

Abstract
It is hypothesized that desensitization of neuronal nicotinic acetylcholine receptors (nAChRs) induced by chronic exposure to nicotine initiates upregulation of nAChR number. To test this hypothesis directly, oocytes expressing α4β2 receptors were chronically incubated (24–48 hr) in nicotine, and the resulting changes in specific [3H]nicotine binding to surface receptors on intact oocytes were compared with functional receptor desensitization. Four lines of evidence strongly support the hypothesis. (1) The half-maximal nicotine concentration necessary to produce desensitization (9.7 nm) was the same as that needed to induce upregulation (9.9 nm). (2) The concentration of [3H]nicotine for half-maximal binding to surface nAChRs on intact oocytes was also similar (11.1 nm), as predicted from cyclical desensitization models. (3) Functional desensitization of α3β4 receptors required 10-fold higher nicotine concentrations, and this was mirrored by a 10-fold shift in concentrations necessary for upregulation. (4) Mutant α4β2 receptors that do not recover fully from desensitization, but not wild-type channels, were upregulated after acute (1 hr) applications of nicotine. Interestingly, the nicotine concentration required for half-maximal binding of α4β2 receptors in total cell membrane homogenates was 20-fold lower than that measured for surface nAChRs in intact oocytes. These data suggest that cell homogenate binding assays may not accurately reflect the in vivo desensitization affinity of surface nAChRs and may account for some of the previously reported differences in the efficacy of nicotine for inducing nAChR desensitization and upregulation.
Keywords: nicotine addiction, Xenopus oocytes, regulation, ion channel, radiolabeled binding, nicotinic receptor subtypes, CNS
Neuronal nicotinic acetylcholine receptors (nAChRs) are ligand-gated cation channels activated by the endogenous neurotransmitter acetylcholine and exogenous drugs, such as nicotine (Role, 1992; Sargent, 1993). The effect of both acute and chronic nicotine on the activity of different nAChR subtypes may be relevant to tolerance, dependence, and withdrawal symptoms associated with nicotine addiction (Wonnacott, 1990; Balfour, 1994; Dani and Heinemann, 1996). One result of chronic exposure to tobacco-related levels of nicotine (Benowitz et al., 1989) is the upregulation of high-affinity α4β2 subunit-containing [3H]nicotine binding sites in the CNS (Marks et al., 1983; Benwell et al., 1988; Flores et al., 1992, 1997; Breese et al., 1997). Such upregulation of receptor number may contribute to the addiction process. The mechanism by which chronic nicotine exposure leads to receptor upregulation is not known.
In addition to upregulation of receptor number, chronic nicotine exposure can be associated with a long-lasting downregulation in receptor responsiveness (Lukas, 1991; Marks et al., 1993; Peng et al., 1994; Hsu et al., 1996). This decrease in function is thought to be, at least in part, a consequence of agonist-induced desensitization (Boyd, 1987). In addition, nicotine-induced receptor desensitization has been hypothesized to be responsible for receptor upregulation (Marks et al., 1983; Schwartz and Kellar, 1985). A straightforward prediction of this hypothesis is that nicotine concentrations that produce receptor desensitization should also induce receptor upregulation. Results of several studies suggest this prediction to be false, because nicotine concentrations necessary to induce desensitization and upregulation can differ by several orders of magnitude (Peng et al., 1994; Bencherif et al., 1995; Whiteaker et al., 1998). However, these conclusions are based on several assumptions. First, that agonist equilibrium binding assays accurately assess the desensitized state(s) of the receptor. This may not be true. Agonist dose-dependencies for equilibrium binding and functional estimates of desensitization can differ by several orders of magnitude (Marks et al., 1996); such differences would account for the apparent nicotine concentration mismatch between measures of desensitization and upregulation. Second, agonist binding measured in standard membrane homogenization assays are assumed to reflect agonist binding to functional intact receptors, although some evidence suggests that this is not true (Whiteaker et al., 1998).
A more direct test of the prediction would be to assess desensitization functionally and compare this result with receptor upregulation measured by agonist binding to intact, cell-surface receptors. In the present study, α4β2 nAChRs expressed in Xenopus oocytes were chronically incubated in nicotine. Nicotine dose–response relationships were constructed for functional receptor desensitization and for receptor upregulation assessed by surface [3H]nicotine binding to intact oocytes. To further examine the hypothesis that nicotine-induced receptor desensitization is responsible for receptor upregulation, we also tested the predictions that nAChRs with lower affinities for functional desensitization will also have a lower affinity for receptor upregulation and that mutant α4β2 nAChRs that fail to recover fully from desensitization will upregulate after the removal of nicotine.
MATERIALS AND METHODS
Xenopus oocyte preparation and cRNA injection.Procedures for preparation of oocytes have been described in detail previously (Quick and Lester, 1994). Briefly, oocytes were defolliculated and maintained at 18°C in incubation medium containing ND96 (96 mm NaCl, 2 mm KCl, 1 mmMgCl2, and 5 mm HEPES, pH 7.4), 1.8 mm CaCl2, 50 μg/ml gentamycin, and 5% horse serum. Subunit cRNAs were synthesized in vitro(Message Machine; Ambion, Austin, TX) from linearized plasmid templates of rat cDNA clones. A mutant α4 subunit (α4 S336A) was created in which a PKC consensus serine phosphorylation site was mutated to alanine (pALTER 1; Promega, Madison, WI). The mutation was verified by sequencing (Fenster et al., 1999). Oocytes were injected with 25 ng of cRNA/subunit/oocyte; α and β subunit cRNAs were injected in 1:1 ratios. All salts and drugs were obtained from Sigma (St. Louis, MO).
Electrophysiology. Whole-cell currents were measured at room temperature (20–25°C), 24–120 hr after injection, with a GeneClamp 500 amplifier (Axon Instruments, Foster City, CA) in a standard two-microelectrode voltage-clamp configuration. Electrodes were filled with 3 m KCl and had resistances of 0.5–2 MΩ. Oocytes were clamped between −40 and −60 mV and superfused continuously in ND96 containing 1.8 mm CaCl2 (ND96+Ca). Nicotine was applied in these solutions. (−)-Nicotine tartrate (nicotine) was prepared from frozen stock solutions. All currents were recorded on a chart recorder and/or on an 80486-based computer with AxoScope software (Axon Instruments) after 50–100 Hz low-pass filtering at a digitization frequency of 200 Hz. Solutions were gravity fed via a six-way manual valve (Rainin Instruments, Woburn, MA) to the oocyte in the recording chamber. Solution exchange considerations are discussed by Fenster et al. (1997).
Criteria for functional data selection. Except for the experiments shown in Figure1B, oocytes with initial nicotinic response amplitudes >3 μA and <50 nA were not included in the data analysis. Additionally, responses were required to be at least twofold greater than the holding current, and the holding current at a given membrane potential was required to be <100 nA. Functional receptor desensitization is usually calculated as the reduction in response amplitude induced by a brief test pulse of agonist after a period of continuous incubation with the desensitizing agent (Katz and Thesleff, 1957: Feltz and Trautman, 1982). However, because of the long incubations (24–48 hr) in the present study, it is necessary to correct this measurement for any time-dependent changes in basal receptor expression–function that occur independent of nicotine exposure. Thus, response amplitudes in control oocytes from the same batch (not incubated in nicotine) were monitored over the same period. For each oocyte incubated in nicotine (nic), the fractional desensitization was calculated as the ratio of the current amplitude (I) at time (t) to the initial current amplitude at t = 0, Δnic =Inic(t)/Inic(t=0). This value was then normalized for changes in control receptor function, Δcon =Icon(t)/Icon(t=0), obtained from control (con) oocytes (see Fig. 3). Thus, the overall estimate of desensitization was calculated as Δnic/Δcon (see Fig. 3). For statistical comparison of mean data, weighted means t tests were performed.
Fig. 1.
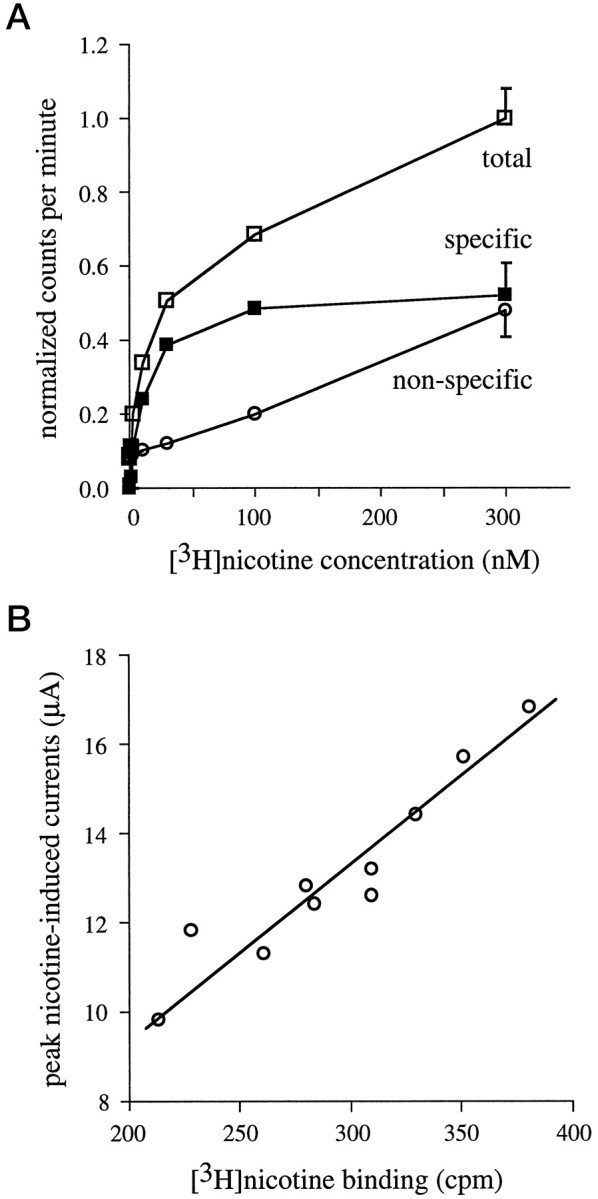
Specific [3H]nicotine binding to intact oocytes. Oocytes were injected with α4 and β2 subunit cRNAs and assayed 3 d after injection. A, [3H]Nicotine binding is saturable. Individual α4β2-expressing oocytes were incubated in various [3H]nicotine concentrations (0, 0.3, 1, 3, 10, 30, 100, and 300 nm) for 60 min. [3H]Nicotine binding was determined in the absence (total [3H]nicotine binding, open squares) or presence (nonspecific [3H]nicotine binding, open circles) of 100 μm nonradiolabeled acetylcholine. Specific [3H]nicotine binding (filled squares) was determined by subtracting nonspecific counts from total counts. Each data point represents the measurement of five to six oocytes; for clarity, the mean ± SEM across all nicotine concentrations is plotted with the symbols at the 300 nmnicotine value. B, Specific [3H]nicotine binding is correlated with nicotine-induced currents. α4β2-expressing oocytes were voltage-clamped at −40 mV, and peak currents were elicited using 100 μm nicotine. Specific surface [3H]nicotine binding was then determined in these same oocytes by incubation in 60 nm[3H]nicotine for 1 hr. The correlation for these two measurements was 0.94.
Fig. 3.
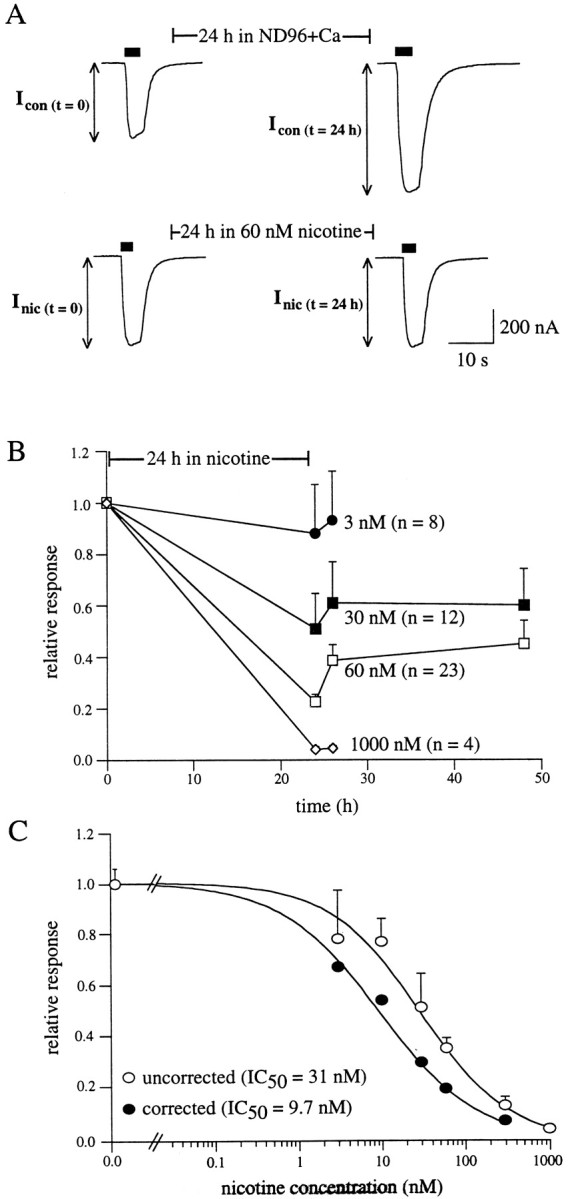
Functional desensitization of α4β2 nAChRs after chronic nicotine incubation. A, Representativetraces measured before and after incubation. Peak currents in α4β2-expressing oocytes induced by a nicotine test pulse (10 μm, 5 sec) were measured. The oocyte was then incubated for 24 hr in ND96+Ca in the absence (top traces) or presence (bottom traces) of 60 nm nicotine. Peak currents were remeasured immediately after removal from 24 hr incubation. To account for changes in basal receptor expression over the 24 hr incubation (top traces), the magnitude of nicotine-induced desensitization was defined as the ratio of the fractional response remaining after nicotine exposure to the fractional response remaining over the same period of time in the absence of nicotine (see Materials and Methods).B, Representative fractional nicotine-induced responses after 24 hr nicotine incubation. α4β2-expressing oocytes were incubated for 24 hr in control media or media containing 3, 30, 60, or 1000 nm nicotine. Measurement of peak currents before and after incubation and calculation of desensitization are described in A. For the 30 and 60 nm conditions, peak responses were also determined 2 and 24 hr after removal from nicotine. C, Inhibition dose–response curves constructed from fractional responses as described in B. Data are shown both uncorrected (open circles) and corrected (filled circles) for the amount of upregulation (i.e., relative increases in [3H]nicotine binding) observed after 24 hr incubation in nicotine at the same concentration. The amount of upregulation at a given concentration is taken from the data in Figure2B. The corrected values were calculated by dividing decreases in receptor function by the amount of upregulation (see Materials and Methods). The solid lines are logistic fits to mean data from which the half-maximal nicotine concentration for desensitization was n = 3–32 oocytes per data point.
[3H]nicotine binding assays. Binding assays were performed on both intact oocytes and on oocyte membranes after homogenization. Binding to intact oocytes was performed essentially as described previously (Chang and Weiss, 1999). Uninjected oocytes or nAChR-injected oocytes were visually inspected to ensure that collagenase treatment (2 mg/ml; Type A; Boehringer Mannheim, Indianapolis, IN) successfully removed follicle cells from around the oocyte membrane. The presence of the follicle layer resulted in high, nonspecific [3H]nicotine labeling that prevented accurate assessment of specific [3H]nicotine binding (our unpublished observations). The oocytes were removed from incubation media and rinsed in ND96+Ca for several minutes. Individual oocytes were next placed in a single well of a 96-well plate containing 40 μl of ND96+Ca. Stocks (5×) of [3H]nicotine [(−)-[N-methyl-3H]nicotine; 82.0 Ci/mmol; DuPont-NEN, Boston, MA] were prepared by dilution in ND96+Ca. The assay (60 min at room temperature) was initiated with the addition of 10 μl of 5× [3H]nicotine to the well, followed by gentle trituration of the media for several seconds so as not to disrupt the integrity of the cell. The assay was terminated in one of two ways: the oocyte was either suspended from the cut end of a pipette tip by light suction or pipetted into the cut end of a pipette tip along with 4 μl of assay solution. In the former case, the oocyte was then submerged sequentially into four different 2.5 ml wells containing ice-cold ND96+Ca. The total wash time for all four wells was 16 sec (4 sec/well). In the latter case, the oocyte was dropped into each wash well and repipetted into the next well. Based on a set of control experiments, the total wash time for all four wells was 18.8 ± 2.1 sec (n = 15 trials). Radioactivity was measured in one of two ways. After the final wash, the oocyte was placed directly into a scintillation vial containing nonaqueous liquid scintillation cocktail (Scintisafe F; Fisher Scientific, Houston, TX). The amount of radioactivity was estimated within 2 min of the wash step. In some experiments after washing, the oocyte was placed in one well of a 96-well plate containing ND96+Ca, and the bound [3H]nicotine was allowed to dissociate (see below; Fig. 2C). The contents of each well were then subjected to liquid scintillation counting. Where compared, these two methods yielded similar results. Oocyte batches in which nonspecific [3H]nicotine binding to uninjected oocytes was higher than 100 cpm were not used in data collection; based on this constraint, ∼30% of the oocyte batches were usable.
Fig. 2.
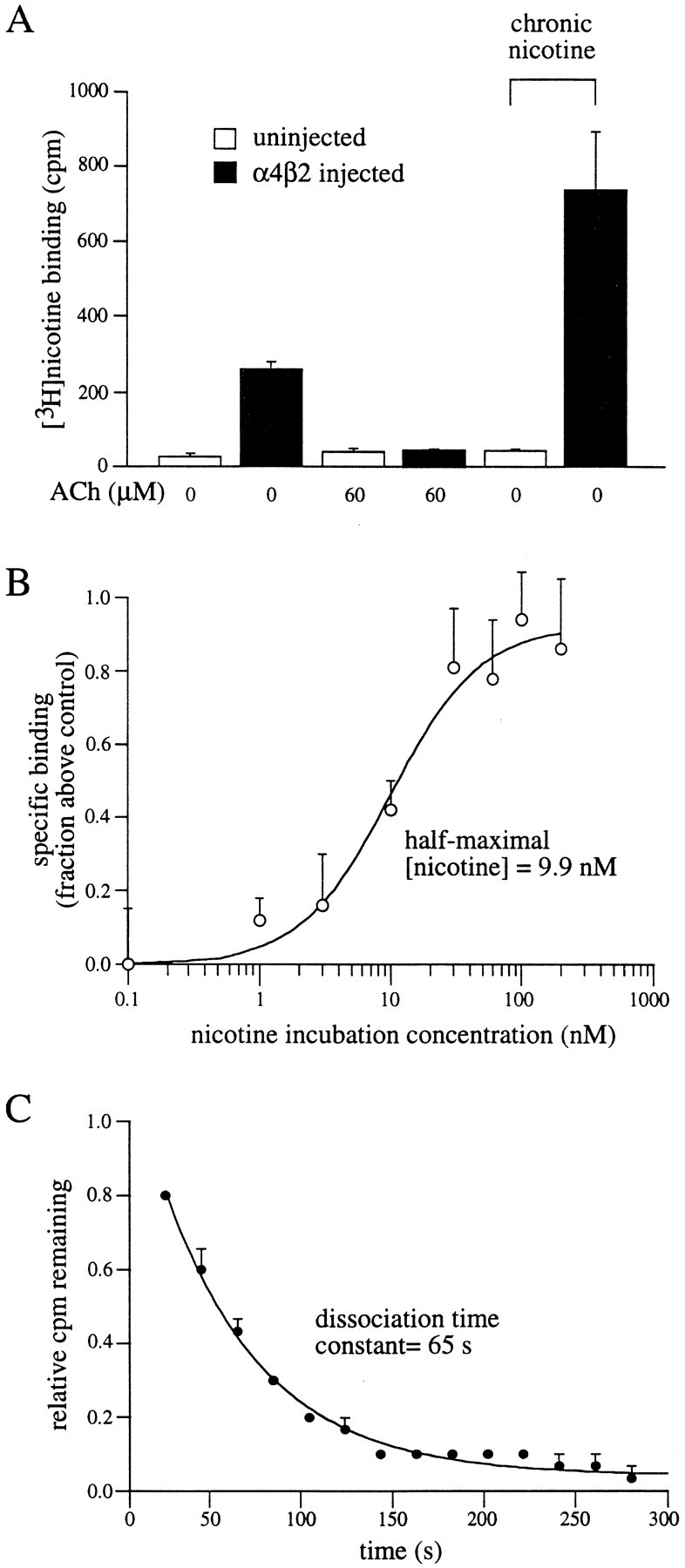
Upregulation of surface α4β2 receptors after chronic nicotine incubation. A, Specific [3H]nicotine binding to intact oocytes is upregulated by nicotine. Oocytes were uninjected (open bars) or injected (filled bars) with α4 and β2 subunit cRNAs. Twenty-four hours later, oocytes were placed in ND96+Ca with (two right-most bars) or without 60 nm nicotine. Surface binding assays were performed 24 hr later using 60 nm [3H]nicotine. Nonradiolabeled ACh (60 μm) was used on some oocytes to determine nonspecific binding. Data are from five oocytes per condition. B, Upregulation is nicotine concentration-dependent. α4β2-expressing oocytes were incubated in various nicotine concentrations for 24 hr and then subjected to [3H]nicotine binding assays using 60 nm[3H]nicotine. A dose–response curve was then constructed from the relative increase in specific [3H]nicotine binding at each nicotine incubation concentration. Data are plotted as the amount of specific [3H]nicotine binding for oocytes incubated in nicotine compared with the specific [3H]nicotine binding for control oocytes not incubated in nicotine (n = 4–9 oocytes per data point).C, Nicotine dissociates rapidly from chronically incubated oocytes. α4β2-expressing oocytes were incubated in 300 nm [3H]nicotine for 24 hr. Dissociation of bound [3H]nicotine was determined as described in Materials and Methods and plotted with respect to time. The solid line is an exponential fit to the data (n = 3 oocytes).
Specific [3H]nicotine binding and channel function. Direct comparison of functional desensitization and receptor upregulation in intact oocytes has three requirements: that we can accurately measure specific [3H]nicotine binding; that the measures of specific binding correlate with functional expression; and that the measured binding is occurring only on surface nAChRs. Control experiments demonstrating measurement of specific [3H]nicotine binding are shown in Figure1A. Oocytes expressing α4β2 nAChRs were incubated for 1 hr in various concentrations of [3H]nicotine. Specific binding was calculated as the total counts per minute minus the nonspecific counts per minute; nonspecific counts per minute was defined as the counts per minute of oocytes measured in the presence of 100 μmunlabeled acetylcholine. These data show that [3H]nicotine binding to intact oocytes is saturable and that nicotinic receptor agonists are competitive with [3H]nicotine binding. Figure 1Bshows that α4β2 nAChRs currents activated by saturating concentrations of nicotine (100 μm) and subsequent [3H]nicotine binding (60 nm) to the same intact oocyte are highly correlated (r = 0.94).
From the slope of the regression fit (37 nA/cpm), it is possible to calculate the number of bound nicotine molecules per functional channel. The number of functional channels can be estimated from the ratio of the measured peak current to the single channel current, assuming that all the channels are open at the peak. The single channel current will be 0.8 pA at −60 mV using a single channel conductance of 13.3 pS (Papke et al., 1989). Thus, there will be ∼4.625 * 104 nAChR channels/cpm. The number of bound nicotine molecules per counts per minute was estimated as 6.12 * 106 from the following:
| Equation 1 |
where SA is the specific activity of [3H]nicotine in Curie per moles, ƒ is the factor for converting disintegrations per second to counts per minute with a counting efficiency of 0.54, and NA is Avagadro’s number. Thus, these data predict 132 molecules of nicotine for each functional channel. Assuming that two molecules of nicotine bind per channel, these data imply that only ∼2% channels are functional. In terms of a single oocyte with ∼300 cpm (Fig.1B), the number of bound [3H]nicotine molecules would be ∼1.8 * 109, corresponding to ∼109total receptors, of which ∼2 * 107 would be functional. Because of some nonspecific binding in this instance, and a likely underestimation of the number of channels as a result of noninstantaneous solution-exchange, this will be a lower limit for the percentage of functional channels. With ∼20% nonspecific binding (Fig. 2A) and assuming only one-third of channels are open at peak, the number of functional channels could be ∼6%. Similar discrepancies between toxin binding and channel conductance have been noted for sodium channels (Ritchie and Rogart, 1977). For nAChRs reported here, the excess silent receptors could represent a reserve pool and/or desensitized receptors (Margiotta et al., 1987;Bencherif et al., 1995). Because α4β2 nAChRs can enter persistently inactive conformations after chronic nicotine treatment (Lukas, 1991;Peng et al., 1994), it is not unreasonable to suggest that such conformations may pre-exist on the cell surface. Because of the strong correlation between the amounts of total binding and currents (Fig.1B), functional and silent receptor pools are likely to be in equilibrium. Thus, despite the apparent excess of binding sites, these data strongly support the suggestion that functional nAChRs are a subpopulation of the receptor pool that is measured in a binding assay.
[3H]nicotine labels surface receptors in intact oocytes. Three sets of experiments were performed to verify that the binding assays were measuring specific binding to surface nAChRs (i.e., to eliminate the contribution of radioactivity associated with internalized [3H]nicotine). Two experiments were designed to test that the nonaqueous scintillation cocktail was only counting external [3H]nicotine. (1) Nonexpressing oocytes were injected with 5000 cpm (as determined by previous scintillation counting of various [3H]nicotine aliquots). Intact oocytes were then subjected to liquid scintillation counting. Some oocytes were then crushed and recounted; others were removed from the nonaqueous scintillation cocktail and placed in aqueous scintillation cocktail (Scintisafe Econo 1; Fisher Scientific). The crushed oocytes and the intact oocytes counted in aqueous scintillation cocktail revealed radioactive emissions of ∼5000 cpm; the intact oocytes measured in nonaqueous scintillation cocktail revealed radioactive emissions of ∼20 cpm (0.004% of total injected counts; n = 6). (2) [3H]Nicotine binding assays were performed on oocytes expressing α4β2 nAChRs using 60 nm[3H]nicotine. Specific binding was determined as described above (Fig. 1A). One subset of oocytes (n = 5) was counted by placing the intact oocyte in nonaqueous scintillation cocktail; the counts from a second subset of oocytes from the same oocyte batch were determined by permitting the radioactivity to dissociate for 2 min into a well containing ND96+Ca and counting the well contents. If a significant amount of [3H]nicotine was internalized and subsequently counted, then the counts from the intact oocyte should be much higher than in the dissociation study. However, adjusting for the rate of dissociation (see below; Fig. 2C), the number of counts measured in the intact oocytes (167 ± 23 cpm; n = 7) was comparable with that counted by the dissociation method (138 ± 30 cpm; n = 4).
Although these experiments suggested that internal radioactivity will not be counted, a potential confound is that any nonspecific internal accumulation of [3H]nicotine during a 1 hr incubation would begin to leak back out of the oocyte between the end of the assay and the scintillation counting step (∼2 min). To examine the contribution of this effect to our measurements, we put a known number of counts of [3H]nicotine (corresponding to 300 nm [3H]nicotine, the highest concentration we used in the experiments described in this paper) into one well of a 96-well plate with one nonexpressing oocyte and determined the amount of [3H]nicotine that accumulated inside of the oocyte during 1 hr. This amount was between 6 and 11% of the starting external value and varied based on the oocyte batch (three oocyte batches, seven oocytes per batch). We then injected other nonexpressing oocytes with [3H]nicotine corresponding to the number of counts internalized in 1 hr and determined the number of counts that emerged from these oocytes in 2 min. At the highest concentrations tested, this amount corresponded to 9% of the injected counts. Thus, the maximum contamination resulting from the re-emergence of internalized counts during an assay would be ∼1%.
These controls demonstrate that, although [3H]nicotine will accumulate inside the oocyte during a 1 hr assay, it will not be detected by counting intact oocytes in nonaqueous scintillation cocktail. Any [3H]nicotine that leaks back out will contribute negligibly to the total number of counts. Thus, ACh-displacable [3H]nicotine binding to intact oocytes will primarily reflect surface membrane nAChR labeling.
Protocols for specific experiments involving intact oocytes.Specific binding of [3H]nicotine to intact oocytes was used to measure three different parameters. (1) To measure the nicotine concentration necessary for half-maximal nAChR upregulation after chronic nicotine incubation, oocytes were chronically incubated (24–48 hr) in various concentrations of unlabeled nicotine. Assays were performed as described above using final [3H]nicotine concentrations of 60 nmfor α4β2 nAChRs and 300 nm for α3β4 nAChRs. (2) To determine the nicotine concentration necessary for half-maximal α4β2 nAChR binding, assays were performed as described above; final [3H]nicotine concentrations used ranged from 0.1 to 300 nm. For both the upregulation and equilibrium binding assays performed on intact oocytes, nonspecific binding was determined in the presence of 1000-fold excess of unlabeled acetylcholine. (3) To estimate the dissociation rate of bound [3H]nicotine, assays were performed as described above using a final [3H]nicotine concentration of 60 nm. At the end of the assay, the oocyte was suspended from the cut end of a pipette tip by light suction, washed sequentially in four different 2.5 ml wells containing ice-cold ND96+Ca, and then sequentially submerged (20 sec/well) into individual wells of a 96-well plate containing ND96+Ca and 60 μm unlabeled ACh (at room temperature). The contents of each well were then subjected to liquid scintillation counting.
Preparation of total homogenized oocyte membranes. For comparison, the concentrations of nicotine required for equilibrium binding and for receptor upregulation were also determined on oocyte membranes after homogenization. For equilibrium binding assays using cell homogenates, oocyte membranes were prepared as described previously (Corey et al., 1994). Briefly, oocyte membranes were isolated by centrifugation in 0.32 m sucrose in TE buffer (50 mm Tris-HCl, pH 7.5, and 1 mm EDTA) containing protease inhibitors (200 mmphenylmethylsulfonylfluoride, 10 μg/ml aprotinin, and 10 μg/ml leupeptin) using 10–15 strokes of a tight-fitting pestle in a chilled Dounce homogenizer. The homogenate was centrifuged twice at 4°C, 5 min, 1000 × g to remove cell debris, and the remaining supernatant fraction was homogenized at 4°C, 30 min, 100,000 ×g. The membrane pellet was resuspended in homogenization buffer (50 mm Tris-HCl, pH 7.5, and 1 mm EDTA). Equilibrium [3H]nicotine binding assays were performed by adding various concentrations of [3H]nicotine (0.1–10 nm) to the resuspended membranes for 1 hr. Specific binding was determined in the presence of excess ACh (60 μm). The assay was terminated by filtration, using Whatman (Clifton, NJ) GFA filters soaked in polyethyleneimine (0.3% w/v). The filters were washed three times and subjected to liquid scintillation counting. Protein content was determined by the method of Bradford. For receptor upregulation, oocytes (16–20 per group) were incubated, before assay, in various concentrations of nicotine (1–300 nm) for 24 hr. Membranes were prepared as described above, and specific binding was estimated using 60 nm [3H]nicotine. Upregulation was expressed as the increase in binding relative to a control group.
RESULTS
α4β2 nAChRs are upregulated after chronic nicotine treatment
To directly compare the dose-dependency for nicotine-induced upregulation of [3H]nicotine binding sites with that of receptor desensitization, we performed binding assays (see Materials and Methods) on intact oocytes with and without chronic nicotine treatment. In a representative example (Fig.2A), oocytes expressing α4β2 receptors showed 9.7-fold higher levels of surface [3H]nicotine binding than uninjected oocytes. To determine the amount of specific surface [3H]nicotine binding, we tested a subset of α4β2-expressing oocytes in the presence of excess unlabeled ACh. Unlabeled ACh reduced [3H]nicotine counts to levels seen in uninjected control oocytes, permitting two conclusions: (1) that the majority of the [3H]nicotine counts were a result of specific binding to nAChRs, and (2) because ACh is a membrane-impermeant agonist (Whiteaker et al., 1998), that the majority of specific binding was associated with receptors on the plasma membrane (see Materials and Methods). Treatment of α4β2-expressing oocytes with 60 nm nicotine for 24 hr before assay resulted in a 2.8-fold increase in [3H]nicotine binding compared with α4β2-expressing oocytes not incubated in nicotine. This increase in binding caused by nicotine incubation was not seen in uninjected oocytes. Together, these data demonstrate that surface α4β2 nAChR expression is upregulated by chronic nicotine incubation. This result is likely attributable to an actual increase in the number of surface receptors because chronic agonist treatment fails to alter the apparent agonist binding affinity (Marks et al., 1983).
Similar [3H]nicotine binding assays were repeated after 24 hr chronic nicotine treatment using nicotine concentrations ranging from 0.1 to 300 nm. Figure 2Bshows a dose–response curve constructed from the relative increases in specific [3H]nicotine binding after chronic nicotine treatment at eight different nicotine concentrations. Half-maximal upregulation of α4β2 nAChR expression was calculated to occur at nicotine incubation concentrations of 9.9 nm.
Chronic incubation using unlabeled nicotine raises a potential confound; that is, the unlabeled nicotine must fully dissociate from the receptor during the time of the binding assay. Otherwise, the number of [3H]nicotine binding sites will be underestimated. To rule out this possibility and to determine the rate at which nicotine dissociates from α4β2 nAChRs, we performed an agonist dissociation assay (Fig. 2C). Oocytes were incubated in 300 nm [3H]nicotine for 24 hr. In intact oocytes, dissociation (as described in Materials and Methods) of [3H]nicotine from these receptors followed a single exponential time course with a time constant of 65 sec (Fig.1C), which is similar to previous estimates (Marks and Collins, 1982; Lippiello et al., 1987). These data show that nicotine fully dissociates from α4β2 receptors within minutes and suggest that estimates of receptor upregulation after chronic nicotine treatment are likely not to be underestimated.
α4β2 nAChRs are desensitized after chronic nicotine treatment
To test the prediction that the concentrations of nicotine necessary for upregulation are similar to the nicotine concentrations necessary for desensitization, we measured desensitization functionally by two-electrode voltage clamp. Functional desensitization was assessed by measuring the fraction of activable receptors remaining after a 24 hr nicotine incubation at concentrations ranging from 3 to 1000 nm. Changes in receptor responsiveness were estimated from whole-cell response amplitudes to nicotine test pulses applied near the EC50 for activation (10–20 μm) (Fenster et al., 1997). Test pulses were administered before and immediately after incubation in nicotine (Katz and Thesleff, 1957; Feltz and Trautmann, 1982). Example test pulses are shown in Figure3A. As illustrated in thetraces from oocytes not treated with nicotine, control responses were often larger when measured 24 hr later (e.g., because of continual protein synthesis). To account for changes in whole-cell receptor responses, which were independent of nicotine and occurred over the incubation time period, the fractional response remaining after 24 hr nicotine incubation was normalized to that of control oocytes not incubated in nicotine (see Materials and Methods).
Chronic incubation with increasing concentrations of nicotine resulted in a dose-dependent decrease in α4β2 receptor function (Fig.3C). The data in Figure 3B also show the effect of removal of nicotine on the relative responses of these oocytes. There was a small increase in peak currents 1–2 hr after removing the oocytes from nicotine incubation but almost no recovery thereafter. This increase in response may represent the return of some of the desensitized receptors to the activable state in the first 2 hr after removal from nicotine incubation. Both the decreases in α4β2 receptor function after 24 hr nicotine incubation and the limited recovery after removal of nicotine are similar to results reported previously (Hsu et al., 1996).
Figure 3C shows a dose–response curve constructed from the relative decrease in α4β2 receptor function after chronic nicotine treatment at six different nicotine concentrations (open circles). Half-maximal desensitization of α4β2 nAChRs was calculated to occur at nicotine incubation concentrations of 31 nm. Thus, it might be concluded that the concentrations of nicotine necessary to induce desensitization are approximately threefold higher than those required for upregulation. However, the calculated relative decrease in receptor function after nicotine exposure does not directly reveal the fraction of receptors that are activatable after incubation, because the total number of α4β2 receptors is upregulated during the 24 hr nicotine incubation. It is therefore necessary to correct for this change in receptor number. We normalized the relative response remaining after nicotine incubation by the amount of receptor upregulation observed at the particular nicotine incubation concentration (Fig. 2B). For example, after chronic incubation with 60 nm nicotine, the relative increase in [3H]nicotine binding (b) was 178 ± 16%, and the fractional response remaining (f) was 0.36 ± 0.04 (i.e., 1.78 of the initial number of receptors were now responsible for 0.36 of the relative response). After correction (f/b), the fraction of activable receptors remaining at the end of the 24 hr treatment with 60 nmnicotine was estimated to be 0.20. After correction of all concentration points, the half-maximal desensitization of α4β2 nAChRs was calculated to occur at nicotine incubation concentrations of 9.7 nm (Fig. 3C, filled circles). The similarity between the half-maximal values for upregulation of specific surface [3H]nicotine binding to intact oocytes (Fig. 2B) and functional desensitization is consistent with the idea that desensitization is a trigger for upregulation.
Upregulation and desensitization of lower affinity nAChRs are correlated
If desensitization is a common trigger for upregulation of many different nAChRs, then an nAChR that has a lower affinity for nicotine-induced desensitization should exhibit a comparably lower affinity for upregulation. To test this hypothesis, we examined the concentration-dependence of nicotine-induced desensitization and upregulation for α3β4 nAChRs, a receptor subtype with ∼10-fold lower affinity for nicotine than α4β2 nAChRs (Fenster et al., 1997). As described for α4β2 nAChRs in Figure 3, functional desensitization was assessed from changes in receptor responsiveness after 24 hr incubation in nicotine at concentrations ranging from 30 nm to 10 μm. Representative tracesfor α3β4-expressing oocytes untreated or treated for 24 hr in 3 μm nicotine are shown in Figure4A. The relative responses remaining after incubation at two different concentrations, 1 and 10 μm, are shown in Figure 4B. These data were normalized to the responses of control oocytes not incubated in nicotine to factor in changes in receptor expression. Also, similar to the results shown for α4β2 nAChRs, little recovery of function from 24 hr nicotine incubation was evident in α3β4-expressing oocytes, even after 24 hr in the absence of nicotine. Similar experiments were repeated at several other nicotine incubation concentrations to obtain a dose–response curve for functional desensitization of α3β4 nAChRs caused by chronic nicotine treatment. The uncorrected half-maximal nicotine concentration for functional α3β4 desensitization was calculated to be 462 nm (Fig. 4C, open circles).
Fig. 4.

Desensitization and upregulation of α3β4 nAChRs. A, Representative traces measured before and after incubation. Experiments are the same as in Figure3A, except that test pulses were performed with 60 μm nicotine and the 24 hr incubation was performed with 3 μm nicotine. B, Representative fractional nicotine-induced responses after 24 hr nicotine incubation. α3β4-expressing oocytes were incubated for 24 hr in control media or media containing 300 nm or 10 μm nicotine. Measurement of peak currents before and after incubation and calculation of desensitization are described in Figure3A. Peak responses were also determined 2 and 24 hr after removal from nicotine. C, Inhibition dose–response curves constructed from fractional responses as described in B. Data are shown both uncorrected (open circles) and corrected (filled circles) for the amount of upregulation (i.e., relative increases in [3H]nicotine binding) observed after 24 hr incubation in nicotine at the same concentration (Fig.3C). The amount of upregulation at a given concentration is taken from the data in Figure 4D. Thesolid lines are logistic fits to mean data from which the half-maximal nicotine concentration for desensitization wasn = 3–15 oocytes per data point. D, Upregulation is nicotine concentration-dependent. [3H]Nicotine binding assays were performed as described in Figure 2B. Data are plotted as the amount of specific [3H]nicotine binding for oocytes incubated in nicotine compared with the specific [3H]nicotine binding for control oocytes not incubated in nicotine (n = 4–9 oocytes per data point).
To determine the levels of nicotine necessary for α3β4 upregulation, we examined specific surface [3H]nicotine binding to α3β4 nAChRs in intact oocytes after 24 hr nicotine incubation using concentrations ranging from 30 nm to 10 μm. An upregulation dose–response curve was constructed from the relative increases in [3H] nicotine binding, and these data are plotted in Figure 4D. The half-maximal nicotine concentration for upregulation was estimated to be 215 nm. As described above for α4β2 nAChRs, we then used the relative increases in α3β4 binding at each nicotine concentration to correct for the relative response measured in the desensitization assays. The correction yielded a half-maximal nicotine concentration for functional α3β4 desensitization of 141 nm (Fig. 4C,filled circles). Thus, α3β4 receptors require ∼10-fold higher nicotine concentrations to induce half-maximal desensitization than α4β2 receptors, and this shift is paralleled by a comparable shift in the nicotine concentrations necessary for upregulation.
Receptors that do not recover from desensitization upregulate after brief applications of nicotine
Previously, we produced a mutant α4 subunit in which a putative PKC phosphorylation site was eliminated (S336A). Briefly, when this subunit is coexpressed with a wild-type β2 subunit, it forms receptors in oocytes that exhibit many functional properties similar to the wild-type α4β2 receptors. For example, (1) dose–response relationships for nicotine-induced activation estimate an EC50 value of 13 μm, which is similar to the estimated EC50 value for wild-type receptors (15 μm) (Fenster et al., 1997); and (2) rates into the desensitized state (induced by 2 min applications of 10 μm nicotine) are the same as for wild-type α4β2 receptors. However, α4S336Aβ2 receptors are different from wild-type receptors in one way that is important for the present studies; whereas wild-type α4β2 receptors recovered fully from desensitization (with a time constant of ∼43 min), α4S336Aβ2 receptors showed <20% total recovery (Fenster et al., 1999). We reasoned that if desensitization is the trigger for upregulation, then we should be able to make specific predictions regarding upregulation in the mutant that would be different from wild-type α4β2 receptors.
Because the mutant α4β2 receptor does not readily return to the activatable state after desensitization, one prediction is that once mutant receptors are desensitized by nicotine, then nicotine will no longer be required to produce upregulation. This would not be true of wild-type receptors, which would recover from desensitization in the absence of nicotine. To test this prediction, we subjected oocytes injected with either wild-type or mutant receptors to periodic nicotine treatment. Specifically, the oocytes were incubated for 1 hr every 12 hr in 60 nm nicotine. The rationale was that 1 hr treatment would desensitize both receptor types, but the wild-type receptors would recover in the absence of nicotine during the subsequent hour; nicotine was added for 1 hr at 12 hr intervals to desensitize any new receptors that had been inserted during the assay. Data obtained from oocytes in the periodically incubated condition were compared with oocytes that were untreated or incubated continuously for 48 hr in 60 nm nicotine. Wild-type nAChRs were not significantly upregulated after periodic treatment. In contrast, the mutant nAChRs were upregulated to levels comparable with that seen with continuous nicotine treatment (Fig.5C).
Fig. 5.
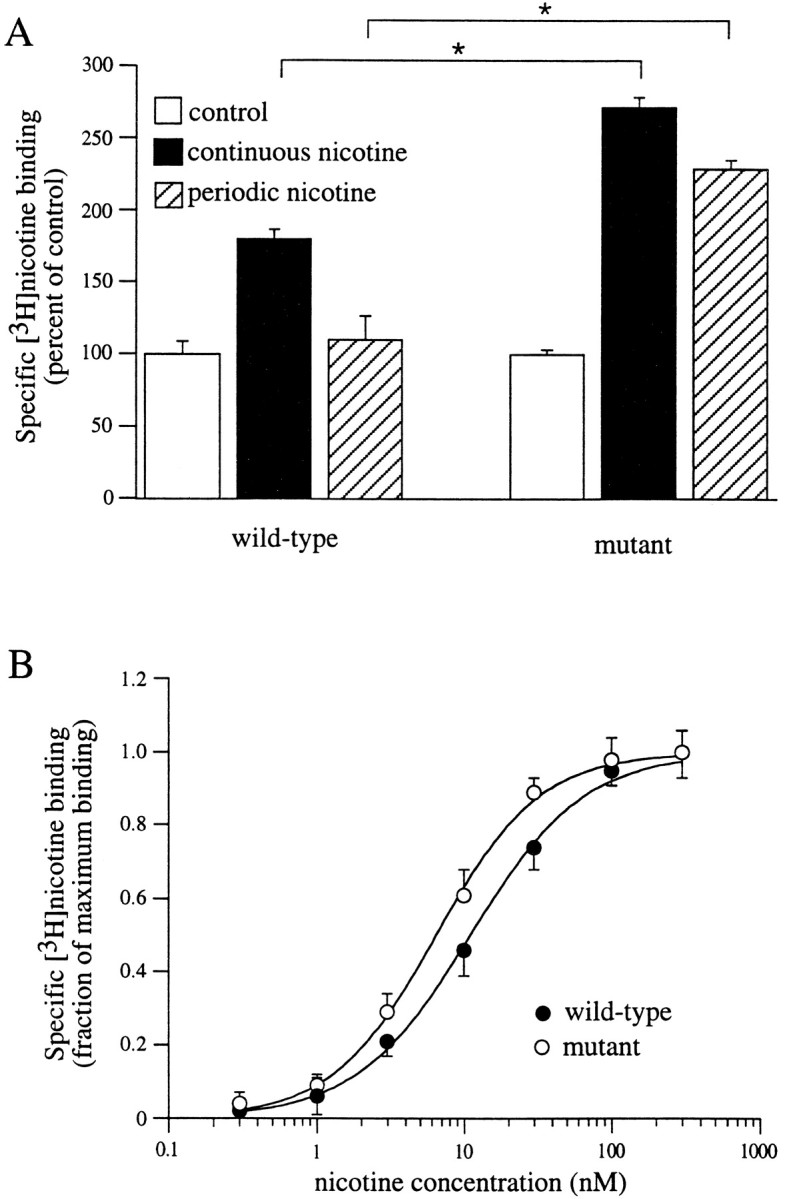
Upregulation and equilibrium binding to mutant (S336A) α4β2 receptors. A, Mutant, but not wild-type, α4β2 receptors are upregulated by periodic nicotine treatment. Expressing oocytes were incubated continuously for 48 hr in ND96+Ca (open bars), continuously for 48 hr in 60 nm nicotine (filled bars), or periodically (hatched bars; 1 hr in 60 nm nicotine every 12 hr for 48 hr). Specific surface [3H]nicotine binding to intact oocytes was then measured for 1 hr using 60 nm[3H]nicotine. Data are from five to seven oocytes per condition. *p < 0.05; unpairedt test. B, Equilibrium binding to intact oocytes expressing wild-type (filled circles) or mutant (S336A; open circles) α4β2 receptors. Assays were performed for 1 hr using [3H]nicotine concentrations from 0.1 to 300 nm. Dose–response curves were constructed from the amount of specific [3H]nicotine binding normalized to maximal [3H]nicotine binding (n = 5–6 oocytes per data point).
A potential explanation for these data are that nicotine remains bound to the mutant receptors and thus the “periodic” treatment is in fact “continuous” treatment. To rule out this possibility, we examined the dissociation rate of [3H] nicotine from α4S336Aβ2 receptors. The dissociation time constant was 69 sec (data not shown), which is similar to that observed for wild-type α4β2 nAChRs (Fig. 2C). The most straightforward interpretation of the periodic treatment data are that, at least in the case of the mutant receptor, the unoccupied desensitized state is sufficient to induce upregulation.
Mutant α4β2 nAChRs have a higher apparent affinity for nicotine
In addition to the finding that periodic treatment with nicotine could upregulate mutant receptors, continuous incubation with 60 nm nicotine for 48 hr resulted in a greater amount of mutant nAChR upregulation (∼150%) compared with wild-type receptor nAChR upregulation (∼80%; Fig. 5A). These data imply that the mutant nAChRs may have a higher affinity for nicotine (i.e., upregulation would occur at lower agonist concentrations). To test this idea, we replotted the [3H]nicotine saturation data presented in Figure 1A for wild-type α4β2 nAChRs measured in intact oocytes (Fig. 5B, filled circles). The concentration of nicotine required for half-maximal binding to wild-type receptors in intact oocytes was estimated to be 11 nm. In comparison, the concentration of nicotine required for half-maximal binding to mutant α4S336Aβ2 receptors in intact oocytes was estimated to be 6 nm. Thus, mutant α4β2 receptors are more readily upregulated than wild-type nAChRs, and this difference is correlated with a shift in equilibrium nicotine binding.
Equilibrium binding to α4β2 nAChRs is altered by membrane homogenization
The present data suggest that upregulation and desensitization are closely related phenomena. Previous estimates of these parameters have not revealed such a close association between the concentration of nicotine necessary for upregulation and that which produces desensitization (Peng et al., 1994; Whiteaker, 1998). In both of these studies, however, estimates of the potency of nicotine for desensitization was obtained from equilibrium [3H]nicotine binding. Therefore, one possibility for the apparent concentration mismatch between upregulation and desensitization is that equilibrium binding studies measure receptors in an altered state(s) from that encountered when measuring functional desensitization. In the present study, the half-maximal value obtained for equilibrium binding of [3H]nicotine to α4β2 nAChRs in intact oocytes (11 nm) (Fig.5B) is comparable with that for desensitization (10 nm) (Fig. 3C). Together with our observation that the number of surface agonist binding sites and functional channels are correlated (Fig. 1B), these data suggest that equilibrium [3H]nicotine binding to intact surface receptors and functional desensitization are likely measuring the same population of α4β2 nAChRs in the same state(s).
Another explanation for large differences between previously observed dose–response curves for receptor upregulation and equilibrium binding is that the majority of binding studies have been performed on membrane homogenates. Such preparations may differ from surface binding in intact preparations because of contributions from nonsurface receptors and/or changes in the biochemical state of the receptor (Wonnacott, 1987). To test this hypothesis, we performed saturation [3H]nicotine binding assays on cell lysates obtained from oocytes expressing α4β2 nAChRs and compared the results with those obtained from the intact oocyte experiments (Fig.6). After membrane homogenization, the dose–response curve for equilibrium nicotine binding was shifted significantly to the left; the concentration of nicotine for half-maximal binding was estimated to be 0.4 nm. These data demonstrate that the apparent affinity of nAChRs for nicotine is altered after membrane homogenization (Whiteaker et al., 1998), implying that equilibrium binding may not always accurately reflectin vivo receptor desensitization. In addition, at near saturating concentrations of [3H]nicotine (60 nm), specific binding to membrane homogenates was approximately sixfold greater than for surface binding to intact oocytes, implying that >80% of the binding in membrane homogenates was to intracellularly localized receptors. Together, the increase in both the apparent affinity and the number of receptors after membrane homogenization argues that most nAChRs in oocytes exist in a high-affinity intracellular pool, with a smaller receptor population on the cell surface that has a lower apparent affinity for nicotine.
Fig. 6.

Equilibrium binding to intact and homogenized oocyte membranes. α4β2-expressing oocytes from the same oocyte batch were subjected to membrane-intact (filled circles) or membrane-homogenate (open circles) [3H]nicotine binding assays. Assays were performed for 1 hr using [3H]nicotine concentrations from 0.1 to 300 nm. Dose–response curves were constructed from the amount of specific [3H]nicotine binding normalized to maximal [3H]nicotine binding (n = 5–6 oocytes per data point).
Upregulation of α4β2 nAChRs in total membrane homogenates
If desensitization of surface α4β2 receptors is the trigger for upregulation of intracellular, as well as surface, receptors (Whiteaker et al., 1998), then the half-maximally effective concentration for upregulation of both populations should be the same. If, however, these populations are regulated independently, then their concentration requirements for upregulation may be different. As we have shown for surface receptors, upregulation occurs at concentrations that induce desensitization. Because intracellular receptors (the majority of binding sites on homogenized membranes) appear to have a higher apparent affinity than surface receptors, it might be predicted that upregulation will occur at lower concentrations of nicotine. However, after chronic (24 hr) exposure to nicotine, we observed that upregulation of specific [3H]nicotine binding to isolated membranes required higher agonist concentrations than those necessary for upregulation of surface receptors. The half-maximal concentration of nicotine for upregulation of intracellular nAChRs was 58 nm (Fig. 7). Thus, as others have observed previously, there is a discrepancy between the concentrations required for half-maximal equilibrium binding and those necessary for upregulation in homogenized membranes (Peng et al., 1994;Bencherif et al., 1995; Warpman et al., 1998; Whiteaker et al., 1998).
Fig. 7.

Upregulation of total α4β2 receptors after chronic nicotine incubation. Specific [3H]nicotine binding to homogenized membranes is upregulated by nicotine. Oocytes were injected with α4 and β2 subunit cRNAs. Twenty-four hours later, oocytes were placed in ND96+Ca in the absence or presence of various concentrations of nicotine for 24 hr. After preparation of homogenized membranes, binding assays were performed using 60 nm [3H]nicotine. The plot shows a dose–response curve constructed from the relative increase in specific [3H]nicotine binding to homogenized membranes at each nicotine incubation concentration. Data are plotted as the amount of specific [3H]nicotine binding for oocytes incubated in nicotine compared with the specific [3H]nicotine binding for control oocytes not incubated in nicotine (n = 16–20 oocytes per data point).
DISCUSSION
In the present study, we provide several lines of evidence to suggest that, for α4β2 nAChRs on the surface of oocytes, receptor upregulation is directly related to receptor desensitization: (1) the half-maximal value for nicotine-induced upregulation is equal to the half-maximal effective concentrations of nicotine required for both functional receptor desensitization and equilibrium binding to surface nAChRs in intact oocytes; (2) the half-maximal value for upregulation of α3β4 nAChRs is ∼10-fold higher than for α4β2 nAChRs, and this shift is mirrored by a 10-fold shift in the half-maximal concentration necessary for functional desensitization; and (3) mutant α4β2 receptors, for which recovery from the desensitized state does not readily occur (Fenster et al., 1999), can be upregulated in the absence of nicotine. Additionally, we find that much lower concentrations of [3H]nicotine are needed for half-maximal equilibrium binding to receptors after membrane homogenization. These latter data may account for some, but not all, of the discrepancies between apparent affinities for equilibrium binding and upregulation observed here and in other systems (Peng et al., 1994;Whiteaker et al., 1998).
α4β2 nAChR equilibrium binding and functional desensitization
Based on the Katz and Thesleff (1957) cyclical model of desensitization, as illustrated below, nAChRs may exist either in activable states R or higher affinity desensitized statesD, where L is equal to the ratio of desensitized to activatable receptorsD/R.

Because the affinity of nicotine is higher for the desensitized state compared with the activatable state (i.e.,Ko ≫ K1) (Katz and Thesleff, 1957; Feltz and Trautmann, 1982), prolonged nicotine exposure should stabilize receptors in the agonist-bound desensitized state AD. Based on this model, the apparent affinity of the desensitized state can be estimated using either measures of functional desensitization or equilibrium binding (Lippiello et al., 1987;Grady et al., 1994). Consistent with this idea, we and others (Higgins and Berg, 1988; Grady et al., 1994) find that apparent affinities for equilibrium binding and functional desensitization are similar.
After chronic exposure to low concentrations of nicotine, our functional assessment of α4β2 nAChR desensitization measures the relative fraction of activatable receptorsR/Rmax remaining (Feltz and Trautmann, 1982):
| Equation 2 |
At equilibrium, [3H]nicotine binding will measure the fraction ƒ of total receptors in the desensitized stateAD:
| Equation 3 |
Both assessments of the potency of nicotine for inducing desensitization will be influenced by the ratio of desensitized to activatable receptors L (Marks et al., 1996); thus, the half-maximal effective concentrations of nicotine that we have calculated are related, but not equal, to the affinity of nicotine for the desensitized state. The true affinity of the desensitized stateK1 can be determined only if an independent estimate of L is obtained (Lippiello et al., 1987). More importantly for the present study, Equations 2 and 3 predict the same half-maximal agonist values for functional desensitization and equilibrium binding if K1 and Lremain constant for both types of measurements. The most straightforward empirical method for doing this is to determine these two half-maximal concentrations under comparable conditions. Measuring both specific radiolabeled binding to surface nAChRs in intact oocytes and functional desensitization in the same oocytes (or oocytes from the same batch) is one such approach.
It has been hypothesized that chronic nicotine-induced upregulation in the number of high affinity (α4β2) binding sites in the CNS is a consequence of receptor desensitization (Marks et al., 1983; Schwartz and Kellar, 1985). From the model, upregulation will be directly related to occupation of the desensitized state by nicotine, and therefore the concentration of nicotine required for upregulation can be predicted from Equation 3. Then, if the desensitization hypothesis is correct, the half-maximal nicotine concentrations for both upregulation and equilibrium [3H]nicotine binding should be the same. Our data for nAChRs expressed on the surface of intact oocytes are consistent with this hypothesis.
Receptor pools and apparent [3H]nicotine equilibrium binding affinities
We observed a >20-fold decrease in the half-maximal [3H]nicotine concentration necessary for α4β2 nAChR equilibrium binding after membrane homogenization compared with that measured for surface receptors in intact oocytes. In addition, membrane homogenization revealed a population of receptors (∼80% of total receptors) not measured during surface binding assays on intact oocytes. From these observations, we may conclude that the majority of nAChRs are intracellular (Whiteaker et al., 1998), with a higher apparent affinity for nicotine than surface receptors. Why then is the surface pool of receptors not detected as a lower affinity component (∼20%) of [3H] nicotine binding to homogenized membranes? Aside from the potential difficulty in distinguishing this component, it may be that membrane homogenization affects the integrity of surface receptors, allowing them to enter higher affinity states.
From cyclical models of desensitization, there are two ways of altering the apparent agonist binding affinity (Marks et al., 1996): a change in the microscopic affinity constant K1 or a change in the ratio of desensitized to activatable receptors L. Because the dissociation rate of nicotine from intact oocytes is similar to that obtained after membrane homogenization (Marks and Collins, 1982), the microscopic affinity of the desensitized stateK1 is likely unaffected by cell lysis. Thus, the effect of membrane homogenization may be in part explained by a shift in L, the initial fraction of receptors in the desensitized state. If this shift favors more receptors in the desensitized state, then the apparent binding affinity will increase and approach the microscopic affinity of the desensitized state (Marks et al., 1996).
We suggest that some of the differences in the apparent affinity of nicotine for receptors on internal and/or homogenized membranes compared with intact surface receptors are a result of altered biochemical regulation. Consistent with this idea, it has been shown that recovery from desensitization of α4β2 nAChRs is enhanced by PKC activation and phosphatase inhibition (Eilers et al., 1997; Fenster et al., 1999). Because recovery from desensitization likely proceeds via the transition from desensitized to activatable receptors, then the allosteric constant L will be increased by phosphorylation, which will in turn lead to a decrease in the apparent binding affinity (Equation 3). In support of this suggestion, we report here that mutant α4β2 receptors have an approximately twofold higher apparent [3H]nicotine binding affinity than wild-type receptors. Because the rate of nicotine dissociation from the desensitized state is not changed in mutant receptors, the increased affinity of mutant channels is predicted from the decrease inL that likely results from the slowed rate of recovery from desensitization in the mutant channel (Fenster et al., 1999).
Based on the above interpretation, we suggest that, in contrast to wild-type α4β2 nAChRs, mutant receptors become trapped in the unbound desensitized state, after chronic exposure to nicotine. It follows that because the mutant receptor can upregulate in the absence of ligand (Fig. 5), the unoccupied desensitized state may be sufficient for inducing upregulation under certain circumstances. This result is consistent with the finding that chronic PKC inhibition, which alone can downregulate α4β2 receptor function (Eilers et al., 1997), presumably by shifting more receptors into the desensitized state (Fenster et al., 1999), can cause upregulation (Golpalakrishnan et al., 1997) in the absence of nicotine.
Is desensitization a general trigger for upregulation of nAChRs?
If upregulation of both surface and intracellularly localized receptors is triggered through a common mechanism, e.g., the interaction of nicotine with cell surface receptors (Whiteaker et al., 1998), then upregulation of both pools should have the same dependency on nicotine concentration. However, the half-maximal concentration of nicotine required for upregulation of the intracellular pool (in homogenized membranes) was higher (∼60 nm) than that necessary for upregulation of surface nAChRs (∼10 nm). These data imply that, unlike surface nAChRs, upregulation of intracellular α4β2 receptors in oocytes is not initiated through an interaction with the desensitized state of surface nAChRs. Moreover, if the apparent binding affinity for nicotine (∼400 pm) in homogenized membranes primarily reflects desensitized intracellular receptors, it is very unlikely that upregulation is mediated via occupation of the desensitized state of this pool of receptors. Based on our suggestion for surface receptors, it may be that intracellular nAChRs are shifted to a higher affinity state by membrane homogenization. However, because there is only a twofold difference in the apparent agonist binding affinities to intracellular chick α4β2 nAChRs between intact M10 cells and isolated membranes (Whiteaker et al., 1998), this seems improbable. Thus, in the case of intracellular receptors, we, like many others, are left to explain why greater than saturating concentrations of nicotine are required for receptor upregulation (Peng et al., 1994; Bencherif et al., 1995;Warpman et al., 1998; Whiteaker et al., 1998). One explanation is that nicotine directly (i.e., in a nonreceptor-mediated manner) interferes with processes that regulate the number of nAChRs. To answer this question, it will be necessary to more fully understand the factors that control the movement of receptors between functional and silent surface pools and intracellular pools.
Conclusions
The overall problem of how chronic nicotine alters the number and function of nAChRs remains unresolved; however, we suggest that desensitization plays an important role in upregulation of the population of receptors on the plasma membrane. Although other factors must be taken into account in vivo (Rowell and Li, 1997), the relationship between desensitization and upregulation should be useful for predicting the long-term consequences of tobacco-related levels of nicotine on different subtypes of nAChRs in the CNS.
Footnotes
This research was supported by United States Public Health Service Grants DA-11940 and NS-31669 (R.A.J.L.) and W. M. Keck Foundation Grant 931360. We thank Drs. Y. Chang and D. S. Weiss for sharing their methods on radiolabeled ligand binding in single oocytes before publication.
Correspondence should be addressed to Robin A. J. Lester, Department of Neurobiology, CIRC 560, University of Alabama at Birmingham, 1719 Sixth Avenue South, Birmingham, AL 35294-0021.
REFERENCES
- 1.Balfour DJ. Neural mechanisms underlying nicotine dependence. Addiction. 1994;89:1419–1423. doi: 10.1111/j.1360-0443.1994.tb03738.x. [DOI] [PubMed] [Google Scholar]
- 2.Bencherif M, Fowler K, Lukas RJ, Lippiello PM. Mechanisms of up-regulation of neuronal nicotinic acetylcholine receptors in clonal cell lines and primary cultures of fetal rat brain. J Pharmacol Exp Ther. 1995;275:987–994. [PubMed] [Google Scholar]
- 3.Benowitz NL, Porchet H, Jacob P., III Nicotinic dependence and tolerance in man: pharmacokinetic and pharmocodynamic investigations. Prog Brain Res. 1989;79:279–287. doi: 10.1016/s0079-6123(08)62487-5. [DOI] [PubMed] [Google Scholar]
- 4.Benwell M, Balfour DJK, Anderson J. Evidence that tobacco smoking increases the density of (−)-[3H] nicotine binding sites in human brain. J Neurochem. 1988;50:1243–1247. doi: 10.1111/j.1471-4159.1988.tb10600.x. [DOI] [PubMed] [Google Scholar]
- 5.Boyd ND. Two distinct kinetic phases of desensitization of acetylcholine receptors of clonal rat PC12 cells. J Physiol (Lond) 1987;389:45–67. doi: 10.1113/jphysiol.1987.sp016646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Breese CR, Marks MJ, Logel J, Adams CE, Sullivan B, Collins AC, Leonard S. Effect of smoking history on [3H]nicotine binding in human postmortem brain. J Pharmacol Exp Ther. 1997;283:7–13. [PubMed] [Google Scholar]
- 7.Chang Y, Weiss DS. Channel opening locks agonist onto the GABAC receptor. Nat Neurosci. 1999;2:219–225. doi: 10.1038/6313. [DOI] [PubMed] [Google Scholar]
- 8.Corey JL, Davidson N, Lester HA, Brecha N, Quick MW. Protein kinase C modulates the activity of a cloned γ-aminobutyric acid transporter expressed in Xenopus oocytes via regulated subcellular redistribution of the transporter. J Biol Chem. 1994;269:14759–14767. [PubMed] [Google Scholar]
- 9.Dani JA, Heinemann S. Molecular and cellular aspects of nicotine abuse. Neuron. 1996;16:905–908. doi: 10.1016/s0896-6273(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 10.Eilers H, Schaeffer E, Bickler PE, Forsayeth JR. Functional deactivation of the major neuronal nicotinic receptor caused by nicotine and a protein kinase-C dependent mechanism. Mol Pharmacol. 1997;52:1105–1112. [PubMed] [Google Scholar]
- 11.Feltz A, Trautmann A. Desensitization at the frog neuromuscular junction: a biphasic process. J Physiol (Lond) 1982;322:257–272. doi: 10.1113/jphysiol.1982.sp014036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fenster CP, Rains M, Noerager B, Quick MW, Lester RAJ. Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J Neurosci. 1997;17:5747–5759. doi: 10.1523/JNEUROSCI.17-15-05747.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fenster CP, Beckman ML, Parker JC, Sheffield EB, Whitworth TL, Quick MW, Lester RAJ. Regulation of α4β2 nicotinic receptor desensitization by calcium and protein kinase C. Mol Pharmacol. 1999;55:432–443. [PubMed] [Google Scholar]
- 14.Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of α4 and β2 subunits and is upregulated by chronic nicotine treatment. Mol Pharmacol. 1992;41:31–37. [PubMed] [Google Scholar]
- 15.Flores CM, Davila-Garcia MI, Ulrich YM, Kellar KJ. Differential regulation of neuronal nicotinic receptor binding sites following chronic nicotine administration. J Neurochem. 1997;69:2216–2219. doi: 10.1046/j.1471-4159.1997.69052216.x. [DOI] [PubMed] [Google Scholar]
- 16.Golpalakrishnan M, Molinari EJ, Sullivan J. Regulation of human α4β2 neuronal nicotine acetylcholine receptors by cholinergic channel ligand and second messenger pathways. Mol Pharmacol. 1997;52:524–534. [PubMed] [Google Scholar]
- 17.Grady SR, Marks MJ, Collins AC. Desensitization of nicotine-stimulated [3H]dopamine release from mouse striatal synaptosomes. J Neurochem. 1994;62:1390–1398. doi: 10.1046/j.1471-4159.1994.62041390.x. [DOI] [PubMed] [Google Scholar]
- 18.Higgins LS, Berg DK. A desensitized form of neuronal nicotinic acetylcholine receptor detected by [3H]-nicotine binding on bovine adrenal chromaffin cells. J Neurosci. 1988;8:1436–1446. doi: 10.1523/JNEUROSCI.08-04-01436.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu Y-N, Amin J, Weiss DS, Wecker L. Sustained nicotine exposure differentially affects α3β2 and α4β2 neuronal nicotinic receptors expressed in Xenopus oocytes. J Neurochem. 1996;66:667–675. doi: 10.1046/j.1471-4159.1996.66020667.x. [DOI] [PubMed] [Google Scholar]
- 20.Katz B, Thesleff S. A study of the “desensitization” produced by acetylcholine at the motor end-plate. J Physiol (Lond) 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lippiello PM, Sears SB, Fernandes KG. Kinetics and mechanism of L-[3H]nicotine binding to putative high affinity receptor sites in rat brain. Mol Pharmacol. 1987;31:392–400. [PubMed] [Google Scholar]
- 22.Lukas RJ. Effects of chronic nicotinic ligand exposure on functional activity of nicotinic acetylcholine receptors expressed by cells of the PC12 rat pheochromocytoma or the TE671/RD human clonal line. J Neurochem. 1991;56:1134–1145. doi: 10.1111/j.1471-4159.1991.tb11403.x. [DOI] [PubMed] [Google Scholar]
- 23.Margiotta JF, Berg DK, Dionne VE. Cyclic AMP regulates the proportion of functional acetylcholine receptors on chicken ciliary ganglion neurons. Proc Natl Acad Sci USA. 1987;84:8155–8159. doi: 10.1073/pnas.84.22.8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marks MJ, Collins AC. Characterization of nicotine binding in mouse brain and comparison with the binding of α-bungarotoxin and quinuclidinyl benzilate. Mol Pharmacol. 1982;22:554–564. [PubMed] [Google Scholar]
- 25.Marks MJ, Burch JB, Collins AC. Effects of chronic nicotine infusion on tolerance development and cholinergic receptors. J Pharmacol Exp Ther. 1983;226:806–816. [PubMed] [Google Scholar]
- 26.Marks MJ, Grady SR, Collins AC. Downregulation of nicotine receptor function after chronic nicotine infusion. J Pharmacol Exp Ther. 1993;266:1268–1276. [PubMed] [Google Scholar]
- 27.Marks MJ, Robinson SF, Collins AC. Nicotinic agonists differ in activation and desensitization of 86Rb+ efflux from mouse thalamic synaptosomes. J Pharmacol Exp Ther. 1996;277:1383–1396. [PubMed] [Google Scholar]
- 28.Papke RL, Boulter J, Patrick J, Heinemann S. Single-channel currents of rat neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes. Neuron. 1989;3:589–596. doi: 10.1016/0896-6273(89)90269-9. [DOI] [PubMed] [Google Scholar]
- 29.Peng X, Gerzanich V, Anand R, Whiting PJ, Lindstrom J. Nicotine-induced increase in neuronal receptors results from a decrease in the rate of receptor turnover. Mol Pharmacol. 1994;46:523–530. [PubMed] [Google Scholar]
- 30.Quick MW, Lester HA. Methods for expression of excitability proteins in Xenopus oocytes. In: Conn PM, editor. Methods in neurosciences. Academic; San Diego: 1994. pp. 261–271. [Google Scholar]
- 31.Ritchie JM, Rogart RB. Density of sodium channels in mammalian myelinated nerve fibers and nature of the axonal membrane under the myelin sheath. Proc Natl Acad Sci USA. 1977;74:211–215. doi: 10.1073/pnas.74.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Role LW. Diversity of primary structure and function of neuronal nicotinic acetylcholine receptors. Curr Opin Neurobiol. 1992;2:254–262. doi: 10.1016/0959-4388(92)90112-x. [DOI] [PubMed] [Google Scholar]
- 33.Rowell PP, Li M. Dose–response relationship for nicotine-induced up-regulation of rat brain nicotinic receptors. J Neurochem. 1997;68:1982–1989. doi: 10.1046/j.1471-4159.1997.68051982.x. [DOI] [PubMed] [Google Scholar]
- 34.Sargent PB. The diversity of neuronal acetylcholine receptors. Annu Rev Neurosci. 1993;16:403–443. doi: 10.1146/annurev.ne.16.030193.002155. [DOI] [PubMed] [Google Scholar]
- 35.Schwartz RD, Kellar KJ. In vivo regulation of [3H]acetylcholine recognition sites in brain by nicotinic cholinergic drugs. J Neurochem. 1985;45:427–433. doi: 10.1111/j.1471-4159.1985.tb04005.x. [DOI] [PubMed] [Google Scholar]
- 36.Warpman U, Friberg L, Gillespie A, Hellström-Lindahl E, Zhang X, Nordberg A. Regulation of nicotinic receptor subtypes following chronic nicotinic agonist exposure in M10 and SH-SY5Y neuroblastoma cells. J Neurochem. 1998;70:2028–2037. doi: 10.1046/j.1471-4159.1998.70052028.x. [DOI] [PubMed] [Google Scholar]
- 37.Whiteaker P, Sharples C, Wonnacott S. Agonist-induced up-regulation of α4β2 nicotine acetylcholine receptors in M10 cells: pharmacological and spatial definition. Mol Pharmacol. 1998;53:950–962. [PubMed] [Google Scholar]
- 38.Wonnacott S. Brain nicotine binding sites. Hum Toxicol. 1987;6:343–353. doi: 10.1177/096032718700600502. [DOI] [PubMed] [Google Scholar]
- 39.Wonnacott S. The paradox of nicotinic acetylcholine receptor upregulation by nicotine. Trends Pharmacol Sci. 1990;11:216–219. doi: 10.1016/0165-6147(90)90242-z. [DOI] [PubMed] [Google Scholar]