP neuronal activity may be a direct result of an increase in STN excitatory input (Chesselet and Delfs, 1996) or alternatively caused by the overriding adaptive mechanism of enkephalin reducing GABA release from both striatopallidal terminals and terminals of intrinsic GP axon collaterals. In keeping with this function of enkephalin, levels of mRNA and protein are increased in the striatum of parkinsonism animals (Augood et al., 1989;Asselin et al., 1994), although a small proportion of pallidal neurons show increased mRNA for enkephalin after 6-hydroxydopamine lesions (Marshall et al., 1999).
δ Presynaptic receptors appear to be preferentially located on the terminals of quiescent cells, possibly type II neurons. In contrast, μ opioid receptors appear to play a role in the modulation of GABA release on pallidopallidal terminals of tonically firing type I or type II GP neurons. The overall physiological function of both these presynaptic opioid receptors will be dependent on the degree of STN excitation, the extent of GP neuronal interconnectivity, and the association between quiescent and tonically firing neurons, which at present is unknown.
Footnotes
This work was supported by The Wellcome Trust Grant 050196/Z/97/Z. We thank Drs. Lacey and Wigmore for helpful discussion and appraisal of this manuscript.
Correspondence should be addressed to Ian M. Stanford, The Department of Pharmacology, Division of Neuroscience, The Medical School, The University of Birmingham, Edgbaston, Birmingham B15 2TT, UK.
REFERENCES
- 1.Abou Khalil B, Young AB, Penney JB. Evidence for the presynaptic localization of opiate binding sites on striatal efferent fibers. Brain Res. 1984;323:21–29. doi: 10.1016/0006-8993(84)90261-0. [DOI] [PubMed] [Google Scholar]
- 2.Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 1990;13:266–271. doi: 10.1016/0166-2236(90)90107-l. [DOI] [PubMed] [Google Scholar]
- 3.Asselin MC, Soghomonian JJ, Cote P, Parent A. Striatal changes in preproenkephalin mRNA levels in parkinsonian monkeys. NeuroReport. 1994;5:2137–2140. doi: 10.1097/00001756-199410270-00037. [DOI] [PubMed] [Google Scholar]
- 4.Augood SJ, Emson P, Mitchell IJ, Boyce S, Clarke CE, Crossman AR. Cellular localisation of enkephalin gene expression in MPTP-treated cynomolgus monkeys. Mol Brain Res. 1989;6:85–92. doi: 10.1016/0169-328x(89)90032-6. [DOI] [PubMed] [Google Scholar]
- 5.Austin MC, Kalivas PW. Enkephalinergic and GABAergic modulation of motor activity in the ventral pallidum. J Pharm Exp Ther. 1990;252:1370–1377. [PubMed] [Google Scholar]
- 6.Bausch SB, Patterson TA, Appleyard SM, Chavkin C. Immunocytochemical localization of δ opioid receptors in the mouse brain. J Chem Neuroanat. 1995;8:175–189. doi: 10.1016/0891-0618(94)00044-t. [DOI] [PubMed] [Google Scholar]
- 7.Bevan MD, Booth PAC, Eaton SA, Bolam JP. Selective innervation of neostriatal interneurons by a subclass of neuron in the globus pallidus. J Neurosci. 1998;18:9438–9452. doi: 10.1523/JNEUROSCI.18-22-09438.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolam JP, Smith Y. The striatum and globus pallidus send convergent synaptic inputs onto single cells in the entopeduncular nucleus of the rat: a double anterograde labelling study combined with post-embedding immunocytochemistry for GABA. J Comp Neurol. 1992;321:456–476. doi: 10.1002/cne.903210312. [DOI] [PubMed] [Google Scholar]
- 9.Bontempi B, Sharp FR. Systemic morphine-induced Fos protein in the rat striatum and nucleus accumbens is regulated by μ opioid receptors in the substantia nigra and ventral tegmental area. J Neurosci. 1997;17:8596–8612. doi: 10.1523/JNEUROSCI.17-21-08596.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chesselet MF, Delfs JM. Basal ganglia and movement disorders: an update. Trends Neurosci. 1996;19:417–422. doi: 10.1016/0166-2236(96)10052-7. [DOI] [PubMed] [Google Scholar]
- 11.Cornwall J, Cooper JD, Phillipson OT. Projections to the rostral reticular thalamic nucleus in the rat. Exp Brain Res. 1990;80:157–171. doi: 10.1007/BF00228857. [DOI] [PubMed] [Google Scholar]
- 12.Davies CH, Davies SN, Collingridge GL. Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. J Physiol (Lond) 1990;424:513–532. doi: 10.1113/jphysiol.1990.sp018080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delfs JM, Kong H, Mester A, Chen Y, Yu L, Reisine T, Chesselet MF. Expression of μ opioid receptor mRNA in rat brain: in situ hybridization study at the single cell level. J Comp Neurol. 1994;345:46–68. doi: 10.1002/cne.903450104. [DOI] [PubMed] [Google Scholar]
- 14.DeLong MR. Activity of pallidal neurons during movement. J Neurophysiol. 1971;34:414–427. doi: 10.1152/jn.1971.34.3.414. [DOI] [PubMed] [Google Scholar]
- 15.DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281–285. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- 16.Dewar D, Jenner P, Marsden CD. Effects of opioid agonist drugs on the in vitro release of 3H-GABA, 3H-dopamine and 3H-5HT from slices of the rat globus pallidus. Biochem Pharmacol. 1987;36:1741–1747. doi: 10.1016/0006-2952(87)90062-1. [DOI] [PubMed] [Google Scholar]
- 17.Ding YQ, Kaneko T, Nomura S, Mizuno N. Immunohistochemical localization of μ-opioid receptors in the central nervous system of the rat. J Comp Neurol. 1996;367:375–402. doi: 10.1002/(SICI)1096-9861(19960408)367:3<375::AID-CNE5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 18.Filion M, Tremblay L. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res. 1991;547:142–151. [PubMed] [Google Scholar]
- 19.Frayne SE, Mitchell IJ, Sharpe PT, Sambrook MA, Crossman AR. Distribution of enkephalin gene expression in the striatum of the parkinsonian primate: implications for dopamine agonist induced dyskinesia. Mol Pharmacol. 1991;1:53–58. [Google Scholar]
- 20.Gandia JA, Delasheras S, Garcia M, Giménez-Amaya JM. Afferent projections to the reticular thalamic nucleus from the globus pallidus and substantia nigra of the rat. Brain Res Bull. 1993;32:351–358. doi: 10.1016/0361-9230(93)90199-l. [DOI] [PubMed] [Google Scholar]
- 21.Gerfen CR, Young WS. Distribution of striatonigral and striatopallidal peptidergic neurons in both patch and matrix compartments: an in situ hybridization histochemistry and fluorescent retrograde tracing study. Brain Res. 1988;460:161–167. doi: 10.1016/0006-8993(88)91217-6. [DOI] [PubMed] [Google Scholar]
- 22.Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJJ, Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- 23.Hazrati LN, Parent A. Projection from the external pallidum to the reticular thalamic nucleus in the squirrel monkey. Brain Res. 1991;550:142–146. doi: 10.1016/0006-8993(91)90418-u. [DOI] [PubMed] [Google Scholar]
- 24.Hazrati LN, Parent A. The striatopallidal projection displays a high degree of anatomical specificity in the primate. Brain Res. 1992;592:213–227. doi: 10.1016/0006-8993(92)91679-9. [DOI] [PubMed] [Google Scholar]
- 25.Hontanilla B, Parent A, Giménez-Amaya JM. Compartmental distribution of parvalbumin and calbindin D-28k in rat globus pallidus. NeuroReport. 1994;5:2269–2272. doi: 10.1097/00001756-199411000-00016. [DOI] [PubMed] [Google Scholar]
- 26.Kincaid AE, Penny JB, Jr, Young AB, Neumans SW. Evidence for a projection from the globus pallidus to entopeduncular nucleus in the rat. Neurosci Lett. 1991;128:121–125. doi: 10.1016/0304-3940(91)90774-n. [DOI] [PubMed] [Google Scholar]
- 27.Kita H, Kitai ST. The morphology of globus pallidus projection neurons in the rat: an intracellular staining study. Brain Res. 1994;636:308–319. doi: 10.1016/0006-8993(94)91030-8. [DOI] [PubMed] [Google Scholar]
- 28.Lavoie B, Parent A, Bedard PJ. Effects of dopamine degeneration on striatal peptide expression in parkinsonian monkeys. Can J Neurol Sci. 1991;18:373–375. doi: 10.1017/s0317167100032467. [DOI] [PubMed] [Google Scholar]
- 29.Maneuf YP, Mitchell IJ, Crossman AR, Brotchie JM. On the role of enkephalin cotransmission in the GABAergic striatal efferents to the globus pallidus. Exp Neurol. 1994;125:65–71. doi: 10.1006/exnr.1994.1007. [DOI] [PubMed] [Google Scholar]
- 30.Mansour A, Fox CA, Burke S, Meng F, Thompson RC, Akil H, Watson SJ. μ, δ, and κ opioid receptor mRNA expression in the rat CNS: an in situ hybridization study. J Comp Neurol. 1994;350:412–438. doi: 10.1002/cne.903500307. [DOI] [PubMed] [Google Scholar]
- 31.Mansour A, Fox CA, Akil H, Watson SJ. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci. 1995;18:22–29. doi: 10.1016/0166-2236(95)93946-u. [DOI] [PubMed] [Google Scholar]
- 32.Marshall JF, Hoover BR, Schiller J 1999 Enkephalin-expressing globus pallidus neurons: characteristics and mRNA regulation after nigrostriatal injury. Mov Disord [Abstract], in press.
- 33.Nambu A, Llinás R. Electrophysiology of globus pallidus neurons in vitro. J Neurophysiol. 1994;72:1127–1139. doi: 10.1152/jn.1994.72.3.1127. [DOI] [PubMed] [Google Scholar]
- 34.Nambu A, Llinás R. Morphology of globus pallidus neurons: its correlation with electrophysiology in guinea pig brain slices. J Comp Neurol. 1997;377:85–94. [PubMed] [Google Scholar]
- 35.Napier TC. Dopamine receptors in the ventral pallidum regulate circling induced by opioids injected into the ventral pallidum. Neuropharmacology. 1992;31:1127–1136. doi: 10.1016/0028-3908(92)90009-e. [DOI] [PubMed] [Google Scholar]
- 36.Obeso JA, Rodriguez MC, DeLong MR. Basal ganglia pathophysiology: a critical review. Adv Neurol. 1997;74:3–18. [PubMed] [Google Scholar]
- 37.Olive MF, Anton B, Micevych P, Evans CJ, Maidment NT. Presynaptic versus postsynaptic localization of μ and δ opioid receptors in dorsal and ventral striatopallidal pathways. J Neurosci. 1997;17:7471–7479. doi: 10.1523/JNEUROSCI.17-19-07471.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parent A. Extrinsic connections of the basal ganglia. Trends Neurosci. 1990;13:254–259. doi: 10.1016/0166-2236(90)90105-j. [DOI] [PubMed] [Google Scholar]
- 39.Parent A, Hazrati L-N. Functional anatomy of the basal ganglia. II. The place of subthalamic nucleus and external pallidum in basal ganglia circuitry. Brain Res Rev. 1995;20:128–154. doi: 10.1016/0165-0173(94)00008-d. [DOI] [PubMed] [Google Scholar]
- 40.Peckys D, Landwehrmeyer GB. Expression of μ, κ, and δ opioid receptor messenger RNA in the human CNS: a 35P in situ hybridization study. Neuroscience. 1999;88:1093–1135. doi: 10.1016/s0306-4522(98)00251-6. [DOI] [PubMed] [Google Scholar]
- 41.Smith Y, Bolam JP. Neurons of the substantia nigra reticulata receive a dense GABA-containing input from the globus pallidus in the rat. Brain Res. 1989;493:160–167. doi: 10.1016/0006-8993(89)91011-1. [DOI] [PubMed] [Google Scholar]
- 42.Smith Y, Bolam JP, Von Krosigk M. Topographical and synaptic organisation of GABA containing pallidosubthalamic projection in the rat. Eur J Neurosci. 1990;2:500–511. doi: 10.1111/j.1460-9568.1990.tb00441.x. [DOI] [PubMed] [Google Scholar]
- 43.Stuart GJ, Dodt HU, Sakmann B. Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflügers Arch. 1993;423:511–518. doi: 10.1007/BF00374949. [DOI] [PubMed] [Google Scholar]
- 44.Travagli RA, Williams JT. Endogenous monoamines inhibit glutamate transmission in the spinal cord of the guinea-pig. J Physiol (Lond) 1996;491:177–185. doi: 10.1113/jphysiol.1996.sp021205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tremblay L, Filion M, Bedard PJ. Responses of pallidal neurons to striatal stimulation in monkeys with MPTP-induced parkinsonism. Brain Res. 1989;498:17–33. doi: 10.1016/0006-8993(89)90395-8. [DOI] [PubMed] [Google Scholar]
- 46.Wilson W, Phelan KD. Dual topographic representation of neostriatum in the globus pallidus of rats. Brain Res. 1982;243:354–359. doi: 10.1016/0006-8993(82)90260-8. [DOI] [PubMed] [Google Scholar]