

Abstract
This study takes advantage of continuing advances in the precision of magnetic resonance imaging (MRI) to quantify hippocampal volumes in a series of human subjects with a history of depression compared with controls. We sought to test the hypothesis that both age and duration of past depression would be inversely and independently correlated with hippocampal volume. A sample of 24 women ranging in age from 23 to 86 years with a history of recurrent major depression, but no medical comorbidity, and 24 case-matched controls underwent MRI scanning. Subjects with a history of depression (post-depressed) had smaller hippocampal volumes bilaterally than controls. Post-depressives also had smaller amygdala core nuclei volumes, and these volumes correlated with hippocampal volumes. In addition, post-depressives scored lower in verbal memory, a neuropsychological measure of hippocampal function, suggesting that the volume loss was related to an aspect of cognitive functioning. In contrast, there was no difference in overall brain size or general intellectual performance. Contrary to our initial hypothesis, there was no significant correlation between hippocampal volume and age in either post-depressive or control subjects, whereas there was a significant correlation with total lifetime duration of depression. This suggests that repeated stress during recurrent depressive episodes may result in cumulative hippocampal injury as reflected in volume loss.
Keywords: hippocampus, depression, age, amygdala, magnetic resonance imaging, MRI, glucocorticoids, neurotoxicity, stereology
Hippocampal volume loss in humans has been associated with depression (Sheline et al., 1996) and aging (Jernigan et al., 1991). One explanation for volume loss in depression might be increased neuronal cell death secondary to glucocorticoid (GC) neurotoxicity (Sapolsky et al., 1986; McEwen, 1992). The pathophysiology of major depression involves impairment in negative feedback control of the hypothalamic–pituitary–adrenal (HPA) axis (Young et al., 1991), resulting in elevated cortisol levels during depressive episodes (Carroll et al., 1981). Prolonged exposure to elevated levels of glucocorticoids reduces hippocampal cell number (Sapolsky et al., 1985) and can induce cultured neurons to undergo apoptosis (Reagan and McEwen, 1997). This same effect has been shown in intact animals. Chronic stress or chronic administration of glucocorticoids to rodents (Watanabe et al., 1992) or nonhuman primates (Sapolsky et al., 1990) results in the degeneration of vulnerable hippocampal neurons, especially CA3 pyramidal cells. Animals exposed to high physiological levels of corticosterone (CORT) exhibited a persistent depletion of hippocampal CORT receptors and evidence of an impaired HPA axis (Sapolsky et al., 1983). Furthermore, a recent study (Lupien et al., 1998) has shown that in human aging, higher cortisol levels correlated longitudinally with greater hippocampal volume loss. In addition to direct neurotoxicity, glucocorticoid exposure may also enhance neuronal vulnerability to other insults, including hypoxia/ischemia (Tombaugh and Sapolsky, 1992), superoxide radical generators (McIntosh and Sapolsky, 1996), and hypoglycemia (Lawrence and Sapolsky, 1994).
Some studies in rodents have found age-related hippocampal neuronal loss based on neuron density studies (Landfield et al., 1981; Meaney et al., 1988; Issa et al., 1990). However, studies using unbiased stereological techniques have not found age-related loss of neurons in the CA1–CA3 hippocampal regions in humans, monkeys, or rodents (West, 1993; Rapp and Gallagher, 1996; Rasmussen et al., 1996). Differences in sampling procedures and counting methods could possibly account for some of the discrepancies between studies. Aged animals have decreases in hippocampal CORT receptors and evidence of an impaired HPA axis (Sapolsky et al., 1983) similar to animals exposed to high physiological levels of CORT. Although both aging and neurotoxic exposure produce loss of vulnerable hippocampal neurons, the combination of neurotoxic exposure and aging may have a synergistic effect through enhanced vulnerability to cell damage (Sapolsky, 1992). The analogy between aging and glucocorticoid exposure-induced neuronal vulnerability raises the question of the nature of the interaction between glucocorticoids and age. Because neuropathological studies of cell loss in Alzheimer’s disease have been associated with volume loss in magnetic resonance imaging (MRI) studies, observable volume loss in MRI studies of depression may also result from cell loss. The present study was intended to use advances in the precision of MRI of the brain to quantify hippocampal volumes in a series of normal and depressed human subjects in mid and late life. We sought to test the hypothesis that both increasing age and increasing duration of depression would be independently associated with reductions in hippocampal volume. In addition we tested whether there would be an interaction between the two variables to produce synergistic effects in reducing hippocampal volume.
MATERIALS AND METHODS
Subjects. Depressed subjects were recruited to the outpatient psychiatry service at Washington University School of Medicine (WUSM), primarily from referrals from other psychiatrists who knew the inclusionary and exclusionary criteria (17/24 subjects) and also from advertisements to the general public (7/24 subjects). Control subjects were recruited primarily from the Aging and Development Project maintained by the Psychology Department at Washington University (13/24 subjects) and also from notices posted at the medical center (11/24 subjects). Subjects were recruited over a 4 year period. Subjects ranged in age from 23 to 86 years (mean = 54; median = 56). Inclusionary criteria were a history of recurrent major depression, with at least one previous episode requiring psychiatric treatment, female gender, right-handedness, and no medical illness potentially affecting the CNS. No subjects with current acute depression were included in the study to eliminate potential confounds in MRI volumetric measurements related to state-dependent changes such as hypercortisolemia. None of the subjects had been acutely depressed within the past 4 months. Only women were selected to decrease the incidence of occult cardiovascular disease and to minimize gender-based brain differences (Aboitiz et al., 1992). A case-control design was used to match patients with a history of depression (post-depressed) for age and educational level within 2 years. Groups were matchedpost hoc for height, a predictor of general brain size (Andreason et al., 1994). Potential subjects were screened by one of us (Y.I.S.) using a questionnaire, medical history, review of medical records, and physical exam to exclude those with medical problems potentially affecting the CNS. Exclusionary conditions included a current or past neurological disorder, head trauma, uncontrolled hypertension, myocardial infarction or ischemia, diabetes, Cushing’s disease, steroid use, and drug/alcohol abuse. All subjects had documented normotension. In addition, subjects who had received more than three courses of electroconvulsive therapy (ECT) were excluded. Subjects gave written informed consent before inclusion in the study.
Clinical assessment was conducted by a psychiatrist (Y.I.S.) experienced in the use of the Diagnostic Interview for Genetic Studies (DIGS). The DIGS is a structured interview with high reliability (Nurnberger et al., 1993) that was used to make the diagnosis of past episodes of recurrent major depression by Diagnostic and Statistical Manual of Mental Disorders-IV criteria and to exclude other psychiatric diagnoses. The DIGS was used to assess both depressed and control subjects. Current antidepressant status and dosage were determined. To determine the presence and severity of any current depressive symptoms, all patients and controls were assessed using the Hamilton Rating Scale for Depression (HAMD).
Estimation of total time depressed. The number of symptoms and duration (days) of each episode were determined using the DIGS, as described above to define episodes of major depression. This was conducted using the Post Life Charting Method (Post et al., 1988) to anchor each episode. The total cumulative duration of depression was then calculated, summing over all episodes. Rigorous diagnostic criteria were used to establish each episode, and whenever possible, corroborating information was obtained from family members or treating psychiatrists, because in depressed populations, self-reporting may underestimate the duration of earlier episodes. Patients with more severe histories of depression, such as those in the current study, have been shown to have greater stability of diagnosis (Rice et al., 1987).
Neuropsychological testing. To determine the functional significance of brain volumetric changes, neuropsychological testing was performed. Subjects were administered the following tests: Wechsler Memory Scale-Revised (WMS-R), Logical Memory I (LMI) and II (LMII) subtests (Wechsler, 1987); Auditory Verbal Learning Test (AVLT) (Rey, 1964); Trail Making Test, parts A and B; and Weschler Adult Intelligence Scale-Revised (WAIS-R), Information and Block Design subtests (Wechsler, 1981). All tests were administered before administration of the dexamethasone suppression test (see below).
The Logical Memory subtest was administered according to the standardized protocol outlined in the WMS-R Manual (1987). Two stories were read aloud to the subject by the examiner. The subject was asked to repeat the story using as many of the same words as they could recall from memory after each story. One point was given for each verbatim or accepted alternate response phrase. After a 30 min delay the subject was asked to recall each story again. Subjects’ responses were recorded on audiotape and transcribed to ensure accurate scoring. Scores were tabulated for each story as well as the total score for the immediate (LMI) and delayed (LMII) trials.
The AVLT was administered using a standard protocol (Lezak, 1995) to present a 12-word list five times followed each time by recall of as many words as possible, then a second 12-word list with recall. After a 30 min delay subjects were asked to recall the first list again. One point was given for each recalled word during each presentation. The sum of the total number of words recalled in presentations one through five was also calculated.
Trails A and B were administered in two parts according to a standard protocol (Lezak, 1995). On part A, subjects were asked to draw a line to connect consecutively numbered circles as quickly as possible, with time necessary to complete the task recorded in seconds. On part B, subjects were asked to draw a line connecting consecutively numbered and lettered circles as quickly as possible, alternating between numbers and letters (1 to A, A to 2, 2 to B, etc). Time to completion was recorded in seconds.
WAIS-R Information was administered according to the WAIS-R Manual (1981). Twenty-nine questions arranged in order of increasing difficulty were read directly from the manual, and the subjects’ responses were recorded. The test was discontinued after five consecutive failures. One point was scored for each correctly answered question. WAIS-R Block Design also was administered according to the WAIS-R Manual (1981). Subjects were presented with designs requiring assembly of four blocks (first five designs) or nine blocks (last four designs). For each design the subject was shown a card with a picture of the design. There was a 1 min time limit on the first five designs and a 2 min time limit on the last four designs. The test was discontinued after two consecutive failed design constructions. Subjects were given points according to time needed for completion of each correctly assembled design, and the total earned points was calculated.
Magnetic resonance imaging. MRI scans were obtained using a Magnetom SP-4000 1.5T imaging system (Siemens Medical Systems, Iselin, NJ), and a standard Siemens 30 cm circularly polarized radio frequency (rf) head coil. Anatomic images consisted of 128 contiguous 1.25-mm-thick sagittal slices and were acquired using magnetization-prepared rapid gradient echo (MPRAGE) acquisition. No sedation was used during scanning. Specific MPRAGE scanning parameters were as follows: TR = 10 msec, TE = 4 msec, inversion time = 300 msec, flip angle = 8, matrix = 256 × 256 pixels, voxel size = 1 × 1 × 1.25 mm, slice thickness = 1.25 mm.
Image preprocessing. A graphics workstation (Sun Sparcstation 20, Sun Microsystems, Mountain View, CA) was used for initial image processing using ANALYZE software (Biomedical Imaging Resource, Mayo Foundation) (Robb, 1990). Images were interpolated to 0.5 mm sections. They were then reoriented to the anterior-posterior commissure plane for standard alignment. To minimize inter-scan variations, MR images underwent gray-scale normalization. Cylindrical regions-of-interest subvolumes inclusive of the amygdala and hippocampus were generated to aid in tissue classification. These subvolumes were analyzed using PeakFit software (Jandel Scientific, San Rafael CA), by the Marquardt-Levenberg algorithm for nonlinear curve fitting (Donald and Marquardt, 1963). The images were then scaled to create high contrast between gray and white matter in an eight-bit format.
Stereological method and reliability. Two raters (Y.I.S., M.S.) measured unilateral hippocampal gray matter volumes after training and assessment standardization with a neuroradiologist (M.H.G.) expert in hippocampal neuroanatomy. Stereological estimation methods, which have been used with precision in microscopy and MR volume determination (Mayhew, 1992) were used to estimate all hippocampal, amygdala, and whole brain volumes. Sampling parameters and grid size were set to yield at least 150 “hits” per measurement, a number that has previously been determined to yield reliable measurements in brain volume determination (Gundersen, 1988). From three-dimensional MRI cubical subvolumes composed of 0.5 × 0.5 × 0.5 mm voxels, coronal slices were sampled every 1.5 mm for hippocampus, every 1.0 mm for total amygdala, and every 0.5 mm for amygdala core nuclei. A rigid grid of points was then superimposed on the images. Grid points falling within the hippocampal, total amygdala, or amygdala core nuclei gray matter (see definitions below) were counted. Measurements were made using MEASURE (courtesy of P. Barta, Johns Hopkins University) in the coronal orientation, but the program displays grid points simultaneously in three orthogonal perspectives: sagittal, axial, and coronal (Fig. 1) to aid in anatomic localization. Volume estimates were calculated by MEASURE based on the number of selected grid points. Raters were blind to subject identity and measured left and right volumes separately. Mean volumes were determined from an average of four measurements of each volume, two measurements each by two independent raters for hippocampal, amygdala core nuclei, and total cerebral volumes. Means for total amygdala volumes, left and right, were determined from an average of two independent measures by the same rater. Intra-rater and inter-rater reliability were determined using the intraclass R. Intra-rater correlation coefficients were calculated for total (0.94 and 0.95), left (0.90 and 0.95), and right (0.95 and 0.98) hippocampal volumes, total (0.97 and 0.96), left (0.94 and 0.93), and right (0.92 and 0.90) amygdala core nuclei volumes, total (0.90), left (0.86), and right (0.88) whole amygdala volumes, and whole brain volumes (0.93 and 0.93). Inter-rater correlation coefficients were calculated for total (0.95), left (0.90), and right (0.95) hippocampal volumes, total (0.89), left (0.90), and right (0.82) amygdala core nuclei volumes, and whole brain volumes (0.92). For hippocampus, 48 pairs of intra-rater measurements and 48 pairs of inter-rater comparisons were made. For amygdala, 48 pairs of intra-rater and inter-rater core amygdala measurements were made, and 48 pairs of intra-rater measurements were made for whole amygdala by one rater. For whole brain, 48 pairs of intra-rater reliability measurements were made, and 28 pairs of inter-rater reliability measurements were made.
Fig. 1.
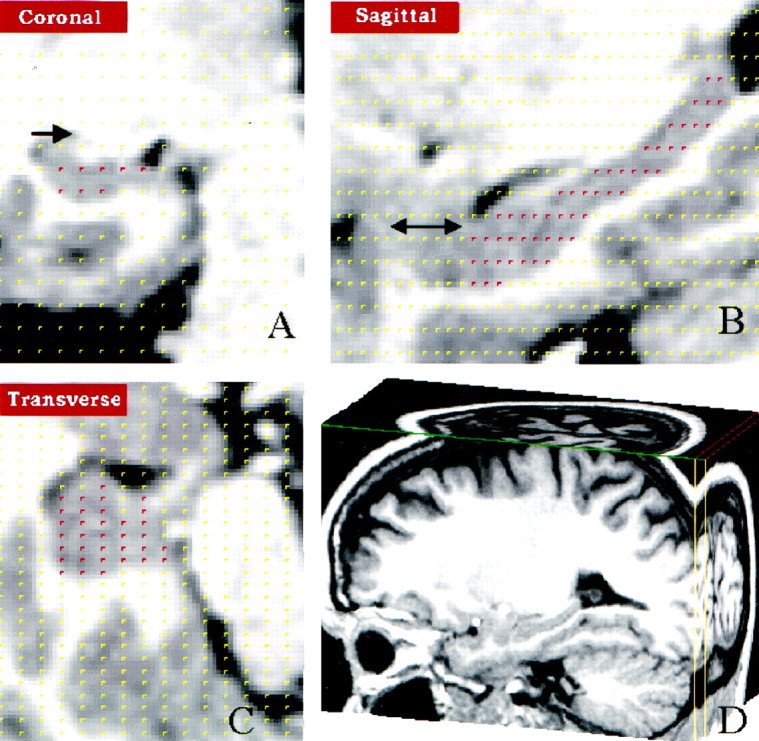
Hippocampal volume measurement using stereology.A, Coronal section through the hippocampus. Points within the hippocampus highlighted in red were selected from a randomly placed 5 × 5 mm2 grid overlying the hippocampus and are simultaneously displayed in (A) coronal, (B) sagittal, and (C) horizontal views. Arrowindicates the caudate (A), and ↔ indicates the amygdala 171 (B). D, Cubic volumes containing the hippocampus were sectioned out from the total brain volume.
Anatomic definition of the hippocampus. Anatomical boundaries were defined by specific rules (Duvernoy, 1988; Bartzokis et al., 1993) as follows: every third coronal slice was assessed, beginning with the posterior-most slice in which the hippocampus was visualized. Orthogonal views were consulted in cases of anatomic uncertainty, although the coronal view retained priority. Posteriorly, the tail of the hippocampus continues as the indusium griseum, a thin strip of gray matter overlying the surface of the corpus callosum. For purposes of measurement, the posterior-most slice for inclusion was defined as the slice where the hippocampus first appeared adjacent to the trigone of the lateral ventricle. Included tissues were an elongated gray matter complex bordered superiorly by the fornix-fimbria white matter junction, inferiorly by parahippocampal gyrus white matter, medially by the subarachnoid spaces of various cisterns (e.g., ambient cistern), and laterally by the CSF-filled lateral ventricle. The gray matter complex included the cornu ammonis (CA), dentate gyrus, and subiculum, (i.e., the head and body of the hippocampus were included). The vertical digitation of the head of the hippocampus, which curves up and medial to the amygdala in coronal sections, was included. Excluded tissues were the fornix-fimbria white matter complex; the alveus, the intraventricular white matter covering of the hippocampus; the white matter of the parahippocampal gyrus; various fluid-filled spaces including ventricles, subarachnoid spaces, and sporadic fluid-density spaces in the hippocampus complex; and the amygdala proper and the white matter border with it, a thin white matter line, discernible in high resolution MR images, which separated the hippocampus from the amygdala.
The subiculum extends medially from the cornu ammonis. The superior component of the CA does not extend medially far enough to aid in demarcating the border of the hippocampus, and the inferior component of the CA is in direct continuity with the subiculum. Because no clear gross anatomic separation exists between the hippocampus, subiculum, presubiculum, or parasubiculum, we adopted a previous procedure to include the subiculum in the measured volume of the hippocampus (Bartzokis et al., 1993).
Anatomic definition of the amygdala core nuclei. As described (Sheline et al., 1998), these structures were defined by the white matter tracts (longitudinal association bundles) surrounding them. Included were the basal nucleus, accessory basal nucleus, and the lateral nucleus (medial portions). Excluded structures were the periamygdaloid areas, the medial nucleus, and the central nucleus.
Anatomic definition of the noncore amygdala. The amygdala was visualized in three planes simultaneously during measurement, and some boundaries were better visualized in one plane than in another. The noncore amygdala was defined by measuring the total amygdala and subtracting the core amygdala. The anterior boundary of the amygdala, visualized in coronal section, was the first section in which the temporal stalk connected to the white matter of the insula. Dorsally, visualized in coronal section, the border was defined in anterior sections by the endorhinal sulcus between the basal forebrain and temporal lobe, and posteriorly in sagittal sections by a horizontal to the posteroinferior edge of the temporal stem with the temporal horn of the lateral ventricle. Ventrally, visualized in sagittal section, the amygdala was bounded by a horizontal line connecting to the ventral/anterior edge of the hippocampus. Posteriorly, seen in sagittal section, the amygdala was bounded by its border with the hippocampus. Medially, seen in coronal section, the amygdala was bounded by subarachnoid space. Laterally, seen in coronal section, the amygdala was bounded by white matter. These boundaries defined the total amygdala. The noncore amygdala was defined as the total amygdala minus the core amygdala.
Total cerebral volume. All brain tissue of the cerebral hemispheres, both gray and white matter, was included in the assessment of total cerebral volume. The midbrain superior to the pons was also included. The superior border of the pons was chosen as the point of demarcation because it is easily recognizable. This volume measurement was made using stereological methods as described above.
Cortisol levels. Blood samples were obtained to measure cortisol concentrations at 8:00 A.M. and 4:00 P.M. on the day before (baseline) and the day after oral administration of 1 mg dexamethasone at 11:00 P.M. (dexamethasone suppression test). Plasma was obtained within 1 hr of obtaining each sample by centrifuging the sample at ≅1000 × g for 15 min. All plasma samples were stored at −20°C until assay by fluorescence polarization immunoassay using the TDX analyzer and a commercially available kit (Abbott Laboratories).
Data analysis. Two-tailed paired t tests were used to compare post-depressed and control subjects on all demographic and MRI measures. The Pearson correlation was used to determine the significance of correlations among and between MRI measures and total cumulative duration of depression. Before using the Pearson correlation, variables were tested to determine whether they met criteria for normal distribution (Kolmogorov–Smirnov test). Pearson correlations were used to determine the correlation between age and hippocampal volumes separately in both the depressed and control samples, as well as in the total sample. A multiple regression with age and depression duration as independent variables was used to test these effects on hippocampal volumes. To examine the effect of age on hippocampal volumes, after removing the effect of depression duration, a regression analysis was conducted using age versus hippocampal volumes corrected for depression duration. This correction factor was derived from the multiple regression analysis with age and depression duration as independent variables and hippocampal volume as the dependent variable. In addition, to determine whether there was an interaction between age and depression status in predicting hippocampal volumes, these variables were combined into a new variable, and a multiple regression analysis was run using all three variables.
RESULTS
Demographic and cortisol data
Table 1 summarizes demographic and cortisol data for the 24 post-depressed subjects and their case-matched controls. The HAMD scores are consistent with the absence of acute current depression. Post-depressed subjects had a mean of 4.8 lifetime episodes of major depression (range 1–18) accounting for a lifetime mean of 1058 d depressed (range 21–3752; median, 624). Five subjects had received electroconvulsive therapy (ECT) previously during the course of their treatment and had elapsed times since last ECT treatment of 34, 30, and 14 years, and 7 and 11 months. Sixteen of the 24 post-depressed subjects were currently receiving antidepressants: selective serotonin reuptake inhibitors in four cases; tricyclic antidepressants in five cases; trazodone in two cases; and maprotiline, moclobemide, mirtazapine, nefazodone, and buproprion in one case each. Two post-depressed patients had previously been treated with neuroleptics: one with chlorpromazine and one with haloperidol. In both cases treatment duration was brief, and neither patient was psychotic at the time of treatment. Cortisol data were obtained from 17 of the total of 24 subject pairs. There was no difference between post-depressed and matched controls in either baseline cortisol levels (p = 0.56) or post-dexamethasone cortisol levels (p = 0.38) (Table 1), as would be expected in subjects who were not acutely depressed.
Table 1.
Demographic and clinical characteristics of subjects with a history of major depression and matched controls
| Variable | Depressed subjects (n = 24) | Matched controls (n = 24) | Paired t test | |||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | t | p | |
| Age (year) | 52.8 | 18.4 | 52.8 | 17.8 | 0.0001 | 0.99 |
| Education (year) | 16.3 | 3.5 | 16.3 | 4.1 | −0.08 | 0.94 |
| Height (cm) | 162.1 | 5.4 | 163.8 | 6.9 | −1.2 | 0.24 |
| Cortisol (μg/l) | ||||||
| Baseline | 21.86a | 11.70 | 21.37b | 10.13 | −0.511-c | 0.62c |
| DST | 1.99a | 1.26 | 1.99b | 1.13 | −0.98c | 0.35c |
| HAMD | 5.9 | 4.8 | 1.3 | 1.5 | 4.7 | 0.0001 |
| Days depressed | 1058 | 1032 | ||||
Cortisol levels based onn = 17.
Cortisol levels based on n = 17.
Calculated from 14 matched pairs.
In this sample, 26 women were postmenopausal and 22 were premenopausal. Of the 26 postmenopausal women, 14 were from the post-depressed group and 12 were controls (χ2 p value = 0.67). In the total sample, 17 women were on hormone replacement therapy. Of women on hormone replacement therapy, 9/17 were post-depressed and 8/17 were controls (χ2 p value = 0.85).
Brain volumetric data
Table 2 summarizes brain volumetric data for post-depressed subjects and matched controls. Mean total, left, and right hippocampal gray matter volumes were significantly smaller for post-depressed subjects than for controls. On average the differences were 9, 10, and 8% for the total, left, and right volumes, respectively (Fig. 2). Mean total, left, and right amygdala core nuclei volumes were significantly smaller in post-depressives than in control subjects; on average 13, 13, and 12% for total, left, and right volumes, respectively (Table 2), whereas amygdala noncore volumes, total amygdala volumes, and total cerebral volumes did not differ. Examining hormone replacement therapy alone as a predictor of total hippocampal volume, there was no statistically significant difference in volume between subjects on hormone replacement therapy (4699 mm3) and those not on therapy (4780 mm3) (p = 0.67). Postmenopausal status alone did not predict total hippocampal volume (4845 mm3 premenopausal vs 4620 mm3 postmenopausal; p = 0.23).
Table 2.
Comparison of subjects with a history of major depression and matched controls on brain volumetric data
| Variable | Depressed (n = 24) | Matched controls (n = 24) | Paired t test | |||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | t | p | |
| Hippocampal gray matter volume (mm3) | ||||||
| Left | 2230 | 323 | 2482 | 305 | −2.9 | 0.008 |
| Right | 2264 | 320 | 2468 | 309 | −2.3 | 0.03 |
| Total | 4496 | 602 | 4951 | 601 | −2.7 | 0.01 |
| Amygdala gray matter volume (mm3) | ||||||
| Left core | 531 | 93 | 610 | 106 | 3.3 | 0.004 |
| Right core | 514 | 97 | 585 | 102 | 2.8 | 0.009 |
| Total core | 1045 | 180 | 1196 | 202 | 3.2 | 0.004 |
| Left noncore | 1081 | 298 | 1111 | 253 | −0.44 | 0.66 |
| Right noncore | 1147 | 341 | 1131 | 241 | 0.24 | 0.81 |
| Total noncore | 2228 | 610 | 2242 | 480 | −0.11 | 0.91 |
| Total cerebral volume (mm3 × 103) | ||||||
| Whole brain | 1075 | 170 | 1078 | 168 | −0.08 | 0.94 |
Fig. 2.

Left and right hippocampal gray matter volumes. The lines connect the hippocampal volumes for each subject pair consisting of subject with a history of major depression (post-depressed) and the age, gender, education, and height-matched case control; the mean and the SDs for each group are also shown.
Correlations between hippocampal gray matter volumes, age, menopausal status, and total time depressed
In the total sample, the correlation between age and total hippocampal volume was r = −0.19 (p = 0.20). For post-depressed subjects the correlation between total hippocampal volume and age wasr = −0.32 (p = 0.13). In control subjects the correlation between age and total gray matter hippocampal volume was r = −0.08 (p = 0.71). Analysis of interaction indicated that there was no interaction between age and depression status (p = 0.43). In contrast, there was a significant inverse correlation between total days depressed and total hippocampal gray matter volume (r = −0.60; p = 0.002) (Fig. 3). This was true for both left hippocampal gray matter volume (r = −0.57,p = 0.003) and right hippocampal gray matter volume (r = −0.55, p = 0.005).
Fig. 3.

Correlation between duration of depression and hippocampal volume. The Pearson correlation between cumulative lifetime total days of major depression was derived from the Diagnostic Interview for Genetic Studies using the Post Life Charting Method (Post et al., 1988) and the total hippocampal gray matter volumes.
To correct for a contribution of depression duration to the relationship between age and hippocampal volume, a regression analysis was conducted using age versus hippocampal volume corrected for depression duration. This analysis showed no significant correlation with age (r = −0.17, p = 0.26) (Fig.4).
Fig. 4.

Correlation between age and total hippocampal gray matter volumes corrected for depression duration. The correction factor was derived from a multiple regression ANOVA using age and depression duration as independent variables and hippocampal gray matter volume as the dependent variable (see Materials and Methods).
A multiple regression analysis did not find a significant relationship (p = 0.61) using hormone replacement therapy, post-menopausal status, and age as independent variables and total hippocampal volume as the dependent variable. Examination of left and right hippocampal volumes separately also did not yield significant relationships.
Correlations between hippocampal gray matter volumes and amygdala core nuclei volumes
The correlation between total hippocampal gray matter volume and total amygdala core nuclei volume for post-depressed subjects wasr = 0.68, p = 0.0001 (Fig.5), and this held for the left (r = 0.65, p = 0.0004) and right sides (r = 0.58, p = 0.002) as well. In control subjects these respective correlations were r = 0.50 (p = 0.01), r = 0.54 (p = 0.006), and r = 0.42 (p = 0.04). In contrast, the correlations in post-depressed subjects between total and both left and right hippocampal gray matter volumes and total (r = −0.11,p = 0.63) (Fig. 5), left (r = 0.15, p = 0.49), and right (r = −0.02,p = 0.93) amygdala noncore nuclei volumes, respectively, were not significant. The respective correlations in control subjects (r = 0.46, p = 0.02;r = 0.45, p = 0.03; andr = 0.42, p = 0.04) with amygdala noncore nuclei volumes were significant.
Fig. 5.

Correlations between hippocampal and amygdala volumes. A, The Pearson correlation between total hippocampal gray matter volume and the total amygdala core nuclei volume; B, the correlation between total hippocampal gray matter volume and total amygdala noncore volume in subjects with a history of major depression.
Neuropsychological data
As shown in Table 3, scores for list learning, total recall and list learning, long delay were significantly decreased in subjects with a history of depression compared with controls. Paragraph recall scores did not achieve significance. Trails A, Trails B, and WAIS information subscale scores were not significantly different between the groups. The WAIS block design subscale score was significantly less in subjects with a history of depression than in control subjects.
Table 3.
Comparison of subjects with a history of major depression and matched controls on neuropsychological variables
| Variable | Depressed subjects (n = 24) | Matched controls (n = 24) | Paired t test | |||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | t | p | |
| List learning (total) | 45.6 | 8.1 | 49.2 | 5.6 | 2.5 | 0.02 |
| List learning (delayed) | 9.4 | 2.4 | 10.6 | 1.6 | 2.5 | 0.02 |
| Trails A | 35.5 | 16.6 | 32.6 | 20.8 | −0.70 | 0.49 |
| Trails B | 85.7 | 51.8 | 69.6 | 32.8 | −1.6 | 0.12 |
| Logical memory | 25.1 | 9.0 | 27.2 | 6.6 | 1.0 | 0.31 |
| WAIS information | 21.4 | 4.9 | 21.6 | 4.1 | 0.17 | 0.87 |
| WAIS block | 29.9 | 10.4 | 33.9 | 10.0 | 2.4 | 0.03 |
Effect of removing subjects who had received ECT
The data analyses comparing hippocampal gray matter volumes between post-depressed and control subjects were rerun after removal of the five subjects who had received ECT and their matched controls. There was still a significant difference in total (t = −2.1, p = 0.04) and left (t = −2.4,p = 0.03) hippocampal gray matter volumes and a trend toward a difference in right (t = −1.8,p = 0.08) hippocampal volumes between post-depressed and control subjects. In post-depressed subjects there was also a significant correlation between days depressed and both left (r = −0.59, p = 0.005) and right (r = −0.53, p = 0.01) hippocampal volumes.
DISCUSSION
The main findings of this study are the lack of a correlation between age and hippocampal volume in medically healthy subjects and the finding of smaller hippocampal volumes in subjects with a history of depression. These subjects also had evidence of poorer verbal memory performance, despite being in remission. Extending our pilot study (Sheline et al., 1996), we report that 24 women with a history of depression, but no medical comorbidity, had bilaterally smaller hippocampal volumes than age-matched controls. Findings of decreased hippocampal volumes have been reported in acutely depressed subjects (Narayan et al., 1998) and in patients suffering from post-traumatic stress disorder (Bremner et al., 1995). We show that post-depressed women exhibited abnormalities on a neuropsychological measure of hippocampal function, the AVLT, suggesting that their reduced hippocampal volumes were the result of functionally significant damage. Furthermore, hippocampal volumes correlated inversely with the cumulative lifetime duration of depression. In contrast to differences in hippocampal volumes and AVLT scores, post-depressed patients did not differ from control subjects in overall brain size or on general intellectual performance as assessed by the WAIS information subscale.
Given age- and depression-related increases in medical conditions, it is critical to know whether patients with changes in brain structure volumes had concurrent physical illnesses that could enhance neuronal vulnerability. In the present study, unlike most previous studies, we selected only subjects with no current or past medical or neurological conditions. An increased incidence of microinfarction has been linked to both chronic hypertension (Kobayashi et al., 1991) and diabetes (Aronson, 1973; Desmond et al., 1993). We excluded patients with any history of substance abuse because alcohol dependence can cause cerebral atrophy (Charness, 1993). Patients with a history of myocardial infarction were excluded because it is a cerebrovascular disease risk factor and produces a large independent increase in the incidence of depression (Carney et al., 1987).
Contrary to our initial hypothesis, there was not a significant age-related hippocampal volume loss in either post-depressed or control subjects. Some previous studies of hippocampal volume in “normal” aging found a decrease starting at age 50 (Jernigan et al., 1991), whereas others did not (Lim et al., 1990). Our sample showed slight (∼1% per decade) nonsignificant decreases in hippocampal volumes with age, more so in the depressed population, although there was no interaction between age and depression status. It is possible that aging per se may not lead to neurodegeneration in the hippocampus but rather may increase vulnerability to other factors causing cell death, and these factors may have differed across study populations. It is possible that by selecting subjects without medical comorbidity we may have created a sample of “supernormals” who do not show a decrease in brain structure volumes with age.
One confounding variable is ECT. Although there has been no direct evidence demonstrating ECT-induced structural brain changes (Devanand et al., 1994), animal electroconvulsive stimulation (ECS) studies suggest that brief kindled seizures may induce selective hippocampal neuronal loss (Cavazos et al., 1994). ECS exposure sufficient to induce kindling, however, far exceeds the human equivalent ECT electrical doses. We did not exclude ECT-treated patients, because they had the most severe histories of depression. A post hoc analysis after removing the five ECT-treated subjects showed a significant difference in total hippocampal gray matter volume (p = 0.04) and a correlation between total days depressed and total hippocampal gray matter volume (p = 0.004). Another confound was ongoing antidepressant therapy in most post-depressed subjects, although there is no known evidence that therapeutic levels are neurotoxic or that antidepressant exposure reduces brain volumes.
Additional potentially confounding variables, which we examinedpost hoc, were menopausal status and hormone replacement therapy. The effects of estrogen on rat hippocampus dendritic spine density (Wooley et al., 1997) and on neuropsychological function in postmenopausal women in some studies (Resnick et al., 1998) make this an important variable to examine. We did not find any significant differences between the groups in either postmenopausal or hormone replacement status, making it unlikely that these factors could account for differences in brain volumes. When we examined the issue of whether there were differences in hippocampal volume between postmenopausal and premenopausal subjects and between subjects who had received hormone replacement therapy versus those who had not, we did not find significant differences. However, we have a small sample size, and it would be interesting to examine this question in a larger study prospectively with concurrent serum hormone level determination.
Our study found significant differences between post-depressed subjects and controls in a neuropsychological measure of hippocampal function, AVLT list learning, total score, and delayed score. In subjects with more severe hippocampal damage caused by mesial temporal sclerosis, the degree of left hippocampal atrophy in patients with left temporal lobe epilepsy was correlated with severity of verbal memory deficits as measured by AVLT total and delayed recall (Kilpatrick et al., 1997). In addition, in nondemented subjects who developed Alzheimer’s disease during longitudinal followup (Tierney et al., 1996), the single biggest neuropsychological predictor was the AVLT score. Subjects in both groups had known hippocampal pathology that correlated with abnormalities in the AVLT. Our finding of abnormalities in AVLT in post-depressed subjects who also had hippocampal volume decreases appears to indicate hippocampal dysfunction. The groups did not differ, however, in logical memory test performance, another measure of hippocampal function (Squire, 1992). Depressed subjects did have lower WAIS block design subscale scores. Bremner et al. (1995) found that post-traumatic stress disorder patients performed more poorly on the Benton Visual Retention task, a test that reflects spatial memory and depends on right hippocampal integrity. Another possibility is that post-depressed subjects performed more poorly on timed tasks. However, both the WAIS block design and Trails A and B are timed tests; there was no difference in performance on Trails A and B between post-depressed and control subjects.
Although further study will be needed to fully elucidate the nature of the observed association between recurrent depression and volume reduction, evidence suggests that the link may be causal—that sustained depression in humans may cause hippocampal damage, resulting in a reduction of tissue volume and memory function. A limitation of the current study is that reliability of the Post Life Charting Method in determining retrospective measures of depression duration has not been established. There is no reason to believe that the data create a biased relationship between depression duration and hippocampal volumes, however, because volume data were obtained independently and blindly. The quantitative correlation between depression duration and hippocampal volume is explained most directly by such a causal relationship in that progressively more severe changes occur in the hippocampus as the length of time depressed, and presumably the length of time with abnormal GC levels, increases. Furthermore, in animals, repeated episodes of stress have been shown to damage the hippocampus. This appears to be mediated by GC-induced neurotoxicity (for review, see Sapolsky et al., 1986; Reagan and McEwen, 1997). In addition, there is evidence in normal elderly humans that long-term exposure to GCs predicts hippocampal atrophy and memory deficits (Lupien et al., 1998).
The relevance to depression of these studies showing that chronically elevated GC levels damage hippocampal neurons depends, however, on the assumption that depression is associated with dysregulation of the glucocorticoid system. Almost 40 years ago, excessive cortisol secretion during depression was first reported (Gibbons and McHugh, 1962). Hypercortisolism and insensitivity to feedback suppression during depression have been extensively investigated to determine the relative contributions of adrenal hypersensitivity to ACTH (Amsterdam et al., 1989), pituitary resistance to GC feedback (Holsboer et al., 1987), pituitary hypersensitivity to CRF and other hormones (Gold et al., 1984), and resistance at the hippocampus (Sapolsky et al., 1991;Young et al., 1991). Although we do not have cortisol levels from previous depressive episodes, many of our subjects had histories of hospitalization, ECT treatment, and multiple episodes, indicating relatively more severe major depression, a predictor of elevated cortisol and dexamethasone suppression test nonsuppression (Whiteford et al., 1987). Taken together, these findings suggest that HPA axis dysregulation in depression can produce repeated episodes of hypercortisolemia, which may result in hippocampal neurotoxicity.
The precise mechanisms whereby chronic glucocorticoid exposure leads to hippocampal cell death are not defined, but enhanced vulnerability to excitotoxicity may be a main factor (Armanini et al., 1990). We have shown a reduction in volumes of the core nuclei of the amygdala in recurrent major depression (Sheline et al., 1998) and now demonstrate a high correlation between hippocampal and amygdala core nuclei volume loss. Because glutamatergic pyramidal cells in the core nuclei of the amygdala are predominant and reciprocally connected with the hippocampus, it may be that overexcitation in one part of this circuit produces damage in connected structures. Furthermore, there was no difference in volumes of noncore amygdala nuclei, which contain primarily nonglutamatergic cells (Price et al., 1987).
In addition to glucocorticoid-induced neurotoxicity, other mechanisms consistent with hippocampal volume loss in recurrent depression include neurotoxic effects of corticotrophin-releasing factor (CRF) and a decrease in trophic factors such as brain-derived neurotrophic factor (BDNF). Elevated CRF levels have been described in both depressed humans (Nemeroff et al., 1984) and animal models of depression and have neurotoxic effects (Wong et al., 1995). Furthermore, CRF antagonists are neuroprotective against seizure-induced excitotoxicity (Maecker et al., 1997). Stress decreases the expression of BDNF (Duman et al., 1997), and BDNF is neuroprotective (Mamounas et al., 1995). These mechanisms—glucocorticoid toxicity, CRF neurotoxicity, and BDNF loss resulting in increased neuronal vulnerability—are not mutually exclusive and could all be operating to alter the vulnerability of hippocampal neurons in depression.
In summary, the present study shows an association between depression and structural changes in the hippocampus. It should be pointed out that volume changes cannot be equated with neuropathological studies directly performing cell counts; only the latter would provide direct evidence for cell loss. This gray matter hippocampal volume loss does not appear to be related to aging but could be related to factors commonly associated with aging, such as medical illnesses, which were not examined in this study. Depression-related volume loss does appear to be cumulative, suggesting that immediate recognition and treatment of depressive episodes is important in preventing cumulative damage that occurs with repeated episodes of depression.
Footnotes
This work was supported by National Institutes of Health Grants MH K07 1370 (Y.I.S.), MH 58444 (Y.I.S.), and MH 54731 (M.A.M), and by the Washington University General Clinical Research Center National Institutes of Health Grant 5M01 RR00036. We thank Dr. Rosalind Neuman for helpful statistical advice.
Correspondence should be addressed to Dr. Yvette I. Sheline, Department of Psychiatry, Box 8134, Washington University School of Medicine, 4940 Childrens Place, St. Louis, MO 63110.
REFERENCES
- 1.Aboitz F, Scheibel A, Zaidel E. Morphometry of the Sylvian fissure and the corpus callosum, with emphasis on sex differences. Brain. 1992;115:1521–1541. doi: 10.1093/brain/115.5.1521. [DOI] [PubMed] [Google Scholar]
- 2.Amsterdam J, Maislin G, Berwish N, Phillips J, Winokur A. Enhanced adrenocortical sensitivity to submaximal doses of cosyntropin in depressed patients. Arch Gen Psychiatry. 1989;46:550–554. doi: 10.1001/archpsyc.1989.01810060072011. [DOI] [PubMed] [Google Scholar]
- 3.Andreasen NC, Flashman L, Flaum M, Arndt S, Swayzee V, O’Leary DS, Ehrhardt JC, Yuh WTC. Regional brain abnormalities in schizophrenia measured with magnetic resonance imaging. JAMA. 1994;272:1763–1769. [PubMed] [Google Scholar]
- 4.Armanini MP, Hutchins C, Stein BA, Sapolsky RM. Glucocorticoid endangerment of hippocampal neurons is NMDA-receptor dependent. Brain Res. 1990;532:7–12. doi: 10.1016/0006-8993(90)91734-x. [DOI] [PubMed] [Google Scholar]
- 5.Aronson S. J. Intracranial vascular lesions in patients with diabetes mellitus. Neuropathol Exp Neurol. 1973;32:183–196. doi: 10.1097/00005072-197304000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Bartzokis G, Mintz J, Marx P, Osborn D, Gutkind D, Chiang F, Phelan CK, Marder SR. Reliability of in vivo volume measures of hippocampus and other brain structures using MRI. Magn Reson Imaging. 1993;11:993–1006. doi: 10.1016/0730-725x(93)90218-3. [DOI] [PubMed] [Google Scholar]
- 7.Bremner J, Randall P, Scott T, Bronen R, Seibyl J, Southwick S, Delaney R, McCarthy G, Charney D, Innis R. MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry. 1995;152:973–981. doi: 10.1176/ajp.152.7.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carney R, Rich M, teVelde A. Major depressive disorder in coronary artery disease. Am J Cardiol. 1987;60:1273–1275. doi: 10.1016/0002-9149(87)90607-2. [DOI] [PubMed] [Google Scholar]
- 9.Carroll R, Feinberg M, Greden J, Tarika J, Albala A, Haskett R, James M, Kronfol A, Lohr N, Steiner M, de Vigne J, Young E. A specific laboratory test for the diagnosis of melancholia. Standardization, validation, and clinical utility. Arch Gen Psychiatry. 1981;38:15–22. doi: 10.1001/archpsyc.1981.01780260017001. [DOI] [PubMed] [Google Scholar]
- 10.Cavazos JE, Das I, Sutula TP. Neuronal loss induced in limbic pathways by kindling: evidence for induction of hippocampal sclerosis by repeated brief seizures. J Neurosci. 1994;14:3106–3121. doi: 10.1523/JNEUROSCI.14-05-03106.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charness ME. Brain lesions in alcoholics. Alcohol Clin Exp Res. 1993;17:2–11. doi: 10.1111/j.1530-0277.1993.tb00718.x. [DOI] [PubMed] [Google Scholar]
- 12.Devanand DP, Dwork AJ, Hutchinson ER, Bolwig TG, Sackeim HA. Does ECT alter brain structure? Am J Psychiatry. 1994;151:957–970. doi: 10.1176/ajp.151.7.957. [DOI] [PubMed] [Google Scholar]
- 13.Donald J, Marquardt W. An algorithm for least-square estimation of nonlinear parameters. J Soc Indust Appl Math. 1963;11:431–441. [Google Scholar]
- 14.Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 15.Duvernoy HM. Human hippocampus: an atlas of applied anatomy. Bergmann Verlag; Munich: 1988. [Google Scholar]
- 16.Gibbons J, McHugh P. Plasma cortisol in depressive illness. J Psychiatry Res. 1962;1:162–171. doi: 10.1016/0022-3956(62)90006-7. [DOI] [PubMed] [Google Scholar]
- 17.Gold P, Chrousos G, Kellner C, Post R, Roy A, Avgerinos P, Schulte H, Oldfield E, Loriaux D. Psychiatric implications of basic and clinical studies with CRF. Am J Psychiatry. 1984;141:619–627. doi: 10.1176/ajp.141.5.619. [DOI] [PubMed] [Google Scholar]
- 18.Gundersen HJG, Bendtsen TF, Korbo N. Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. Acta Pathol Microbiol Scand. 1988;96:379–394. doi: 10.1111/j.1699-0463.1988.tb05320.x. [DOI] [PubMed] [Google Scholar]
- 19.Holsboer F, Gerken A, Stalla G, Muller O. Blunted aldosterone and ACTH release after human CRH administration in depressed patients. Am J Psychiatry. 1987;144:229–231. doi: 10.1176/ajp.144.2.229. [DOI] [PubMed] [Google Scholar]
- 20.Issa AM, Rowe W, Gauthier S, Meaney MJ. Hypothalamic-pituitary-adrenal activity in aged, cognitively impaired and cognitively unimpaired rats. J Neurosci. 1990;10:3247–3254. doi: 10.1523/JNEUROSCI.10-10-03247.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jernigan TL, Archibald SL, Berhow MT, Sowell ER, Foster DS, Hesselink JR. Cerebral structure on MRI, Part I: localization of age-related changes. Biol Psychiatry. 1991;29:55–67. doi: 10.1016/0006-3223(91)90210-d. [DOI] [PubMed] [Google Scholar]
- 22.Kilpatrick C, Murrie V, Cook M, Andrews D, Desmond P, Hopper J. Degree of left hippocampal atrophy correlates with severity of neuropsychological deficits. Seizure. 1997;6:213–218. doi: 10.1016/s1059-1311(97)80008-8. [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi S, Okada K, Yamashita K. Incidence of silent lacunar lesion in normal adults and its relation to cerebral blood flow and risk factors. Stroke. 1991;22:1379–1383. doi: 10.1161/01.str.22.11.1379. [DOI] [PubMed] [Google Scholar]
- 24.Landfield PW, Baskin RK, Pitler TA. Brain aging correlates: retardation by hormonal-pharmacological treatments. Science. 1981;214:581–584. doi: 10.1126/science.6270791. [DOI] [PubMed] [Google Scholar]
- 25.Lawrence MS, Sapolsky RM. Glucocorticoids accelerate ATP loss following metabolic insults in cultured hippocampal neurons. Brain Res. 1994;646:303–306. doi: 10.1016/0006-8993(94)90094-9. [DOI] [PubMed] [Google Scholar]
- 26.Lezak MD. Neuropsychological assessment, Ed 3. Oxford UP; New York: 1995. [Google Scholar]
- 27.Lim KO, Zipursky RB, Murphy GM, Jr, Pfefferbaum A. In vivo quantification of the limbic system using MRI: effects of normal aging. Psychiatry Res. 1990;35:15–26. doi: 10.1016/0925-4927(90)90005-q. [DOI] [PubMed] [Google Scholar]
- 28.Lupien SJ, de Leon M, deSanti S, Convit A, Tarshish C, Nair NPV, Thakur M, McEwen BS. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci. 1998;1:69–73. doi: 10.1038/271. [DOI] [PubMed] [Google Scholar]
- 29.Maecker H, Desai A, Dash R, Rivier J, Vale W, Sapolsky R. Astressin, a novel and potent CRF antagonist, is neuroprotective in the hippocampus when administered after a seizure. Brain Res. 1997;744:166–170. doi: 10.1016/s0006-8993(96)01207-3. [DOI] [PubMed] [Google Scholar]
- 30.Mamounas LA, Blue ME, Siuciak JA, Altar C. BDNF promotes the survival and sprouting of serotonergic axons in the rat brain. J Neurosci. 1995;15:7929–7939. doi: 10.1523/JNEUROSCI.15-12-07929.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mayhew TM. A review of recent advances in stereology for quantifying neural structure. J Neurocytol. 1992;21:313–328. doi: 10.1007/BF01191700. [DOI] [PubMed] [Google Scholar]
- 32.McEwen BS. Re-examination of the glucocorticoid hypothesis of stress and aging. In: Swaab D, Hofman M, Mirmiran M, Ravid R, van Leeuwen F, editors. Progress in brain research. Elsevier; New York: 1992. pp. 356–383. [DOI] [PubMed] [Google Scholar]
- 33.McIntosh LJ, Sapolsky RM. Glucocorticoids increase the accumulation of reactive oxygen species and enhance adriamycin-induced toxicity in neuronal culture. Exp Neurol. 1996;141:201–206. doi: 10.1006/exnr.1996.0154. [DOI] [PubMed] [Google Scholar]
- 34.Meaney MJ, Aitkin DH, van Berkel C, Bhatnagar S, Sapolsky RM. Effect of neonatal handling on age-related impairments associated with the hippocampus. Science. 1988;239:766–768. doi: 10.1126/science.3340858. [DOI] [PubMed] [Google Scholar]
- 35. Narayan M, Anderson E, Miller H, Staib L, Charney D, Bremner J. 1998. Hippocampal volume in MDD. Abstract presented at the annual meeting of the American Psychiatric Association, May 1998, Toronto, Canada
- 36.Nemeroff CB, Widerlov E, Walleus H, Karlsson I, Eklund K, Kilts CD, Loosen PT, Vale W. Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science. 1984;226:1342–1344. doi: 10.1126/science.6334362. [DOI] [PubMed] [Google Scholar]
- 37. Nurnberger J, Blehar MC, Kaufmann CA, York-Cooler C, Simpson SG, Harkavy-Friedman J, Severe JB, Malaspina D, Reich T, collaborators from the NIMH Genetics Initiative Diagnostic interview for genetic studies. Rationale, unique features, and training. Arch Gen Psychiatry 11 1993. 849 859; 863–864. [DOI] [PubMed] [Google Scholar]
- 38.Post RM, Roy-Byrne PP, Uhde T. Graphic representation of the life course of illness in patients with affective disorder. Am J Psychiatry. 1988;145:844–848. doi: 10.1176/ajp.145.7.844. [DOI] [PubMed] [Google Scholar]
- 39.Price J, Russchen F, Amaral D. The limbic region. II: The amygdaloid complex. In: Bjorkland A, Hokfelt T, Swanson L, editors. Handbook of chemical neuroanatomy, Vol 5. Elsevier; New York: 1987. pp. 279–388. [Google Scholar]
- 40.Rapp PR, Gallagher M. Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. Proc Natl Acad Sci USA. 1996;93:9926–9930. doi: 10.1073/pnas.93.18.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rasmussen T, Schliemann T, Sorensen JC, Zimmer J, West MJ. Memory impaired aged rats: no loss of principal hippocampal and subicular neurons. Neurobiol Aging. 1996;17:143–147. doi: 10.1016/0197-4580(95)02032-2. [DOI] [PubMed] [Google Scholar]
- 42.Reagan LP, McEwen BS. Controversies surrounding glucocorticoid-mediated cell death in the hippocampus. J Chem Neuroanat. 1997;13:149–167. doi: 10.1016/s0891-0618(97)00031-8. [DOI] [PubMed] [Google Scholar]
- 43.Resnick S, Maki K, Golski S, Kraut M, Zonderman A. Effects of estrogen replacement therapy on PET cerebral blood flow and neuropsychological performance. Horm Behav. 1998;34:171–182. doi: 10.1006/hbeh.1998.1476. [DOI] [PubMed] [Google Scholar]
- 44.Rey A. L’examen clinique en psychologie. Presses Universitaires de France; Paris: 1964. [Google Scholar]
- 45.Rice J, Endicott J, Knesevich M, Rochberg N. The estimation of diagnostic sensitivity using stability data: an application to major depressive disorder. J Psychiat Res. 1987;21:337–345. doi: 10.1016/0022-3956(87)90080-x. [DOI] [PubMed] [Google Scholar]
- 46.Robb RA. A software system for interactive and quantitative analysis of biomedical images. In: Hohne KH, Fuchs M, Pizer SM, editors. 3D imaging in medicine, Vol F. Nato ISI Series; 1990. pp. 333–361. [Google Scholar]
- 47.Sapolsky RM. Stress, the aging brain and the mechanisms of neuron death, pp 305–339. MIT; Cambridge, MA: 1992. Is this relevant to the human? [Google Scholar]
- 48.Sapolsky RM, Krey LC, McEwen BS. Corticosterone receptors decline in a site-specific manner in the aged rat brain. Brain Res. 1983;289:235–240. doi: 10.1016/0006-8993(83)90024-0. [DOI] [PubMed] [Google Scholar]
- 49.Sapolsky RM, Krey LC, McEwen BS. Prolonged glucocorticoid exposure reduces hippocampal neuron number: implications for aging. J Neurosci. 1985;5:1222–1227. doi: 10.1523/JNEUROSCI.05-05-01222.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocrinol Rev. 1986;7:284–301. doi: 10.1210/edrv-7-3-284. [DOI] [PubMed] [Google Scholar]
- 51.Sapolsky RM, Uno H, Rebert CS, Finch CE. Hippocampal damage associated with prolonged glucocorticoid exposure in primates. J Neurosci. 1990;10:2897–2902. doi: 10.1523/JNEUROSCI.10-09-02897.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sapolsky RM, Zola-Morgan S, Squire L. Inhibition of glucocorticoid secretion by the hippocampal formation in the primate. J Neurosci. 1991;11:3695–3704. doi: 10.1523/JNEUROSCI.11-12-03695.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA. 1996;93:3908–3913. doi: 10.1073/pnas.93.9.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sheline YI, Gado M, Price JL. Amygdala core nuclei volumes are decreased in recurrent major depression. NeuroReport. 1998;9:2023–2028. doi: 10.1097/00001756-199806220-00021. [DOI] [PubMed] [Google Scholar]
- 55.Squire LR. Declarative and nondeclarative memory: multiple brain systems supporting learning and memory. J Cognit Neurosci. 1992;4:232–243. doi: 10.1162/jocn.1992.4.3.232. [DOI] [PubMed] [Google Scholar]
- 56.Tierney MC, Szalai JP, Snow WG, Fisher RH, Nores A, Nadon G, Dunn E, St. George-Hyslop PH. Prediction of probable Alzheimer’s disease in memory-impaired patients: a prospective longitudinal study. Neurology. 1996;46:661–665. doi: 10.1212/wnl.46.3.661. [DOI] [PubMed] [Google Scholar]
- 57.Tombaugh GC, Sapolsky RM. Corticosterone accelerates hypoxia- and cyanide-induced ATP loss in cultured hippocampal astrocytes. Brain Res. 1992;588:154–158. doi: 10.1016/0006-8993(92)91356-j. [DOI] [PubMed] [Google Scholar]
- 58.Watanabe Y, Gould E, McEwen BS. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 1992b;588:341–345. doi: 10.1016/0006-8993(92)91597-8. [DOI] [PubMed] [Google Scholar]
- 59.West MJ. Regionally specific loss of neurons in the aging human hippocampus. Neurobiol Aging. 1993;14:287–293. doi: 10.1016/0197-4580(93)90113-p. [DOI] [PubMed] [Google Scholar]
- 60.Wechsler D. WAIS-R manual. The Psychological Corporation; New York: 1981. [Google Scholar]
- 61.Wechsler D. Wechsler Memory Scale-Revised Manual. The Psychological Corporation; San Antonio, TX: 1987. [Google Scholar]
- 62.Whiteford H, Peabody C, Csernansky J, Warner M, Berger P. Elevated baseline and postdexamethasone cortisol levels. A reflection of severity or endogeneity? J Affective Disord. 1987;12:199–202. doi: 10.1016/0165-0327(87)90027-9. [DOI] [PubMed] [Google Scholar]
- 63.Wong M, Loddick S, Bongiorno P, Gold P, Rothwell N, Licinio J. Focal cerebral ischemia induces CRH mRNA in rat cerebral cortex and amygdala. NeuroReport. 1995;6:1785–1788. doi: 10.1097/00001756-199509000-00019. [DOI] [PubMed] [Google Scholar]
- 64.Wooley C, Weiland N, McEwen B, Schwartzkroin P. Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor-mediated synaptic input: correlation with dendritic spine density. J Neurosci. 1997;17:1848–1859. doi: 10.1523/JNEUROSCI.17-05-01848.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Young EA, Haskett RF, Murphy-Weinberg V, Watson SJ, Akil H. Loss of glucocorticoid fast feedback in depression. Arch Gen Psychiatry. 1991;48:693–698. doi: 10.1001/archpsyc.1991.01810320017003. [DOI] [PubMed] [Google Scholar]