

Abstract
Central sensitization, the hyperexcitability of spinal processing that often accompanies peripheral injury, is a major component of many persistent pain states. Here we report that the neurotrophin, brain-derived neurotrophic factor (BDNF), is a modulator of excitability within the spinal cord and contributes to the mechanism of central sensitization. BDNF, localized in primary sensory neuron cell bodies and central terminals, potentiates nociceptive spinal reflex responses in an in vitro spinal cord preparation and induces c-fos expression in dorsal horn neurons. NMDA receptor-mediated responses, known as a major contributor to central sensitization, were significantly enhanced by exogenous BDNF. Systemic NGF treatment, a procedure that mimics peripheral inflammatory states, raises BDNF levels in sensory neurons and increases nociceptive spinal reflex excitability. This increased central excitability is reduced by trkB-IgG, a BDNF “antagonist.” We also show directly that inflammatory pain-related behavior depends on BDNF release in vivo. Thus behavioral nociceptive responses induced by intraplantar formalin and by intraplantar carageenan are significantly attenuated by trkB-IgG. Hence BDNF is appropriately localized and regulated in inflammatory states and is sufficient and necessary for the expression of central sensitization in the spinal cord. We propose that BDNF may function as a modulator of central sensitization in pathological states, and our results suggest that pharmacological antagonism of BDNF may prove an effective and novel analgesic strategy for the treatment of persistent inflammatory pain states.
Keywords: BDNF, sensory neuron, spinal cord, nociception, hyperalgesia, NMDA
A key mechanism that contributes to many persistent pain states is the increased excitability of sensory processing in the dorsal horn of the spinal cord that results in greater firing of spinal output systems to given noxious inputs and to normally innocuous stimuli. This is the phenomenon of central sensitization, now widely recognized as a major etiological factor in the generation of referred pain syndromes and secondary hyperalgesia. One possible mechanism contributing to central sensitization is the enhanced responsiveness of the NMDA receptor after its phosphorylation (Chen and Huang 1992). The factors leading to this altered state of phosphorylation, however, are not clear. Tachykinins such as substance P, produced and released by nociceptive sensory neurons within the spinal dorsal horn, have been implicated. The results, however, of clinical trials with tachykinin receptor antagonists have been very disappointing, whereas mice null mutant for either substance P or its receptor display a rather modest phenotype in terms of pain-related behavior (Cao et al., 1998; Zimmer et al., 1998). It is a strong possibility, therefore, that other novel, centrally acting mediators are associated with central sensitization. There is now compelling evidence from another area of the CNS, the hippocampus, that brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family of growth factors, is a modulator of central neuronal activity. Evidence now links the activity-dependent release of BDNF to the expression of hippocampal long-term potentiation (LTP), a characteristic activity-dependent increase in synaptic potency within the CNS (Kang and Schumann, 1995). LTP is significantly impaired in brain slices obtained from BDNF null mutant mice (Korte et al., 1995;Patterson et al., 1996) and is reduced in normal mice by the BDNF-scavenging protein trkB-IgG (Figurov et al., 1996). Evidence is now rapidly accumulating that BDNF may play a role in the spinal cord similar to that in the hippocampus. Unlike other neurotrophins BDNF is constitutively present in sensory neurons (Ernfors et al., 1990; Apfel et al., 1996; Michael et al., 1997). It undergoes anterograde axonal transport (Zhou and Rush, 1996; Michael et al., 1997; Fawcett et al., 1998; Tonra et al., 1998) and is localized within the spinal dorsal horn, where it is associated with synaptic vesicles (Michael et al., 1997). The majority of dorsal root ganglion (DRG) cells expressing BDNF have trkA receptors (i.e., respond to NGF), and NGF promotes the expression of BDNF in these cells (Apfel et al., 1996; Cho et al., 1997; Michael et al., 1997). There is now abundant evidence that NGF is ubiquitously expressed in inflamed tissues, and its upregulation is a key component in the generation of persistent inflammatory hyperalgesia (McMahon et al., 1995). NGF itself is proinflammatory and will induce a sensitization of the sensory nervous system to noxious stimuli at several different levels. A rapid peripheral sensitization of nociceptor responses to thermal stimuli may be detected within minutes of peripheral administration of NGF, and behavioral and electrophysiological studies have shown that sequestration of endogenous NGF produces decreased nociceptor activity (Lewin and Barde, 1996). It is likely that NGF has a major influence on nociceptive sensory systems other than its peripheral sensitizing effects. First, NGF is retrogradely transported by nociceptors and is known to have a major effect on gene expression in those cells (Lewin and Barde, 1996). Second, later components of the hyperalgesia both induced by NGF injection and as a result of peripheral inflammation appear to have a major central component (Lewin et al., 1994, Thompson et al., 1995). BDNF expression is dramatically enhanced in nociceptors after NGF treatment and peripheral injury (Apfel et al., 1996; Cho et al., 1997;Michael et al., 1997; Tonra et al., 1998). In this paper we suggest that BDNF is a major modulator of spinal excitability, and its NGF-mediated upregulation may play a key role in persistent inflammatory pain states.
Some of this work has been presented previously in abstract form (Bennett et al., 1996a; Dassan et al., 1998).
MATERIALS AND METHODS
In vitro spinal cord experiments
Isolated hemisected spinal cords were prepared from 12- to 14-d-old rat pups. Animals were deeply anesthetized by Enflurane inhalation, and the spinal column containing all thoracic, lumbar, and sacral segments was rapidly removed and submerged in oxygenated, cooled Krebs’ solution, (in mm: NaCl, 138.0; KCl, 1.35; NaHCO3, 21.0; NaH2PO4, 0.58; MgCl2, 1.16; CaCl2, 1.26; Glucose, 10.0) at 4°C. The spinal cord was subsequently removed from the spinal column and hemisected in oxygenated Krebs’ solution at 4°C. Spinal cords were placed in a perspex recording chamber (1.0 ml approximate volume) and superfused at 10 ml/min with the same modified oxygenated Krebs’ solution at room temperature (18–22°C). A recovery period of at least 2 hr was left between removal of the cord and recording. Isolated spinal cords were prepared in an identical manner either from naïve animals or from pups treated 24 hr earlier with NGF (1.0 mg/kg, i.p.). Afferent-evoked reflex responses were recorded with a close-fitting glass suction electrode from the L5 ventral root after stimulation of the ipsilateral L5 dorsal root in all experiments (L5 DR-VRP). The L5 dorsal root was stimulated via a glass suction electrode with constant current stimuli at a distance of ∼20 mm from the dorsal root entry zone. Stable DC recordings of the evoked ventral root potentials can be recorded for several hours by this method. Responses were conventionally amplified, measured, and plotted using a Gould (Valley View, OH) DSO 420 digital oscilloscope. The electrical thresholds for activation of afferent fiber populations have previously been determined (Thompson et al., 1992, 1994), and the L5 DR-VRP has been well characterized (Thompson et al., 1992). Briefly, single 500 μA, 500 μsec electrical pulses to the L5 dorsal root evokes a very long duration ventral root potential (VRP), which consist of a short latency A-fiber-mediated monosynaptic component, which is largely resistant to antagonists acting at the NMDA receptor site and a further very long duration prolonged component. This prolonged component consists of both NMDA and neurokinin receptor-sensitive components, has its peak amplitude at a latency of ∼2.0 sec, and is only present at stimulus strengths sufficient to activate unmyelinated high-threshold C-fibers. The following parameters of the VRP were measured: (1) the peak amplitude of the A-fiber-evoked response measured within a 200 msec window after stimulation; and (2) prolonged VRP integrated area, measured between 0 and 8.0 sec latency. The overwhelming majority of the VRP area is composed of activity evoked by C-fiber activity. We have previously shown that this prolonged VRP area is a faithful measure of the degree of central synaptic excitability (Thompson et al., 1994). Amplitudes and areas were normalized to mean values obtained from a series of control preparations run in the absence of compounds and expressed as a percentage of mean control values. One-way ANOVA was performed followed by Tukey’s post hoctest. A value of p < 0.05 was indicative of significance.
NMDA was dissolved in Krebs’ solution from frozen aliquots and held in a secondary reservoir that could be connected to the recording chamber at an identical flow rate. A fixed volume (3.3 ml) was superfused in a bolus for analysis of NMDA-induced depolarizations. The amplitude of NMDA-evoked responses was expressed as a percentage of the maximal response obtained after superfusion with 100 μmNMDA. All neurotrophins were prepared from frozen aliquots and dissolved to their final concentrations in Krebs’ solution just before application to the recording chamber from separate reservoirs without any change in the bath flow rate and recirculated for a minimum of 30 min.
Immunohistochemistry and animal surgery
BDNF. Postnatal rats (12 d old) were treated with systemic NGF (1 mg/kg; n = 4) or saline (n = 4), and a control naïve group were untreated (n = 4). After 24 hr, rats were deeply anesthetized with pentobarbitone (80 mg/kg, i.p.) and transcardially perfused with 50 ml of saline followed by 300 ml of paraformaldehyde (4% in 0.1 m phosphate buffer). L4 and L5 DRG and lumbar spinal cord tissue was dissected, post-fixed in 4% paraformaldehyde (2 hr at 4°C), and transferred to 20% sucrose (overnight at 4°C). Tissue was then blocked in OCT embedding compound (BDH, Poole, Dorset, UK) on dry ice. Transverse sections through the DRG (15 μm thickness) and cord (20 μm thickness) were cut on a cryostat and thaw-mounted onto Superfrost Plus microscope slides (BDH). Sections were immunostained for BDNF using indirect tyramide signal amplification (DuPont NEN, Boston, MA). Sections were incubated with the following (all reagents were diluted in PBS containing 0.2% Triton X-100): normal goat serum (10%, 1 hr), rabbit polyclonal anti-BDNF (a gift from Amgen, Thousand Oaks, CA; 1:2000, 38 hr), biotin-conjugated goat anti-rabbit antibody (Vector Laboratories, Burlingame, CA; 1:400, 1 hr 30 min), avidin–biotin complex (Vector; 1:5 with no Triton X-100 in the buffer, 30 min), biotinyl tyramide (DuPont NEN; 1:75, 8 min), and FITC-conjugated extra-avidin (Sigma, St. Louis, MO; 1:500, 2 hr). All slides were coverslipped with PBS-glycerol (1:3) containing 2.5% 1,4-diazabicyclo-(2.2.2)-octane (antifading agent, Sigma) and visualized under a Leitz (Wetzlar, Germany) fluorescent microscope. To determine the degree of BDNF immunoreactivity in the dorsal horn, images of three randomly chosen sections per animal were captured at 20× objective magnification by a Grundig (Fürth, Germany) FA87 digital camera with integrating frame store and imported into Visilog software. A threshold level was set, which was kept constant for the NGF, saline, and naïve groups. Four boxes (27 × 27 μm) were placed over the image within lamina I and II of the dorsal horn. The area occupied by BDNF-immunoreactive terminals was then calculated.
c-fos. Intrathecal cannulae were implanted for delivery of BDNF as previously described in adult male Wistar rats (Bennett et al., 1996b). Animals were allowed to recover for at least 48 hr so that c-fos activation after surgery would subside. Animals received a bolus injection of 5 μg of BDNF (n = 3), neurotrophin-3 (NT-3; n = 3), or NGF (n = 3) in 10 μl of saline, another group of animals received saline only (n = 4). Three hours after this bolus injection animals were perfused transcardially with heparinized saline followed by 400 ml of 4% paraformaldehde. Another group of BDNF-treated animals were perfused 24 hr after BDNF administration (n = 3). The lumbar enlargement (L3–L5) of the spinal cord was identified and post-fixed in paraformaldehyde for 1 hr before being transferred to 20% sucrose overnight. The entire lumbar enlargement of the spinal cord was cut serially at a thickness of 20 μm, and every fifth section slide was mounted. For c-fos staining sections were incubated for 30 min in 10% normal goat serum and then transferred to a rabbit anti-c-fos primary antiserum (1:100,000, AB5; Oncogene Science, Uniondale, NY) for 24 hr. Primary antibody was subsequently revealed using 1:400 biotinylated goat anti-rabbit IgG (Vector) and the Vectastain ABC system (Vector). Sections were then developed with a solution containing 0.05% 3,3′-diamenobenzidine, 0.04% (NH4)2SO4.NiSO4, and 0.01% H2O2 in 0.1 m phosphate buffer, pH 7.3. To maintain consistency control and treated sections were immunostained simultaneously. To analyze c-fos immunostaining all sections in a given series were examined. The number of c-fos immunoreactive nuclei within the superficial and deep dorsal horn was counted in each section, and an average of the number of nuclei per section was calculated for each animal.
Behavior
Subcutaneous injection of dilute formalin (50 μl, 2% formaldehyde in saline) was used as a model of acute peripheral injury. Two groups of animals were prepared with indwelling intrathecal cannulae. In one group (n = 8), the BDNF-sequestering antibody trkB-IgG (0.16 mg/ml, 10 μl plus 10 μl saline flush) was acutely delivered 30 min before behavioral tests by bolus injection (20 μl total). In the second group (n = 8), saline (20 μl) was used as control. Cannulae were implanted at least 24 hr before behavioral testing. One half of the animals in each group received NGF 24 hr before acute behavioral testing (NGF 1.0 mg/kg, i.p.). The other half from each group received saline control. On the day of testing, animals were placed in a perspex box and given 30 min to habituate to the testing environment. Animals were then lightly anesthetized with halothane, and compounds were injected through the indwelling cannula. A 30 min recovery period was allowed between administration of compounds and formalin injection. Nociceptive behavior was then scored for 55 min. Behaviors were timed separately in 5 min bins and weighted based on the following scale: licking or biting the injected paw was given a score of 2 (T2); elevation of the injected paw off the testing floor surface was given a score of 1 (T1); and full weight bearing of the injected paw was given a score of 0. Pain scores were then calculated for each individual 5 min time bin according to the equation: score = T1 + 2(T2)/300 sec. One-way ANOVA was followed by Dunnetts’ test. p < 0.05 was regarded as significant.
Intraplantar injection of carageenan (50 μl, 2%) was used a model of more persistent inflammatory injury. Two groups of animals were again prepared with indwelling intrathecal cannulae. Cannulae were implanted at least 24 hr before behavioral testing. In one group (n = 5), trkB-IgG (0.16 mg/ml, 10 μl plus 10 μl saline flush) was delivered 30 min before each behavioral test by bolus injection (20 μl total). In the second group (n = 5), saline (20 μl) was used as control. Thermal and mechanical thresholds of the treated paws were assessed 3, 6, and 24 hr after carageenan injection.
In all behavioral experiments the experimenter was blind to all treatments. Differences between trkB-IgG- and saline-treated groups were assessed by two-way ANOVA with repeated measures.p < 0.05 was regarded as significant.
RESULTS
BDNF selectively enhances afferent-evoked spinal reflex activityin vitro
Neurotrophins do not penetrate the spinal cord after systemic delivery. To determine the effect of BDNF on spinal neuron activity, exogenous BDNF was delivered to an isolated spinal cord preparation. High-intensity (500 μA, 500 μsec) electrical stimulation of the L5 dorsal root in this system evokes a very prolonged reflex response, which may be recorded from the corresponding ipsilateral ventral spinal root. This reflex response, the prolonged VRP, has several distinct components distinguishable by their threshold, latency, and pharmacological profile (Thompson et al., 1992). The early component is a short duration wave evoked by fast-conducting, myelinated A-fibers (Fig. 1a, dashed arrow). The late component is a prolonged response, only present at stimulus strengths sufficient to activate unmyelinated nociceptive C-fibers (Fig. 1a, solid arrow). Under control conditions both components remain constant for several hours. BDNF superfusion (2–1000 ng/ml, 30 min) significantly increased the C-fiber-mediated component of the reflex response (189.7 ± 28.3% of control value, 200 ng/ml, 1.0 hr after initiation of superfusion; n = 7;p < 0.01, ANOVA; Fig. 1a,c). The increase in the C-fiber component was sustained, outlasting the duration of BDNF administration (173 ± 30.6%, 200 ng/ml; p < 0.01, ANOVA 1.5 hr after initiation of BDNF administration). A small increase in the non-nociceptive (A-fiber) component was also observed, which did not reach significance (146 ± 18.8% of control, 200 ng/ml, at 1.0 hr; Fig. 1d). The BDNF-induced increase in excitability was completely eliminated by previous exposure of the spinal cord to trkB-IgG (500 ng/ml; n = 3; Fig.1c,d). The effect of BDNF on afferent-evoked responses was dose-dependent, although the maximal effect was not determined (Fig.1b). Acute superfusion of spinal cord preparations with NGF (200 ng/ml; n = 3) or NT-3 (600 ng/ml;n = 3) for 30 min did not have any effect on A- or C-fiber evoked reflex excitability at any time point after superfusion (Fig. 1e,f).
Fig. 1.
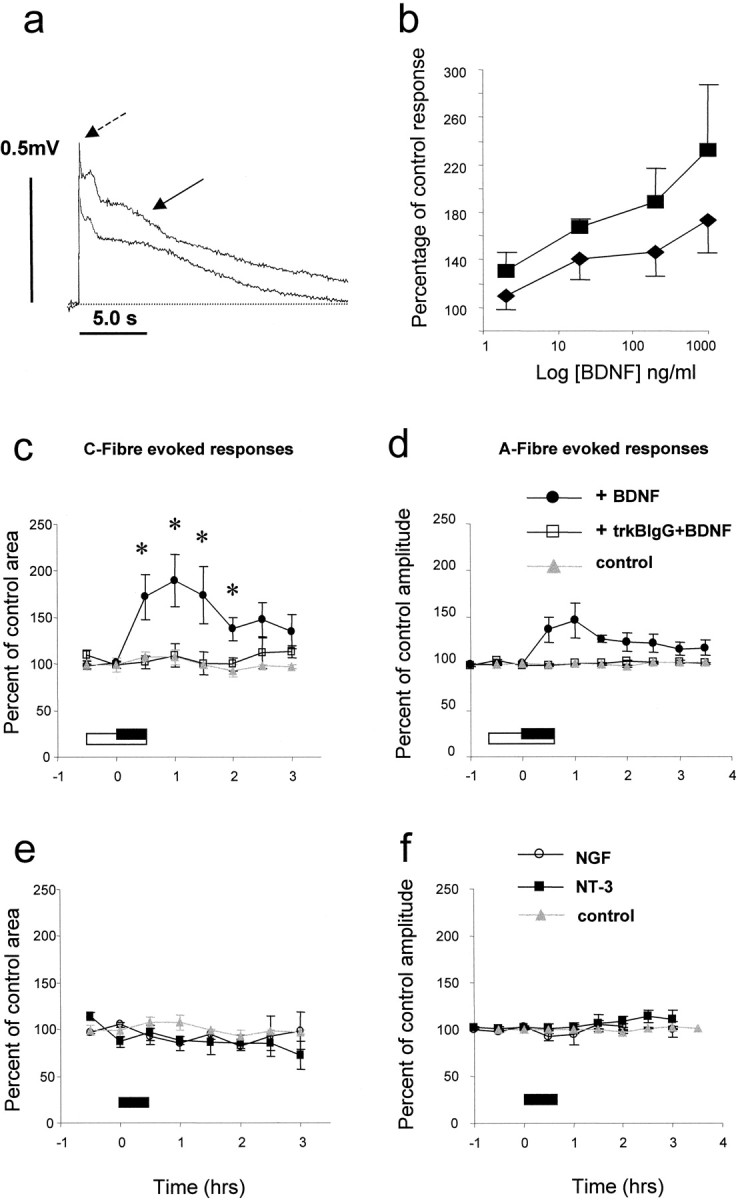
Effect of superfusion of BDNF, NGF, and NT-3 on afferent-evoked spinal reflex activity recorded in vitro. c, e, C-fiber-evoked responses. d, f, A-fiber-evoked responses. All responses were evoked in preparations from naïve animals. Compounds were superfused in the bathing medium for the periods indicated by filledand open bars. a, Example VRP responses showing a significant increase in the C-fiber-mediated component of the reflex response after BDNF (arrow). The short latency A-fiber-evoked component is also indicated by the dashed arrow. c, Summarized data. BDNF (200 ng/ml) was superfused either alone for 30 min (filled bar) or together with trkB-IgG (500 ng/ml) (open bar). BDNF superfusion alone was followed by a significant increase in the size of the C-fiber-evoked VRP. Pre- and co-administration of trkB-IgG along with BDNF prevented the BDNF-induced response. d, BDNF superfusion alone induced a nonsignificant and transient rise in the size of the A-fiber-evoked response. Pre- and co-administration of trkB-IgG along with BDNF prevented the small increase in amplitude. The effect of BDNF on afferent-evoked responses was dose-dependent (b); however, the maximal response was not established. NGF (200 ng/ml; e, f) or NT-3 (600 ng/ml; e, f) did not have any effect on either C- or A-fiber-evoked VRP responses.
We have determined the role of endogenous BDNF in spinal excitability by examining the effect of superfusion of the BDNF sequestering antibody trkB-IgG on A- and C-fiber evoked reflex responses. We have also determined the circumstances under which endogenous BDNF contributes to spinal excitability by assessing reflex activity in spinal cords prepared from naïve animals and from animals pretreated 24 hr earlier in vivo with NGF (1.0 mg/kg). This procedure enhances BDNF levels and mimics the situation found in inflammatory states. In spinal cords prepared from naïve animals, neither the A- nor C-fiber evoked reflex responses were affected by trkB-IgG (Fig.2b,c; 30 min trkB-IgG, 500 ng/ml; n = 3). In spinal cords prepared from NGF-pretreated animals it was noted that the baseline reflex excitability was raised compared with equivalent responses from naïve animals; this increase was not significant (Fig.2b). The C-fiber-evoked response from these preparations, however, was significantly reduced by 30 min superfusion with trkB-IgG (49.5 ± 5.1% of pre trkB-IgG superfusion level, 500 ng/ml; Fig.2a,b; p < 0.01; n = 3). A-fiber responses were not significantly affected by 30 min superfusion with trkB-IgG. Neither was the amplitude of A-fiber evoked responses in naïve and NGF-pretreated preparations significantly different (Fig. 2c).
Fig. 2.

Effect of superfusion of trkB-IgG on afferent-evoked spinal reflex activity recorded in vitrofrom naïve preparations and those pretreated in vivo with NGF. b, trkB-IgG (500 ng/ml) was superfused for the period indicated by the filled bar. In preparations from NGF-pretreated animals there was a trend for the C-fiber component of the reflex to be greater than in control preparations (b, filled circles). After trkB-IgG superfusion there was a sustained and significant depression of the C-fiber-evoked reflex component (b, filled circles). In naïve preparations the C-fiber component was unaffected by trkB-IgG (b, open squares).a, example VRP response showing a significant depression of the C-fiber-mediated component of the reflex response (arrow) after trkB-IgG superfusion in an NGF-pretreated preparation. c, In both naïve preparations and those taken from NGF-pretreated animals the A-fiber component of the response was unaffected by trkB-IgG superfusion. All responses were normalized with respect to mean control responses from naïve preparations.
BDNF enhances NMDA-induced depolarizations in the rat spinal cordin vitro
Superfusion of isolated spinal cords with brief pulses of NMDA (20 sec, 3.3 ml, 20–100 μm) induced short duration depolarizing responses recorded from the ventral roots. These depolarizations were dose-dependent and transient, returning to baseline potential within 3–4 min (Fig.3a,b). After establishment of baseline NMDA-evoked responses, spinal cords were superfused for 30 min with BDNF (200 ng/ml). NMDA-evoked responses were repeated immediately after BDNF superfusion. Responses to 100 μm NMDA were unaffected by previous exposure to BDNF and were considered to represent the maximal response of the tissue. Submaximal responses, however, expressed as a percentage of this maximal response were significantly increased after BDNF superfusion (Fig. 3a,b). The mean amplitude of ventral root depolarization evoked by 50 μm NMDA after BDNF superfusion was 63.3 ± 4.6% of maximal response, compared with 35.5 ± 0.8% of maximal response before BDNF (p < 0.05, paired ttest).
Fig. 3.
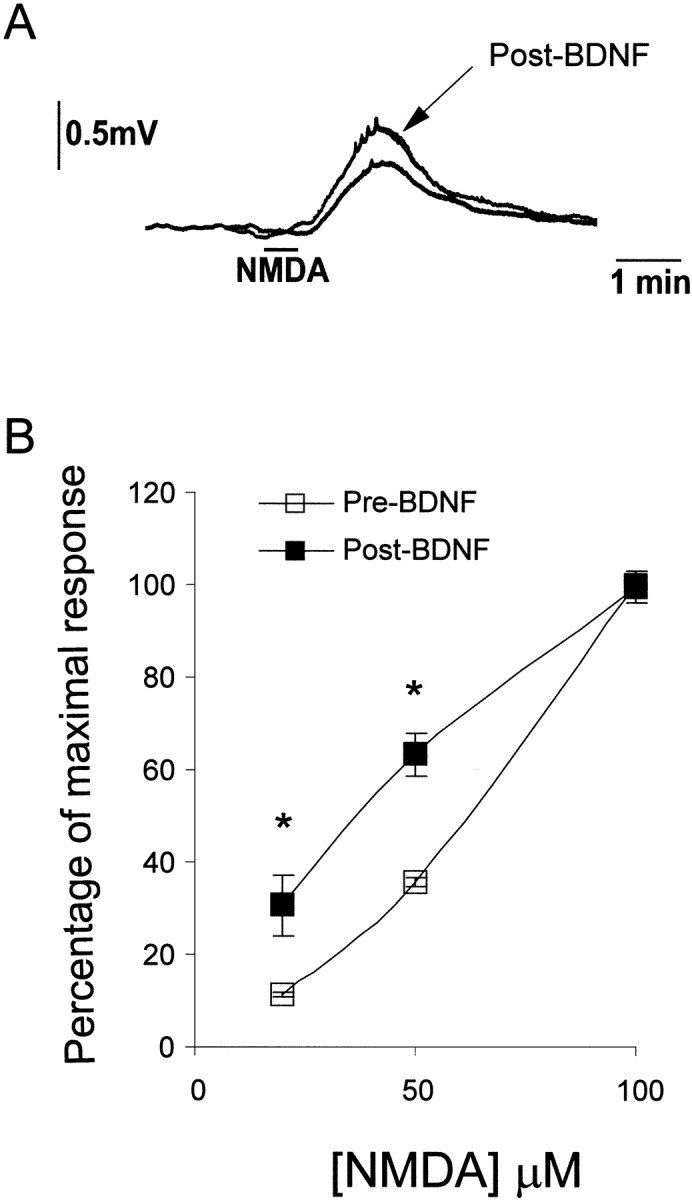
NMDA-evoked ventral root responses are enhanced by pretreatment of spinal cords in vitro with BDNF. A leftward shift of the NMDA-induced ventral root potential dose–response curve was observed after 30 min of superfusion of spinal cords with BDNF (200 ng/ml; n = 3). Responses to 100 μm NMDA were unaffected by previous exposure to BDNF and were considered to represent maximal response of the tissue. Submaximal responses were, however, enhanced by BDNF pretreatment and have been expressed as a percentage of the control maximum response to 100 μm NMDA. Typical, depolarizing ventral root responses to 50 μm NMDA before and after BDNF superfusion are shown in A.
NGF pretreatment induces BDNF upregulation in spinal dorsal horn of neonatal rats
Our results on the effect of trkB-IgG on spinal cord reflex excitability suggest that BDNF has a minor role under normal circumstances but may function as a synaptic modulator when its levels are raised, as in conditions of peripheral inflammation or experimentally, after NGF pretreatment. In adult spinal cord and DRG, BDNF expression is closely linked to NGF levels. Here we demonstrate that pretreatment with NGF significantly increases the level of BDNF immunoreactivity (BDNF-IR) within the spinal cord of neonatal animals used in the present study. Figure4 shows BDNF-IR within the spinal cord 24 hr after NGF (Fig. 4A, b; 1.0 mg/kg, i.p.) or vehicle administration (Fig. 4A, a; saline, i.p.). Image analysis showed a fourfold increase in the intensity of BDNF-IR in the superficial dorsal horn in NGF-treated animals compared with saline controls (652.8 ± 59.5 vs 158.8 ± 51.7, arbitrary units;p < 0.01, Students unpaired t test). A corresponding increase in the number of sensory neuron profiles expressing BDNF-IR in the DRG was also observed (Fig. 4A, c,d). These results demonstrate that under conditions when trkB-IgG significantly reduces nociceptive reflex activity in vitro, BDNF levels are raised within the spinal dorsal horn.
Fig. 4.
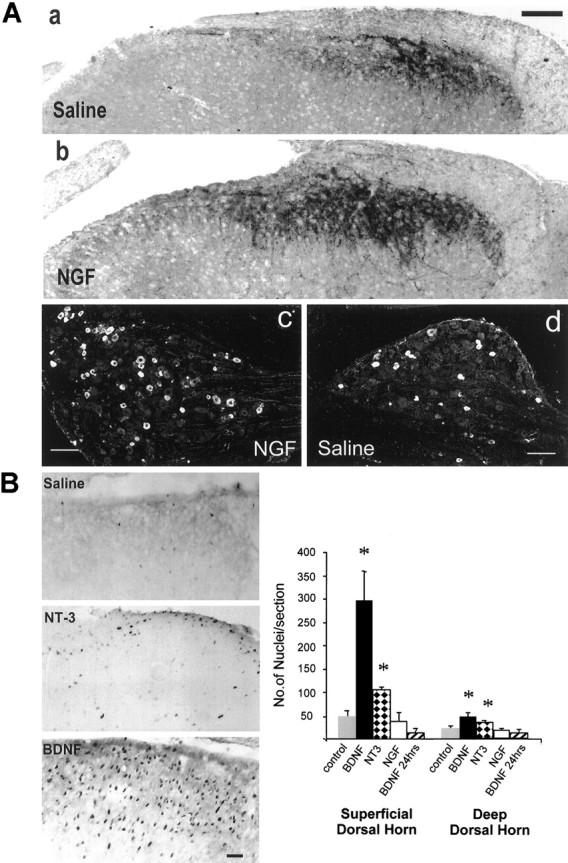
Dark-field and bright-field photomicrographs showing BDNF immunoreactivity within the spinal dorsal horn (A, a, b) and DRG (A, c, d) of 12–14-d-old rats and c-fos immunoreactivity within the superficial dorsal horn of adult rats (B).A, a, d, BDNF immunoreactivity in animals treated 24 hr earlier with systemic vehicle injection. BDNF levels in the spinal cord are low, and a few small-diameter sensory neuronal profiles express BDNF-IR within the lumbar ganglia. A, b, c, Animals treated 24 hr earlier with NGF (1 mg/kg, i.p.). A significant increase in the level of BDNF-IR is observed within the dorsal horn, and increased numbers of sensory neuronal profiles contain BDNF within the DRG. B, c-fos immunoreactivity within the dorsal horn of animals treated with intrathecal saline, NT-3 (5 μg), or BDNF (5 μg) for 3 hr. Positively stained cells demonstrate an intense black reaction product within the nucleus. There are a few scattered fos-IR nuclei within the dorsal horn of saline-treated animals, and NT-3 produces a small increase in the number of fos-IR nuclei. BDNF application however, results in a large increase in the number of fos-IR nuclei especially within the superficial laminae of the dorsal horn. The accompanying graph displays counts of the number of fos-IR nuclei within the superficial and deep dorsal horn of saline- and neurotrophin-treated animals. Three hours after BDNF administration (5 μg) there is a large, significant (p < 0.05, unpaired t test) increase in the number of fos-IR nuclei within the superficial dorsal horn and a smaller but still significant increase in the deep dorsal horn. The number of fos-IR nuclei has returned to baseline 24 hr after BDNF treatment. NT-3 administration (5 μg) also produces a significant increase in the number of fos-IR nuclei within the superficial and deep dorsal horn, whereas NGF (5 μg) has no effect. Scale bars: A, 100 μm; B, 50 μm. Plates a andb in A were photographed as dark-field images and digitally inverted without alteration of contrast.
BDNF induces c-fos expression within the superficial dorsal horn
As a further measure of the ability of BDNF to produce altered postsynaptic function in the dorsal horn, we have tested the ability of BDNF to induce c-fos, an immediate early gene expressed in dorsal horn neurons in response to noxious stimulation. Three hours after intrathecal administration of BDNF (5 μg) to adult rats, a large increase in the number of c-fos-immunoreactive cells per section was observed compared with treatment with saline (Fig.4B). This increase was particularly marked in the superficial dorsal horn (p < 0.05, unpairedt test), and there was also a small but significant increase in the deep dorsal horn (p < 0.05, unpairedt test). Twenty-four hours after BDNF administration c-fos expression within the superficial dorsal horn was no longer elevated. NT-3 (5 μg) produced a very much smaller but still significant increase in the number of fos-immunoreactive nuclei within the superficial and deep dorsal horn (p < 0.05, unpaired t test; Fig. 4B), whereas NGF (5 μg) had no significant effect. These experiments show that BDNF may induce altered postsynaptic gene expression through activation of inducible systems after trk receptor activation and phosphorylation.
Endogenous BDNF contributes to nociceptive behavioral responses
The preferential effect of trkB-IgG on C-fiber-evoked responses observed in vitro suggests a role for BDNF in spinal nociceptive processing. We have directly tested this by assessing the effect of trkB-IgG on nociceptive behavioral responses in adult rats. The effect of intrathecal administration of trkB-IgG on two models of peripheral injury was determined.
First, we have assessed the effect of trkB-IgG on the behavioral responses to injection of dilute formalin into the rat hindpaw. Subcutaneous injection of dilute formalin (50 μl, 2% formaldehyde in saline) into the plantar surface of the hindpaw evokes a characteristic, biphasic, nociceptive behavioral response that reflects acute and prolonged nociceptor discharge phases. The second phase of the formalin response is also known to depend in part on the induction of central sensitization (Coderre and Melzack 1992). In these experiments we have assessed the circumstances under which BDNF may act as a synaptic modulator by measuring the effects of intrathecal trkB-IgG on responses to formalin in naïve animals and in animals in which we have mimicked an inflammation-induced upregulation of BDNF by pretreatment 24 hr earlier with NGF. In naïve animals intrathecal trkB-IgG (10 μl, 0.16 mg/ml plus 10 μl flush) 30 min before formalin injection had no significant effect on either phase of the nociceptive response when compared with the effect of saline alone (Fig. 5a, i). In animals pretreated 24 hr earlier with NGF (1.0 mg/kg), there was a trend (nonsignificant) for the second phase of the nociceptive response to intraplantar formalin injection to be higher than in naïve controls (Fig. 5a, ii). Intrathecal delivery of trkB-IgG (10 μl, 0.16 mg/ml) 30 min before formalin injection in these animals significantly attenuated the nociceptive response (Fig. 5a, ii, b), compared with saline controls.
Fig. 5.

Effect of intrathecal trkB-IgG or vehicle on behavioral nocifensive responses to subcutaneous injection of dilute formalin. Substances were delivered intrathecally 30 min before formalin injection. Experiments were performed on naïve animals (a, i) or on animals pretreated 24 hr earlier with NGF (a, ii; 1 mg/kg, i.p.). In naïve animals trkB-IgG had no effect on either phase of the formalin response, and both phases were indistinguishable from saline-treated controls (a, i, b). In NGF-pretreated animals, however, the second phase of the formalin response was significantly reduced by intrathecal trkB-IgG (a, ii, b). The formalin response was suppressed in terms of both amplitude of response and duration of effect.
Second, we have assessed the contribution of BDNF to spinal synaptic transmission in a more prolonged model of inflammatory pain. We have measured the effect of trkB-IgG on behavioral responses evoked after intraplantar injection of 2% carageenan. Mechanical thresholds for hindpaw withdrawal (von Frey hairs) and the latency of hindpaw withdrawal from a noxious thermal stimulus were measured 3 and 24 hr after intraplantar injection of carageenan (2%, 50 μl). Either TrkB-IgG (10 μl, 0.16 mg/ml plus 10 μl flush) or saline (20 μl) was delivered as a bolus injection via an indwelling intrathecal catheter 30 min before each behavioral test. A significant difference between the effect of TrkB-IgG and saline was observed on the latency of hindpaw withdrawal to a noxious thermal stimulus at both time points after carageenan injection (p < 0.05, two-way ANOVA with repeated measures; Fig.6A). There was a trend for the mechanical withdrawal threshold in the TrkB-IgG-treated animals to be higher than saline-treated controls; however, this did not reach significance (Fig. 6B).
Fig. 6.
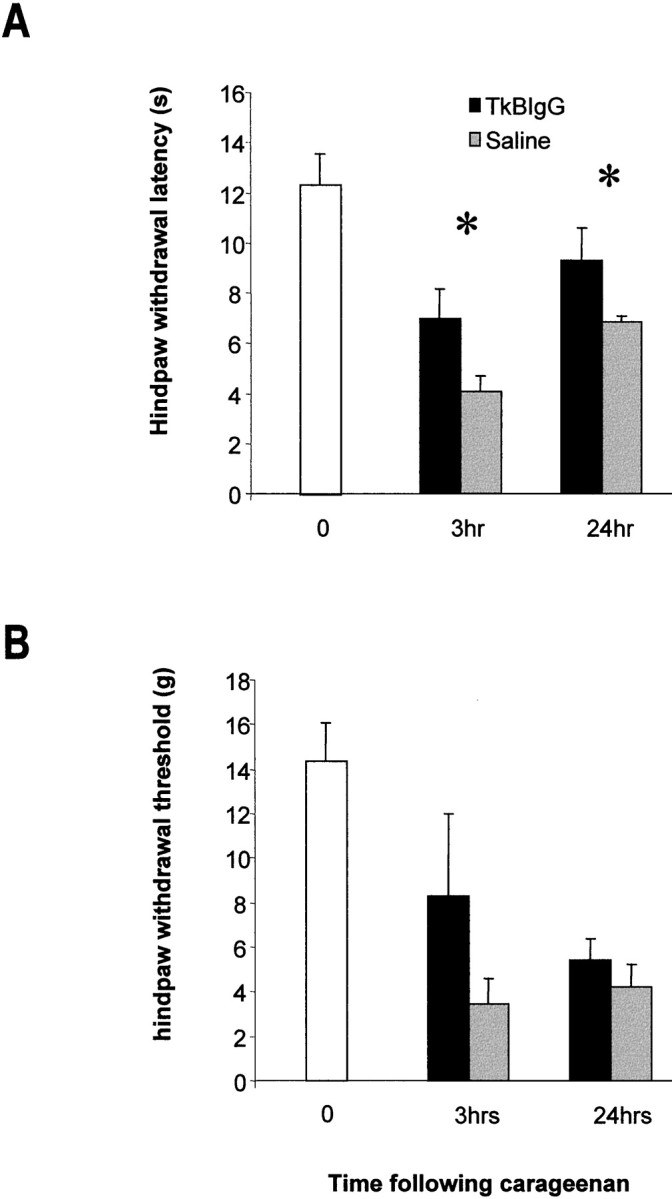
Effect of intrathecal trkB-IgG (10 μl, 0.16 mg/ml plus 10 μl flush) or saline vehicle (20 μl) on hindpaw thermal withdrawal latencies and mechanical thresholds after intraplantar injection of carageenan (50 μl, 2%). Thermal and mechanical thresholds for withdrawal of the treated hindpaw were measured 3 and 24 hr after intraplantar injection. Compounds were administered 30 min before each testing point. A, A significant difference was present between the effect of saline and trkB-IgG on thermal withdrawal latencies at both time points (p < 0.05, two-way repeated measures ANOVA). B, There was a trend for the mechanical thresholds for paw withdrawal to be higher in those animals treated with trkB-IgG; however, this was not significant.
DISCUSSION
Our results suggest that the neurotrophin BDNF may be an endogenous modulator within the spinal dorsal horn that regulates synaptic activity in an NMDA-dependent manner. The principal findings are as follows: (1) exogenous BDNF application to an isolated spinal cord induces a significant increase in nociceptive spinal reflex activity; (2) the BNDF-sequestering antibody trkB-IgG is effective in reducing spinal nociceptive reflex activity when endogenous levels of BDNF are enhanced after pretreatment with NGF or after peripheral inflammation; (3) it is likely that the effect of BDNF application to the in vitro spinal cord is mediated via enhancement of NMDA receptor-mediated responses; (4) BDNF also induces induction of cell signaling cascades within dorsal horn neurons and subsequent c-fos expression; and (5) in inflammatory models the behavioral nociceptive responses may be modulated by endogenous BDNF.
The ventral root potential measured in the current study in vitro is routinely used as an accurate, albeit indirect, measure of spinal excitability (Thompson et al., 1994). Here we have presented evidence for a slowly incrementing increase in reflex activity after BDNF superfusion. There are three possible sites of action of BDNF in our experiments: (1) Ventral horn. Motoneurons express the catalytic isoform of trkB (Koliatsos et al., 1993). Several observations, however, suggest that motoneurons are not the major site of activity of BDNF in this study. First, the enhancement of the monosynaptic A-fiber component of the VRP response by BDNF was minor compared with its effect on the polysynaptic C-fiber-evoked component. Second, there is no effect of the BDNF antagonist trkB-IgG on A-fiber-evoked responses. In contrast, trkB-IgG significantly and specifically reduced C-fiber responses after NGF pretreatment (a procedure known to increase BDNF expression only in trkA-expressing sensory neurons; Michael et al., 1997). (2) Sensory neurons. A significant number of sensory neurons express trkB (McMahon et al., 1994). These trkB-expressing neurons have medium to large cells bodies, likely to have myelinated axons, for the most part to be recruited at low stimulus intensity levels and subserve non-nociceptive functions. NGF does not increase BDNF in these trkB-expressing cells. It is unlikely therefore that the primary action of BDNF in our study is mediated via alterations in the excitability of the primary sensory neurons. (3) Dorsal horn. The present evidence suggests that the effect of BDNF on spinal signaling in the in vitro preparation is predominantly confined to the superficial region of the spinal dorsal horn. Here it fulfils several of the requirements of a neuromodulator. It is present within the central terminals of nociceptive afferents (Zhou and Rush, 1996; Michael et al., 1997; this study), where it is associated with dense-core vesicles (Michael et al., 1997). Functional trkB receptors are present in this region of the spinal cord (Bradbury et al., 1998). Moreover, levels of BDNF rise dramatically within the superficial spinal cord after peripheral injury or administration of NGF (Cho et al., 1997; Michael et al., 1997; Ernfors et al., 1990; this study). Furthermore, we have now demonstrated that functional antagonism of endogenously released BDNF in the in vitro cord significantly attenuates spinal reflex activity.
Our data are consistent with the role of BDNF as a neuromodulator in another region of the CNS. In the hippocampus (Kang and Schumann, 1995), BDNF is necessary for the full expression of one of the most widely studied forms of synaptic plasticity, long term potentiation. Here we also find that BDNF is both sufficient and necessary for the expression of central sensitization within the spinal cord. There may also be direct parallels between the mechanism involved in both the spinal cord and the hippocampus. Both LTP and central sensitization require prolonged alterations in synaptic strength. In the hippocampus this appears to be achieved at least partly through phosphorylation of the NMDA receptor. In particular, phosphorylation of the NR1 and NR2B NMDA subunits occurs acutely in the presence of BDNF (Suen et al., 1997), and BDNF will in fact induce expression of NR2A and NR2B NMDA receptor subunits in this system (Small et al., 1998). The NR2 subunits of the NMDA receptor define its pharmacological and biophysical properties (Sucher et al., 1996), and in the hippocampus BDNF potentiates NMDA responses via a threefold increase in NMDA receptor open time (Levine et al., 1998). It is interesting to note, however, that under certain circumstances, receptor phosphorylation may not be required for BDNF-induced augmentation of NMDA-mediated responses but that BDNF may have a direct effect at the glycine modulatory site (Jarvis et al., 1997). It appears that AMPA responses are not influenced by BDNF (Levine et al., 1998). These data are entirely compatible with the present findings. NMDA-evoked depolarizations within the isolated spinal cord were significantly potentiated, and we have also demonstrated that BDNF produces a selective amplification of C-fiber-mediated, nociceptive-evoked responses. Synaptic responses mediated by C-fiber activation are mediated to a large extent by NMDA receptor activation (Thompson et al., 1990, 1992). Indeed, synaptic responses are boosted in an activity-dependent manner as a result of NMDA receptor activation, the phenomenon of central sensitization (Woolf and Thompson, 1990; Woolf, 1991). A-fiber responses are largely AMPA-mediated and were not influenced by BDNF superfusion in the present study.
The signal pathway responsible for BDNF-mediated enhancement of NMDA responses is not apparent from the present experiments. Previous studies have demonstrated under in vitro conditions that a tyrosine phosphorylation-dependent enhancement of NMDA receptor activity occurs via Src and Fyn (Wang and Salter, 1994; Suzuki and Okamura-Noji, 1995; Kohr and Seeburg, 1996). The present findings indicate that in addition to modulating NMDA receptor ion channel activity, BDNF receptor tyrosine phosphorylation may also serve to link signal transduction pathways in the postsynaptic cell as evidenced by robust c-fos expression in the dorsal horn.
In the present study we have demonstrated a key role for endogenous BDNF in the modulation of spinal excitability both in vitroand in behavioral responses in two models of inflammatory pain. In particular, these experiments have provided key evidence for the circumstances under which endogenous BDNF is likely to function as a central modulator. Therefore in naïve animals, trkB-IgG, a potent BNDF-sequestering molecule, was without significant effect on either spinal reflex excitability in vitro or the acute behavioral responses to formalin injection. BDNF is present, albeit in limited amounts, within primary afferent terminals in these animals. It is likely, however, that under such circumstances, BDNF does not play a major role in the modulation of nociceptive spinal reflex responses. In contrast, after systemic pretreatment with NGF, nociceptive spinal reflex activity both in vitro and in vivo was significantly attenuated by the BDNF-sequestering antibody. These results may be directly correlated to the NGF-dependent upregulation of BDNF that has been demonstrated both in the present study and elsewhere (Apfel et al., 1996; Michael et al., 1997). It is likely therefore that after NGF pretreament, BDNF plays a much more prominent role as a central synaptic mediator, and this contribution is revealed by the use of the sequestering antibody.
There is now extensive and compelling evidence that NGF expression is increased in a large variety of inflammatory states, including Freund’s adjuvant- and carageenan-induced inflammation (Donnerer et al., 1992; Safieh-Garabedian et al., 1995) and experimentally induced cystitis (Oddiah et al., 1998). The ability to block many of the sensory abnormalities that accompany these inflammatory states by NGF antagonism has led to the widely held belief that NGF is in fact a major inflammatory mediator (McMahon, 1996; McMahon et al., 1997). Several reports have demonstrated that the sensory hyperalgesia associated with experimental inflammation is dependent on endogenously produced NGF (Lewin et al., 1994; Woolf et al., 1994; Dmitrieva et al., 1997), and indeed exogenous NGF is a potent algogen capable of inducing hyperalgesia in both humans and rats (Lewin et al., 1994; Petty et al., 1994, Andreev et al., 1995). The mechanism of NGF action is now considered to have both peripheral and central components (Lewin et al., 1994; McMahon, 1996). Because exogenous or inflammation-derived NGF does not cross the blood–brain barrier, the effect of NGF on central spinal nociceptive processes is likely to be mediated by sensitization of primary afferent fibers or by changes in gene expression of responsive afferent fibers. The levels of both substance P and calcitonin gene-related peptide are regulated by NGF. These sensory neuropeptides are well studied in relation to the processing of nociceptive information in the spinal cord, and there is previous evidence that NGF treatment will augment spinal reflex activity in a manner dependent on neurokinin receptor activation (Thompson et al., 1995). In addition, it has been reported that the central component of NGF-induced hyperalgesia is highly dependent on NMDA receptor activation with a particular selectivity toward the accompanying thermal hyperalgesia (Lewin et al., 1994). This is consistent with our present data whereby the thermal hyperalgesia that developed after peripheral carageenan injection was more reliably inhibited by trkB IgG delivery than the mechanical sensitivity, suggesting a BDNF-mediated modulation of NMDA receptor function. Our present results therefore strongly suggest that the upregulation of BDNF is a major contributor to the central mechanism of NGF- and inflammation-induced hyperalgesia.
In summary, our data suggest that BDNF is a key mediator of central sensitization, a phenomenon that is responsible for persistent spinal excitability changes that underlie postinjury pain hypersensitivities. Our data also suggest that trkB receptor blockade, allied to current strategy against the NMDA receptor complex, may substantially augment the clinical treatment of persistent inflammatory pain states.
Footnotes
This work was supported by the Medical Research Council (UK), the Special Trustees of St. Thomas’ Hospital, and the Physiological Society (London, UK). We thank Dr. Q. Yan for the gift of the BDNF antibody used in this study.
Correspondence should be addressed to Stephen W. N. Thompson, Neuroscience Research Centre, Division of Physiology, St. Thomas’ Hospital Campus, Lambeth Palace Road, London SE1 7EH, UK.
REFERENCES
- 1.Andreev NY, Dmitrieva N, Koltzenburg M, McMahon SB. Peripheral administration of NGF in the adult rat produces a thermal hyperalgesia that requires the presence of sympathetic postganglionic neurones. Pain. 1995;63:109–115. doi: 10.1016/0304-3959(95)00024-M. [DOI] [PubMed] [Google Scholar]
- 2.Apfel SC, Wright DE, Wiideman AM, Dormia C, Snider WD, Kessler JA. Nerve growth factor regulates the expression of brain-derived neurotrophic factor mRNA in the peripheral nervous system. Mol Cell Neurosci. 1996;7:134–142. doi: 10.1006/mcne.1996.0010. [DOI] [PubMed] [Google Scholar]
- 3.Bennett DLH, French J, Priestley JV, McMahon SB. The effects of BDNF on cfos and NOS expression in dorsal horn neurones of the spinal cord. Soc Neurosci Abstr. 1996a;22:396.15. [Google Scholar]
- 4.Bennett DLH, French J, Priestley JV, McMahon SB. NGF but not NT-3 or BDNF prevents the A fibre sprouting into laminae II of the spinal cord that occurs following axotomy. Mol Cell Neurosci. 1996b;8:211–220. doi: 10.1006/mcne.1996.0059. [DOI] [PubMed] [Google Scholar]
- 5.Bradbury EJ, King V, Simmons LJ, Priestley JV, McMahon SB. NT-3 but not BDNF prevents atrophy and death of axotomised spinal cord projection neurons. Eur J Neurosci. 1998;10:3058–3068. doi: 10.1046/j.1460-9568.1998.00307.x. [DOI] [PubMed] [Google Scholar]
- 6.Cao YQ, Mantyh PW, Carlson EJ, Gillespie AM, Epstein CJ, Basbaum AI. Primary afferent tachykinins are required to experience moderate to intense pain. Nature. 1998;392:390–394. doi: 10.1038/32897. [DOI] [PubMed] [Google Scholar]
- 7.Chen L, Huang LM. PKC reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature. 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- 8.Cho HJ, Kim SY, Park MJ, Kim DS, Kim JK, Chu MY. Expression of mRNA for brain-derived neurotrophic factor in the dorsal root ganglion following peripheral inflammation. Brain Res. 1997;74:358–362. doi: 10.1016/S0006-8993(97)00048-6. [DOI] [PubMed] [Google Scholar]
- 9.Coderre TJ, Melzack R. The contribution of excitatory amino acids to central sensitization and persistent nociception after formalin-induced tissue injury. J Neurosci. 1992;12:3665–3670. doi: 10.1523/JNEUROSCI.12-09-03665.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dassan P, Trevedi P, McMahon SB, Shelton D, II, Jones M, Swanson G, Thompson SWN. BDNF induces a prolonged increase in spinal reflex activity in vitro. Soc Neurosci Abstr. 1998;24:154.13. [Google Scholar]
- 11.Dmitrieva N, Shelton D, Rice A, McMahon S. The role of NGF in a model of visceral inflammation. Neuroscience. 1997;78:449–459. doi: 10.1016/s0306-4522(96)00575-1. [DOI] [PubMed] [Google Scholar]
- 12.Donnerer J, Schuligoi R, Stein C. Increased content and transport of substance P and CGRP in sensory nerves innervating inflamed tissue: evidence for a regulatory function of NGF in vivo. Neuroscience. 1992;49:693–698. doi: 10.1016/0306-4522(92)90237-v. [DOI] [PubMed] [Google Scholar]
- 13.Ernfors P, Wetmore C, Olson L, Persson H. Identification of cells in rat brain and peripheral tissues expressing mRNA for members of the nerve growth factor family. Neuron. 1990;5:511–526. doi: 10.1016/0896-6273(90)90090-3. [DOI] [PubMed] [Google Scholar]
- 14.Fawcett JP, Bamji SX, Causing CG, Aloyz R, Ase AR, Reader TA, McLean JH, Miller FD. Functional evidence that BDNF is an anterograde neuronal trophic factor in the CNS. J Neurosci. 1998;18:2808–2821. doi: 10.1523/JNEUROSCI.18-08-02808.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 16.Jarvis CR, Xiong Z-G, Plant JR, Churchill D, Lu W-Y, MacVicar BA, MacDonald JF. Neurotrophin modulation of NMDA receptors in cultured murine and isolated rat neurons. J Neurophysiol. 1997;78:2363–2371. doi: 10.1152/jn.1997.78.5.2363. [DOI] [PubMed] [Google Scholar]
- 17.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 18.Kohr G, Seeburg PH. Subtype specific regulation of recombinant NMDA receptor channel by protein tyrosine kinases of the src family. J Physiol (Lond) 1996;492:445–452. doi: 10.1113/jphysiol.1996.sp021320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koliatsos VE, Clatterbuck RE, Winslow JW, Cayouette MH, Price DL. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron. 1993;10:359–367. doi: 10.1016/0896-6273(93)90326-m. [DOI] [PubMed] [Google Scholar]
- 20.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levine ES, Crozier RA, Black IB, Plummer MR. BDNF modulates hippocampal synaptic transmission by increasing NMDA receptor activity. Proc Natl Acad Sci USA. 1998;95:10235–10238. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewin GR, Barde YA. Physiology of the neurotrophins. Annu Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- 23.Lewin GR, Reuff A, Mendell LM. Peripheral and central mechanisms of NGF-induced hyperalgesia. Eur J Neurosci. 1994;6:1903–1912. doi: 10.1111/j.1460-9568.1994.tb00581.x. [DOI] [PubMed] [Google Scholar]
- 24.McMahon SB. NGF as a mediator of inflammatory pain. Philos Trans R Soc Lond B Biol Sci. 1996;351:431–440. doi: 10.1098/rstb.1996.0039. [DOI] [PubMed] [Google Scholar]
- 25.McMahon SB, Armanini MP, Ling LH, Philips HS. Expression and co-expression of trk receptors in subpopulations of adult primary sensory neurons projecting to identified peripheral targets. Neuron. 1994;12:1161–1171. doi: 10.1016/0896-6273(94)90323-9. [DOI] [PubMed] [Google Scholar]
- 26.McMahon SB, Bennett DL, Priestley JV, Shelton DL. The biological effects of endogenous nerve growth factor on adult sensory neurons revealed by a trkA-IgG fusion molecule [see comments]. Nat Med. 1995;1:774–780. doi: 10.1038/nm0895-774. [DOI] [PubMed] [Google Scholar]
- 27. McMahon SB, Bennett DL, Michael G, Priestley JV. Neurotrophic factors and Pain. Proceedings of the eighth World Congress on Pain. Progress in brain research and management, Vol 8 Jensen TS, Weisenfeld-Hallin S. 1997. Seattle: International Association for the Study of Pain.
- 28.Michael GJ, Averill S, Nitkunan A, Rattray M, Bennett DL, Yan Q, Priestley JV. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. J Neurosci. 1997;17:8476–8490. doi: 10.1523/JNEUROSCI.17-21-08476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oddiah D, Anand P, McMahon SB, Rattray M. Rapid increase of NGF, BDNF and NT-3 mRNAs in inflamed bladder. NeuroReport. 1998;9:1455–1458. doi: 10.1097/00001756-199805110-00038. [DOI] [PubMed] [Google Scholar]
- 30.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:137–145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 31.Petty BG, Petty BG, Cornblath DR, Adornato BT, Chaudhry V, Flexner C, Wachsman M, Sinicropi D, Burton LE, Peroutka SJ. The effect of systemically administered recombinant human nerve growth factor in healthy human subjects. Ann Neurol. 1994;36:244–246. doi: 10.1002/ana.410360221. [DOI] [PubMed] [Google Scholar]
- 32.Safieh-Garabedian B, Poole S, Allchornce A, Winter J, Woolf CJ. Contribution of interleukin-1 beta to the inflammation-induced increase in NGF levels and inflammatory hyperalgesia. Br J Pharmacol. 1995;115:1265–1275. doi: 10.1111/j.1476-5381.1995.tb15035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Small DL, Murray CL, Mealing GEA, Poulter MO, Buchan AM, Morley P. BDNF induction of MNDA receptor subunit NR2A expression in cultured rat cortical neurons. Neurosci Lett. 1998;252:211–214. doi: 10.1016/s0304-3940(98)00587-4. [DOI] [PubMed] [Google Scholar]
- 34.Sucher NJ, Awobuluyi M, Choi YB, Lipton SA. NMDA receptors: from genes to channels. Trends Pharmacol Sci. 1996;17:348–355. [PubMed] [Google Scholar]
- 35.Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY, Black IB. Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-d-aspartate receptor subunit 1. Proc Natl Acad Sci USA. 1997;94:8191–8195. doi: 10.1073/pnas.94.15.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki T, Okumura-Noji K. NMDA receptor subunits 1 (NR2A) and 2 (NR2B) are substrates for fyn in the postsynaptic density fraction isolated from the rat brain. J Neurosci Res. 1995;37:506–514. doi: 10.1006/bbrc.1995.2662. [DOI] [PubMed] [Google Scholar]
- 37.Thompson SWN, King AE, Woolf CJ. Activity-dependent changed in rat ventral horn neurones in vitro summation of prolonged afferent evoked postsynaptic depolarizations produce a d-APV sensitive windup. Eur J Neurosci. 1990;2:638–649. doi: 10.1111/j.1460-9568.1990.tb00453.x. [DOI] [PubMed] [Google Scholar]
- 38.Thompson SWN, Gerber G, Sivilotti LG, Woolf CJ. Long duration ventral root potentials in the neonatal rat spinal cord in vitro: the effects of ionotropic and metabotropic excitatory amino acid receptor antagonists. Brain Res. 1992;595:87–97. doi: 10.1016/0006-8993(92)91456-o. [DOI] [PubMed] [Google Scholar]
- 39.Thompson SWN, Dray A, Urban L. Injury-induced plasticity of spinal reflex activity: NK1 receptor activation and enhanced A- and C-fibre mediated responses in the rat spinal cord in vitro. J Neurosci. 1994;14:3672–3687. doi: 10.1523/JNEUROSCI.14-06-03672.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson SWN, Dray A, McCarson KE, Krause JE, Urban L. Nerve growth factor induces mechanical allodynia associated with neurokinin-1 receptor activation and novel A-fibre evoked spinal reflex activity in the rat. Pain. 1995;62:219–231. doi: 10.1016/0304-3959(94)00271-F. [DOI] [PubMed] [Google Scholar]
- 41.Tonra JR, Curtis R, Wong V, Cliffer KD, Park JS, Timmes A, Nguyen T, Lindsay RM, Acheson A, DiStefano PS. Axotomy upregulates the anterograde transport and expression of brain-derived neurotrophic factor by sensory neurons. J Neurosci. 1998;18:4374–4383. doi: 10.1523/JNEUROSCI.18-11-04374.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- 43.Woolf CJ. Generation of acute pain: central mechanisms. Br Med Bull. 1991;47:523–533. doi: 10.1093/oxfordjournals.bmb.a072490. [DOI] [PubMed] [Google Scholar]
- 44.Woolf CJ, Thompson SWN. The induction and maintenance of central sensitization is dependent on NMDA receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain. 1990;44:293–300. doi: 10.1016/0304-3959(91)90100-C. [DOI] [PubMed] [Google Scholar]
- 45.Woolf CJ, Safieh-Garabedian B, Ma QP, Crilly P, Winter J. NGF contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience. 1994;62:327–331. doi: 10.1016/0306-4522(94)90366-2. [DOI] [PubMed] [Google Scholar]
- 46.Zhou XF, Rush RA. Endogenous brain-derived neurotrophic factor is anterogradely transported in primary sensory neurons. Neuroscience. 1996;74:945–953. doi: 10.1016/0306-4522(96)00237-0. [DOI] [PubMed] [Google Scholar]
- 47.Zimmer A, Zimmer AM, Baffi J, Usdin T, Reynolds K, Konig M, Palkovits M, Mezey E. Hypoalgesia in mice with a targeted deletion of the tachykinin 1 gene. Proc Natl Acad Sci USA. 1998;95:2630–2635. doi: 10.1073/pnas.95.5.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]