

Abstract
Alternative splicing of the avian trkB receptor generates an extracellular deletion (ED) isoform missing 11 amino acids from the neurotrophin-binding domain of the full-length (FL) receptor. When expressed in fibroblasts, the ED isoform exhibited restricted neurotrophin specificity compared with that of the FL receptor. Brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4) activated the FL receptor, as determined by tyrosine phosphorylation. However, only BDNF was capable of significant activation of the ED isoform, although to a reduced level. Because positively charged residues in NT-3 are important for binding to trkB, two negatively charged aspartate residues within the 11 amino acid motif of FL trkB were mutated to examine the role of electrostatic interactions on ligand binding. As found for the ED isoform, the FL mutated receptor displayed a similar loss of NT-3- and NT-4-mediated activation, in addition to a diminished responsiveness to BDNF. Because of these profound effects on ligand specificity, reverse transcription-PCR was used to understand the expression of the FL and ED receptor isoforms at the level of single neurons. The predominant expression pattern of either FL or ED isoforms in single embryonic DRG neurons establishes the existence of two subpopulations exhibiting differential responsiveness to trkB ligands, indicating that regulated splicing of the extracellular domain of trkB may serve as a mechanism to restrict neuronal responsiveness to the neurotrophins.
Keywords: trkB, alternative splicing, dorsal root ganglia, neurotrophin specificity, single-cell RT-PCR, chicken
Members of the neurotrophin gene family, including nerve growth factor, brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4), exert pleiotropic effects on neurons during nervous system development and in the adult (Korsching, 1993; Lindsay, 1994; Snider, 1994; Lo, 1995; Thoenen, 1995) and require stringent regulatory mechanisms to function properly. Neuronal responsiveness to the neurotrophins is determined not only by restricted expression and regulated release of the neurotrophins but also by the expression pattern of neurotrophin receptors and components of the signaling complex. The trk receptors, a family of receptor tyrosine kinases including trkA, trkB, and trkC, serve as the principal signal-transducing receptors for the neurotrophins (Barbacid, 1994). Neurons are capable of expressing a surprising diversity of trk receptor structural isoforms that arise via alternative splicing (Barbacid, 1994). The complex splicing patterns of the trk receptors suggest alternative splicing as an important additional means of regulating neuronal responsiveness to the neurotrophins.
Alternative splicing affects both the extracellular ligand-binding domains and the intracellular signal-transducing domains of the trks (Barbacid, 1994). Isoforms with small deletions within the extracellular domain have been described for trkA, trkB, and trkC (Meakin et al., 1992; Shelton et al., 1995; Garner et al., 1996). The extracellular deletion isoform of chick trkB (ED trkB) is a naturally occurring splice variant lacking 11 amino acids in the extracellular domain (Garner et al., 1996). The deletion occurs between the second immunoglobulin-like domain and the transmembrane domain of trkB and results from the alternative splicing of exon 9 (Strohmaier et al., 1996). Because Ig-like domain swapping has been shown to confer ligand specificity of the receptor (Perez et al., 1995; Urfer et al., 1995), a deletion in close proximity to this domain is likely to affect ligand specificity. In this study, comparison of the ligand specificity of the full-length (FL) and ED receptor isoforms revealed that ED trkB exhibits a more restricted neurotrophin specificity than does FL trkB. Although NT-3, NT-4, and BDNF activate FL receptors, only BDNF is capable of robust but decreased activation of ED receptors. The effect of the naturally occurring 11 amino acid deletion can be mimicked by mutation of two aspartate residues within the 11 amino acid motif of the full-length receptor, consistent with a role of electrostatic interactions in the binding of nonpreferred ligands to trkB.
The existence of trkB isoforms with distinct ligand specificities suggests that trkB-expressing (trkB+) neurons may regulate their neurotrophin responsiveness by alternative splicing within the extracellular domain. For this reason, we sought to determine whether there are distinct subpopulations of trkB+ neurons expressing either FL or ED trkB exclusively. Single-cell reverse transcription (RT)-PCR analysis of neurons of the embryonic avian dorsal root ganglia (DRG) revealed that ED and FL trkBs are expressed in predominantly distinct populations of neurons. Our results indicate alternative splicing within the extracellular domain of trkB as a mechanism for regulating the neurotrophin responsiveness of trkB-expressing neurons.
MATERIALS AND METHODS
Receptor expression in chicken embryonic fibroblasts
The cDNAs for chick FL and ED trkB subcloned previously into theEcoRI site of pBluescript SK+(BSK+) were cut with NotI downstream of the cDNA inserts. A ClaI site was engineered at the site of the NotI cut by means of an adaptor oligo (Hughes et al., 1987). The cDNAs were then excised from BSK+ with ClaI and subcloned into theClaI site of a replication-competent avian-specific retroviral construct, RCAS-A-BP (Hughes et al., 1987).
To generate the mutated full-length construct (designated FL*), we subcloned the cDNA for FL trkB into the HindIII site of BSK+, lacking the BamHI site in the multiple-cloning site. The clone was then cut with BamHI to cut the FL cDNA at the BamHI site located within the 11 amino acid motif and then blunt-ended with mung bean nuclease, followed by digestion of a downstream BspE1 site. In a separate reaction, two oligos, one encoding mutations of Asp residues to Ala, were used to amplify a 100 bp fragment of the extracellular domain spanning the BamHI and BspE1 sites. The upstream oligo encoding the double mutation was 5′-GCGCCAGCCGTTTATGAATATGAAACC-3′. The downstream oligo was 5′-GGAAACATCCGGAGAAGTGAT-3′. The double mutation also converted theBamHI site within the 11 amino acid motif to aKas1 site. The PCR product was cut with Kas1, blunted with Klenow, and cut with BspE1. This fragment was then cloned into the prepared FL construct in BSK+. Mutated clones were identified by Kas1 and BspE1 digestion, verified by sequencing, and subcloned into RCAS-A-BP (Hughes et al., 1987).
Retroviral constructs containing the cDNAs for FL, ED, and FL* trkB were electroporated into wild-type chicken embryonic fibroblasts (CEFs; United States Department of Agriculture, Avian Disease and Oncology, Ann Arbor, MI). Transfected CEFs were grown in M199:DMEM (1:1; Life Technologies, Gaithersburg, MD) supplemented with 3% fetal bovine serum, 2% chick serum, 1% glutamine, and 50 U/ml pen-strep (Life Technologies) in a 37°C, 5% CO2 atmosphere.
Stimulation and cell lysis
CEFs expressing FL, ED, or FL* trkB were grown to confluency on 100 mm tissue culture plates. For single-dose experiments, cells were stimulated for 5 min at 37°C with 50 ng/ml BDNF, NT-3, or NT-4 or remained untreated as a negative control. For dose–response experiments, cells were stimulated with 100, 50, 25, 10, 5, or 0 ng/ml BDNF or NT-3 for 5 min at 37°C. After stimulation, the medium was removed, and cells were washed briefly with ice-cold PBS containing 0.1 mm sodium orthovanadate. Cells were lysed for 30 min at 4°C in 1.5 ml of ice-cold lysis buffer (9.1 mm sodium diphosphate, 1.7 mm sodium monophosphate, 150 mm NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS, pH 7.25) supplemented with 20 μg/ml aprotinin, 1 mm PMSF, 1 mm sodium orthovanadate, and 1 μg/ml leupeptin. Lysates were collected and passed six times through a 22-gauge needle to ensure disruption of cells. Cellular debris was removed from the lysate by centrifugation at 15,000 ×g for 5 min, and the supernatant was assayed for total protein content using the Micro-BCA Protein Assay Kit (Pierce, Rockford, IL).
Wheat germ agarose precipitations and electrophoresis
Because the extracellular domain of trkB is highly glycosylated, FL, ED, and FL* trkB receptors were precipitated from cell lysates using wheat germ lectin conjugated to agarose. Precipitations were performed by adding 100 μg of wheat germ agarose (WGA) (Pharmacia, Piscataway, NJ) to lysate containing 0.25 mg of total protein. Receptors were precipitated overnight at 4°C with constant rocking. Precipitates were spun down at 5000 × g, and the WGA pellets were washed five times with 10 ml of lysis buffer supplemented with 0.1 mm sodium orthovanadate. Pellets were then resuspended in 50 μl of 2× sample buffer (6% SDS, 6% β-mercaptoethanol, and 1% bromophenol blue) and boiled for 5 min. Samples were spun briefly to pellet out the agarose, and the supernatant was loaded onto a 4–12% Tris-tricine gradient gel (Novex Electrophoresis, San Diego, CA) for separation of proteins. Proteins were then transferred to polyvinylidene fluoride membrane using the Novex XCell II Mini-Cell electroblotting system.
Immunoblotting
Blots were incubated in blocking buffer (6% BSA in TBS and 0.02% Tween) for 1 hr. For anti-phosphotyrosine blots, membranes were incubated with monoclonal anti-phosphotyrosine (4G10; Upstate Biotechnology, Lake Placid, NY; diluted 1:2000 in blocking buffer) for 1 hr, washed three times for 10 min each in TBS and Tween, and then incubated in an HRP-conjugated anti-mouse secondary antibody (diluted 1:5000 in 3% BSA, TBS, and Tween) for 1 hr. After incubation in secondary antibody, membranes were washed four times for 15 min each in TBS and Tween and analyzed using the ECL detection system (Amersham, Arlington Heights, IL). To verify receptor expression, blots were subsequently stripped in stripping buffer (100 mmβ-mercaptoethanol, 2% SDS, and 62.5 mm Tris-HCl, pH 6.7) for 30 min at 50°C and reprobed with an antibody raised against the extracellular domain of chick trkB [anti-BEC; generously provided by F. Lefcort, Montana State University (Von Bartheld et al., 1996)] diluted 1:1000 in blocking buffer. This antibody recognizes the FL, ED, and FL* receptors. Band intensities were quantified by laser densitometry on a SciScan 5000 laser densitometer. Multiple exposures were obtained for each blot, and densitometry measurements were made on films from shorter exposures with subsaturating band intensities. Phosphotyrosine (PY) signals were normalized to the level of receptor expression by dividing the raw PY densitometry reading by the corresponding raw BEC densitometry reading. Background phosphorylation (i.e., the normalized PY signal for the unstimulated condition) was then subtracted from each normalized experimental PY signal to examine only ligand-dependent receptor phosphorylation.
Single-cell RT-PCR
DRG dissociation and isolation of single cells.Fertilized White Leghorn chicken eggs were obtained from Squire Valleevue Farm (Gates Mills, OH) and staged according to the method ofHamburger and Hamilton (1951). Ten thoracic DRG per embryonic day 10 (E10) chick embryo were dissected out into HBSS and dissociated in 5 ml of 0.25% trypsin (prepared in Ca2+- and Mg2+-free PBS; Life Technologies) at 37°C for 15 min. Trypsinization was terminated by addition of 10 ml of serum-containing medium (M199:DMEM, supplemented with 3% fetal bovine serum, 2% chick serum, 1% glutamine, and 50 U/ml pen-strep; Life Technologies). Cells were pelleted by centrifugation at 1000 rpm for 5 min at 4°C, resuspended in 1 ml of fresh medium, and then gently triturated 15× through each of three increasingly smaller-bored, fire-polished Pasteur pipets. One-tenth of the cell suspension was diluted into 12 ml of fresh medium in a 60 mm dish. Cells were preplated for 1 hr to remove non-neuronal cells from suspension. Individual neurons were identified under the microscope as large, process-bearing cells. A total of 160 single DRG neurons were picked (in an approximate volume of 0.5 μl each) using a microcapillary pipet attached to a Picospritzer II (General Valve, Fairfield, NJ) and transferred to 0.5 ml PCR reaction tubes containing 1 μl of PBS supplemented with 1 U/μl RNase inhibitor (Perkin-Elmer, Emeryville, CA). Cells were immediately frozen on dry ice. Single CEF cells exogenously expressing either FL or ED trkB were grown up as described above and were picked to serve as positive control samples for single-cell RT-PCR. In addition, single wild-type (WT) CEF cells lacking expression of trkB isoforms were picked to serve as negative control samples. Cells were immediately frozen on dry ice.
RT-PCR. To isolate RNA from single cells, we thawed cells on ice and resuspended the cells in 14 μl of RT master mix lacking RNase inhibitor and reverse transcriptase (final concentration of RT components, 5 mm MgCl2, 1× PCR II buffer, 1 mm each dNTP, and 2.5 μm random hexamers; GeneAmp RNA PCR kit, Perkin-Elmer). Cells were then lysed in a thermal cycler preheated to 99°C for 2 min. After lysis, samples were immediately placed on ice, and 200 U of murine leukemia virus reverse transcriptase and 1 U/μl RNase inhibitor were added to give a total RT reaction volume of 20 μl. Reverse transcription was performed for 1 hr at 42°C, followed by two times 40 cycles of PCR amplification using nested primer pairs for chick trkB. The final concentration of PCR components was 2 mmMgCl2, 1× PCR II buffer, 200 μm each dNTP, 2.5 U/100 μl Taq DNA polymerase, 0.5 mmupstream primer, and 0.5 mm downstream primer (GeneAmp RNA PCR kit, Perkin-Elmer). Both rounds of PCR were hot-started by addition of Taq when the reactions reached 94°C. The entire RT reaction was amplified during the first round of 40 cycles. The second round of 40 cycles was set up using 1 μl of the first-round reaction. The first-round upstream trkB primer was 5′-AAAATACATGTTATCAATCA-3′. The first-round downstream trkB primer was 5′-AGTCTTCACTGCTACCAA-3′. The second-round upstream trkB primer was 5′-GGACAACCCTACCCACCTGA-3′. The second-round downstream trkB primer was 5′-TTCTCCCAGTTCTCTTTTGA-3′.
Reaction products were digested with either BspE1 orBamHI to distinguish between the isoforms further. Uncut and digested products were separated on a 1.5% agarose gel and transferred to GeneScreen Plus (DuPont NEN, Wilmington, DE) by the downward alkaline Southern transfer technique (Chomczynski, 1992). Blots were hybridized overnight (5× SSC, 1% SDS, and 100 μg/ml salmon sperm DNA; 20°C below melting temperature) with 32P-end-labeled oligonucleotide probes complementary to (1) nucleotides 1289–1312 within the trkB transmembrane domain (TM oligo), (2) nucleotides 1081–1104 within the trkB extracellular domain (5′ oligo), (3) nucleotides 1148–1180 [11 amino acid (aa) motif oligo], (4) nucleotides 1405–1428 of J1 trkB (J1 oligo), (5) nucleotides 1423–1446 of J2 trkB (J2 oligo 1), and (6) nucleotides 1395–1417 of J2 trkB (J2 oligo 2). Final posthybridization washes were performed in 0.1× SSC plus 0.1% SDS at 7°C below melting temperature, and blots were placed under x-ray film.
Controls for contamination. Special precautions were taken to avoid potential contamination problems. All RT-PCR reactions were set up in a template-free UV-irradiated isolation hood in a separate laboratory. Nevertheless, analysis of 4 of the 160 DRG neurons revealed amplification of a contaminating FL* mutant cDNA. Contamination of these four cell samples was likely introduced at the level of single-cell isolation rather than at the level of reverse transcription or PCR, because FL*-expressing, retroviral-secreting CEF cells were being cultured at the time of DRG neuron isolation. Because reverse transcription and PCR were set up using master mixes, cDNA contamination at either of these steps could have resulted in systematic, widespread contamination. The detection of contaminating FL* in only 4 out of 160 DRG neuron samples argues against systematic contamination of master mixes. Furthermore, the large number of negative DRG samples (145 out of 160), the consistent detection of the correct isoforms in FL and ED CEF-positive control lanes, and the consistent lack of bands in wild-type CEF-negative control lanes suggest the absence of contamination with other trkB cDNAs. Because FL and ED CEF-positive control cells and wild-type CEF-negative control cells were isolated and handled under conditions identical to those for the single DRG neurons, it follows that any widespread contamination would likely have appeared as inappropriate isoform expression in positive or negative control samples, which was never seen.
TA subcloning and sequencing
To identify PCR products further, we subcloned newly amplified products using a dual promoter TA cloning kit (Invitrogen, San Diego, CA). PCR products were ligated into the pCRII TA-cloning vector, transformed into One Shot competent cells, and cultured in the presence of X-gal and IPTG. Between 10 and 20 white colonies were picked for each ligation, digested with EcoRI to drop out the subcloned insert, and further digested with BspE1 to distinguish between multiple PCR products. Selected clones were sequenced in both directions using the SP6 and T7 promoters. Sequencing was performed by the Molecular Biology Core Laboratory (Case Western Reserve University, Cleveland, OH) on an ABI Prism automated sequencer (model 377; Perkin-Elmer).
RESULTS
Extracellular deletion in the trkB juxtamembrane domain alters responsiveness of trkB to neurotrophins BDNF, NT-3, and NT-4
Signal transduction via the trk receptors is initiated by neurotrophin-induced receptor dimerization and autophosphorylation of specific tyrosine residues, resulting in activation of the receptor’s intrinsic kinase activity. Therefore, tyrosine phosphorylation of trk receptors serves as an indicator of neurotrophin binding and receptor activation. To examine the effects of the naturally occurring extracellular deletion on trkB neurotrophin specificity, we expressed ED and FL trkB isoforms exogenously in CEF cells and stimulated the isoforms with 50 ng/ml BDNF, NT-3, or NT-4. FL trkB isoforms in CEF cells were activated in response to all three neurotrophins (Fig.1A). FL trkB was maximally responsive to BDNF (arbitrarily set at 100% of the maximal response), whereas NT-3 and NT-4 elicited somewhat lower levels of responsiveness (Fig. 1B; 76 ± 11 and 76 ± 12% of the response to BDNF, respectively). In contrast, ED trkB was differentially responsive to the three neurotrophins (Fig.1A). BDNF elicited the greatest response (Fig.1B; 54 ± 13% of the maximal response). Responsiveness of ED to NT-4 was dramatically reduced (Fig.1B; 18 ± 6% of the maximal response), and responsiveness to NT-3 was nearly eliminated (Fig.1B; 3 ± 2% of the maximal response).
Fig. 1.

ED trkB is differentially responsive to neurotrophin stimulation. A, CEF cells expressing either FL or ED trkB were stimulated with 50 ng/ml BDNF, NT-3, or NT-4 or remained untreated as a negative control. TrkB isoforms were precipitated from cells, separated by SDS-PAGE, and immunoblotted with anti-PY antibody 4G10 (top), followed by stripping and reprobing with anti-BEC (bottom). Neurotrophin treatments are indicated across the top. The molecular weight on the left refers to the size of the mature FL trkB isoform. Because of the small size difference between FL and ED, the two isoforms are not readily distinguished on the basis of size. Bands shown are within the linear range of film exposure. Scanning and reproducing the original films tended to heighten contrast, darkening bands and whitening background.B, Anti-PY and anti-BEC band intensities were quantified by laser densitometry. Multiple exposures were obtained for each blot, and densitometry measurements were made on films from shorter exposures with subsaturating band intensities. Anti-PY signals were normalized to the level of receptor expression, and background phosphorylation [unstimulated condition (unstim)] was subtracted out. Normalized anti-PY intensities for FL and ED trkB after neurotrophin stimulation are shown here as the percent of the maximal response, with the FL response to BDNF arbitrarily set as the 100% maximal response.
Comparison of the dose responsiveness of FL and ED trkB to BDNF and NT-3 stimulation further underscored the differential responsiveness of ED trkB to neurotrophin stimulation and the near lack of response to NT-3 (Fig. 2). Maximal stimulation of ED trkB occurred at a concentration of 50 ng/ml BDNF (Fig.2A,B; 93 ± 7% of the maximal response). In contrast, stimulation with 50 ng/ml NT-3 elicited only a 7 ± 2% of the maximal response (Fig. 2B). Although increasing the concentration of NT-3 to 100 ng/ml increased the responsiveness to 16 ± 8%, 100% maximal responsiveness of ED was not achieved at any concentration of NT-3 tested (Fig.2B). In addition, the ratio of anti-PY signal to anti-BEC signal was much greater at high concentrations (50–100 ng/ml) of BDNF than at high concentrations of NT-3 (Fig. 2A; 5.6-fold compared with 1.1-fold, respectively). Unlike ED, FL trkB activation exhibited similar dose responsiveness after stimulation with BDNF and NT-3 (Fig. 2C). Both elicited maximal phosphorylation at concentrations of 50 ng/ml and had similar EC50 values (∼30 ng/ml).
Fig. 2.

ED trkB has decreased affinity for NT-3.A, Dose–response blot of ED trkB after stimulation with BDNF and NT-3. CEF cells expressing ED trkB were stimulated with 0, 5, 10, 25, 50, or 100 ng/ml BDNF or NT-3. ED trkB receptors were precipitated from cell lysates, separated by SDS-PAGE, and immunoblotted with anti-phosphotyrosine antibody 4G10, followed by stripping and reprobing with an antibody against the extracellular domain of avian trkB. Top, Anti-PY signal.Bottom, Level of receptor expression (anti-BEC). Neurotrophin treatments are indicated across the top. The molecular weight on the left refers to the size of the mature trkB ED isoform. Bands shown are within the linear range of film exposure. Scanning and reproducing the original films tended to heighten contrast, darkening bands and whitening background.B, Comparison of dose responsiveness of ED trkB to BDNF and NT-3 stimulation. Anti-PY and anti-BEC band intensities were quantified by laser densitometry. Multiple exposures were obtained for each blot, and densitometry measurements were made on films from shorter exposures with subsaturating band intensities. Anti-PY signals were normalized to the level of receptor expression, and background phosphorylation [unstimulated condition (unstim)] was subtracted out. Normalized anti-PY intensities for ED trkB after BDNF or NT-3 stimulation are shown here as the percent of the maximal response, with the ED response to 100 ng/ml BDNF arbitrarily set as the 100% maximal response. The EC50 value for BDNF stimulation of the ED was 25 ng/ml. For NT-3 concentrations of 5, 10, 25, and 50 ng/ml, error bars are not appreciably wider than data points.C, Comparison of dose responsiveness of FL trkB to BDNF and NT-3 stimulation. CEF cells expressing FL trkB were stimulated with 0, 5, 10, 25, 50, or 100 ng/ml BDNF or NT-3. Normalized anti-PY intensities for FL trkB after BDNF or NT-3 stimulation are shown here as the percent of the maximal response, with the FL response to 100 ng/ml BDNF arbitrarily set as the 100% maximal response. The EC50 value for both BDNF and NT-3 stimulation is 30 ng/ml.
Quantification also revealed an overall decreased responsiveness of ED compared with FL. The response of ED to BDNF was only approximately one-half (54 ± 13%) of the FL response to BDNF, whereas the responses to NT-3 and NT-4 were reduced to 3 ± 2 and 18 ± 6%, respectively (Fig. 1B). ED was less responsive than was FL at every concentration of BDNF tested (Fig.3). Despite the decreased responsiveness of ED, responses for both FL and ED were approaching saturation at 50 ng/ml BDNF. Furthermore, comparison of the EC50 values for the two curves indicated that both ED and FL were half-maximally activated by 25 ng/ml BDNF.
Fig. 3.

ED trkB exhibits decreased responsiveness to BDNF stimulation despite equal affinity of BDNF for FL and ED trkB. CEF cells expressing either FL or ED trkB were stimulated with 0, 5, 10, 25, 50, or 100 ng/ml BDNF. TrkB isoforms were precipitated from cell lysates, separated by SDS-PAGE, and immunoblotted with anti-phosphotyrosine antibody 4G10, followed by stripping and reprobing with an antibody against the extracellular domain of avian trkB. Anti-PY and anti-BEC band intensities were quantified by laser densitometry. Anti-PY signals were normalized to the level of receptor expression, and background phosphorylation was subtracted out. Normalized anti-PY intensities are shown here as the percent of the maximal response, with the FL response to 100 ng/ml BDNF arbitrarily set as the 100% maximal response. The EC50 value for BDNF stimulation of both FL and ED trkB was 25 ng/ml. For BDNF concentrations of 5 and 10 ng/ml, error bars are not appreciably wider than data points.
Mutation of aspartate residues within the 11 amino acid motif of FL trkB mimics the effects of the deletion
The differential responsiveness of ED trkB to the neurotrophins implicates the existence of distinct binding determinants for the neurotrophins within the extracellular domain of FL trkB. Extensive mutational and chimeric analysis of the neurotrophins and the trk receptors has revealed determinants on both the factors and the receptors that are critical for binding and conferring specificity of ligand–receptor interactions. Interestingly, two positively charged residues (R31 and H33) within NT-3 are critical for the binding of NT-3 to its nonpreferred trk receptors, trkA and trkB (Ryden and Ibanez, 1996). Positively charged residues within the neurotrophins may interact electrostatically with negatively charged residues within the receptors. The 11 amino acid motif of FL trkB contains two negatively charged aspartate residues at positions 390 and 392 (Fig.4). To test the potential role of D390 and D392 in neurotrophin binding, we mutated these two residues to neutral alanines to generate a mutant trkB isoform designated FL* (Fig.4).
Fig. 4.

Sequence comparison of FL, ED, and FL* trkB isoforms. Top, The sequence of avian FL trkB is shown with the sequence of the 11 amino acid extracellular deletion motif indicated. Asterisks indicate the two aspartate (D) residues that were mutated in the FL receptor to generate the FL* mutant. Bottom, The sequence of the naturally occurring ED isoform is shown.
The double D→A mutation was surprisingly good at mimicking the effects of the naturally occurring 11 amino acid extracellular deletion (compare Figs. 1, 5). Like ED, the mutant isoform was differentially responsive to neurotrophin stimulation, exhibiting the greatest responsiveness to BDNF and the least responsiveness to NT-3 (Fig. 5A). Responsiveness to NT-4 was decreased to only 10 ± 2% of the maximal response, whereas responsiveness to NT-3 was decreased to 5 ± 1% (Fig.5B). The overall decrease in neurotrophin responsiveness was also recapitulated in the FL* mutant. Although FL* exhibited greater responsiveness to BDNF than to the other neurotrophins, the response of FL* trkB to BDNF was only 67% of the FL response to BDNF (data not shown).
Fig. 5.
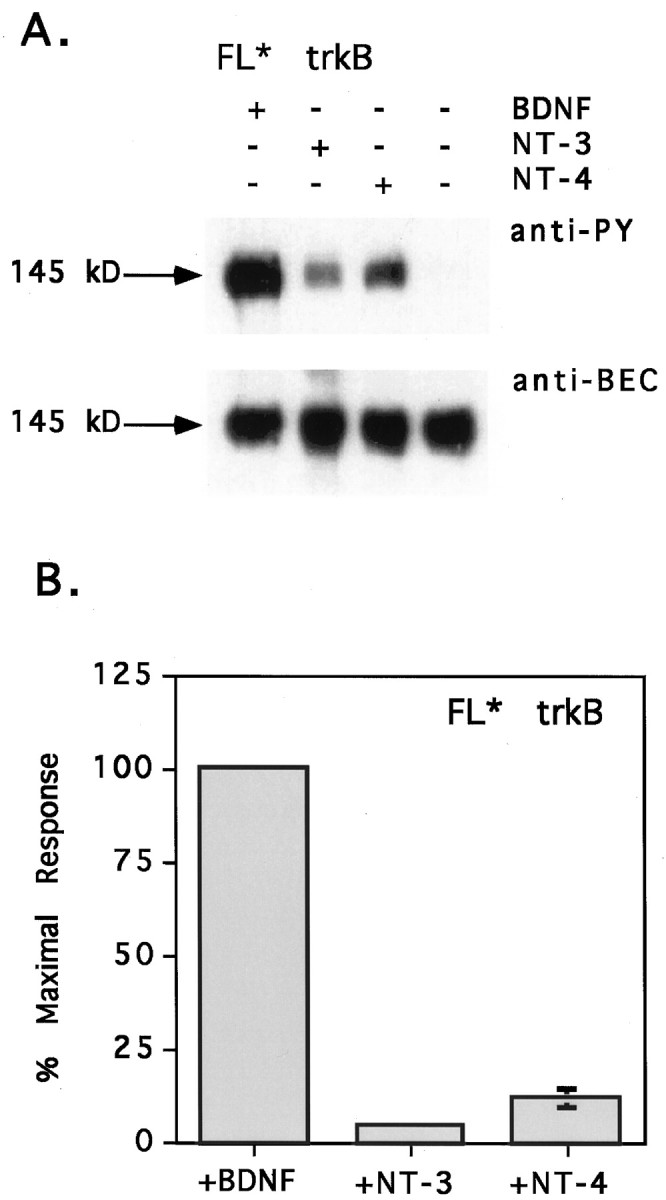
The double aspartate mutation closely mimics the effects of the extracellular deletion. A, CEF cells expressing FL* trkB were stimulated with 50 ng/ml BDNF, NT-3, or NT-4 or remained untreated as a negative control. TrkB isoforms were precipitated from cells, separated by SDS-PAGE, and immunoblotted with anti-PY antibody 4G10 (top), followed by stripping and reprobing with anti-BEC (bottom). Neurotrophin treatments are indicated across the top. The molecular weight on the left refers to the size of the FL* trkB isoform. Bands shown are within the linear range of film exposure. Scanning and reproducing the original films tended to heighten contrast, darkening bands and whitening background. B, Anti-PY and anti-BEC band intensities were quantified by laser densitometry. Multiple exposures were obtained for each blot, and densitometry measurements were made on films from shorter exposures with subsaturating band intensities. Anti-PY signals were normalized to the level of receptor expression, and background phosphorylation was subtracted out. Normalized anti-PY intensities are shown here as the percent of the maximal response, with the FL* response to BDNF arbitrarily set as the 100% maximal response. For NT-3 stimulation, the error bar is not appreciably wider than the data point.
FL and ED trkB are expressed in predominantly distinct populations of neurons in avian DRG
The existence of two trkB isoforms with distinct neurotrophin specificities raises the possibility that neurotrophin specificities of trkB-expressing neurons are dependent on which isoform is expressed. For this reason, we sought to determine whether there are distinct subpopulations of trkB+ neurons expressing either FL or ED trkB exclusively. Previous RT-PCR analysis of total RNA from E4.5, E7.5, and E11.5 avian DRG revealed expression of both FL and ED isoforms in a ratio of 60 FL:40 ED (A. S. Garner, F. Lefcort, and T. H. Large, unpublished observations). To determine whether FL and ED trkB were expressed in distinct, nonoverlapping populations of avian DRG neurons, single-cell RT-PCR was performed (as schematically depicted in Fig. 6).
Fig. 6.

Single-cell RT-PCR strategy (not drawn to scale). The cDNAs for FL and ED trkB are shown schematically at thetop with start ATG, the extracellular domain (EC), the 11 amino acid motif (11 aa), the transmembrane domain (TM), the intracellular juxtamembrane domain (JM), the tyrosine kinase domain (TK), and theBspE1 digestion site indicated. Reverse-transcribed cDNAs from single cells were subjected to two times 40 cycles of PCR amplification using nested primer pairs (indicated by dashed arrows) to give rise to FL and/or ED products of 586 or 553 bp, respectively. Second-round amplification products were digested withBspE1 to give products of the sizes shown. Digested products were separated on a 1.5% agarose gel, transferred to nitrocellulose, and probed with several different oligo probes. The 5′ and TM oligo probes (designated probes 1,3, respectively) label any trkB product. The 5′ oligo probe labels the 205 bp BspE1 digestion fragment, whereas the TM oligo probe labels the 381 bp fragment containing the transmembrane domain. The 11 aa motif oligo probe (designated probe 2) specifically labels the 205 bp fragment of FL trkB.
Of the 160 DRG neurons analyzed, 10 expressed trkB isoforms, as verified by hybridization to an oligo probe against a portion of the trkB transmembrane domain (Fig.7B, TM oligo). PCR products from one cell (142) did not blot with the TM oligo, indicating amplification of a false-positive product from this cell. To distinguish between multiple trkB isoforms in the 10 trkB+ cells, we digested PCR reaction products with BspE1 and blotted the products with several oligo probes (as depicted in Fig.6). By the use of these criteria, the 10 trkB+ cells could be categorized further into one of four classes, differing in their expression of trkB receptor isoforms: (1) cells expressing FL trkB exclusively, (2) cells expressing ED trkB exclusively, (3) cells coexpressing FL and ED, and (4) cells coexpressing FL with a juxtamembrane insertion variant, termed J1 trkB (Garner et al., 1996). Representative examples of each of the four neuronal classes identified are shown in Figure 8 along with positive and negative controls.
Fig. 7.

Amplification of trkB products from single DRG neurons. Reverse transcription of RNA from single DRG neurons, FL and ED CEF-positive control cells, and WT CEF-negative control cells was followed by two rounds of PCR amplification using nested trkB-specific primer pairs. PCR products were separated on a 1.5% agarose gel.A, Ethidium bromide staining of PCR amplification products. DRG neuron samples are labeled by cell number across thetop. FL CEF and ED CEF lanes correspond to amplification products from FL and ED CEF-positive control cells. The WT CEF lane corresponds to amplification from a WT CEF-negative control cell. FL trkB migrates at the size of 586 bp, whereas ED trkB migrates at the size of 553 bp.B, TM oligo probing of PCR products. PCR products were transferred to nitrocellulose and probed with a probe against the transmembrane domain of trkB (TM oligo). Subsequent sequencing of PCR products allowed for identification of the trkB isoforms expressed. The isoform identity for each cell sample is indicated below each lane.
Fig. 8.
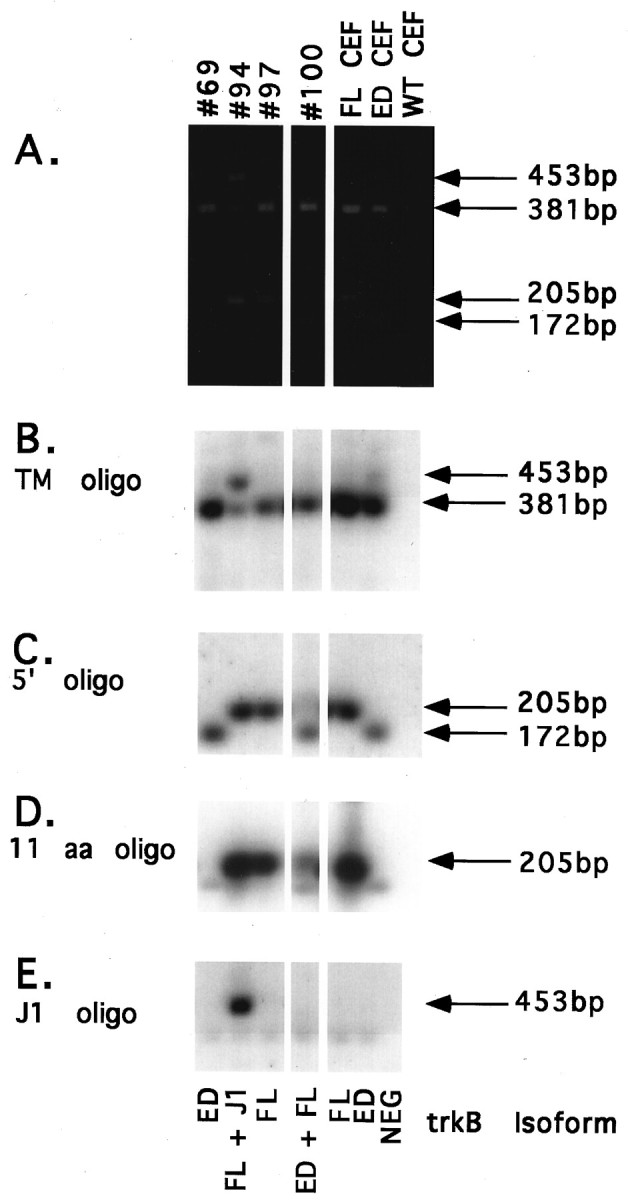
Identification of trkB isoforms in single DRG neurons. Amplification products from single trkB+ DRG neurons, FL and ED CEF-positive control cells, and a WT CEF-negative control cell were digested with BspE1, separated on a 1.5% agarose gel, transferred to nitrocellulose, and probed with several oligo probes.A, Ethidium bromide staining ofBspE1-digested PCR products. DRG neuron samples are labeled by cell number across the top. Positive controls are labeled FL CEF and ED CEF. The negative control is labeled WT CEF. Sizes of FL fragments (381 and 205 bp), ED fragments (381 and 172 bp), and J1 fragments (453 and 205 bp) are indicated to the right.B, TM oligo probing. Sizes of the TM-containing fragments of FL and ED (381 bp) and J1 (453 bp) are indicated to theright. C, 5′ oligo probing. Sizes of the FL and J1 fragment (205 bp) and the ED fragment (172 bp) are indicated to the right. D, 11 aa motif oligo probing. The size of the FL and J1 fragments is indicated to theright. E, J1 oligo probing. The size of the J1 fragment is indicated to the right. Sequencing of PCR products allowed for verification of trkB isoform identity from each cell, as indicated below each lane.
The first class of DRG neurons expressed FL trkB exclusively and is represented here by cell 97. The BspE1 digestion pattern of cell 97 (Fig. 8A) was representative of 6 of the 10 trkB+ cell samples (43, 45, 97, 101, 126, and 147). The digestion fragments comigrated with fragments of the BspE1-digested FL CEF-positive control, indicating expression of FL trkB in these six cells. The TM oligo again verified the digested products as trkB products, labeling the 381 bp TM-containing fragment (Fig.8B; see cell 97). Similarly, an oligo probe against a 24 bp sequence near the 5′ end of all trkB PCR products (Fig. 6) labeled the 205 bp fragment in these six samples (Fig. 8C; see cell 97). The 205 bp fragment also hybridized to an oligo against the sequence encoding the 11 amino acid motif (Fig. 8D; see cell 97), indicating expression of the FL trkB isoform. TA subcloning of PCR products confirmed FL expression in these six cells. A seventh cell (18) can also be categorized as a class 1 neuron. Although two trkB+ products are evident in this cell sample (Fig. 7), sequencing of the TA-subcloned products revealed that both products represent FL trkB. The larger PCR product arose from amplification with the second-round 5′ PCR primer and the first-round 3′ PCR primer carried over from the first round of PCR. Amplification with this primer pair results in a 667 bp FL trkB product.
The second class of DRG neurons expressed ED exclusively and was comprised of one cell, 69. The BspE1 digestion pattern of cell 69 was similar to that of the ED CEF-positive control (Fig.8A). Again, probing with both the TM and 5′ oligos verified the PCR products from this cell as trkB products (Fig.8B,C). Unexpectedly, the smaller 172 bp fragment also hybridized weakly to the 11 amino acid motif probe (Fig. 8D). However, weak hybridization to the 11 amino acid motif probe was also seen for the ED CEF-positive control (Fig. 8D). Therefore, we suspect that binding to the 172 bp fragment was nonspecific. The absence of specific labeling by the 11 amino acid motif probe was consistent with the ED-likeBspE1 digestion pattern seen by ethidium bromide staining. Finally, sequencing of TA-subcloned PCR products confirmed expression of ED in this cell.
The third class of DRG neurons coexpressed FL and ED and was comprised of one cell, 100. Although the BspE1 digestion pattern of cell 100 suggested exclusive expression of ED trkB (Fig.8A), probing with the 5′ oligo revealed the presence of a less abundant 205 bp fragment (Fig. 8C) that had not been apparent by ethidium bromide staining. The 205 bp band hybridized to the 11 amino acid motif probe (Fig. 8D), indicating the expression of FL trkB in this cell. TA subcloning allowed for the isolation of two distinct PCR products from cell 100. The most abundant subclones (12 out of 14) exhibited an ED-likeBspE1 digestion pattern, whereas a less abundant subclone (2 out of 14) exhibited a FL-like BspE1 digestion pattern (data not shown). Sequencing of these subclones confirmed the identity of the two forms as ED and FL trkB.
The fourth class of DRG neurons coexpressed FL and a J1 insertion isoform of trkB and was comprised of one cell, 94. The BspE1 digestion pattern contained fragments that comigrated with FL CEF-positive control bands as well as a larger fragment that migrated at the approximate size (453 bp) of a BspE1-digested J1 or J2 trkB isoform (Fig. 8A). The TM oligo verified the digested products as trkB products, labeling both the 381 bp TM-containing fragment and the larger 453 bp fragment. Expression of FL trkB was verified by intense labeling of the 205 bp fragment with the ED oligo probe (Fig. 8D). To determine the identity of the 453 bp fragment, we probed the blot with oligos complementary to sequences within the J1 and J2 insertions. The J1 oligo intensely labeled the 453 bp band (Fig. 8E), indicating expression of J1 trkB in this cell. In contrast, no J2-specific labeling was seen (data not shown). TA subcloning allowed for the isolation of two distinct products that were subsequently identified as FL and J1 trkB by sequencing.
DISCUSSION
The naturally occurring trkB extracellular deletion restricts activation by NT-3 and NT-4 and decreases responsiveness to BDNF, the preferred ligand. Decreased activation by BDNF may reflect a decrease in binding affinity or an altered conformation of the ligand-bound receptor that is less efficient at dimerization and/or activation. A reduced BDNF affinity for ED can be ruled out, because dose–response curves demonstrated similar EC50 values for ED and FL activation (Fig. 3) and binding results are nearly identical for the two isoforms (Strohmaier et al., 1996). Because receptor activation is a multistep process, it is conceivable that BDNF efficiently binds ED but is inefficient at promoting subsequent events. For example, the deletion may change the receptor conformation sufficiently to decrease the rate of dimerization or kinase activation. An alternative explanation is that subtle conformational changes in the ED dimer reduce the number of cytoplasmic phosphorylation sites per activated ED receptor. An analysis of the phosphotyrosines in activated ED is critical for distinguishing between the two models. It is interesting to consider that if activated ED receptors indeed display altered tyrosine phosphorylation, they may also couple to different signal transduction pathways and cellular responses (Minichiello et al., 1998).
The greatly reduced activation of ED by NT-3 and NT-4 suggests that the 11 amino acid motif provides a critical part of the binding sites for these neurotrophins. This is consistent with the finding that the second immunoglobulin-like domain, immediately N-terminal to the deletion, is crucial for neurotrophin binding and specificity (Urfer et al., 1995). The absence of ED activation by NT-3 is presumably not caused by a gross structural perturbation, because BDNF is still capable of ED activation. The decreased ratio of anti-PY to anti-BEC signals (Fig. 2A) suggests that there may be a decrease in the number of tyrosines phosphorylated per activated receptor. However, the dramatically decreased affinity of NT-3 for ED trkB shown here by dose–response analysis and elsewhere by binding assays (Strohmaier et al., 1996) suggests that the disparity between BDNF- and NT-3-induced phosphorylation of ED is most likely attributable to differences in receptor occupancy. The fact that the BDNF affinity for ED is unchanged suggests that the binding site for BDNF and the binding sites for NT-3 and NT-4 are nonequivalent, as has been suggested previously for the NT-4 and BDNF binding domains on mammalian trkB (Klein et al., 1992).
The effects of the extracellular deletion on ligand selectivity were recapitulated by the double D→A mutation within the 11 amino acid motif of FL trkB. Extensive mutational and chimeric analysis has revealed that specific neurotrophin binding to preferred trk receptors is primarily determined by residues within variable loop II of the neurotrophins (Ibanez et al., 1993; Ilag et al., 1994). In contrast, binding of NT-3 to its nonpreferred receptors (i.e., trkA and trkB) requires a positively charged R-X-H motif within variable loop I (Ryden and Ibanez, 1996). A similar motif (R-X-R) occurs in variable loop I of NT-4 (Ryden et al., 1995). The existence of a complementary negatively charged D-X-D motif within the 11 amino acid motif of FL trkB suggested that binding of NT-3 and NT-4 to trkB is mediated via electrostatic interactions. Consistent with this hypothesis, mutation of D-X-D to neutral A-X-A resulted in the loss of activation by NT-3 and dramatically reduced activation by NT-4. The ability of NT-4 to promote phosphorylation of the mutant (FL*) receptor, although at a greatly reduced level, suggests that maximal activation of trkB by NT-4 requires synergistic interaction of multiple ligand–receptor contacts, as has been suggested from mutational analysis of NT-4 (Ilag et al., 1994). Unlike NT-3 and NT-4, BDNF lacks positive charges within variable loop I but contains positive charges in spatially close variable loop V. Interestingly, these residues are dispensable for BDNF binding to trkB but are important for BDNF-induced phosphorylation (Ibanez et al., 1993), consistent with the decreased activation of FL* and a model that electrostatic interactions are involved in BDNF activation of trkB.
Importantly, the altered ligand specificity of ED and FL* was observed in a fibroblast background, a permissive cellular environment for neurotrophin activation of trk receptors (Ip et al., 1993). A similar extracellular deletion in trkA also restricts receptor activation, but only in neuron-like pheochromocytoma 12 (PC12) cells expressing p75 (Barker et al., 1993; Benedetti et al., 1993; Clary and Reichardt, 1994). Therefore, there are at least two potential means of restricting trkB activation: (1) alternative splicing within the extracellular domain and (2) expression in a neuronal environment that is likely to involve regulation by p75. Although the ligand specificity of ED has not been examined in cells coexpressing p75, FL isoforms expressed in PC12 cells are not significantly activated by NT-3 or NT-4 (Ip et al., 1993) (A. S. Garner and T. H. Large, unpublished observations). Interestingly, binding of NT-3 and NT-4 to p75 requires the same loop I residues suggested here to be critical for trkB binding (Ryden et al., 1995). Lack of FL activation by NT-3 and NT-4 in PC12 cells may, therefore, be the result of these factors binding more effectively to p75.
The existence of trkB isoforms with distinct neurotrophin specificities suggests that splicing within the extracellular domain serves as a mechanism to regulate neurotrophin responsiveness of trkB+ neurons. Examination of trkB isoform expression in single DRG neurons has revealed predominantly exclusive expression of FL or ED. Although 16% of chick thoracic DRG neurons are trkB+ between E9 and E18 (Williams and Ebendal, 1995), only 6.25% of E10 DRG neurons examined were trkB+, presumably because single-cell RT-PCR is an inefficient process. In this study, 80% of trkB-expressing DRG neurons were FL+, while 20% were ED+. In previous RT-PCR analysis from E4.5, E7.5, and E11.5 total chick DRG RNA, ∼60% of trkB transcripts represented FL, while 40% represented ED (Garner, Lefcort, and Large, unpublished observations). Because predominantly large-diameter neurons were selected in this study, the unexpectedly high number of FL+ neurons may reflect differences in the size distribution of FL- and ED-expressing neurons. Although the percentage of ED+ neurons is likely to be underestimated by the single-cell analysis, it was not our intent to determine the relative expression of trkB isoforms within DRG but to determine the extent of coexpression within a single cell. One example of FL and ED coexpression was seen, although ED was more abundant in this cell. Although the significance of coexpression is not clear, it is conceivable that this neuron was in the process of switching splicing pattern, serving to either expand or restrict neurotrophin specificity. Coexpression of FL with the J1 juxtamembrane insertion isoform (Garner et al., 1996) was also seen. Although the function of J1 remains unknown, the insertion does not appear to alter the ligand specificity of trkB (K. L. Boeshore and T. H. Large, unpublished observations). The cytoplasmic localization of the insertion close to the binding sites for SHC and FRS2 activation (Obermeier et al., 1993; Peng et al., 1995; Kouhara et al., 1997) suggests that J1 may have altered signaling capabilities.
Expression of FL and ED within distinct populations of DRG neurons predicts the existence of two subpopulations of trkB+ neurons differing in their ability to respond to the neurotrophins. Such differential responsiveness may be responsible, in part, for generating appropriate connections between DRG neurons and target tissues that may express one or more trkB ligands. Neurons expressing FL could be supported by targets expressing BDNF, NT-4, and possibly NT-3, because recent examination of NT-3(−/−) mice has suggested that NT-3 supports a subpopulation of trkB+ DRG neurons in vivo via activation of trkB (Farinas et al., 1998). Expression of the more restricted ED isoform presumably places neurons projecting to NT-3- and NT-4-expressing targets at a selective disadvantage. In addition to the DRG, other neuronal populations exhibiting differential responsiveness to the principal trkB ligands in vivo have been described. Within the nodose-petrosal ganglion complex, BDNF and NT-4 support survival of primarily nonoverlapping populations of neurons (Conover et al., 1995; Liu et al., 1995; Erickson et al., 1996). Injection of BDNF, but not NT-4, into the developing optic tectum of Xenopusresults in increased branching of optic axons (Cohen-Cory and Fraser, 1995). Similarly, BDNF and NT-4 have layer-specific effects on dendritic branching in ferret visual cortex (McAllister et al., 1995), and delivery of NT-4, but not BDNF, to ferret visual cortex prevents shrinkage of LGN neurons accompanying monocular deprivation (Riddle et al., 1995). Populations supported by both BDNF and NT-4 in vivo must have access to both factors and be able to respond to them. These cells presumably express the less restrictive FL isoform. However, cells requiring either BDNF or NT-4 exclusively, as determined by gene knock-out, may have access in vivo to only BDNF or NT-4 or, alternatively, may express more restricted receptor isoforms. Finally, effects mediated exclusively by either BDNF or NT-4 may also reflect differences in the downstream-signaling pathways activated by the two factors, because recent evidence (Minichiello et al., 1998) suggests that BDNF- and NT-4-induced signaling pathways mediated via trkB are not equivalent.
Footnotes
This work was supported by National Institutes of Health Grants EY-11373 and EY-08885. We thank Nell Malec for help in single DRG neuron isolation. We also thank Drs. Karl Herrup, David Katz, Frances Lefcort, and Vance Lemmon for critical reading of this manuscript.
Correspondence should be addressed to Dr. Kristen L. Boeshore, Department of Neurosciences, School of Medicine, Case Western Reserve University, 2109 Adelbert Road, Cleveland, OH 44106-4975.
Dr. Large’s present address: Sphinx Pharmaceuticals, Research Triangle Park, NC 27709
REFERENCES
- 1.Barbacid M. The trk family of neurotrophin receptors. J Neurobiol. 1994;25:1386–1403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- 2.Barker PA, Lomen HC, Gensch EM, Meakin SO, Glass DJ, Shooter EM. Tissue-specific alternative splicing generates two isoforms of the trkA receptor. J Biol Chem. 1993;268:15150–15157. [PubMed] [Google Scholar]
- 3.Benedetti M, Levi A, Chao MV. Differential expression of nerve growth factor receptor leads to altered binding affinity and neurotrophin responsiveness. Proc Natl Acad Sci USA. 1993;90:7859–7863. doi: 10.1073/pnas.90.16.7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chomczynski P. One hour downward alkaline capillary transfer for blotting of DNA and RNA. Anal Biochem. 1992;201:134–139. doi: 10.1016/0003-2697(92)90185-a. [DOI] [PubMed] [Google Scholar]
- 5.Clary DO, Reichardt LF. An alternatively spliced form of the nerve growth factor receptor TrkA confers an enhanced response to neurotrophin 3. Proc Natl Acad Sci USA. 1994;91:11133–11137. doi: 10.1073/pnas.91.23.11133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen-Cory S, Fraser SE. Effects of brain-derived neurotrophic factor on optic axon branching and remodelling in vivo. Nature. 1995;378:192–196. doi: 10.1038/378192a0. [DOI] [PubMed] [Google Scholar]
- 7.Conover JC, Erickson JT, Katx DM, Bianchi LM, Poueymirou WT, McClain J, Pan L, Helgren M, Ip NY, Boland P, Friedman B, Wiegand S, Vejsada R, Kato AC, DeChiara TM, Yancopoulos GD. Neuronal deficits, not involving motor neurons, in mice lacking BDNF and/or NT4. Nature. 1995;375:235–238. doi: 10.1038/375235a0. [DOI] [PubMed] [Google Scholar]
- 8.Erickson JT, Conover JC, Borday V, Champagnat J, Barbacid M, Yancopoulos GD, Katz DM. Mice lacking brain-derived neurotrophic factor exhibit visceral sensory losses distinct from mice lacking NT-4 and display a severe developmental deficit in control of breathing. J Neurosci. 1996;16:5361–5371. doi: 10.1523/JNEUROSCI.16-17-05361.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farinas I, Wilkinson GA, Backus C, Reichardt LF, Patapoutian A. Characterization of neurotrophin and trk receptor functions in developing sensory ganglia: direct NT-3 activation of trkB neurons in vivo. Neuron. 1998;21:325–334. doi: 10.1016/s0896-6273(00)80542-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garner AS, Menegay HJ, Boeshore KL, Xie XY, Voci JM, Johnson JE, Large TH. Expression of trkB receptor isoforms in the developing avian visual system. J Neurosci. 1996;16:1740–1752. doi: 10.1523/JNEUROSCI.16-05-01740.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamburger V, Hamilton H. A series of normal stages in the development of the chick embryo. J Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- 12.Hughes SH, Greenhouse JJ, Petropoulos CJ, Sutrave P. Adaptor plasmids simplify the insertion of foreign DNA into helper-independent retroviral vectors. J Virol. 1987;61:3004–3012. doi: 10.1128/jvi.61.10.3004-3012.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ibanez CF, Ilag LL, Murray-Rust J, Persson H. An extended surface of binding to trk tyrosine kinase receptors in NGF and BDNF allows the engineering of a multifunctional pan-neurotrophin. EMBO J. 1993;12:2281–2293. doi: 10.1002/j.1460-2075.1993.tb05882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ilag LL, Lennerberg P, Persson H, Ibanez CF. Role of variable B-hairpin loops in determining biological specificities in neurotrophin family. J Biol Chem. 1994;269:19941–19946. [PubMed] [Google Scholar]
- 15.Ip NY, Stitt TN, Tapley P, Klein R, Glass DJ, Fandl J, Greene LA, Barbacid M, Yancopoulos GD. Similarities and differences in the way neurotrophins interact with the trk receptors in neuronal and non-neuronal cells. Neuron. 1993;10:137–149. doi: 10.1016/0896-6273(93)90306-c. [DOI] [PubMed] [Google Scholar]
- 16.Klein R, Lamballe F, Bryant S, Barbacid M. The trkB tyrosine protein kinase is a receptor for neurotrophin-4. Neuron. 1992;8:947–956. doi: 10.1016/0896-6273(92)90209-v. [DOI] [PubMed] [Google Scholar]
- 17.Korsching S. The neurotrophic factor concept: a reexamination. J Neurosci. 1993;13:2739–2748. doi: 10.1523/JNEUROSCI.13-07-02739.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the ras/MAPK signaling pathway. Cell. 1997;89:693–702. doi: 10.1016/s0092-8674(00)80252-4. [DOI] [PubMed] [Google Scholar]
- 19.Lindsay RM. Neurotrophins and receptors. Prog Brain Res. 1994;103:3–14. [PubMed] [Google Scholar]
- 20.Liu X, Ernfors P, Wu H, Jaenisch R. Sensory but not motor neuron deficits in mice lacking NT4 and BDNF. Nature. 1995;375:238–241. doi: 10.1038/375238a0. [DOI] [PubMed] [Google Scholar]
- 21.Lo DC. Neurotrophic factors and synaptic plasticity. Neuron. 1995;15:979–981. doi: 10.1016/0896-6273(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 22.McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 23.Meakin SO, Suter U, Drinkwater CC, Welcher AA, Shooter EM. The rat trk protooncogene product exhibits properties characteristic of the slow nerve growth factor receptor. Proc Natl Acad Sci USA. 1992;89:2374–2378. doi: 10.1073/pnas.89.6.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minichiello L, Casagranda F, Tatche RS, Stucky CL, Postigo A, Lewin GR, Davies AM, Klein R. Point mutation in trkB causes loss of NT4-dependent neurons without major effects on diverse BDNF responses. Neuron. 1998;21:335–345. doi: 10.1016/s0896-6273(00)80543-7. [DOI] [PubMed] [Google Scholar]
- 25.Obermeier A, Lammer R, Wiesmuller KH, Jung G, Schlessinger J, Ullrich A. Identification of trk binding sites for Shc and phosphatidylinositol 3′-kinase and formation of a multimeric signaling complex. J Biol Chem. 1993;268:22963–22966. [PubMed] [Google Scholar]
- 26.Peng X, Greene LA, Kaplan DR, Stephens RM. Deletion of a conserved juxtamembrane sequence in trk abolishes NGF-promoted neuritogenesis. Neuron. 1995;15:395–406. doi: 10.1016/0896-6273(95)90043-8. [DOI] [PubMed] [Google Scholar]
- 27.Perez P, Coll PM, Hempstead BL, Martin-Zanca D, Chao M. NGF binding to the trk tyrosine kinase receptor requires the extracellular immunoglobulin-like domains. Mol Cell Neurosci. 1995;6:97–105. doi: 10.1006/mcne.1995.1010. [DOI] [PubMed] [Google Scholar]
- 28.Riddle DR, Lo DC, Latz LC. MT-4 mediated rescue of lateral geniculate neurons from effects of monocular deprivation. Nature. 1995;378:189–191. doi: 10.1038/378189a0. [DOI] [PubMed] [Google Scholar]
- 29.Ryden M, Ibanez CF. Binding of neurotrophin-3 to p75LNGFR, TrkA, and TrkB mediated by a single functional epitope distinct from that recognized by trkC. J Biol Chem. 1996;271:5623–5627. doi: 10.1074/jbc.271.10.5623. [DOI] [PubMed] [Google Scholar]
- 30.Ryden M, Murray-Rust J, Glass D, Ilag LL, Trupp M, Yancopoulos GD, McDonald NQ, Ibanez CF. Functional analysis of mutant neurotrophins deficient in low-affinity binding reveals a role for p75LNGFR in NT-4 signalling. EMBO J. 1995;14:1979–1990. doi: 10.1002/j.1460-2075.1995.tb07190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shelton DL, Sutherland J, Gripp J, Camerato T, Armanini MP, Phillips HS, Carroll K, Spencer SD, Levinson AD. Human trks: molecular cloning, tissue distribution, and expression of extracellular domain immunoadhesions. J Neurosci. 1995;15:477–491. doi: 10.1523/JNEUROSCI.15-01-00477.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 33.Strohmaier C, Carter BD, Urfer R, Barde YA, Dechant G. A splice variant of the neurotrophin receptor trkB with increased specificity for brain-derived neurotrophic factor. EMBO J. 1996;15:3332–3337. [PMC free article] [PubMed] [Google Scholar]
- 34.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 35.Urfer R, Tsoulfas P, O’Conell L, Shelton DL, Parada LF, Presta LG. An immunoglobulin-like domain determines the specificity of neurotrophin receptors. EMBO J. 1995;14:2785–2805. doi: 10.1002/j.1460-2075.1995.tb07279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Von Bartheld CS, Williams R, Lefcort F, Clary DO, Reichardt LF, Bothwell M. Retrograde transport of neurotrophins from the eye to the brain in chick embryos: roles of the p75NTR and trkB receptors. J Neurosci. 1996;16:2995–3008. doi: 10.1523/JNEUROSCI.16-09-02995.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams R, Ebendal T. Neurotrophin receptor expression during development of the chick spinal sensory ganglion. NeuroReport. 1995;6:2277–2282. doi: 10.1097/00001756-199511270-00003. [DOI] [PubMed] [Google Scholar]