

Abstract
Transient forebrain ischemia produced by four-vessel occlusion (4-VO) triggers the delayed death of CA1 neurons in the hippocampus, resulting in behavioral deficits of spatial learning performance. We demonstrate that CA1 neuronal loss induced by 4-VO (12 min) is preceded by a selective and marked elevation of catalytically active caspase-3 in these neurons, indicative of apoptosis. Virally mediated overexpression of the anti-apoptotic gene X chromosome-linked inhibitor of apoptosis protein (XIAP) prevented both the production of catalytically active caspase-3 and degeneration of CA1 neurons after transient forebrain ischemia. CA1 neurons protected in this manner appeared to function normally, as assessed by immunohistochemical detection of the neuronal activity marker nerve growth factor inducible-A and by spatial learning performance in the Morris water maze. These findings indicate that caspase-3 activation is a key event in ischemic neuronal death and that blockade of this event by XIAP overexpression permits CA1 neurons to survive and operate properly after an ischemic insult.
Keywords: XIAP, caspase-3, DNA fragmentation, water maze, apoptosis, NGFI-A
Accumulating evidence indicates that apoptosis contributes to neuronal cell death in a variety of neurodegenerative contexts (Choi, 1996; Rinkenberger and Korsmeyer, 1997). Recently, a provocative link between developmental apoptosis and neurodegenerative disease has been shown for acute spinal muscular atrophy (SMA) by the identification of a candidate gene encoding neuronal apoptosis inhibitor protein (NAIP) (Roy et al., 1995). Disruption of NAIP function is thought to precipitate SMA by permitting unrestrained developmental apoptosis in a variety of sensory and motor systems. In support of this hypothesis, we have observed a striking overlap between neuronal populations lost in SMA and the distribution of NAIP immunoreactivity in the rodent CNS (Xu et al., 1997b). We have also demonstrated that treatments that elevate NAIP expression prevent CA1 neuron loss after transient forebrain ischemia (Xu et al., 1997a).
The molecular mechanisms by which NAIP blocks apoptosis are at least partly dependent on three conserved domains, ∼45–60 amino acids in length, known as baculoviral-inhibitor of apoptosis repeats (BIRs), which are shared by all four members of the inhibitor of apoptosis protein (IAP) family [NAIP, X chromosome-linked IAP (XIAP), human IAP-1 (HIAP-1), and HIAP-2] (Liston et al., 1996). Overexpression of each member of the IAP family or just the BIR domains found in these proteins is sufficient to block apoptosis in a variety of cell lines produced by several different triggers (Liston et al., 1996; Deveraux et al., 1997; Roy et al., 1997). Recent evidence suggests that the IAPs may prevent apoptosis by inhibiting caspase-3 activity (Deveraux et al., 1997; Roy et al., 1997).
Proteolytic activation of the cysteine protease caspase-3 appears to be a key event in the execution of programmed cell death (apoptosis) in the CNS (Johnson et al., 1996; Alnemri, 1997; Nicholson and Thornberry, 1997). The lack of programmed cell death in caspase-3 knock-out mice suggests that this protease may participate in neuronal apoptosis during CNS development (Kuida et al., 1996). Recent evidence suggests caspase-3 may participate in ischemic–excitotoxic brain damage (Yakovlev et al., 1997; Chen et al., 1998; Endres et al., 1998; Namura et al., 1998). In the present study, we examined the production of conformationally active caspase-3 and DNA fragmentation in degenerating CA1 neurons after transient cerebral ischemia to study the relationship between ischemia-induced caspase-3 activation and apoptosis. Given that XIAP has been shown to be a potent inhibitor of both caspase-3 activation and apoptosis, we examined the effects of adenovirally mediated XIAP overexpression on the these events in CA1 neurons after a brief episode of forebrain ischemia. The functional status of CA1 neurons 7–14 d after transient global ischemia was assessed at both the cellular and behavioral levels by immunohistochemical detection of the neuronal activity marker nerve growth factor inducible-A (NGFI-A) in CA1 neurons and measurement of spatial learning performance in the Morris water maze, respectively. Previous studies have shown that a brief period of global forebrain ischemia severely suppresses both of these measures of normal CA1 synaptic activity (Auer et al., 1989; Kiessling et al., 1993; Dragunow et al., 1994; Olsen et al., 1994; McGahan et al., 1998).
MATERIALS AND METHODS
Transient forebrain ischemia model. All animal procedures conformed to the Guide for the Care and Use of Experimental Animals (Olfert et al., 1993) endorsed by the Medical Research Council of Canada. Transient forebrain ischemia was produced using published modifications of the four-vessel occlusion (4-VO) method (Pulsinelli and Buchan, 1988). Adult male Wistar rats (250–275 gm; Charles River, Montréal, Canada) were anesthetized using 1.5% halothane 1 d before ischemia. The alar foraminae of the first cervical vertebra were then approached through a dorsal midline neck incision. The vertebral arteries were permanently occluded by electrocauterization. The carotid arteries were isolated through a ventral midline neck incision, and silk ligatures were placed loosely around them. The incisions were then closed with topical xylocaine jelly. The next day, rats were randomly divided into two groups and subjected to either 12 min of forebrain ischemia (experimental) or manipulation without carotid occlusion (sham). Forebrain ischemia was achieved by lifting the common carotid arteries using the silk ligatures placed around them and occluding the vessels with microaneurysm clips (#160–863; George Tiemann & Co., Plainview, NY). Brain temperature was measured indirectly via a thermocouple probe placed in the temporalis muscle. Body temperature was maintained between 36 and 37°C by external warming. Ischemia was terminated by removal of the microaneurysm clips. Animals that convulsed after reperfusion were omitted from the study, along with those that did not develop fully dilated pupils or failed to lose righting reflex during ischemia. In the case of sham-treated animals, the carotids were exposed but not occluded.
Stereotaxic injection of adenoviral constructs. Viral constructs have been described previously by Liston et al. (1996). Adenoviral vectors (3 μl injected over 10 min; 1 × 106/μl particles) were injected into the dorsal hippocampus using a 28 gauge needle at the following coordinates (in mm): anteroposterior, −3.6; mediolateral, ±2.2 (left and right sides); and dorsoventral, −3.2 from bregma (Paxinos and Watson, 1986).
Antibodies, immunohistochemistry, cell counting, and in situ end labeling. The antibody for catalytically active caspase-3 was raised in rabbits against the (p17/p12)2 conformer corresponding to the catalytically competent enzyme. The specificity of this antisera has been confirmed by immunoprecipitation of active caspase from multiple cell lines induced to undergo apoptosis by a variety of triggers (Rasper et al., 1998). Fresh frozen sections (12-μm-thick) were incubated with PBS containing 0.1% Triton X-100. Next, the sections were incubated with primary antibody (1:2000) for 48 hr at 4°C. After three washes with PBS (5 min each), the sections were incubated with CY3-labeled donkey anti-rabbit IgG (1:500; Amersham, Buckinghamshire, UK) for 2 hr at room temperature. For double immunofluorescence labeling, biotinylated acetyl-aspartyl-glutamyl-valyl-aspart-1-aldehyde (DEVD-CHO) (1:1000; Biomol, Plymouth Meeting, PA) and FITC-labeled streptavidin (1:50; Amersham) were coincubated with the primary and secondary antibodies, respectively. Immunohistochemical detection of XIAP (1:1000), β-galactosidase (1:1000), neuronal nuclei (NeuN) (1:200), or NGFI-A (1:15000) were performed on free-floating sections (12-μm-thick) cut from brain tissue that had been fixed with paraformaldehyde as described previously (Xu et al., 1997a). The XIAP antibody is an affinity-purified rabbit polyclonal antisera raised against recombinant human XIAP. The specificity of this antibody has been confirmed by Western blotting (Li et al., 1998). A monoclonal antibody for β-galactosidase was obtained from Promega (Madison, WI). NeuN was detected using a monoclonal antibody (also referred to as A60; generously provided by Dr. R. J. Mullen, University of Utah School of Medicine, Salt Lake City, UT) (Mullen et al., 1992). NGFI-A was detected using a polyclonal rabbit antibody (generously provided by Dr. R. Bravo, Bristol-Myers Squibb Pharmaceutical Research Institute, Princeton, NJ). Ischemia-induced DNA fragmentation was assessed using in situ end labeling (ISEL) as described by Xu et al. (1997a). Cell counts were performed manually on randomly selected sections by an observer who was unaware of the treatment conditions. Cells that displayed fragmented DNA or immunoreactivity for NeuN, NGFI-A, or active caspase-3 were counted in the medial, intermediate, and lateral regions of CA1 (400 μm each in length) of the hippocampus using computer-assisted image analysis. Sections cut at levels 2.8, 3.6, and 4.0 mm posterior to bregma (two sections per level) were analyzed for each animal. Neuronal density (cells/100 μm of CA1 layer) was calculated by averaging these values.
Morris water maze. The task for all of the animals in each trial consisted of finding a hidden clear plastic platform (∼18 cm in diameter) that was placed 50 cm away from the wall of the water maze (170 cm in diameter, 90 cm in depth) and 3 cm below the water. The platform remained in the same position for all sessions and trials. The starting quadrant was randomized every day, with all animals using the same order. The animals were faced toward the pool wall before being released. The time for them to reach the hidden platform was recorded. The animals were allowed to rest 60 sec on the platform between trials. If an animal failed to reach the platform in 120 sec, it was manually guided to the platform. Before surgery, all animals were pretrained. Each animal received four trials per day for four consecutive days. These animals then underwent stereotaxic injections of virus, followed by 4-VO surgery a week later. One week after 4-VO surgery, all the animals were examined as in the pretraining, except that on the last day they were tested with the platform raised 2 cm above the surface of the water and wrapped in black. This raised platform session was performed to determine whether factors other than learning and memory were responsible for deficits in spatial navigation.
Statistical analysis. A one-way ANOVA was performed on the cell count and behavioral data. If the analysis was significant, multiple comparisons were performed using Tukey’s test.
RESULTS
Transient cerebral ischemia selectively activates caspase-3 in CA1 neurons
To investigate the role of caspase-3 in ischemia-induced CA1 neuronal degeneration, an antibody that selectively recognizes conformationally mature caspase-3 [(p17/p12)2 tetramer] (Rasper et al., 1998) was used to detect catalytically active caspase-3 in hippocampal tissue sections from animals subjected to a brief episode of global ischemia (12 min) produced by 4-VO. Catalytically active caspase-3 was not present in CA1 neurons of animals subjected to sham 4-VO surgery (Fig.1A) or killed 3 hr after 4-VO (data not shown). Significant levels of (p17/p12)2 immunoreactivity were apparent in CA1 pyramidal neurons 24 hr after transient forebrain ischemia. At this time point, (p17/p12)2 immunoreactivity was confined to the cytoplasm of most CA1 neurons; however, a small number of CA1 neurons situated close to midline displayed intense nuclear labeling (Fig.1B). Nuclear labeling appeared to increase in medial portions of the CA1 region at 48 hr (Fig. 1C). In contrast, (p17/p12)2 immunoreactivity in lateral aspects of the CA1 region remained moderate and was primarily confined to the cytoplasm of pyramidal neurons (Fig. 1D). By 72 hr, intense nuclear labeling was frequently observed in pyramidal neurons located throughout the entire CA1 subfield (Fig.1E,F). However, few (p17/p12)2-positive CA1 neurons were seen 120 hr (5 d) after transient forebrain ischemia (data not shown). At no time point was caspase-3 activation detected in CA2 or CA3 sectors of the dorsal hippocampus. Similar results were obtained in adjacent sections using biotinylated DEVD-CHO to label conformationally mature caspase-3. Moreover, double staining demonstrated that the same CA1 neurons were labeled with the (p17/p12)2-specific antibody (Fig.1G) and biotinylated DEVD-CHO (Fig. 1H), suggesting that both methods specifically label catalytically active caspase-3. Together, these findings indicate that caspase-3 activation precedes the delayed death of CA1 neurons after an episode of transient forebrain ischemia.
Fig. 1.
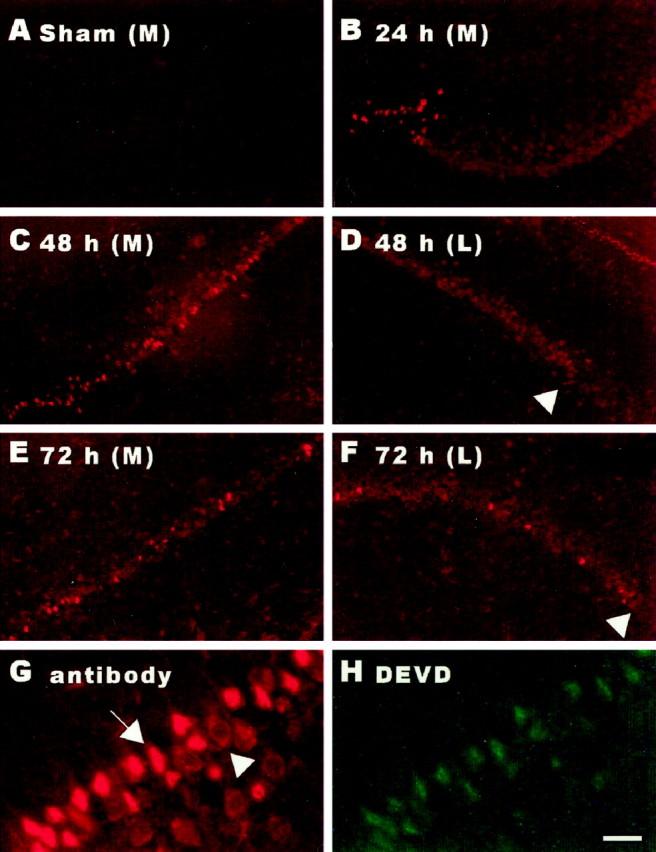
Cerebral ischemia activates caspase-3 in CA1 neurons. Brain sections from animals exposed to 12 min of 4-VO were processed for immunohistochemical detection of conformationally active caspase-3. Catalytically active caspase-3 was not detected in the hippocampal CA1 subfield of animals exposed to sham 4-VO (A). At 24 hr after 4-VO, caspase-3 activation was seen in CA1 pyramidal neurons. The majority of CA1 neurons displayed moderate labeling, whereas a small population of neurons situated close to midline exhibited intense nuclear staining (B). At 48 hr, a larger number of neurons in the medial CA1 were intensely labeled (C). Neurons in the lateral CA1 remained moderately labeled (D,arrowhead, CA2). By 72 hr, intense nuclear labeling was frequently observed throughout the entire CA1 subfield (E, medial CA1; F, lateral CA1;F, arrowhead, CA2). Immunoreactivity for conformationally active caspase-3 was restricted to the CA1 subfield (D, F,arrowheads, CA2). The (p17/p12)2tetramer-specific antibody (G) and biotinylated DEVD (H) labeled the same neurons. Note moderate cytoplasmic (arrowhead) versus intense nuclear labeling (arrow) (G). Scale bar:A–F, 200 μm; G, H, 50 μm.
Activation of caspase-3 precedes DNA fragmentation
The relationship between caspase-3 activation and apoptosis in CA1 neurons after transient forebrain ischemia was assessed by examining the appearance of fragmented DNA in CA1 neurons by in situend labeling in tissue sections adjacent to those processed for catalytically active caspase-3 (Fig. 2). ISEL staining was first observed in medial CA1 neurons 48 hr after transient cerebral ischemia (Fig. 2C). ISEL was not seen in lateral aspects of the CA1 region until 72 hr after 4-VO (Fig.2E,F). At 120 hr, intense ISEL was detected in the majority of CA1 neurons (Fig.2G,H). Although the time course for DNA fragmentation clearly occurs later than that for conformationally mature caspase-3, the two events compliment each other both temporally and spatially.
Fig. 2.
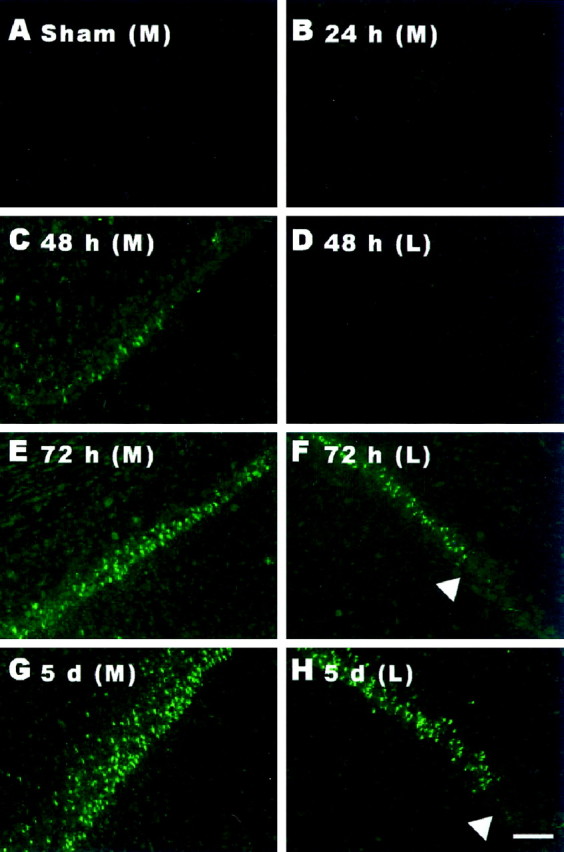
Cerebral ischemia causes DNA fragmentation in CA1 neurons. Ischemia-induced DNA fragmentation was assessed usingin situ end labeling as described by Xu et al. (1997a). Labeling was not seen in the CA1 subfield of animals exposed to sham 4-VO (A) or those examined 24 hr after 4-VO (B). At 48 hr after 4-VO, ISEL was detected in a small group of CA1 pyramidal neurons located proximal to midline (C). No labeling was seen in lateral CA1 (D). By 72 hr, ISEL was frequently observed in neurons of both medial (E) and lateral aspects of CA1 (F, arrowhead, CA2). At 5 d, the majority of neurons in the CA1 region displayed intense labeling (G, medial; H, lateral; H,arrowhead, CA2). ISEL was restricted to the CA1 region (H). Scale bar, 200 μm.
XIAP overexpression reduces ischemia-induced caspase-3 activation and apoptosis in CA1 neurons
XIAP blocks apoptosis induced by a variety of triggers in several different cell lines (Liston et al., 1996). At least part of this anti-apoptotic activity appears to be mediated by the inhibition of conformationally mature caspase-3 (Deveraux et al., 1997). We have recently shown that NAIP, another member of the human IAP family, attenuates the loss of CA1 neurons after a brief period of cerebral ischemia (Xu et al., 1997a). These observations suggest that IAPs may reduce ischemic damage by blocking caspase-3 activation in CA1 neurons. To test this hypothesis, we used an adenoviral construct capable of overexpressing XIAP in CA1 neurons. This adenoviral construct was injected unilaterally into the dorsal hippocampus. To control for effects of the adenoviral construct that were unrelated to XIAP overexpression, an adenoviral construct encoding bacterial β-galactosidase (lacZ) was injected into the contralateral hippocampus. Virally mediated overexpression of lacZ and XIAP was confirmed by immunohistochemical detection of β-galactosidase and human XIAP, respectively (Fig.3A,B). All of the animals were subjected to 4-VO (12 min) 7 d after stereotaxic surgery. In the dorsal hippocampus, which received the lacZ construct, a large number of (p17/p12)2-positive CA1 neurons (17 ± 3/100 μm) were detected 48 hr after the ischemia insult (Fig. 3C). In contrast, very few (p17/p12)2-immunoreactive neurons (2 ± 1/100 μm) were present in the contralateral hippocampus, which had been injected with the XIAP construct (Fig. 3D). These results suggest that XIAP overexpression attenuates ischemia-induced caspase-3 activation.
Fig. 3.
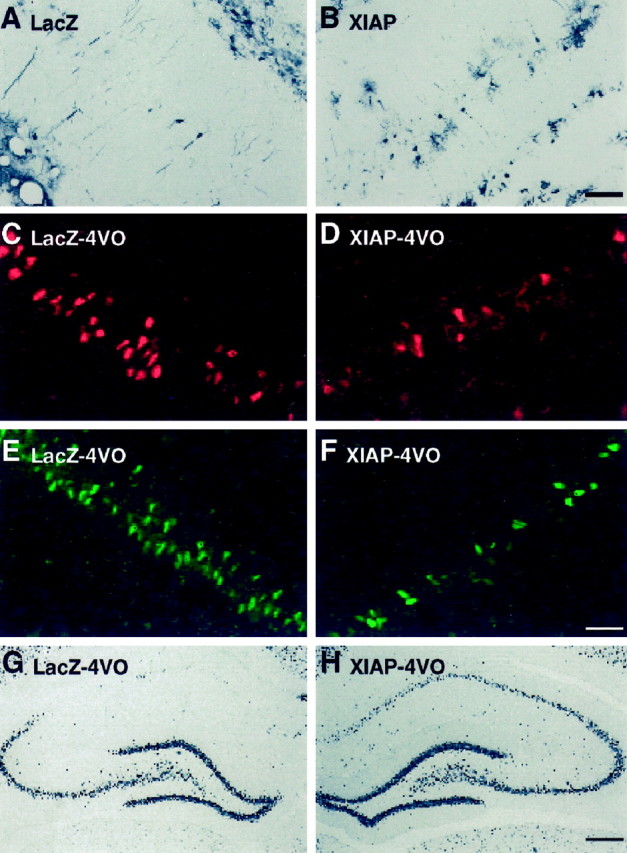
Attenuation of ischemia-induced caspase-3 activation and apoptotic death in CA1 neurons by XIAP overexpression. Animals received intrahippocampal injections of adenoviral constructs containing either lacZ (left side) or XIAP (right side) and were subjected to 4-VO or sham surgery 5 d later. Virally mediated overexpression of lacZ and XIAP was confirmed by immunohistochemical detection of β-galactosidase and human XIAP, respectively. The majority of immunoreactive cells in the CA1 layer appeared to be pyramidal neurons. However, a small number of glial cells were also infected with the viral constructs. Scale bar (in B):A, B, 75 μm. In animals that received intrahippocampal injections of the adenoviral constructs followed by sham 4-VO (n = 4), approximately the same number of CA1 neurons were immunoreactive for β-galactosidase (9 ± 2) and human XIAP (10 ± 3). Levels of catalytically active caspase-3 were assessed in CA1 neurons by immunohistochemical detection of the (p17/p12)2 tetramer 48 hr after 4-VO (n= 4) (C, D). In the hippocampus injected with the lacZ construct, a large number of neurons were stained in the medial aspect of CA1 (C). In contrast, very few immunoreactive neurons were present in the contralateral hippocampus that had been injected with the XIAP construct (D). DNA fragmentation was examined 5 d after 4-VO in animals injected with these viral constructs. A larger number of ISEL-positive CA1 neurons were observed in the lacZ-injected side (E). In contrast, considerably fewer ISEL-positive neurons were detected in the XIAP-injected side (F). Scale bar (in F):C–F, 75 μm. Immunohistochemistry for NeuN was performed to stain surviving neurons 7 d after 4-VO (12 min). A small number of neurons was observed in the CA1 subfield injected with the lacZ construct (G), whereas the majority of neurons in the XIAP-injected side remained NeuN-positive (H). Scale bar (inH): G, H, 400 μm.
We next determined whether XIAP overexpression also reduces ischemia-induced DNA fragmentation in CA1 neurons. Six animals that had received intrahippocampal injections of the lacZ (left side) and XIAP (right side) constructs were subjected to 4-VO (12 min) and examined for ISEL 5 d later. A larger number of ISEL-positive CA1 neurons (16 ± 2/100 μm) were observed in the lacZ-injected side compared with the XIAP-injected side (3 ± 1/100 μm) (Fig.3E,F). Consequently, the ability of XIAP to block caspase-3 activation was associated with a reduction of DNA fragmentation in CA1 neurons.
Sections adjacent to those used for ISEL labeling were processed for immunohistochemical detection of NeuN, a neuron-specific vital marker (Mullen et al., 1992). In agreement with the inhibitory effects of XIAP overexpression on caspase-3 activation and DNA fragmentation, there was a dramatic reduction in the loss of CA1 neurons after a brief episode of transient forebrain ischemia (Fig.3G,H). Cell counts revealed that considerably more CA1 neurons remained in the XIAP-injected side compared with the contralateral side, which had received the lacZ construct. Only 4 ± 1 neurons/100 μm length of the CA1 region were NeuN-positive in the lacZ-injected side. A consistent loss of CA1 neurons was observed throughout the medial to lateral extent of the dorsal hippocampus. In comparison, 16 ± 2 neurons were detected in the CA1 region (100 μm length) of the XIAP-injected side. These findings indicate that XIAP overexpression produced a fourfold elevation of neuronal survival after global ischemia.
XIAP overexpression attenuates ischemia-induced behavioral and cellular deficits
To assess the functional status of CA1 neurons that survived the ischemic episode, we examined spatial learning performance using the Morris water maze. In agreement with previous reports (Olsen et al., 1994), we observed that exposure to a brief period of global forebrain ischemia significantly impaired spatial learning in animals that received bilateral intrahippocampal injections of the lacZ construct (Fig. 4A). In contrast, animals injected bilaterally with the XIAP construct and subjected to transient forebrain ischemia (XIAP–4-VO) exhibited the same short latencies as sham-operated animals (lacZ–sham). Accordingly, the performance of XIAP–4-VO animals was superior to that of the lacZ–4-VO group. Tissue sections from the dorsal hippocampus of each animal were processed for immunohistochemical detection of NeuN after the water maze task. Consistent with the behavioral results, adenovirally mediated XIAP overexpression significantly reduced the loss of CA1 neurons 14 d after an episode of transient global ischemia (Fig. 4B). This finding suggests that CA1 neurons protected from ischemic injury by XIAP overexpression are functionally normal.
Fig. 4.

XIAP overexpression prevents deficits in spatial learning and neuronal activity after 4-VO. Spatial learning performance in the Morris water maze (A). Three groups, composed of nine animals each, were pretrained to find a submerged platform over the course of 4 d (4 trials/d). All three groups of animals displayed similar latencies to reach the platform (pretraining). Next, two of the three groups received bilateral intrahippocampal injections of either the lacZ (LacZ-4VO) or XIAP (XIAP-4VO) adenoviral constructs and were exposed to transient global ischemia (12 min) 7 d later. The third group, which received bilateral intrahippocampal injections of the lacZ virus and were subjected to sham 4-VO, served as controls (LacZ-sham). Compared with the lacZ-sham group, animals in the lacZ–4-VO group took considerably longer to find the submerged platform over the course of 3 d (testing) (p < 0.01). In contrast, XIAP–4-VO animals exhibited the same short latencies as the lacZ-sham group (p > 0.05). Furthermore, the performance of XIAP–4-VO animals was superior to that of the lacZ–4-VO group (p < 0.01). All three groups performed equally on the raised platform test (cued) (p > 0.05). Quantification of NeuN- and NGFI-A-immunoreactive CA1 neurons (B, C). Cell counts of NeuN-immunoreactive neurons revealed that the average neuronal density for the lacZ–sham, lacZ–4-VO, and XIAP–4-VO groups was 24 ± 1, 2.6 ± 0.7, and 14 ± 2 (cells/100 μm), respectively (B). Although fewer in number, similar results were obtained for NGFI-A-immunoreactive neurons (C). The average density of NGFI-A-immunoreactive neurons for the lacZ–sham, lacZ–4-VO, and XIAP–4-VO groups was 16 ± 1.3, 1 ± 0.5, and 11 ± 2, respectively. Statistical analysis revealed that, for both NeuN and NGFI-A immunoreactivity, significantly more labeled neurons were present in the XIAP–4-VO than lacZ–4-VO group (p < 0.01). ★p < 0.01 relative to lacZ–sham and XIAP–4-VO; *p < 0.01 relative to lacZ–sham and lacZ–4-VO. Immunohistochemical detection of NGFI-A (D). Top, LacZ–sham;middle, lacZ–4-VO; bottom, XIAP–4-VO.
To address the functional status of CA1 neurons at the cellular level, we examined expression of the neuronal activity marker NGFI-A. We have demonstrated previously that similar results are obtained with NeuN-, NGFI-A-, and Nissl-stained sections (Xu et al., 1997a; McGahan et al., 1998; our unpublished observations). Basal expression of NGFI-A is driven by natural synaptic activity (Worley et al., 1991) and is severely depressed in CA1 neurons after an episode of global forebrain ischemia (Kiessling et al., 1993; Dragunow et al., 1994;Olsen et al., 1994; McGahan et al., 1998). In keeping with performance in the Morris water maze, XIAP overexpressing CA1 neurons that survived the ischemic insult displayed considerably higher levels of NGFI-A than animals injected with the lacZ construct (Fig.4C,D). The ability of XIAP overexpression to maintain high basal levels of NGFI-A expression in CA1 neurons after the ischemic insult is further evidence that these neurons function properly.
DISCUSSION
Caspase-3 mediates ischemic neuronal apoptosis of CA1 neurons
We have demonstrated that catalytically active caspase-3 is generated in CA1 neurons that are undergoing ischemic cell death. This finding is in agreement with recent reports demonstrating cleavage of caspase-3 and increased enzymatic activity of this protease in ischemic brain tissue after transient cerebral ischemia (Chen et al., 1998;Namura et al., 1998). Furthermore, our observation that ischemia-induced caspase-3 activation precedes DNA fragmentation in vulnerable CA1 neurons suggests that these events may be linked. The pathway by which caspase-3 activation leads to DNA fragmentation in neurons is unclear. However, a caspase-3-activated deoxyribonuclease, termed CAD, has been identified, which causes the degradation of chromosomal DNA into nucleosomal units characteristic of apoptosis (Enari et al., 1998). In normal cells, CAD activity is suppressed by binding to its inhibitor ICAD. Catalytically active caspase-3 cleaves ICAD, permitting CAD to enter the nucleus and commence DNA fragmentation (Sakahira et al., 1998). Several nuclear enzymes responsible for genomic integrity, such as poly(ADP-ribose) polymerase and DNA-dependent protein kinase, are also cleaved by caspase-3 (Nicholson et al., 1995). The preferential localization of conformationally mature caspase-3 in the nuclei of degenerating CA1 neurons is therefore compatible with a role for this protease in DNA fragmentation.
XIAP attenuates caspase-3-mediated ischemic apoptosis of CA1 neurons
X chromosome inhibitor of apoptosis is a member of the human inhibitor of apoptosis family. XIAP overexpression prevents apoptosis induced by a variety of triggers in several different cell lines (Liston et al., 1996). We have shown previously that virally mediated overexpression of another member of the IAP family, NAIP, in the rat hippocampus attenuates ischemic neuronal loss in CA1 by blocking apoptosis (Xu et al., 1997a). The anti-apoptotic property of the IAPs has been attributed to the ability of these proteins to selectively inhibit the activity of group II caspases (3 and 7) (Deveraux et al., 1997; Roy et al., 1997). In keeping with this proposal, we have demonstrated that attenuation of ischemia-induced CA1 neuronal loss by overexpression of XIAP in the hippocampus is associated with a reduction of caspase-3 activation and DNA fragmentation. Because XIAP inhibits caspase-3 activity, it is therefore likely that XIAP overexpression may prevent ischemic neuronal apoptosis by blocking caspase-3 activation.
In contrast to XIAP, peptide inhibitors such as DEVD-CHO and benzyloxycarbonyl-Val-Ala-Asp(ome)-fluoromythylketone discriminate poorly between group I (caspase-1, -4, and -5), II (caspase-2, -3, and -7), and III (caspase-6, -8, and -9) caspases (Nicholson and Thornberry, 1997). As a result, it is unclear whether the neuroprotective properties of these compounds stem from the inhibition of a single or multiple caspase subtypes. The ability of XIAP, a selective group II inhibitor, to reduce CA1 injury highlights the importance of caspase-3 as a mediator of ischemic neuronal death and suggests that small molecules that selectively inhibit this protease should be neuroprotective.
XIAP overexpression attenuates ischemia-induced cellular and behavioral deficits
CA1 neurons of the hippocampus play a key role in spatial learning, as assessed by the Morris water maze. Consequently, ischemic damage to the hippocampus causes deficits in spatial learning (Olsen et al., 1994). Consistent with this finding, we have observed that the loss of CA1 neurons produced by a brief episode of transient cerebral ischemia was associated with impaired performance in the Morris water maze. XIAP overexpression, which reduced the injurious effects of cerebral ischemia on CA1 neurons, preserved spatial navigation in the Morris water maze. Indeed, the performance of these animals was comparable with that of sham-treated rats. These findings suggest that neurons protected from ischemic injury by XIAP overexpression retain normal physiological function.
Neuronal expression of the immediate early gene NGFI-A in CA1 neurons is driven by natural synaptic activity. This has lead to the suggestion that NGFI-A expression may be a marker for neuronal activity in the brain (Worley et al., 1991). Similar to spatial learning performance, a behavioral measurement of neuronal function, expression of NGFI-A in CA1 neurons was severely depressed by a brief episode of cerebral ischemia in CA1 neurons. Overexpression of XIAP in these neurons attenuated ischemia-induced reduction in NGFI-A expression. This finding provides evidence that XIAP overexpression maintains cellular homeostatsis after an experimental stroke.
In summary, the present study provides evidence that CA1 neurons undergoing delayed cell death after an episode of transient forebrain ischemia generate catalytically active caspase-3. Caspase-3 activation preceded DNA fragmentation in CA1 neurons, suggesting that this enzyme participates in the execution of ischemia-induced neuronal apoptosis. In support of this proposal, blockade of caspase-3 activation by overexpression of XIAP, a potent caspase-3 inhibitor, attenuated ischemic neuronal death in the CA1 region. Moreover, XIAP overexpression appeared to maintain normal neuronal function after an episode of cerebral ischemia. These findings suggest that therapeutic strategies based on XIAP elevation or small molecule inhibitors of caspase-3 may be useful in the treatment of stroke and possibly other acute neurodegenerative disorders.
Footnotes
This work was supported by Medical Research Council of Canada Grant MT-11539 and Heart and Stroke Foundation of Ontario Grant NA-2938.
Correspondence should be addressed to G. S. Robertson, Department of Pharmacology, Merck Frosst Centre for Therapeutic Research, 16711 Trans Canada Highway, Kirkland, Quebec H9H 3L1, Canada.
REFERENCES
- 1.Alnemri ES. Mammalian cell death proteases: a family of highly conserved aspartate specific cysteine proteases. J Cell Biochem. 1997;64:33–42. doi: 10.1002/(sici)1097-4644(199701)64:1<33::aid-jcb6>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 2.Auer RN, Jensen ML, Whishaw IQ. Neurobehavioral deficit due to ischemic brain damage limited to half of the CA1 sector of the hippocampus. J Neurosci. 1989;9:1641–1647. doi: 10.1523/JNEUROSCI.09-05-01641.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi DW. Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol. 1996;6:667–672. doi: 10.1016/s0959-4388(96)80101-2. [DOI] [PubMed] [Google Scholar]
- 5.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 6.Dragunow M, Beilharz E, Sirimanne E, Lawlor P, Williams C, Bravo R, Gluckman P. Immediate-early gene protein expression in neurons undergoing delayed death, but not necrosis, following hypoxic-ischaemic injury to the young rat brain. Brain Res Mol Brain Res. 1994;25:19–33. doi: 10.1016/0169-328x(94)90274-7. [DOI] [PubMed] [Google Scholar]
- 7.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 8.Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gomez-Isla T, Hyman BT, Moskowitz MA. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J Cereb Blood Flow Metab. 1998;18:238–247. doi: 10.1097/00004647-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Johnson EM, Jr, Deckwerth TL, Deshmukh M. Neuronal death in developmental models: possible implications in neuropathology. Brain Pathol. 1996;6:397–409. doi: 10.1111/j.1750-3639.1996.tb00872.x. [DOI] [PubMed] [Google Scholar]
- 10.Kiessling M, Stumm G, Xie Y, Herdegen T, Aguzzi A, Bravo R, Gass P. Differential transcription and translation of immediate early genes in the gerbil hippocampus after transient global ischemia. J Cereb Blood Flow Metab. 1993;13:914–924. doi: 10.1038/jcbfm.1993.114. [DOI] [PubMed] [Google Scholar]
- 11.Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Kim JM, Liston P, Miyazaki T, Mackenzie AE, Korneluk RG, Tsang BK. Expression of inhibitor of apoptosis proteins (IAPs) in rat granulosa cells during ovarian follicular development and atresia. Endocrinology. 1998;139:1321–1328. doi: 10.1210/endo.139.3.5850. [DOI] [PubMed] [Google Scholar]
- 13.Liston P, Roy N, Tamai K, Lefebvre C, Baird S, Cherton-Horvat G, Farahani R, McLean M, Ikeda JE, MacKenzie A, Korneluk RG. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature. 1996;379:349–353. doi: 10.1038/379349a0. [DOI] [PubMed] [Google Scholar]
- 14.McGahan L, Hakim AM, Nakabeppu Y, Robertson GS. Ischemia-induced CA1 neuronal death is preceded by elevated FosB and jun expression and reduced NGFI-A and JunB levels. Brain Res Mol Brain Res. 1998;56:146–161. doi: 10.1016/s0169-328x(98)00039-4. [DOI] [PubMed] [Google Scholar]
- 15.Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- 16.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 18.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Raju SM, Smulson ME, Yamin T-T, Yu VL, Miller DK. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 19. Olfert ED, Cross BM, McWilliam AA. 1993. Guide to the care and use of experimental animals. Ottawa, Canada: Canadian Council on Animal Care
- 20.Olsen GM, Scheel-Kruger J, Moller A, Jensen LH. Does neuronal damage of CA1 relate to spatial memory performance of rats subjected to transient forebrain ischemia? Acta Neurol Scand. 1994;89:204–209. doi: 10.1111/j.1600-0404.1994.tb01662.x. [DOI] [PubMed] [Google Scholar]
- 21.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; New York: 1986. [DOI] [PubMed] [Google Scholar]
- 22.Pulsinelli WA, Buchan AM. The four-vessel occlusion rat model: method for complete occlusion of vertebral arteries and control of collateral circulation. Stroke. 1988;19:913–914. doi: 10.1161/01.str.19.7.913. [DOI] [PubMed] [Google Scholar]
- 23.Rasper D, Vaillancourt J, Hadano S, Houtzager V, Seiden I, Keen S, Tawa P, Xanthoudakis S, Nasir J, Martindale D, Koop B, Peterson E, Thornberry N, Huang J-Q, MacPherson D, Black S, Hornung F. Cell death attenuation by ’Usurpin’, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ. 1998;5:271–288. doi: 10.1038/sj.cdd.4400370. [DOI] [PubMed] [Google Scholar]
- 24.Rinkenberger JL, Korsmeyer SJ. Errors of homeostasis and deregulated apoptosis. Curr Opin Genet Dev. 1997;7:589–596. doi: 10.1016/s0959-437x(97)80004-4. [DOI] [PubMed] [Google Scholar]
- 25.Roy N, Mahadevan MS, McLean M, Shutler G, Yaraghi Z, Farahani R, Baird S, Besner-Johnston A, Lefebvre C, Kang X, Salih M, Aubry H, Tamai K, Guan X, Ioannou P, Crawford TO, de Jang PJ, Surh L, Ikeda J-E, Korneluk RG, MacKenzie A. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell. 1995;80:167–178. doi: 10.1016/0092-8674(95)90461-1. [DOI] [PubMed] [Google Scholar]
- 26.Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 1997;16:6914–6925. doi: 10.1093/emboj/16.23.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- 28.Worley PF, Christy BA, Nakabeppu Y, Bhat RV, Cole AJ, Baraban JM. Constitutive expression of zif268 in neocortex is regulated by synaptic activity. Proc Natl Acad Sci USA. 1991;88:5106–5110. doi: 10.1073/pnas.88.12.5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu DG, Crocker SJ, Doucet JP, St-Jean M, Tamai K, Hakim AM, Ikeda JE, Liston P, Thompson CS, Korneluk RG, MacKenzie A, Robertson GS. Elevation of neuronal expression of NAIP reduces ischemic damage in the rat hippocampus. Nat Med. 1997a;3:997–1004. doi: 10.1038/nm0997-997. [DOI] [PubMed] [Google Scholar]
- 30.Xu DG, Korneluk RG, Tamai K, Wigle N, Hakim A, Mackenzie A, Robertson GS. Distribution of neuronal apoptosis inhibitory protein-like immunoreactivity in the rat central nervous system. J Comp Neurol. 1997b;382:247–259. [PubMed] [Google Scholar]
- 31.Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]