

Abstract
Brain-derived neurotrophic factor (BDNF) promotes long-term potentiation (LTP) at hippocampal CA1 synapses by a presynaptic enhancement of synaptic transmission during high-frequency stimulation (HFS). Here we have investigated the mechanisms of BDNF action using two lines of BDNF knockout mice. Among other presynaptic impairments, the mutant mice exhibited more pronounced synaptic fatigue at CA1 synapses during high-frequency stimulation, compared with wild-type animals. Quantitative analysis of CA1 synapses revealed a significant reduction in the number of vesicles docked at presynaptic active zones in the mutant mice. Synaptosomes prepared from the mutant hippocampus exhibited a marked decrease in the levels of synaptophysin as well as synaptobrevin [vesicle-associated membrane protein (VAMP-2)], a protein known to be involved in vesicle docking and fusion. Treatment of the mutant slices with BDNF reversed the electrophysiological and biochemical deficits in the hippocampal synapses. Taken together, these results suggest a novel role for BDNF in the mobilization and/or docking of synaptic vesicles to presynaptic active zones.
Keywords: BDNF, knockout mice, hippocampus, synaptic fatigue, vesicle docking, synaptobrevin, synaptophysin
Neurotrophins were originally identified as a family of protein factors that regulate neuronal survival and differentiation. Recent studies have indicated that neurotrophins also play an important role in the development and function of synapses (Lo, 1995; Thoenen, 1995; Bonhoeffer, 1996; Lu and Figurov, 1997). The function of brain-derived neurotrophic factor (BDNF) in synaptic transmission and plasticity in the hippocampus has been the focus of extensive investigation. Both expression and release of BDNF in the hippocampus are enhanced by neuronal activity and excitatory synaptic transmission (Zafra et al., 1990; Isackson et al., 1991; Patterson et al., 1992; Castren et al., 1993; Goodman et al., 1996; Canossa et al., 1997). Acute application of exogenous BDNF enhances neuronal activity and synaptic transmission in embryonic hippocampal neurons in primary cultures (Knipper et al., 1994; Lessmann et al., 1994; Levine et al., 1995), suggesting that BDNF is capable of modulating synaptic function in the hippocampus. However, whether BDNF enhances excitatory synaptic transmission in acute hippocampal slices remains controversial (Kang and Schuman, 1995; Figurov et al., 1996;Patterson et al., 1996; Tanaka et al., 1997; Frerking et al., 1998;Gottschalk et al., 1998; Huber et al., 1998). Much more consistent results have been obtained in elucidating the role of BDNF in the hippocampal long-term potentiation (LTP). Application of exogenous BDNF facilitates LTP induction in neonatal hippocampal slices (Figurov et al., 1996), in which the endogenous BDNF levels are low (Maisonpierre et al., 1990; Friedman et al., 1991). In contrast, treatment with TrkB-IgG, a fusion protein that scavenges endogenous BDNF, reduces the magnitude of LTP in adult hippocampus, in which the endogenous BDNF levels are high (Figurov et al., 1996; Yan et al., 1997).
In addition to pharmacological approaches, the role of BDNF in hippocampal synaptic plasticity has also been studied using BDNF knockout mice. Although the homozygous (−/−) mice exhibit growth retardation, sensory deficits, and impairments in coordination of movement, there is no obvious neuronal loss in the hippocampus (Ernfors et al., 1994; Jones et al., 1994). The heterozygous (+/−) mice show no signs of behavioral abnormalities. Two independent lines of BDNF knockout mice, however, have shown a severe impairment in hippocampal LTP in both −/− and +/− mice (Korte et al., 1995; Patterson et al., 1996). Moreover, +/− mice showed the same degree of impairment as the −/− mice, consistent with the idea that a critical level of BDNF in the hippocampus is important for LTP. This result also suggests that the observed defects in hippocampal LTP are unlikely to be caused by growth retardation, neuronal death, or dysfunction in other regions of the brain. The impairment in hippocampal LTP can be restored after incubation with recombinant BDNF for a few hours (Patterson et al., 1996), or by virus-mediated BDNF gene transfer (Korte et al., 1996), suggesting that a mutation of the BDNF gene per se, rather than cumulative developmental abnormalities, is responsible for impaired LTP in the BDNF knockout mice.
The mechanism by which BDNF regulates hippocampal synaptic plasticity remains unclear, although several lines of evidence support a presynaptic locus of action. Our previous study demonstrated that BDNF treatment significantly attenuates synaptic fatigue during a high-frequency tetanus in immature hippocampal slices (Figurov et al., 1996). Furthermore, manipulations known to reflect presynaptic function, such as paired-pulse facilitation (PPF), synaptic responses to high-frequency stimulation (HFS), and changes in extracellular Ca2+ concentration, revealed a presynaptic modulation of high-frequency transmission by BDNF (Gottschalk et al., 1998). To elucidate the cellular and molecular mechanisms of BDNF action, we have now investigated the properties of hippocampal CA1 excitatory synapses in BDNF knockout mice. We characterized the properties of hippocampal CA1 synapses, using electrophysiological approaches. We also performed a quantitative structural analysis of excitatory synapses on dendritic spines in the CA1 region. Furthermore, we determined the expression levels of several synaptic vesicle proteins in hippocampal synaptosomes from BDNF knockout mice. Finally, we investigated whether the synaptic deficits in BDNF knockout mice can be rescued by treatment of the mutant slices with recombinant BDNF. Our results suggest that impairments in high-frequency transmission in the mutant mice are likely caused by reduced synaptic vesicle docking at the active zones of CA1 synapses, possibly attributable to a decrease in synaptophysin and synaptobrevin levels at nerve terminals.
MATERIALS AND METHODS
BDNF knockout mice. Our BDNF knockout mice colony was raised from two pairs of BDNF +/− mice in C57BL/6 background (Ernfors et al., 1994), purchased from Jackson Laboratories (Bar Harbor, ME). These mice, referred to as BL/6/BDNF, have a low rate of pregnancy and a small number of pups per litter. Moreover, the nursing females are poor caretakers of their pups, resulting in a high rate of mortality, particularly for homozygotes. To raise the large number of mice necessary for biochemical studies, and to obtain enough littermates of the same gender from all three genotypes, we generated a new line of mice by crossing BL/6/BDNF +/− with CD1 mice. A BL/6/BDNF +/− male was crossed with a CD1 female. The F1 +/− mice were crossed among themselves. The F2 +/− were then used as founders to generate all of the mice used in the present study (referred to as CD1/BDNF). The CD1/BDNF mice behave similarly to BL/6/BDNF knockout animals (Ernfors et al., 1994; Jones et al., 1994). The −/− mice were usually smaller than the +/+ and +/− littermates and died within 1 month. They also showed defects in coordination of movement and balance, ataxia, spinning during periods of hyperactivity, and recurrent episodes of freezing seizures. Heterozygous mice were fertile and showed no overt abnormalities. Within each experiment (electrophysiology, electron microscopy, or biochemistry), only same gender, age-matched littermates were used. In experiments using all three genotypes, littermates of postnatal day (P) 25–44 were chosen because −/− animals did not live beyond that period. In experiments in which only +/+ and +/− genotypes were used, 2- to 3-month-old animals were chosen.
Electrophysiological recording. Transverse hippocampal slices (400 μm) were prepared from +/+, +/−, and −/− CD1/BDNF littermates (young adult, P25–44), or from +/+ and +/− BL/6/BDNF littermates (2–3 months old). The slices were maintained in an interface chamber for both recovery (2 hr) and recording, and exposed to an artificial atmosphere of 95% O2/5% CO2, as described previously (Figurov et al., 1996). Perfusion medium [artificial CSF (ACSF), 34°C] contained (in mm): NaCl 124, KCl 3.0, CaCl2 2.5, MgCl2 1.5, NaHCO3 26, KH2PO4 1.25, glucose 10, ascorbic acid 2, pH 7.4. Field EPSPs were evoked in CA1 stratum radiatumby stimulation of Schaffer collaterals with twisted bipolar nichrome electrodes and recorded with ACSF-filled glass pipettes (<5 MΩ) using an Axoclamp-2B amplifier. Test stimuli consisted of monophasic 200 μsec pulses of constant current delivered by stimulus isolation units. Basal synaptic transmission was monitored by alternating, low-frequency stimulation (two per minute) of two separate pathways via two stimulating electrodes (S1 and S2) positioned on both sides of the recording electrode. Only slices exhibiting EPSPs of 2–3 mV in amplitude without superimposed population spikes were used. Stimulus intensity was adjusted to evoke EPSPs of ∼1.3 mV. Input–output curves were obtained by plotting the stimulus voltages against the slopes of EPSPs. LTP was induced by 2 × 1 sec trains at 100 Hz separated by 20 sec using the same test stimulus intensity. Synaptic fatigue was induced by a 1 sec train at 100 Hz. Because the 100th EPSP was often too small to measure accurately, we used the ratio of the slope of the 40th to that of the first EPSP to measure synaptic fatigue. In addition, single exponential equations were fitted to the plots of EPSP slopes versus stimulus number using IGOR Pro program. The responses to paired-pulse stimulation at different interpulse intervals (7–50 msec) were used to measure PPF. The mean slopes of the EPSPs during the first 3 min after the LTP-inducing tetanus were used to measure post-tetanic potentiation (PTP). EPSPs were digitized (10 kHz), filtered at 1 kHz (eight-pole Bessel Filter), analyzed on-line, and stored on magnetic media using acquisition systems DaQSys 1.0 and Process 2.0 (University of Geneva), and custom-developed software (provided by Dr. T. Inoue, University of Tokyo). For recovery experiments, BDNF was added directly into the interface chamber (final concentration 2 nm), and +/− or −/− slices were incubated in BDNF for 3 hr.
Electron microscopy. Eight adult CD1/BDNF littermates (two +/+, three +/−, and three −/−, from three different litters) and two adult BL/6/BDNF littermates (one +/+ and one +/−) were used for electron microscopy analysis. Mice were anesthetized with ether and perfused through the heart with 1% paraformaldehyde and 1.25% glutaraldehyde in 100 mm cacodylate buffer, pH 7.4, for the first 5 min, and then with 2% paraformaldehyde and 2.5% glutaraldehyde in the same buffer for an additional 5 min. After 1 hr, the brains were removed and placed in the last perfusion fixative for an additional 2 hr. Hippocampi were dissected, and transverse sections were made at 400 μm thickness. These hippocampal sections were rinsed in 100 mm cacodylate buffer, post-fixed (1 hr) with 1% OsO4 in 100 mm cacodylate buffer, pH 7.4, rinsed in acetate buffer, pH 5, and stained en bloc with 1% uranyl acetate (12–15 hr; 4°C). After a rinse in acetate buffer and dehydration in a series of ethanol dilutions, slices were rinsed in propylene oxide and flat-embedded in Araldite. Semithin sections (0.5 μm) stained with toluidine blue were used to locate and trim CA1stratum radiatum. Ultrathin sections (60–80 nm) were cut and stained with uranyl acetate and lead citrate.
Only complete profiles of nonperforated asymmetric synapses (identified by the more electron-dense postsynaptic density) on dendritic spines were photographed in a JEOL 100× electron microscope operated at 80 kV at a final magnification of 50,000×. Perforated synapses and symmetrical synapses on shafts were excluded from the sampling. Dendritic spines were identified by their shape and size, their continuity with a dendritic shaft, or their lack of microtubules and mitochondria. Synaptic vesicles were counted only when their plasma membranes were clearly defined for at least one-half their circumference (Heuser and Reese, 1973). A total of 247 asymmetric synapses on CA1 dendritic spines from 3 +/+ mice, 315 synapses from 4 +/− mice, and 257 synapses from 3 −/− mice were analyzed. Photographic negatives were scanned at 300 dpi to make digital images. The quantitative analysis was performed after a blind protocol: the person who did the analysis did not know the genotypes of the mice. Digital image analysis was performed using NIH Image. The parameters measured in each synapse were area of the presynaptic terminal, length of the active zone, thickness of the postsynaptic density, total number of small (∼50 nm) synaptic vesicles per terminal, and number of docked vesicles. The docked vesicles are defined as those located up to one-vesicle-diameter (∼50 nm) distance from the active zone, according to criteria developed by Reese and colleagues (Dickinson-Nelson and Reese, 1983), because these vesicles have been shown to be depleted during sustained, repetitive synaptic activity. To avoid the bias of analyzing the larger synapses in the single sections, only synapses with terminals and active zones of equal magnitude were included in the comparisons between all genotypes.
The number of docked vesicles per active zone length (DV) and the active zone length (L) in the two-dimensional single sections are listed in Table 1. From these numbers, it was possible to estimate the total number of docked vesicles per whole active zone (TDV) as follows:TDV =A/(0.866 · ADV).
Table 1.
Morphological analysis of CA1 synapses in BDNF mutant mice
| DV/μm | L(μm) | DV | A(μm2) | ADV(μm2) | TDV | |
|---|---|---|---|---|---|---|
| CD1/BDNF | ||||||
| +/+ | 3.84 ± 0.11 | 0.86 ± 0.03 | 3.2 ± 0.12 | 0.58 | 0.07 | 9.3 |
| +/− | 2.49 ± 0.07* | 0.93 ± 0.02 | 2.2 ± 0.8* | 0.68 | 0.17 | 4.5 |
| −/− | 2.15 ± 0.06* | 0.92 ± 0.02 | 1.9 ± 0.7* | 0.66 | 0.22 | 3.4 |
| BL/6/BDNF | ||||||
| +/+ | 1.86 ± 0.12 | 0.71 ± 0.033 | 3.37 ± 0.21 | 0.39 | 0.04 | 10.3 |
| +/− | 1.40 ± 0.09* | 0.81 ± 0.032 | 2.40 ± 0.18* | 0.51 | 0.11 | 5.2 |
DV, Number of docked vesicles per active zone;L, active zone length (μm); DV/μm, number of docked vesicles per micrometer of active zone length,DV/μm = DV/L; A, total active zone area (μm2), A = 1/4 · π · L2; ADV, active zone area per docked vesicle, ADV = (L/DV)2; TDV, total number of docked vesicles per whole active zone, TDV =A/(0.866 · ADV). DV/μm,L, and DV are average numbers measured from 247–315 single sections of electron micrographs (mean ± SEM). Data from +/− and −/− were compared with those of +/+, and statistical differences were assessed by ANOVA test. *p< 0.005.
The active zone was assumed to be a flat circular disk of surface areaA = 1/4 · π · L2 (Mayhew, 1979). The average active zone area per docked vesicle (ADV), assuming it as a square, was estimated as (L/DV)2. Because the docked vesicles are clustered in the active zone in a hexagonal array (Gray, 1963), ADV must be adjusted by a factor √ 3/2 = 0.866. The estimated values of A, ADV, and TDVfor CD/BDNF and BL/6/BDNF mice are also presented in Table 1.
Biochemical analysis of synaptic proteins. BL/6/BDNF or CD1/BDNF mice were anesthetized with halothane, and their hippocampi were dissected and frozen in liquid nitrogen. To determine the expression levels of synaptic proteins in the whole hippocampus in BL/6/BDNF mice, frozen hippocampi from +/+ and +/− were homogenized in 1% SDS. Equal amounts of homogenate proteins were separated on an SDS-acrylamide gel, transferred to a Immobilon P membrane (Millipore), and probed with antibodies against the following synaptic proteins: synaptotagmin, synaptophysin, synaptobrevin [vesicle-associated membrane protein (VAMP-2)], SNAP-25, and syntaxin-1. All antibodies were monoclonal IgG or IgM from Chemicom, except for the those against synaptobrevin and synaptotagmin, which were polyclonal antibodies (Dr. M. Takahashi, Mitsubishi Institute). Both monoclonal and polyclonal antibodies were used for synaptophysin. Specific synaptic proteins were detected by 125I-labeled IgG/M and quantified by phosphoimaging and Storm programs (Molecular Dynamics).
Biochemical analysis of synaptic proteins in the nerve terminals was performed only on CD1/BDNF +/+ and +/− animals, because of the difficulties in obtaining sufficient numbers of −/− animals. Generally, hippocampi from four to seven animals of the same genotype, same litter, and preferably same sex were used to prepare synaptosomes. Even in the CD1 genetic background, we normally obtained only one to three −/− animals per litter. The −/− mice are only about half the size of the +/+ and +/− littermates, and therefore very little hippocampal tissues can be obtained from the −/− mice. Furthermore, the −/− mice cannot live beyond 30 d after birth, and often we had to make a decision to kill them around P20 or we lost them. Because of these practical difficulties, we only prepared synaptosomes from +/+ and +/− mice using the differential and discontinuous Ficoll gradient centrifugation method (Sheng et al., 1996). Briefly, each group of hippocampal tissue was homogenized in 10 ml cold Syn buffer (Mannitol 300 mm, EDTA 1 mm, pH 7.4). Aliquots of the homogenates were taken to determine the levels of synaptic proteins in the whole hippocampus. The remaining homogenates were centrifuged at 5000 × g for 10 min. The supernatants were centrifuged at 25,000 × g for 30 min. The pellets were then resuspended in Syn buffer, loaded on Ficoll-gradient tubes, and centrifuged at 32,000 × g at 4°C for 90 min. The proteins in the interfaces between 8 and 12% and 12 and 16% Ficoll gradient were collected, diluted in Syn buffer in a ratio of 1:4, and then centrifuged for 20 min at 25,000 × g. Pellets were resuspended in 500 μl 1× P Buffer (KCl 5.4 mm, MgSO4 0.8 mm, glucose 5.5 mm, HEPES 50 mm, choline chloride 130 mm, BSA 1 mm, and 0.01% CHAPS). Synaptosomes were then solubilized with 3.5 ml buffer (NaCl 150 mm, HEPES 10 mm, and 1.5% Chaps), stirred at 4°C for 1.5 hr, and further centrifuged at 12,000 × g for 1 hr. Supernatants were then ready for SDS-PAGE analysis. Equal amounts of proteins (50 μg) from either whole hippocampus or hippocampal synaptosomes were separated by 10–20% SDS-Tricine gradient gel electrophoresis (Novex) and immunoblotted with specific primary antibodies. The immunoreactive bands were visualized by enhanced chemoluminescence (ECL) (Amersham). The ECL signal intensities were quantified using NIH Image based on standard curves of these synaptic proteins.
All data are presented as mean ± SEM (N = number of animals; n = number of experiments). Statistical significance was determined by either Student’s t test or ANOVA, followed by Fischer protected least significant difference as apost hoc test. # = p < 0.05; * =p < 0.005.
RESULTS
To obtain a large number of mutant mice for detailed electrophysiological, biochemical, and structural studies, and to minimize the potential interference of genetic background to our observations, we generated a new line of BDNF knockout mice by crossing one BDNF line (C57BL/6 background, called BL/6/BDNF) (Ernfors et al., 1994) with an outbreed mouse line (CD1). This new CD1/BDNF line showed several characteristics similar to other BDNF knockout lines (see Materials and Methods) (Ernfors et al., 1994; Jones et al., 1994). Moreover, these mice exhibited the same defects in tetanus-induced LTP in the hippocampus as those reported previously (Korte et al., 1996;Patterson et al., 1996). Although 75% of the slices from +/+ young adult mice (P25–44) exhibited LTP (>20% increase over baseline), only 43 and 44% of the slices derived from +/− and −/− littermates showed LTP, respectively (number of animals: N = 5, 5, 4; number of recordings: n = 35, 21, 27). Furthermore, in those +/− and −/− slices that expressed LTP, there was a significant reduction in the magnitude of the potentiation, when compared with the +/+ slices (potentiation: 83% in +/+,n = 25, 45% in +/−, n = 9; 44% in −/−, n = 12; ANOVA, p < 0.05). Basal synaptic transmission was not affected by the mutation of the BDNF gene in our CD1/BDNF line; no difference was found in the input–output curves obtained from wild-type and mutant hippocampal slices (input–output curve slopes: 1.2 ± 0.12 in +/+, n= 14, and 1.2 +/− 0.08 in +/−, n = 14; ttest, p = 0.6949). To further study the cellular and molecular mechanisms underlying BDNF action in the hippocampus, we examined the properties of CA1 synapses using both CD1/BDNF and BL/6/BDNF lines whenever possible.
BDNF knockout mice show significant synaptic fatigue
Exogenous BDNF attenuates synaptic fatigue induced by a train of HFS in neonatal hippocampus where endogenous BDNF level is low, whereas the BDNF scavenger TrkB-IgG fusion protein reduces synaptic responses to the tetanus in adult hippocampus where endogenous BDNF level is high (Figurov et al., 1996). If BDNF levels are important for hippocampal synapses to follow HFS, synapses in the BDNF mutant mice should exhibit more pronounced synaptic fatigue. Indeed, adult slices prepared from both +/− and −/− CD1/BDNF mice showed more pronounced synaptic fatigue in CA1 synapses during HFS compared with +/+ mice (Fig.1a,b). The percentage of the 40th EPSP slope over the first EPSP slope in the HFS trains (from here on termed “response to HFS”) was 63 ± 3% in +/+, 43 ± 2% in +/−, and 41 ± 2% in −/− (ANOVA, p < 0.005); N (number of animals) = 8, 8, and 6 for +/+, +/−, and −/−, respectively. A similar pattern in response to HFS was also observed in BL/6/BDNF mice (Fig. 1c). HFS induced a mild synaptic fatigue in +/+ adult BL/6/BDNF slices but much more pronounced fatigue in slices from +/− littermates (+/+, 74 ± 3%; +/−, 42 ± 3%; p < 0.05).
Fig. 1.
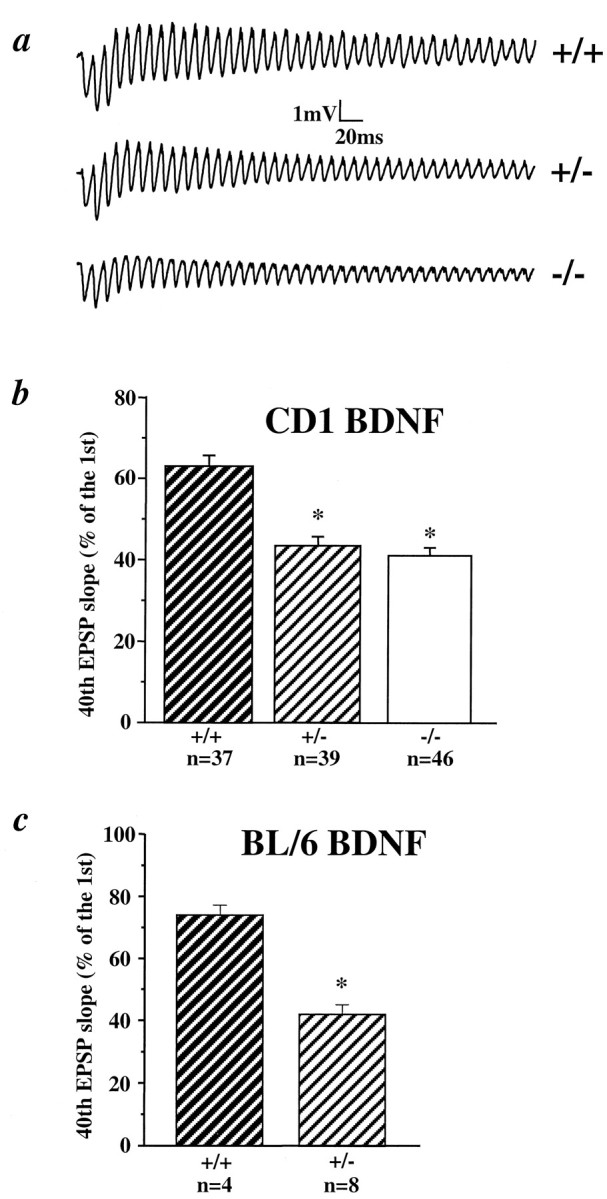
Pronounced synaptic fatigue in CA1 synapses during high-frequency stimulation in BDNF knockout mice. a, Examples of EPSPs elicited by a train of HFS (100 Hz) in hippocampal slices from CD1/BDNF mice. Note the significant synaptic fatigue in +/− and −/− mice. b, Summary of synaptic fatigue in CD1/BDNF mice. The slope of the 40th EPSP in the train is presented as the percentage of the first EPSP slope.n = number of recordings. c, Summary of synaptic fatigue in BL/6/BDNF mice.
To better visualize the difference observed in the synaptic fatigue between the mutant CD1/BDNF mice, we plotted EPSP slopes against the number of stimuli in the train and compared the integrated areas under the plots. Statistically significant differences were observed not only between +/+ and mutants (ANOVA, p < 0.001), but also between +/− and −/− synapses (ANOVA, p = 0.0243) (Fig. 2a). Furthermore, when the decay phases of the normalized EPSP slope/stimulus number plots were fitted with a single exponential equation, we found that the rates of synaptic fatigue were different among the three genotypes (Fig.2b). The decay constants for +/+, +/−, and −/− synapses were 26.5, 18.2, and 13.7 stimuli, respectively. Again, the −/− synapses showed a faster rate of fatigue as compared with the +/−. Interestingly, there was an initial facilitation in the first nine EPSPs for +/+ synapses, whereas in +/− and −/− synapses this facilitation ended at seventh and fourth EPSP, respectively. Thus, the mutation of the BDNF gene leads to severe defects in synaptic transmission at high frequencies.
Fig. 2.

Overall changes in synaptic responses during HFS in CA1 synapses of CD1/BDNF mice. a, EPSP slopes were plotted against the number of the stimulus in the 100 Hz train for each of the synapses, and the integrated areas under the plots were calculated for each synapse and compared among three genotypes. *, Significantly different; ANOVA test with post hoc PLSD Fischer’s test. n = 8 for each genotype.b, Rate of synaptic fatigue in +/+, +/−, and −/− synapses. Normalized EPSP slopes were plotted against the number of the stimulus during HFS. The plot for each genotype was fitted with a single exponential curve, and rate constants were obtained.n = 8 for each genotype.
BDNF knockout mice have impairments in presynaptic transmitter release
Substantial evidence indicates that synaptic fatigue is a presynaptic phenomenon (Zucker, 1989; Dobrunz and Stevens, 1997). Our previous work demonstrated that the effect of BDNF on high-frequency transmission in the neonatal rat hippocampus is mediated, at least in part, by presynaptic mechanisms (Gottschalk et al., 1998). Because BDNF has been shown to attenuate GABA receptor-mediated synaptic responses in hippocampal slices (Tanaka et al., 1997; Frerking et al., 1998), we determined whether the pronounced synaptic fatigue seen in the BDNF mutant mice results from elevated GABAergic inhibition. Slices treated with the GABAA and GABAB receptor antagonists (10 μm bicuculline, 0.1 mm phaclofen, respectively) exhibited the same degree of synaptic fatigue as compared with untreated ones (+/−: 45 ± 2%, n = 6; +/− plus antagonists: 47 ± 3%, n = 9; ttest, p = 0.4). Moreover, the pronounced synaptic fatigue in the mutant mice was not caused by an enhanced desensitization of postsynaptic glutamate receptors. Synaptic responses to HFS recorded from +/− slices treated with and without the desensitization blocker cyclothiazide (0.1 mm) showed no significant difference (the ratio of +/− to +/− plus cyclothiazide = 1.07, n = 6 and 9, respectively;t test, p > 0.5). Thus, the pronounced synaptic fatigue in the mutant slices may result from an impairment in transmitter release properties during HFS, rather than from an elevated GABAergic inhibition or an enhanced desensitization of glutamate receptors.
To characterize the presynaptic deficits in more detail, we examined PTP, a phenomenon thought to be caused by enhanced presynaptic transmitter release (for review, see Zucker, 1989). The magnitude of PTP, defined as a percentage increase of the average EPSP slope 3 min after HFS over a 20 min baseline, appeared to correlate with the levels of BDNF in the hippocampus of CD1/BDNF mice (Fig.3). PTP was significantly reduced in slices from +/− mice (186 ± 14%) and −/− mice (142 ± 7%), as compared with the ones from +/+ animals (226 ± 12%).
Fig. 3.

Impairment of post-tetanic potentiation (PTP) in CD1/BDNF mice. a, Examples of EPSPs before and 1 min after tetanus in different genotypes (separated by double slash). b, Summary of PTP in +/+, +/−, and −/− mice. EPSP slopes monitored within a 3 min time window after tetanus (2 × 1 sec, 100 Hz) were averaged, and PTP is expressed as a percentage of the baseline EPSP slope (collected during the 20 min before tetanus). *, Significantly different from +/+; ANOVA, p < 0.005. n = 5, 5, and 4 for +/+, +/−, and −/−, respectively.
Another frequently used parameter to monitor presynaptic changes is PPF, measured as the ratio of EPSP slopes in response to two successive stimulation pulses (Creager et al., 1980). Unlike the defects in synaptic fatigue and PTP, a significant reduction in PPF was observed at the shorter interpulse intervals (7 and 10 msec) only in −/− but not in +/− (Fig. 4). Thus, different assays for presynaptic properties may have different sensitivities to BDNF levels. These results indicate that presynaptic transmitter release is impaired in BDNF mutant mice.
Fig. 4.

Impairment of paired-pulse facilitation (PPF) at short interpulse intervals in CD1/BDNF mice. a, Sample EPSP traces during PPF at 10 and 50 msec interpulse intervals. b, Plot of PPF at different interpulse intervals. *, Significantly different from +/+; ANOVA,p < 0.005. n = 5, 5, and 4 for +/+, +/−, and −/−, respectively.
Synapses in BDNF knockout mice have fewer docked vesicles
Our electrophysiological studies suggest that the primary defect in the CA1 synapses of BDNF mutant mice is in the synaptic responses to HFS, or synaptic fatigue. To determine how BDNF regulates synaptic fatigue during HFS, we examined the morphology of CA1 synapses in BDNF mutant mice. The general appearance of presynaptic terminals, spines, and postsynaptic densities at excitatory synapses on CA1 dendritic spines was similar between wild-type and mutant animals (Fig.5). However, quantitative analysis revealed that there was a dramatic reduction in the number of synaptic vesicles docked at the active zones in both +/− and −/− mice (Figs.5, 6a). Docked vesicles are defined as those located up to one vesicle diameter (∼50 nm) distance from the active zone. This definition includes vesicles in the active zone that are discharged during sustained, repetitive synaptic activity (Dickinson-Nelson and Reese, 1983). The mean numbers of docked vesicles per active zone length were 3.2 ± 0.12 in +/+, 2.2 ± 0.8 in +/−, and 1.9 ± 0.7 in −/− (ANOVA,p < 0.005, Table 1). On the basis of these numbers, we estimated 9.3 docked vesicles per whole active zone in +/+ mice (see Materials and Methods). In contrast, +/− mice had 4.5 vesicles per active zone, whereas −/− animals exhibited 3.4 docked vesicles per active zone (Table 1). The number of reserve pool vesicles (those located beyond ∼50 nm from the active zone) was not significantly different among the three genotypes (Fig.6b) (ANOVA, p = 0.1768). To avoid the bias of analyzing the larger synapses in the single sections, only synapses with terminals and active zones of equal magnitude were included in the comparisons between all genotypes. Therefore, in our sample of synapses on CA1 dendritic spines, presynaptic terminal area and active zone length were similar in all three genotypes (data not shown). Presynaptic terminal area and active zone length were also similar in all three genotypes (data not shown). Significant differences in the number of docked vesicles per active zone length were also observed between a pair of +/+ and +/− mice from the BL/6/BDNF mice (3.37 ± 0.21 in +/+, n = 46; 2.40 ± 0.18 in +/−,n = 58; t test, p < 0.005). The total number of docked vesicles per whole active zone estimated from those measurements was 10.3 in +/+ and 5.2 in +/− hippocampus (Table 1). Synaptic fatigue is known to result from a gradual depletion of the readily releasable pool of quanta or their structural correlate, the docked vesicles, during HFS (Zucker, 1989; Dobrunz and Stevens, 1997). Thus, impairments in the docking of synaptic vesicles may explain why more pronounced synaptic fatigue is observed during HFS in BDNF knockout mice.
Fig. 5.
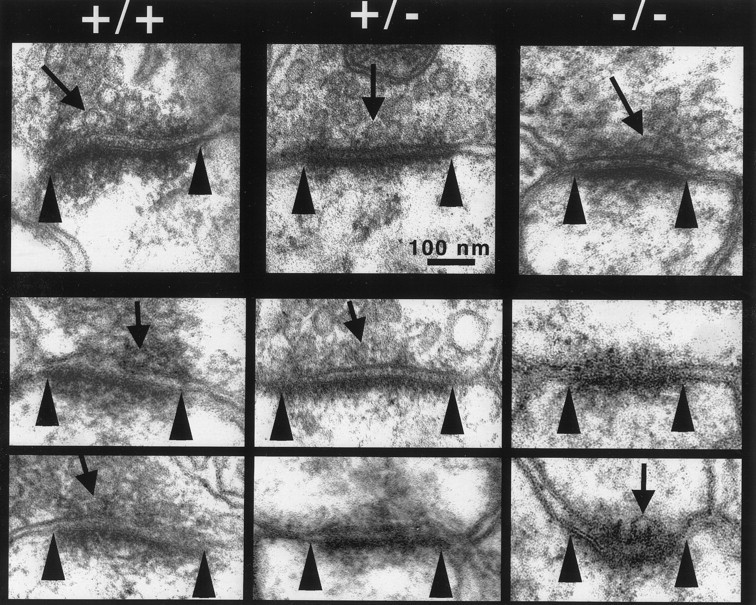
Electron micrographs of excitatory synapses on CA1 dendritic spines in hippocampal stratum radiatum from +/+, +/−, and −/− CD1/BDNF mice. Examples of presynaptic terminals and adjacent dendritic spines displaying normal gross structural features in the three genotypes. Arrows point to a representative example of a morphologically docked vesicle as is defined in the text; arrowheads mark the edges of the active zone/postsynaptic density complexes. Note that +/− and −/− mice have fewer docked vesicles at the active zones.
Fig. 6.
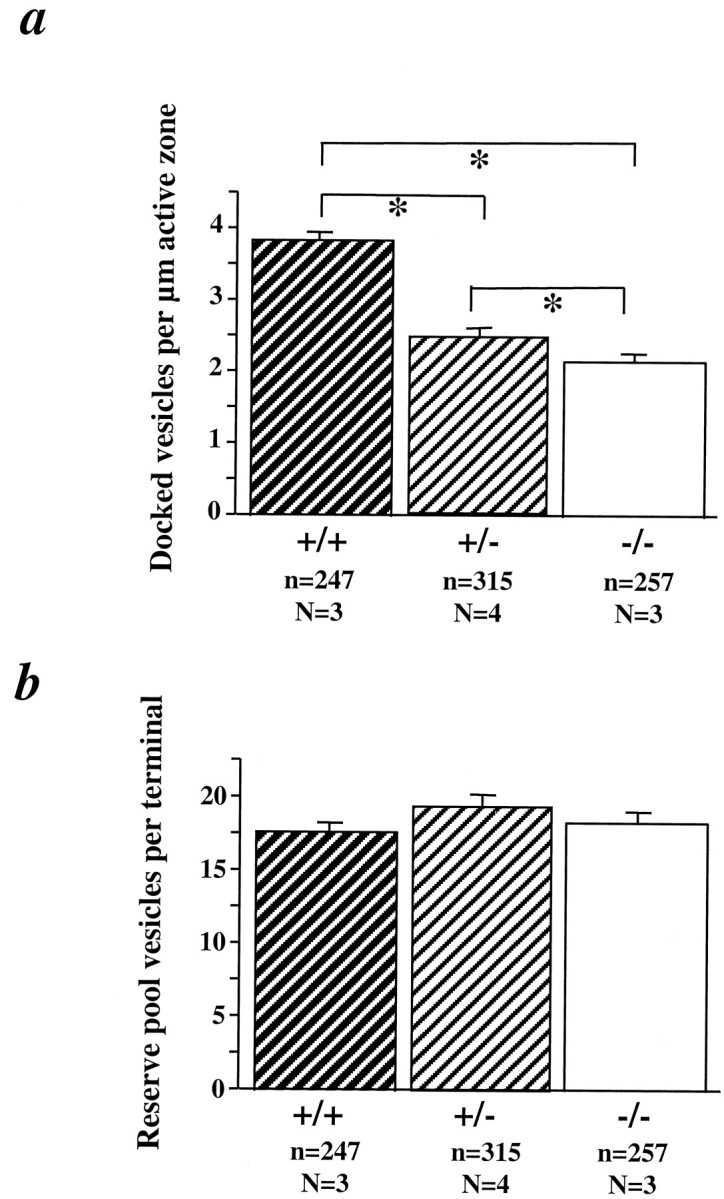
Quantitative analysis of synaptic vesicles in excitatory synapses in BDNF mutant mice. a, Number of docked vesicles per micrometer of active zone length in the three genotypes. Data from CD1/BDNF and those from BL/6/BDNF mutant mice were combined. b, Number of reserve pool vesicles per presynaptic terminal in the three genotypes. n = number of synapses; N = number of animals.
Synaptobrevin and synaptophysin are reduced in hippocampal synapses from BDNF knockout mice
To examine the potential molecular mechanism(s) underlying the regulation of synaptic vesicle mobilization and/or docking by BDNF, we measured in the mutant hippocampus the levels of several proteins of the SNARE complex involved in vesicle docking and fusion (Sudhof, 1995;Calakos and Scheller, 1996). First, we determined the levels of synaptic proteins in homogenates of whole hippocampi prepared from BL/6/BDNF knockout mice. These included proteins on the synaptic vesicles (v-SNAREs), such as synaptobrevin (also called VAMP-2) and synaptotagmin, as well as proteins on the presynaptic plasma membrane (t-SNAREs), such as syntaxin-1 and SNAP-25. We also measured the levels of synaptophysin, a major integral membrane protein on synaptic vesicles. Western blot analysis of the whole hippocampal homogenates showed no differences in the levels of any of the synaptic proteins we examined in three pairs of +/+ and +/− adult hippocampi from BL/6/BDNF mice (Fig. 7a). We did not analyze the −/− because adult littermates of the same gender from the three genotypes of the BL/6/BDNF line were difficult to obtain.
Fig. 7.
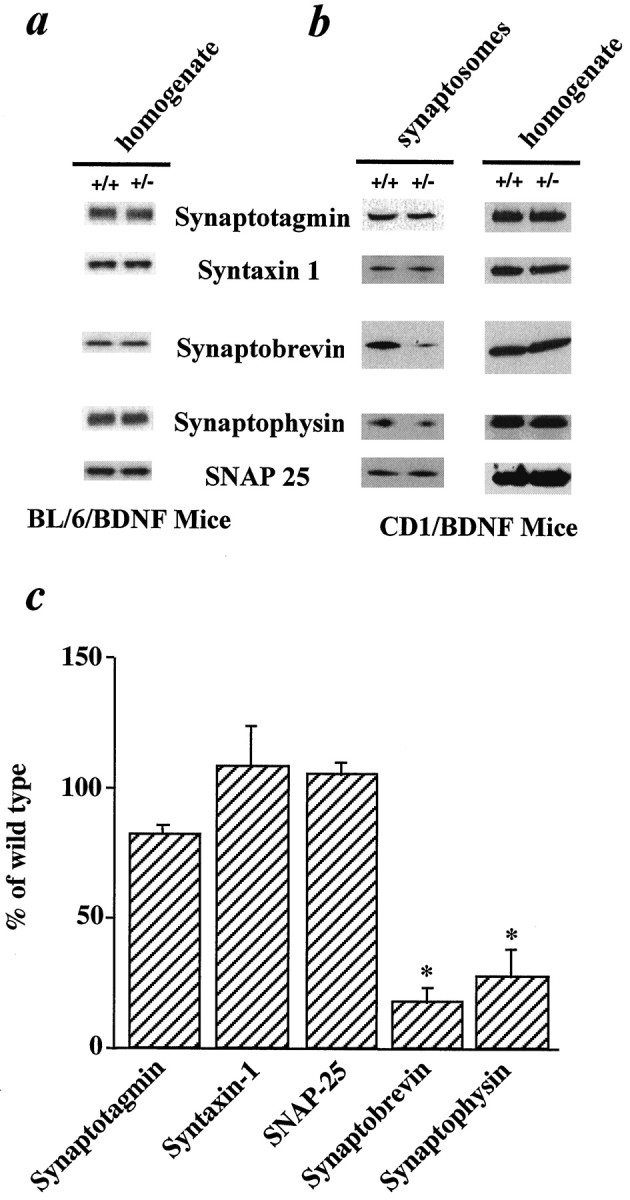
Analysis of synaptic proteins in homogenates of whole hippocampal preparations and in hippocampal synaptosomes from BDNF knockout mice. a, Synaptic proteins detected in whole hippocampal homogenates from BL/6/BDNF mice. The hippocampi were dissected from a pair of +/+ and +/− mice, homogenized, and subject to Western blot analysis using antibodies against specific synaptic proteins. b, Synaptic proteins detected in whole hippocampal preparations (right, homogenate) and synaptosomes (left) from CD1/BDNF mice. Similar Western blot analysis was performed. Note the significant reduction in the levels of synaptobrevin (VAMP-2) and synaptophysin only in synaptosomal preparations, but not of other synaptic proteins. Because synaptotagmin, SNAP25, and syntaxin all show the same levels in the same gel, there is no need to include loading controls in this figure.c, Relative levels of the synaptic proteins in the synaptosomes, quantified by scanning the blots and comparing the bands with standard curves. In each experiment, hippocampal synaptosomes were prepared from a large litter of CD1/BDNF mice (2–3 months old), with three to four +/+ and three to four +/− of the same gender, and Western blots using the same synaptosomal preparation were repeated several times. Four such experiments were performed, and each experiment was repeated two to three times. Data are pooled and presented as a percentage of the wild-type levels.
Because synaptic proteins are distributed throughout hippocampal neurons, including cell bodies, axons, and nerve terminals, significant differences in the distribution of synaptic proteins at presynaptic terminals might be masked by their expression in other parts of the cell. To investigate the potential changes in the levels of synaptic proteins at nerve terminals, synaptosomal fractions from hippocampi of mutant mice were prepared using Ficoll-gradient methods (Sheng et al., 1996). Because of the difficulties in obtaining large numbers of −/− hippocampi for synaptosomal preparations (see Materials and Methods), only CD1/BDNF +/+ and +/− mice were used in the subsequent experiments. Similar to what was observed in BL/6/BDNF mice, no differences were found in the levels of the synaptic proteins in the homogenates of whole hippocampi prepared from CD1/BDNF mice (Fig.7b, right). In synaptosomes prepared from +/− hippocampi, however, a polyclonal antibody detected an 82% decrease in the level of synaptobrevin, an integral synaptic vesicle protein implicated in vesicle docking and/or fusion (Fig. 7b,left, c). In addition, a monoclonal antibody detected a 72% reduction in the amount of another vesicle protein, synaptophysin. Similar result was obtained using a polyclonal antibody against synaptophysin (data not shown). The level of the vesicle protein synaptotagmin was similar in synaptosomes prepared from +/+ and +/− mice (Fig. 7b, left), consistent with the observation that the number of synaptic vesicles in the reserve pool was not reduced in BDNF mutant mice (Fig. 6b). The levels of presynaptic membrane proteins, such as syntaxin-1 and SNAP-25, were also unchanged in the mutant mice (Fig. 7b,c). Similar results were consistently observed in four independent synaptosomal preparations, and pooled data are shown in Figure 7c. Taken together, these results suggest that selective reduction in synaptobrevin and synaptophysin levels occurs at nerve terminals in the hippocampus of BDNF mutant mice.
Treatment with BDNF reverses the synaptic defects in BDNF mutant mice
If the synaptic deficits were attributable to a direct outcome of the mutation of the BDNF gene rather than indirect consequences of the lack of BDNF on hippocampal circuitry, application of BDNF to the mutant slices should reverse the deficits. We incubated the adult mutant slices with recombinant BDNF (2 nm) for 3 hr and then analyzed the physiological and biochemical properties of the hippocampal synapses. In CD1/BDNF mice, synaptic fatigue in the +/− and −/− slices was significantly attenuated after BDNF treatment (Fig. 8a). In fact, the response to the HFS seen in the +/− and −/− slices after BDNF treatment returned to the levels seen in the +/+ slices. Similar results were obtained in BL/6/BDNF mice (+/−: 42%, n= 8, n = 2; +/− plus BDNF: 79%, n = 15, n = 2). In synaptosomes prepared from +/− slices, we reproduced the selective reduction in the levels of synaptobrevin and synaptophysin seen in those from whole hippocampus. Furthermore, incubation of these slices with BDNF for 3 hr reversed the decrease in the synaptosomal levels of synaptobrevin and synaptophysin (Fig.8b,c). Taken together, these data suggest that the synaptic deficits seen in the BDNF mutant mice are not attributable to indirect, developmental consequences of BDNF knockout. Rather, they reflect an acute requirement for BDNF in high-frequency transmission in the hippocampus. We could not examine the vesicle docking in mutant slices treated with BDNF, because it was extremely difficult to obtain the good structural preservation needed to quantify docked vesicles in the acute slices after 3 hr in the interface recording chamber. Interestingly, treatment of the +/+ slices with BDNF also elicited a 50.4% increase in responses to HFS (response to HFS: 63.0 ± 2.6% in control, n = 37, and 94.8 ± 5.1% in BDNF-treated slices, n = 29), and a 151 ± 39% and 48 ± 12% increase in the levels of synaptophysin and synaptobrevin in hippocampal synaptosomes, respectively. These results imply that vesicle docking in CA1 synapses is not saturated even in wild-type animals, and BDNF has a general capacity to modulate this process.
Fig. 8.
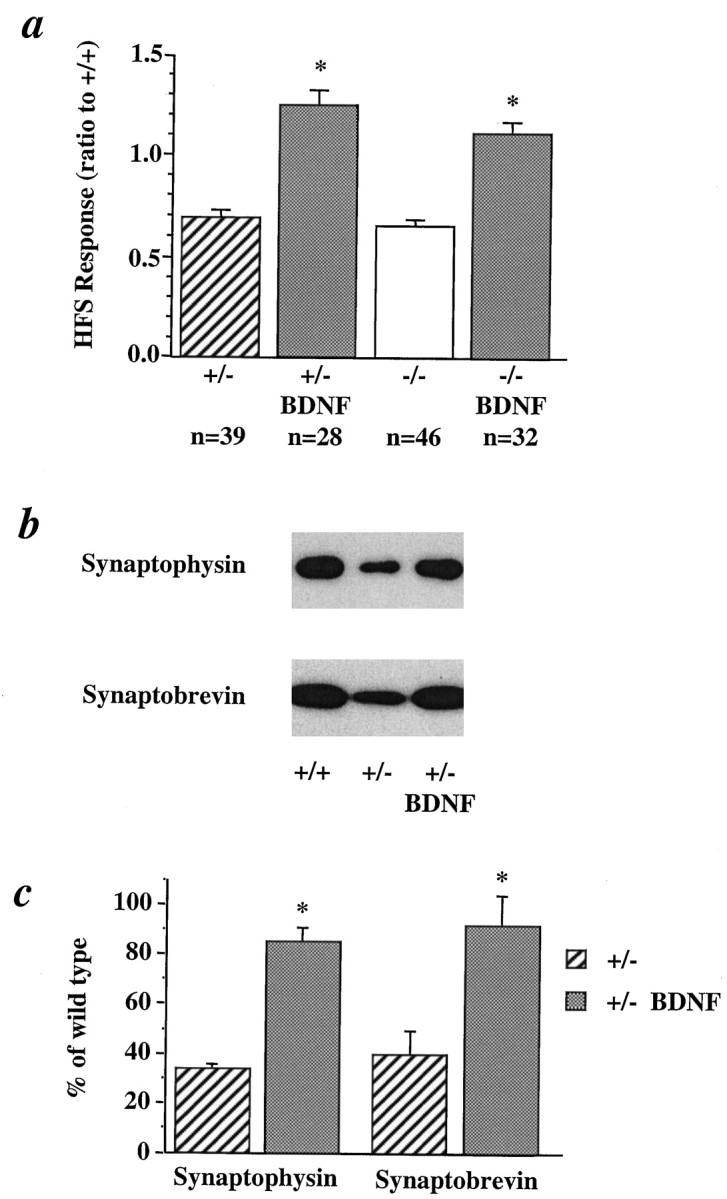
Effect of acute exposure to BDNF on synaptic fatigue and the levels of synaptobrevin and synaptophysin in BDNF mutant slices. a, Synaptic fatigue was induced by a train of 1 sec, 100 Hz stimulation in hippocampal slices from either +/− or −/− CD1/BDNF mice. The slices were then treated with recombinant BDNF (2 nm) for 3 hr. HFS responses in +/− and −/− slices before and after BDNF treatment were normalized to that in +/+ slices (n = 21). N = 3, 2, and 2 for +/+, +/−, and −/−, respectively. b, Examples of Western blots showing the levels of synaptobrevin and synaptophysin in synaptosomes from +/+ and +/− slices and +/− slices plus BDNF. Slices from +/− CD1/BDNF mice were treated with BDNF (2 nm) for 3 hr. Synaptosomes were prepared and synaptic proteins were detected the same way as described in Figure 7. Note that the same levels of actin were detected in all three lanes, suggesting that equal amounts of proteins are loaded. c, Quantification of the relative levels of synaptobrevin and synaptophysin in synaptosomes prepared from +/− slices treated with and without BDNF. Three experiments using independent synaptosomal preparations were performed. Each experiment was repeated two to three times, and data were pooled and presented as a percentage of the wild-type levels.
DISCUSSION
Our study suggests that BDNF may regulate high-frequency transmission by facilitating synaptic vesicle mobilization and/or docking, possibly through the regulation of the distribution of specific synaptic proteins at nerve terminals. Gene targeting experiments often suffer from interference by the complexity of the genetic background of the mice used. For example, although two independent lines of BDNF knockout mice exhibit the same impairment in hippocampal LTP (Korte et al., 1995; Patterson et al., 1996), only one of the lines shows defects in basal synaptic transmission (Patterson et al., 1996). This discrepancy in basal synaptic transmission between the two mutant lines has been attributed to the difference in the genetic background of the two mouse lines, rather than to the effect of BDNF mutation. To minimize the influence of the genetic background on our observations, we have used CD1/BDNF as well as BL/6/BDNF knockout mice whenever possible. Knockout mice from both lines exhibit more severe synaptic fatigue, impairments in LTP, and fewer docked vesicles in CA1 synapses. These results suggest that the described impairments in the knockout mice were not caused by alterations in genetic background but were attributable to the mutation of the BDNF gene.
Our previous study demonstrated that in neonatal rat, BDNF modulation of CA1 synapses is primarily mediated through presynaptic mechanisms (Gottschalk et al., 1998). Here we show that PPF and PTP, two forms of short-term synaptic plasticity that reflect changes in presynaptic properties, were also impaired in the BDNF mutant mice. These results further support the notion that BDNF acts presynaptically. The defects in vesicle docking may account for the severe synaptic fatigue seen in the BDNF knockout mice. The absolute difference in the number of docked vesicles per active zone between +/− and −/− mutant mice was small (2.2 vs 1.9), although it was statistically significant. This small difference may not be detected by our electrophysiological analysis of synaptic fatigue, using the ratio of the 1st and 40th EPSP slopes. A detailed analysis of the time course of responses to HFS indicates that indeed there was a difference in the rate of fatigue at CA1 synapses between +/− and −/− mice. However, the defect in PPF at short interpulse intervals (<20 msec) was observed only in −/− mice. Although this is consistent with our previous finding that exogenous BDNF enhances PPF only at intervals of 20 msec or less (Gottschalk et al., 1998), it is unclear why PPF is more resistant to BDNF reduction. One possibility is that cumulative effects of multiple pulses are required to reveal the small difference in docked vesicles between +/− and −/− mice, and two pulses are not sufficient. Alternatively, BDNF may modulate other presynaptic properties (such as Ca2+ channels), in addition to vesicle docking. In support of the latter view, slices from −/− mice showed more severe impairment in PTP than those from +/− animals. Synaptic fatigue, PPF, and PTP all have different intracellular mechanisms (Zucker, 1989) and therefore may have different sensitivities to BDNF levels.
BDNF and the synaptic vesicle cycle
The synaptic vesicle cycle involves multiple steps, including transport of vesicles to nerve terminals, vesicle docking, a maturation process called “priming,” Ca2+-triggered vesicle fusion, endocytosis, and the formation of new vesicles (Sudhof, 1995;Calakos and Scheller, 1996). In principle, modulation of any of these steps could potentially alter the synaptic responses to repetitive stimulation. Because the attenuation of synaptic fatigue at CA1 synapses by exogenous BDNF occurs only when the frequency of stimulation is >50 Hz (Gottschalk et al., 1998), it is unlikely that the modulation of vesicle transport or endocytosis would contribute to the observed effect of BDNF. The time required for vesicle endocytosis has been estimated to be 1 sec (Ryan et al., 1993), much too slow to contribute to events as fast as 10–20 msec.
Our experiments suggest that BDNF modulates the vesicle docking step. In CA1 excitatory synapses from normal mice (C57BL/6), the mean number of morphologically docked vesicles per active zone is 10.3 based on serial reconstruction (Schikorski and Stevens, 1997), whereas that of releasable quanta is 8.1 based on physiological measurements (Dobrunz and Stevens, 1997). Thus, synaptic vesicles docked at active zones in CA1 synapses represent the readily releasable quanta, as postulated by Katz’s classical theory of synaptic transmission (Katz, 1969). In agreement with the above assessment, we estimated that +/+ CA1 synapses contain 10.3 docked vesicles per active zone in BL/6/BDNF line and 9.3 in CD1/BDNF line, based on measurements from single section electron micrographs (see Materials and Methods). Substantial evidence suggests that synaptic fatigue during sustained high-frequency transmission results from a depletion of the readily releasable pool of quanta or its morphological correlate, the docked vesicles (Highstein and Bennnet, 1975; Heuser and Reese, 1979; Zucker, 1989; Dobrunz and Stevens, 1997; Schikorski and Stevens, 1997). During 100 Hz stimulation, the rate of fusion (every 10 msec) far exceeds that of vesicle transport, endocytosis, or mobilization of vesicles from the reserve pool to the releasable pool (Ryan et al., 1993). Thus the number of docked vesicles is expected to become the rate-limiting factor (Highstein and Bennnet, 1975). We found that CA1 synapses from +/− CD1/BDNF mice had 4.5 docked vesicles per active zone (5.2 in +/− from BL/6/BDNF mice), whereas in −/− animals this number was only 3.4. A selective reduction of the docked vesicle pool in the BDNF knockout mice resulted in more pronounced fatigue.
A reduction in the number of docked vesicles should not affect basal synaptic transmission elicited by low-frequency stimulation (one every 30 sec). According to the measurements by Stevens and Wang (1995), an action potential will deplete at most one quantum (or docked vesicle) at hippocampal synapses. On the basis of our calculation, there are three to five docked vesicles per active zone in the BDNF mutant CA1 synapses. Unless high-frequency repetitive stimulation is applied, docked vesicles should not be depleted in the CA1 synapses of the knockout animals. Indeed, we found no difference in the input–output curves generated from the wild-type and mutant mice. Consistent with this idea, BDNF rescues synaptic fatigue in neonatal rats only when the CA1 synapses are stimulated at a frequency >50 Hz (Gottschalk et al., 1998). These data suggest that the number of docked vesicles determines the degree of synaptic fatigue during HFS and that BDNF modulates high-frequency transmission, but not basal synaptic strength, by regulating vesicle docking.
BDNF and the molecular mechanisms of vesicle docking
The selective reduction in synaptobrevin without affecting synaptotagmin on the synaptic vesicles in the mutant hippocampus is consistent with the observation that the total number of vesicles was not reduced in the mutant CA1 synapses. Moreover, the levels of t-SNAREs syntaxin-1 and SNAP-25 were unaltered, suggesting normal docking sites on the presynaptic membrane. Synaptic vesicles with less synaptobrevin may fail to dock to the presynaptic active zone, leading to the reduction of docked vesicles and pronounced synaptic fatigue. Synaptophysin, a major integral membrane protein on synaptic vesicles, was also downregulated in the BDNF knockout animals. Synaptophysin is complexed with synaptobrevin in nerve terminals (Calakos and Scheller, 1994; Washbourne et al., 1995), and deletion of synaptophysin in mice results in a decrease in synaptobrevin (McMahon et al., 1996). It is unclear which one (or both) is the prime target of BDNF regulation. The finding that BDNF +/− mice exhibited lower levels of synaptobrevin and synaptophysin only in synaptosomes but not in the whole hippocampus suggests that BDNF may facilitate the incorporation of these proteins into the synaptic vesicles, rather than regulating their synthesis.
Substantial biochemical evidence supports the notion that synaptobrevin is required for vesicle docking (Sollner et al., 1993; Calakos et al., 1994; Hayashi et al., 1994; Pevsner et al., 1994). However, cleavage of synaptobrevin by tetanus toxin in the squid giant synapse orDrosophila neuromuscular junction caused a slight increase, rather than a decrease, in the numbers of docked vesicles. This is caused by an accumulation of vesicles after fusion is blocked by the toxin (Hunt et al., 1994; Broadie et al., 1995). One interpretation is that docking after tetanus toxin treatment is mediated by synaptotagmin (Schiavo et al., 1995; Reist et al., 1998). The second possibility is that the toxin could cleave the N-terminal portion of synaptobrevin that mediates fusion, whereas the remaining part of the molecule can still mediate docking. Indeed, the SNARE proteins remain capable of forming heterotrimeric complexes after cleavage of synaptobrevin by tetanus toxin, suggesting that docking is not affected (Hayashi et al., 1994; Pellegrini et al., 1995). Our present study provides new evidence supporting the role of synaptobrevin in vesicle docking. We found that basal synaptic transmission at low frequency was normal in the mutant mice. Conceivably, a reduction of synaptobrevin in the nerve terminals would lead to an impairment in docking without affecting the total number of synaptic vesicles in nerve terminals.
Acute and developmental role of BDNF in the hippocampus
Several studies have demonstrated a role for BDNF in the development of CNS synapses (Cabelli et al., 1995; McAllister et al., 1995). In particular, a recent study showed a marked reduction in axonal branches, synaptic density, and total number of synaptic vesicles in the hippocampus of trkB −/− mice (Martinez et al., 1998). It is therefore important to determine whether the deficits we observed in high-frequency transmission, vesicle docking, and the levels of synaptophysin and synaptobrevin reflect developmental consequences of BDNF knockouts or an acute requirement for BDNF for ongoing synaptic function in the hippocampus. In marked contrast to trkB −/− mice, the morphology of the presynaptic terminals of CA1 synapses in the BDNF knockouts is normal, suggesting that BDNF mutation does not affect the formation of hippocampal synapses. Moreover, acute treatment of the mutant slices with BDNF completely restored the impairment in high-frequency transmission. In synaptosomes prepared from mutant slices, the reduction in the levels of synaptobrevin and synaptophysin was reversed after incubation with BDNF for 3 hr. It seems unlikely that cumulative developmental abnormalities can be reversed within 3 hr of BDNF treatment. For technical reasons, we could not examine whether acute treatment of the mutant slices with BDNF can rescue the decrease in the number of docked vesicles at CA1 synapses at the electron microscopic level. Consistent with our findings, the defects in LTP in slices from BDNF mutant mice can also be restored by acute BDNF treatment (Patterson et al., 1996) or by virus-mediated BDNF gene transfer (Korte et al., 1996). Moreover, we found that application of BDNF to wild-type slices also elicited enhancement in high-frequency transmission and an increase in terminal synaptobrevin and synaptophysin. Thus, BDNF may have a general capacity to modulate vesicle docking in CA1 synapses. Taken together, these results suggest that BDNF plays an important role in the ongoing synaptic function above and beyond its presumptive role in the development of CA1 synapses in the hippocampus.
Footnotes
We express our gratitude to Drs. Tom Reese, Rodolfo Llinás, Story Landis, Chris McBain, Elaine Neale, and Serena Dudek for helpful discussions and critical comments on this manuscript. We also thank Dr. T. Inoue for providing acquisition and analysis software, Dr. M. Takahashi for antibodies against synaptotagmin and synaptobrevin, and D. Abebe, J. Chludzinski, M. F. O’Connell, and Nicole Tartaglia for technical assistance.
Correspondence should be addressed to Dr. Bai Lu, Unit on Synapse Development and Plasticity, National Institute of Child Health and Human Development, National Institutes of Health, Building 49, Room 5A38, 49 Convent Drive, MSC4480, Bethesda, MD 20892-4480.
Dr. Pozzo-Miller’s present address: Department of Neurobiology, University of Alabama at Birmingham, CIRC 429-A2, 1719 Sixth Avenue South, Birmingham AL 35294-0021.
REFERENCES
- 1.Bonhoeffer T. Neurotrophins and activity-dependent development of the neocortex. Curr Opin Neurobiol. 1996;6:119–126. doi: 10.1016/s0959-4388(96)80017-1. [DOI] [PubMed] [Google Scholar]
- 2.Broadie K, Prokop A, Bellen HJ, O’Kane CJ, Schulze KL, Sweeney ST. Syntaxin and synaptobrevin function downstream of vesicle docking in Drosophila. Neuron. 1995;15:663–673. doi: 10.1016/0896-6273(95)90154-x. [DOI] [PubMed] [Google Scholar]
- 3.Cabelli RJ, Horn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- 4.Calakos N, Scheller RH. Vesicle-associated membrane protein and synaptophysin are associated on the synaptic vesicle. J Biol Chem. 1994;269:24534–24537. [PubMed] [Google Scholar]
- 5.Calakos N, Scheller RH. Synaptic vesicle biogenesis, docking, and fusion: a molecular description. Physiol Rev. 1996;76:1–29. doi: 10.1152/physrev.1996.76.1.1. [DOI] [PubMed] [Google Scholar]
- 6.Calakos N, Bennett MK, Peterson KE, Scheller RH. Protein-protein interactions contributing to the specificity of intracellular vesicular trafficking. Science. 1994;263:1146–1149. doi: 10.1126/science.8108733. [DOI] [PubMed] [Google Scholar]
- 7.Canossa M, Griesbeck O, Berninger B, Campana G, Kolbeck R, Thoenen H. Neurotrophin release by neurotrophins: implications for activity-dependent neuronal plasticity. Proc Natl Acad Sci USA. 1997;94:13279–13286. doi: 10.1073/pnas.94.24.13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castren E, Pitkanen M, Sirvio J, Parsadanian A, Lindholm D, Thoenen H, Riekkinen PJ. The induction of LTP increases BDNF and NGF mRNA but decreases NT-3 mRNA in the dentate gyrus. NeuroReport. 1993;4:895–898. doi: 10.1097/00001756-199307000-00014. [DOI] [PubMed] [Google Scholar]
- 9.Creager R, Dunwiddie T, Lynch G. Paired pulse and frequency facilitation in the CA1 region of the in vitro rat hippocampus. J Physiol (Lond) 1980;299:409–424. doi: 10.1113/jphysiol.1980.sp013133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dickinson-Nelson A, Reese TS. Structural changes during transmitter release at synapses in the frog sympathetic ganglion. J Neurosci. 1983;3:42–52. doi: 10.1523/JNEUROSCI.03-01-00042.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 12.Ernfors P, Lee K-F, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- 13.Figurov A, Pozzo-Miller L, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 14.Frerking M, Malenka RC, Nicoll RA. Brain-derived neurotrophic factor (BDNF) modulates inhibitory, but not excitatory, transmission in the CA1 region of the hippocampus. J Neurophysiol. 1998;80:3383–3386. doi: 10.1152/jn.1998.80.6.3383. [DOI] [PubMed] [Google Scholar]
- 15.Friedman WJ, Olson L, Persson H. Cells that express brain-derived neurotrophic factor mRNA in the developing postnatal rat brain. Eur J Neurosci. 1991;3:688–697. doi: 10.1111/j.1460-9568.1991.tb00854.x. [DOI] [PubMed] [Google Scholar]
- 16.Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996;7:223–228. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- 17.Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gray EG. Electron microscopy of presynaptic organelles of the spinal cord. J Anat. 1963;97:101–106. [PMC free article] [PubMed] [Google Scholar]
- 19.Hayashi T, McMahon H, Yamasaki S, Binz T, Hata Y, Sudhof TC, Niemann H. Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 1994;13:5051–5061. doi: 10.1002/j.1460-2075.1994.tb06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heuser JE, Reese TS. Synaptic-vesicle exocytosis captured by quick freezing. In: Schmitt FO, Worden FG, editors. The neurosciences: fourth study program. MIT; Cambridge, MA: 1979. pp. 573–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Highstein SM, Bennnet MVL. Fatigue and recovery of transmission at the Mauthner fiber-giant fiber synapse of the hatchet-fish. Brain Res. 1975;98:229–242. doi: 10.1016/0006-8993(75)90003-7. [DOI] [PubMed] [Google Scholar]
- 23.Huber KM, Sawtell NB, Bear MF. Brain-derived neurotrophic factor alters the synaptic modification threshold in visual cortex. Neuropharmacology. 1998;37:571–579. doi: 10.1016/s0028-3908(98)00050-1. [DOI] [PubMed] [Google Scholar]
- 24.Hunt JM, Bommert K, Charlton MP, Kistner A, Habermann E, Augustine GJ, Betz H. A post-docking role for synaptobrevin in synaptic vesicle fusion. Neuron. 1994;12:1269–1279. doi: 10.1016/0896-6273(94)90443-x. [DOI] [PubMed] [Google Scholar]
- 25.Isackson PJ, Huntsman MM, Murray KD, Gall CM. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron. 1991;6:937–948. doi: 10.1016/0896-6273(91)90234-q. [DOI] [PubMed] [Google Scholar]
- 26.Jones KR, Farinas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 28.Katz B. The release of neural transmitter substances. Liverpool UP; Liverpool: 1969. [Google Scholar]
- 29.Knipper M, da Penha Berzaghi M, Blochl A, Breer H, Thoenen H, Lindholm D. Positive feedback between acetylcholine and the neurotrophins nerve growth factor and brain-derived neurotrophic factor in the rat hippocampus. Eur J Neurosci. 1994;6:668–671. doi: 10.1111/j.1460-9568.1994.tb00312.x. [DOI] [PubMed] [Google Scholar]
- 30.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T. Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci USA. 1996;93:12547–12552. doi: 10.1073/pnas.93.22.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lessmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurons. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- 33.Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lo DC. Neurotrophic factors and synaptic plasticity. Neuron. 1995;15:979–981. doi: 10.1016/0896-6273(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 35.Lu B, Figurov A. Role of neurotrophins in synapse development and plasticity. Rev Neurosci. 1997;8:1–12. doi: 10.1515/revneuro.1997.8.1.1. [DOI] [PubMed] [Google Scholar]
- 36.Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 37.Martinez A, Alcantara S, Borrell V, Del Rio JA, Blasi J, Otal R, Campos N, Boronat A, Barbacid M, Silos-Santiago I, Soriano E. TrkB and TrkC signaling are required for maturation and synaptogenesis of hippocampal connections. J Neurosci. 1998;18:7336–7350. doi: 10.1523/JNEUROSCI.18-18-07336.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayhew TM. Stereological approach to the study of synapse morphometry with particular regard to estimating number in a volume and on a surface. J Neurocytol. 1979;8:121–138. doi: 10.1007/BF01175556. [DOI] [PubMed] [Google Scholar]
- 39.McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 40.McMahon HT, Bolshakov VY, Janz R, Hammer RE, Siegelbaum SA, Sudhof TC. Synaptophysin, a major synaptic vesicle protein, is not essential for neurotransmitter release. Proc Natl Acad Sci USA. 1996;93:4760–4764. doi: 10.1073/pnas.93.10.4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patterson S, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- 42.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 43.Pellegrini LL, O’Connor V, Lottspeich F, Betz H. Clostridial neurotoxins compromise the stability of a low energy SNARE complex mediating NSF activation of synaptic vesicle fusion. EMBO J. 1995;14:4705–4713. doi: 10.1002/j.1460-2075.1995.tb00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pevsner J, Hsu SC, Braun JE, Calakos N, Ting AE, Bennett MK, Scheller RH. Specificity and regulation of a synaptic vesicle docking complex. Neuron. 1994;13:353–361. doi: 10.1016/0896-6273(94)90352-2. [DOI] [PubMed] [Google Scholar]
- 45.Reist NE, Buchanan J, Li J, DiAntonio A, Buxton EM, Schwarz TL. Morphologically docked synaptic vesicles are reduced in synaptotagmin mutants of Drosophila. J Neurosci. 1998;18:7662–7673. doi: 10.1523/JNEUROSCI.18-19-07662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ryan TA, Reuter H, Wendland B, Schweizer FE, Tsien RW, Smith SJ. The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron. 1993;11:713–724. doi: 10.1016/0896-6273(93)90081-2. [DOI] [PubMed] [Google Scholar]
- 47.Schiavo G, Gmachl MJ, Stenbeck G, Sollner TH, Rothman JE. A possible docking and fusion particle for synaptic transmission. Nature. 1995;378:733–736. doi: 10.1038/378733a0. [DOI] [PubMed] [Google Scholar]
- 48.Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sheng Z, Rettig J, Cook T, Catterall WA. Calcium dependent interaction of neuronal N-type calcium channels with presynaptic fusion proteins. Nature. 1996;379:451–454. doi: 10.1038/379451a0. [DOI] [PubMed] [Google Scholar]
- 50.Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 51.Stevens CF, Wang Y. Facilitation and depression at single central synapses. Neuron. 1995;14:795–802. doi: 10.1016/0896-6273(95)90223-6. [DOI] [PubMed] [Google Scholar]
- 52.Sudhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 53.Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–596. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 55.Washbourne P, Schiavo G, Montecucco C. Vesicle-associated membrane protein-2 (synaptobrevin-2) forms a complex with synaptophysin. Biochem J. 1995;305:721–724. doi: 10.1042/bj3050721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yan Q, Rosenfeld RD, Matheson CR, Hawkins N, Lopez OT, Bennett L, Welcher AA. Expression of brain-derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience. 1997;78:431–448. doi: 10.1016/s0306-4522(96)00613-6. [DOI] [PubMed] [Google Scholar]
- 57.Zafra F, Hengerer B, Leibrock J, Thoenen H, Lindholm D. Activity-dependent regulation of BDNF and NGF mRNA in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J. 1990;9:3545–3550. doi: 10.1002/j.1460-2075.1990.tb07564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]