

Abstract
Apolipoprotein (apo) E isoforms are key determinants of susceptibility to Alzheimer’s disease. The apoE4 isoform is the major known genetic risk factor for this disease and is also associated with poor outcome after acute head trauma or stroke. To test the hypothesis that apoE3, but not apoE4, protects against age-related and excitotoxin-induced neurodegeneration, we analyzed apoE knockout (Apoe−/−) mice expressing similar levels of human apoE3 or apoE4 in the brain under control of the neuron-specific enolase promoter. Neuronal apoE expression was widespread in the brains of these mice. Kainic acid-challenged wild-type or Apoe−/− mice had a significant loss of synaptophysin-positive presynaptic terminals and microtubule-associated protein 2-positive neuronal dendrites in the neocortex and hippocampus, and a disruption of neurofilament-positive axons in the hippocampus. Expression of apoE3, but not of apoE4, protected against this excitotoxin-induced neuronal damage. ApoE3, but not apoE4, also protected against the age-dependent neurodegeneration seen in Apoe−/− mice. These differences in the effects of apoE isoforms on neuronal integrity may relate to the increased risk of Alzheimer’s disease and to the poor outcome after head trauma and stroke associated with apoE4 in humans.
Keywords: apolipoprotein E, Alzheimer’s disease, apoE transgenic mice, excitotoxicity, apoE knockout mice, neurodegeneration
Apolipoprotein (apo) E is a 34 kDa protein that participates in the transport of plasma lipids and in the redistribution of lipids among cells (Mahley, 1988). Of the three common apoE isoforms in humans (Utermann et al., 1977), apoE4 is a major risk factor for Alzheimer’s disease (AD) (Corder et al., 1993;Strittmatter et al., 1993; Mayeux et al., 1995; Farrer et al., 1997) and for poor outcome after acute head injury (Nicoll et al., 1996;Teasdale et al., 1997) or stroke (Slooter et al., 1997). The most common isoform, apoE3, differs from apoE4 by only a single amino acid (Weisgraber et al., 1981; Rall et al., 1982).
A role for apoE in neural injury and repair processes was established well before this molecule was implicated in AD (Elshourbagy et al., 1985; Ignatius et al., 1986; Pitas et al., 1987; Mahley, 1988). More recent studies in Apoe−/− mice further suggested that apoE helps protect the brain against acute injury (Chen et al., 1997) and maintain neuronal integrity during aging (Masliah et al., 1995). Other studies have not detected age-related neurological abnormalities in Apoe−/− mice (Anderson et al., 1998; Fagan et al., 1998).
The first suggestion that apoE was involved in AD came from the immunohistochemical localization of apoE in two hallmark lesions of AD brains: amyloid plaques and neurofibrillary tangles (Namba et al., 1991). Additional studies suggested that apoE4 may contribute to these lesions through pathogenic interactions with the Aβ peptide of amyloid plaques (Wisniewski and Frangione, 1992; Ma et al., 1994; Sanan et al., 1994; Strittmatter et al., 1994) and the tau protein of neurofibrillary tangles (Strittmatter et al., 1994).
Another mechanism by which apoE might be involved in neurodegenerative processes is by isoform-specific effects on neurite extension and cytoskeletal stability (Mahley, 1988; Mahley et al., 1995; Weisgraber and Mahley, 1996). Addition of apoE3 to neuronal cultures stimulates neurite outgrowth, stabilizes microtubules, and is associated with an accumulation of cytoplasmic apoE, whereas apoE4 does not have these effects (Nathan et al., 1994, 1995; Bellosta et al., 1995b; Ji et al., 1998).
The above observations raise the possibility that repair and remodeling of neurons in response to injury proceed more effectively in the presence of apoE3 than apoE4, and that this is a reason why apoE4 acts as a susceptibility factor for age-related neurodegenerative diseases such as AD. To test this apoE injury/repair hypothesis in vivo, we expressed apoE3 and apoE4 at comparable levels in the CNS of Apoe−/− mice using the neuron-specific enolase (NSE) promoter and studied the isoform-specific effects on neurodegeneration associated with aging and excitotoxicity in these mice. The rationale for neuronal targeting was based on a number of observations. Apolipoprotein E immunoreactivity in neurons was reported in human AD brains (Han et al., 1994; Bao et al., 1996;Metzger et al., 1996) and in rat brain after ischemia (Horsburgh and Nicoll, 1996). Neuronal expression of apoE mRNA was detected in human brain (Xu et al., 1999). These data indicate that apoE could exert critical effects within neurons. NSE-driven expression of human apoE results in the secretion of human apoE isoforms into the culture medium of transfected neuronal cells (Bellosta et al., 1995b). Also, human apoE isoforms exert similar effects on cultured neuronal cells, whether they are added to the culture medium in purified form (Nathan et al., 1994), expressed in neuronal cells via stable transfection (Bellosta et al., 1995b), or secreted from cocultured astrocytes (Sun et al., 1998). Our comparison of NSE-apoE3 and NSE-apoE4 mice revealed that apoE3 protects the CNS against excitotoxin-induced and age-related neurodegeneration, whereas apoE4 does not.
MATERIALS AND METHODS
Animals. One hundred seventy-eight 3- to 9-month-old mice, weighing 25–35 gm, were studied. Mice were kept under a 12 hr light/dark cycle with free access to sterile water and food (PicoLab Rodent Diet 20, #5053, PMI Nutrition International, St. Louis, MO). Four genotypes were analyzed: wild-type mice,Apoe−/− mice, NSE-apoE3 mice, and NSE-apoE4 mice. NSE-apoE transgenic mice on theApoe−/− background were generated as follows. NSE-apoE transgenes were injected individually into one-cell embryos (ICR strain) by standard procedures; transgenic lines were established from transgenic founders. NSE-apoE3 and NSE-apoE4 lines with matching cerebral levels of transgene expression were selected and crossed with Apoe−/− mice (Piedrahita et al., 1992) provided by Dr. Nobuyo Maeda (University of North Carolina, Chapel Hill, NC). After elimination of wild-typeApoe alleles in two generations of breedings among the resulting offspring, transgenic mice were crossed withApoe−/− mice (C57BL/6J-Apoetm1Unc) from Jackson Laboratories (Bar Harbor, ME) to generate NSE-apoE3 and NSE-apoE4 mice that were at least 75% C57BL/6J. Crosses of NSE-apoE3 or NSE-apoE4 with C57BL/6J-Apoetm1Unc mice from Jackson Laboratories also yielded nontransgenic Apoe−/−littermates (n = 27). Comparison of the latter mice with age-matched C57BL/6J-Apoetm1Unc mice from Jackson Laboratories (n = 23) revealed no significant differences in any of the variables examined (data not shown). Therefore, these two cohorts of mice were combined (Apoe−/− mice) in our statistical analyses.
Genotyping of transgenic mice. Mice transgenic for NSE-apoE3 or NSE-apoE4 were identified by Southern blot analysis of genomic tail DNA using a DNA probe for human APOE (Bellosta et al., 1995a). NSE-apoE3 and NSE-apoE4 mice were differentiated by PCR. Because the human APOE intron 3 was included in the NSE-apoE4 but not in the NSE-apoE3 construct, the amplicon generated with intron 3-spanning primers (forward primer: nucleotides 3158–3175; reverse primer: nucleotides 3815–3834, GenBank accession numberM10065) was 670 base pairs (bp) in NSE-apoE4 mice and 100 bp in NSE-apoE3 mice. Proteinase K-digested tail tissue (1:100 dilution, 2 μl) was subjected to touchdown PCR (Hecker and Roux, 1996) in a total reaction volume of 25 μl with each primer (0.2 μm), dNTPs (dATP, dCTP, dGTP, dTTP, 200 μm each), and 0.15 μl of AmpliTaq GoldR DNA polymerase (Perkin-Elmer, Norwalk, CT). The reaction was run on a GeneAmp PCR System 9600 thermocycler (Perkin-Elmer). PCR products were analyzed on 1.5% agarose gels. To determine the Apoe knockout status of the mice, total plasma cholesterol levels were measured with a cholesterol measurement kit (Sigma, St. Louis, MO). Analysis of brain mRNA by RNase protection assay (RPA) (see below) confirmed the absence of mouse apoE mRNA in all mice with cholesterol levels >30 mg/dl (data not shown).
Kainic acid injections. Kainic acid crosses the blood–brain barrier and induces excitotoxic CNS injury, particularly in the hippocampus and neocortex (Strain and Tasker, 1991; Masliah et al., 1997). Kainic acid (Sigma) was dissolved in saline (0.9%) and injected intraperitoneally at 18 or 25 mg/kg body weight in one dose. Within ∼15 min, all mice developed seizures. Seizure activity was assessed as described (Schauwecker and Steward, 1997). There were no differences in kainic acid-induced seizures across groups of mice with respect to overall incidence, time period between injection and seizure onset, intensity, or duration of seizures (data not shown). This suggests that brain penetration of kainic acid was similar in all the mice. Control animals were injected with saline and did not develop seizures. Mice were killed 6 d after the injection of kainic acid or saline.
Tissue preparation. Mice were anesthetized with chloral hydrate and flush-perfused transcardially with 0.9% saline. Brains were removed and divided sagittally. One hemibrain was post-fixed in phosphate-buffered 4% paraformaldehyde, pH 7.4, at 4°C for 48 hr for vibratome sectioning; the other was snap frozen and stored at −70°C for RNA or protein analysis. Postmortem brain tissues from humans with or without AD were obtained from Dr. Eliezer Masliah (University of California, San Diego, CA) and from Dr. Tom M. Hyde (National Institute of Mental Health, Bethesda, MD).
RNA extraction and analysis. Total RNA was isolated from tissues with TRI-Reagent (Molecular Research Center, Cincinnati, OH) or Tripure (Boehringer Mannheim, Indianapolis, IN). RNA was analyzed by solution hybridization RPA with antisense riboprobes complementary to human apoE mRNA [nucleotides 281–469 of APOE cDNA (GenBank accession number M12529)] or β-actin mRNA [nucleotides 480–559 of mouse β-actin cDNA (GenBank accession number M18194)]. Because the apoE riboprobe also protects a smaller fragment of endogenous mouse apoE mRNA sequence, both human and mouse apoE mRNAs could be identified with this probe. The RPAs were performed essentially as described (Bordonaro et al., 1994). Briefly, sample RNA (10 μg) hybridized to32P-labeled antisense riboprobes was digested with 300 U/ml RNase T1 (Life Technologies, Gaithersburg, MD) and 0.5 μg/ml RNase A (Sigma) in 100 μl of digestion buffer, followed by protein digestion with 10 mg/ml proteinase K (Sigma). RNA was isolated with 4m guanidine thiocyanate and precipitated in isopropanol. Samples were separated on 5–6% acrylamide/8 m urea Tris-borate EDTA gels, and the dried gels were exposed to XAR or Biomax MS film (Kodak, Rochester, NY). Levels of specific transcripts were estimated by quantitating probe-specific signals with a phosphorimager (FUJI-BasIII, Fuji, Tokyo, Japan); β-actin signals were used to correct for differences in RNA content/loading (Johnson et al., 1995).
Analysis of CSF. After methoxyflurane overdose and exsanguination by cardiac puncture, CSF was obtained from nine NSE-apoE3, nine NSE-apoE4, and eightApoe−/− mice. We modified the procedure described by Carp et al. (1971) by using a 25 gauge needle attached to silicon tubing (0.012 inch internal diameter) and piercing the dura mater tangentially. Slight negative pressure was exerted with a tuberculin syringe to start the flow. From each adult mouse, ∼10 μl of CSF was obtained from the cisterna magna with little or no contaminating blood. The CSF was centrifuged in a desktop centrifuge to remove contaminating cells, kept at 4°C, and used within 3 d for Western blotting and quantitation of apoE. Equal volumes of CSF from the different cohorts of mice were loaded on the gels.
Western blot analysis. Brain homogenates from hemibrains were prepared with a triple detergent lysis buffer (Sambrook et al., 1989) and protease inhibitors [phenylmethylsulfonyl fluoride (100 μg/ml), aprotinin (1 μg/ml), and complete inhibitor (2×, catalog no. 1836145, Boehringer Mannheim)]. Insoluble material was removed by centrifugation. The protein concentration in the supernatant was determined with a modified Bradford method (Pierce, Rockford, IL), and sample protein concentrations were equalized with lysis buffer. SDS loading buffer was added, and the samples were heated to 95°C for 5 min. To quantitate apoE in brain tissue and CSF, samples and purified apoE standards (provided by Dr. Karl Weisgraber, Gladstone Institute of Neurological Disease) were separated by SDS-PAGE, electrotransferred to nitrocellulose membranes (Bio-Rad, Hercules, CA), and blocked with PBS containing 5% dried milk and 0.05% Tween. The blots were incubated in polyclonal goat anti-human apoE antibody (1:1000; Calbiochem, San Diego, CA) or in polyclonal rabbit anti-mouse apoE antibody (1:1000; provided by Dr. Jan Borén, Gladstone Institute of Cardiovascular Disease). The bound primary antibodies were detected with horseradish peroxidase-conjugated species-specific antibodies (Amersham, Arlington, IL). Immunodetection was performed with SuperSignal Ultra (Pierce) or ECL (Amersham) according to the manufacturer’s instructions, and the blots were exposed to x-ray film (Biomax MR, Kodak). For semiquantitative assessments of apoE, known quantities of purified human plasma apoE3 or apoE4 [prepared as described by Rall et al. (1986) and provided by Dr. Karl Weisgraber] or recombinant mouse apoE (provided by Dr. Li-Ming Dong, Gladstone Institute of Cardiovascular Disease) were run as standards on the same gels. For quantitation, exposures of Western blots with densities within the linear range of the film were scanned, and the density of the bands was determined by inflection point analysis with Advanced Quantifier software (BioImage, Ann Arbor, MI).
Immunohistochemistry. Post-fixed tissues were cut into 40-μm-thick sections with a vibratome and incubated in 0.3% H2O2 in PBS for 20 min to quench endogenous peroxidase activity. To facilitate penetration of antibodies, sections used for immunoperoxidase staining were preincubated for 4 min in 1 μg/ml proteinase K in a buffer containing 250 mm NaCl, 25 mm EDTA, 50 mm Tris/HCl, pH 8. To block nonspecific reactions, all sections were incubated for 1 hr in 15% normal donkey serum (Jackson ImmunoResearch, West Grove, PA) in PBS or for 7 min in Superblock (Scytec, Logan, UT), followed by a 1 hr incubation in PBS with the primary antibody: polyclonal goat anti-human apoE (Calbiochem) diluted 1:4000 (immunofluorescent staining) or 1:10,000 (immunoperoxidase staining) to detect human apoE, or polyclonal rabbit anti-rat apoE diluted 1:1000 (gift from Dr. Karl Weisgraber) to detect murine apoE. Sections were then washed twice in PBS and incubated for 1 hr with the secondary antibody: fluorescein isothiocyanate (FITC)–(Jackson ImmunoResearch) or biotin-coupled (Vector, Burlingame, CA) anti-goat to detect antigen-bound anti-human apoE or FITC-coupled anti-rabbit (Vector) to detect antigen-bound anti-rat apoE. After three washes in PBS, immunofluorescently labeled sections were mounted in VectaShield (Vector) and viewed with a MRC-1024 laser scanning confocal system (Bio-Rad) mounted on an Optiphot-2 microscope (Nikon, Tokyo, Japan). For immunoperoxidase staining, secondary antibody binding was detected with the ABC-Elite kit (Vector).
The intensity of human apoE immunolabeling of neurons in brains of NSE-apoE mice was determined on immunofluorescently labeled sections with the MRC-1024 system and Lasersharp (Bio-Rad) software. A 10 μm line was drawn through the cytoplasm of five randomly selected neocortical neurons per animal. The intensity of the pixels across this line was determined, and the mean pixel intensity per line was calculated.
Double-labeling for human apoE and cell-specific markers was performed essentially as described above except that sections from transgenic animals were incubated with anti-microtubule-associated protein 2 (MAP-2) antibody (1:40 dilution; Boehringer Mannheim) together with anti-human apoE, and sections from wild-type animals were incubated with anti-glial fibrillary acidic protein (GFAP) antibody (1:500 dilution, Boehringer Mannheim) together with anti-rat apoE. To detect primary antibody binding, sections were incubated for 1 hr in a mixture of secondary antibodies (1:100 dilution; Jackson ImmunoResearch): an FITC-conjugated donkey anti-goat (to detect anti-human apoE), an FITC-conjugated donkey anti-rabbit (to detect anti-rat apoE), and a Cy5-conjugated donkey anti-mouse (to detect anti-MAP-2 or anti-GFAP). After three 10 min washes in PBS, sections were mounted under glass coverslips with VectaShield (Vector) and viewed by confocal microscopy as described above. The Cy5 and FITC channels were viewed individually, and the resulting images were pseudocolored in red (Cy5) or green (FITC) with Adobe Photoshop (version 4.0, Adobe Systems, San Jose, CA). Omission of primary antibodies or incubation of sections with mismatched primary and secondary antibodies resulted in no signal. To exclude the possibility that the signals collected in the FITC channel originated from emission light from the Cy5 fluorophore and vice versa, sections labeled with FITC-conjugated secondary antibodies were imaged in the Cy5 channel, and sections labeled with Cy5-conjugated secondary antibodies were imaged in the FITC channel. No signals were detected under these control conditions.
Semiquantitative assessment of neurodegenerative changes.Brain sections immunolabeled for MAP-2 (a marker of neuronal cell bodies and dendrites) or synaptophysin (a marker of presynaptic terminals) were analyzed semiquantitatively with a Bio-Rad MRC-1024 laser scanning confocal microscope, mounted on a Nikon Optiphot-2 microscope and running Lasersharp software, essentially as described (Masliah et al., 1992; Toggas et al., 1994). Neuronal integrity was assessed in the neocortex and in the stratum radiatum of the hippocampus (CA1 subfield) in four sections per animal (two for each marker). Binding of primary antibodies (Boehringer Mannheim) was detected with an FITC-labeled secondary antibody (Vector). Sections were assigned code numbers to ensure objective assessment, and codes were not broken until analysis was complete. For each mouse, we obtained four to eight confocal images (three to four per section) of the neocortex and two to four confocal images (one to two per section) of the hippocampal CA1 subfield, each covering an area of 210 × 140 μm. The iris and gain levels were adjusted to obtain images with a pixel intensity within a linear range. Four scans were averaged (Kalman filter) to obtain each final image. Each final image was processed sequentially in Lasersharp with the following edge-enhancement filters: C7a (for images of MAP-2-labeled sections); C9a, C3b, and C7a (for images of the neocortex on synaptophysin-labeled sections); and C9a, C7a, C3b, and C9a (for images of the hippocampus on synaptophysin-labeled sections). Digitized images were transferred to a Macintosh computer and analyzed with NIH Image. The area of the neuropil occupied by MAP-2-immunolabeled dendrites or by synaptophysin-immunolabeled presynaptic terminals was quantified and expressed as a percentage of the total image area as described (Masliah et al., 1992). This approach for the semiquantitative assessment of neurodegeneration has been validated in various experimental models of neurodegeneration (Toggas et al., 1994; Masliah et al., 1995) and in diseased human brains (Masliah et al., 1992).
To assess further the integrity of neuronal structures, brain sections were immunolabeled with an antibody against phosphorylated neurofilaments in neuronal axons (1:3000 diluted SM31; Sternberger Monoclonals, Lutherville, MD). Antigen-bound antibody was detected with an FITC-labeled anti-mouse secondary antibody (Vector), diluted 1:75, and imaged with a laser scanning confocal microscope.
ELISA measurement of synaptophysin in neocortical tissue.Neocortical tissue from each hemibrain was homogenized with a Kontes motorized pestle (Fisher Scientific, Pittsburgh, PA) in 0.8 ml ice-cold homogenization buffer (1.85 mmNaH2PO4, 8.4 mmNa2HPO4, 150 mm NaCl, 5 mm benzamidine, 3 mm EDTA, 1 mmMgSO4, 0.05% sodium azide, pH 8), and sonicated for 30–60 sec. Homogenates were centrifuged at 2400 × gfor 10 min at 4°C. The supernatant was then ultracentrifuged (100,000 × g for 1 hr at 4°C). The resulting pellet (particulate fraction) was resuspended in 300–400 μl of homogenization buffer, and the protein concentration was determined by a Bradford assay (Bio-Rad) per manufacturer’s instructions.
For ELISA measurements of synaptophysin, wells of tissue culture plates (Costar, Corning, NY) were coated for 14–16 hr at 4°C with neocortical particulate fractions (0.5 μg of protein). Nonspecific binding sites were blocked with 2% donkey serum (Jackson) in PBS for 30 min at room temperature. The anti-synaptophysin antibody (Dako, Carpinteria, CA), diluted 1:5000 in PBS with 0.5% donkey serum, was applied for 90 min at room temperature. After three 10 min washes with PBS containing 0.05% Tween 20, plates were incubated for 90 min at room temperature with horseradish peroxidase-conjugated anti-rabbit antibody (Amersham) diluted 1:4000 in PBS. After another three 10 min washes with PBS containing 0.05% Tween 20, the reaction was developed with o-phenylenediamine dihydrochloride peroxidase substrate tablets (Sigma) per manufacturer’s instructions. The reaction was stopped after 15 min by adding 25% H2SO4, and the absorbance was measured at 492 nm with an ELISA reader. Absorbance values for wells in which the incubation with anti-synaptophysin antibody was omitted were subtracted from the values obtained. Triplicate absorbance values were obtained for the neocortical particulate fraction of each animal and averaged. In preliminary experiments, the linear range of the ELISA was 0.1–1 μg of protein from neocortical particulate fractions (data not shown). Examination of the particulate fraction of mouse liver, an organ lacking synaptophysin by Western blotting, showed no absorbance (data not shown). This result confirmed the specificity of the ELISA.
Statistical analyses. Quantitative data are expressed as mean ± SEM. Differences between means were assessed by unpaired two-tailed Student’s t test. Differences among means were evaluated by one-way ANOVA followed by Dunnett’s or Tukey-Kramerpost hoc test. The null hypothesis was rejected at the 0.05 level.
RESULTS
Generation of transgenic mice
The rat NSE promoter directs pan-neuronal expression of fusion gene constructs in the CNS of transgenic mice (Forss-Petter et al., 1990). Neuro-2a cells stably transfected with minigenes encoding human apoE3 or human apoE4 placed downstream of the NSE promoter secrete human apoE into the cell culture medium (Bellosta et al., 1995b). These NSE-apoE3 and NSE-apoE4 transgenes (Fig.1) were used to generate mice expressing apoE3 or apoE4 in the brain. Nine NSE-apoE3 and 12 NSE-apoE4 transgenic founder mice were identified by Southern blot analysis. Two lines of transgenic mice that showed comparable levels and distribution of apoE3 versus apoE4 in the brain were selected and crossed onto theApoe knockout (Apoe−/−) background as described in Materials and Methods.
Fig. 1.

NSE-apoE transgenes. The rat NSE promoter (Forss-Petter et al., 1990) was used to direct the expression of two distinct human apoE minigenes in neurons (Bellosta et al., 1995a). From 5′ to 3′, the apoE3 minigene consists of part of the untranslated exon 1 (from the AvrII site), the first intron, and the first 6 bp of exon 2 from the human APOE gene, a fragment of human apoE cDNA contributing the entire apoE coding region, and a genomic segment representing exon 4 noncoding sequence and 112 bp of 3′ untranslated region including the polyadenylation signal. The apoE4 minigene was similar in structure but also included the third intron of the human APOE gene. The sequences of the coding regions of both transgenes were compared to ensure that the only difference between them was the base change in exon 4 encoding cysteine (C) in apoE3 and arginine (R) in apoE4 at amino acid position 112.1, 2, 3, and4 indicate exons of the human APOEgene.
Four groups of mice were analyzed in detail: wild-type mice,Apoe−/− mice, andApoe−/− mice heterozygous for the NSE-apoE3 transgene (NSE-apoE3 mice) or the NSE-apoE4 transgene (NSE-apoE4 mice).
Tissue-specific distribution of apoE3 and apoE4
Human apoE expression in NSE-apoE3 and NSE-apoE4 mice, determined by RPA, was found primarily in neural tissues and gonads (Fig.2A), as observed previously for NSE-driven expression of an indicator gene (Forss-Petter et al., 1990). Immunoblotting showed no human apoE in the plasma of NSE-apoE mice, and plasma lipoprotein cholesterol levels in the NSE-apoE mice were similar to those in nontransgenicApoe−/− littermate controls (data not shown). Plasma apoE is derived almost exclusively from the liver, with little, if any, contribution from the CNS (Linton et al., 1991).
Fig. 2.
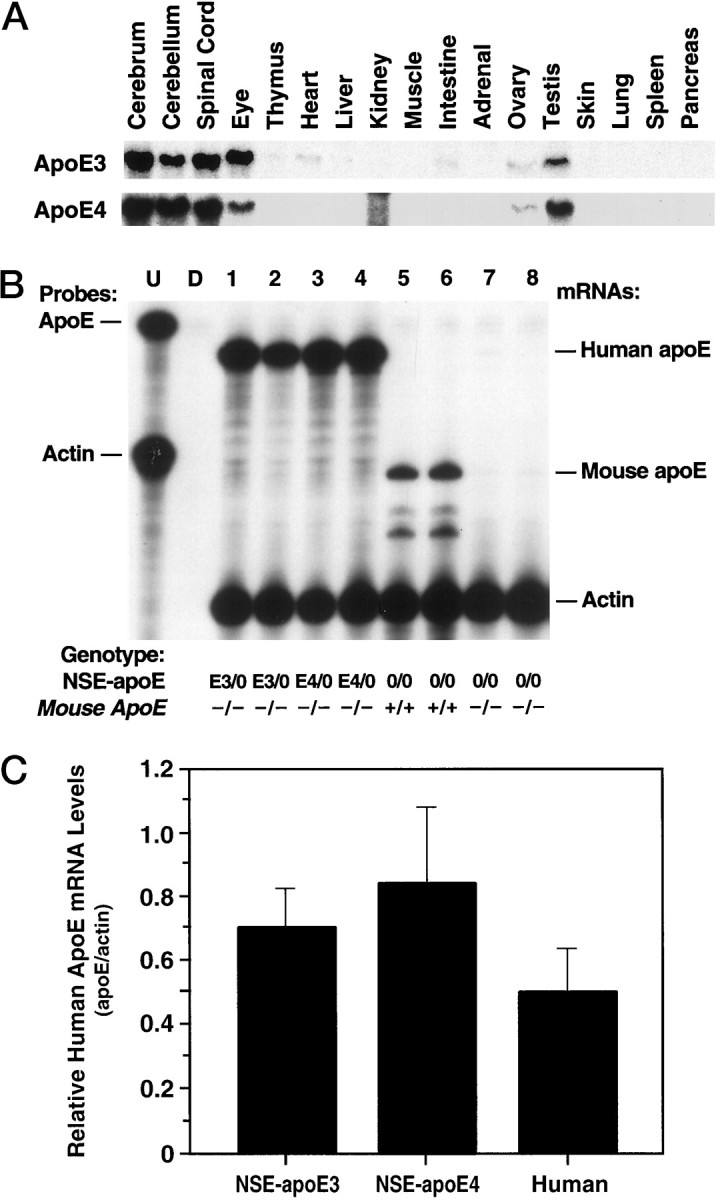
Expression of NSE-apoE transgenes at the RNA level. Total RNA was extracted from different tissues, and steady-state apoE mRNA levels were determined by RPA with an apoE antisense riboprobe that allows differentiation of human from mouse apoE transcripts. A, Representative autoradiograph revealing similar expression patterns of human apoE mRNA across different organs and CNS regions in NSE-apoE3 (top panel) and NSE-apoE4 (bottom panel) mice. Note the predominant expression in CNS, eyes, and gonads. The signal in the kidney lane in the bottom panel is an artifact caused by incomplete RNase digestion of the sample. B, Comparison of cerebral apoE mRNA levels in NSE-apoE mice and controls (n = 2/group). RNA was extracted from entire hemibrains of 7- to 9-month-old NSE-apoE3 (lanes 1, 2), NSE-apoE4 (lanes 3, 4), wild-type (lanes 5, 6), and Apoe−/−(lanes 7, 8) mice and analyzed by RPA. Theleftmost lane shows signals of undigested (U) radiolabeled probes; the other lanes contained the same riboprobes plus either tRNA (D; no specific hybridization) or brain RNA samples, digested with RNases. As outlined in Materials and Methods, the apoE riboprobe protects a larger fragment of human apoE mRNA and a smaller fragment of mouse apoE mRNA (protected mRNAs are indicated on theright). Note the similar levels of human apoE mRNA expression in brains of NSE-apoE3 and NSE-apoE4 mice. Comparable results were obtained in additional cohorts of 7- to 9-month-old mice (n = 9) and in corresponding groups of 3- to 4-month-old mice (n = 6) (data not shown).C, Semiquantitative comparison of human apoE mRNA levels in brain tissues of NSE-apoE mice and humans. Signals from RPAs on total RNA extracted from entire hemibrains of NSE-apoE3 (n = 4) or NSE-apoE4 mice (n = 6) or from the midfrontal gyrus of humans without dementia (n = 9) were quantitated by phosphorimager analysis essentially as described (Rockenstein et al., 1995). No statistically significant differences were identified when the three groups were compared by ANOVA or when the two groups of transgenic mice were compared by unpaired, two-tailed Student’s ttest.
ApoE mRNA and protein levels in the brain and CSF
NSE-apoE3 and NSE-apoE4 mice showed similar steady-state levels of human apoE mRNA in their brains (Fig. 2B,C). These levels were similar to those found in human frontal cortex (Fig.2C). Human apoE protein levels, assessed by Western blot analysis, were also similar in human and transgenic mouse brains (Fig.3A); no apoE was detected inApoe−/− mice (data not shown). Although apoE is produced exclusively by neurons in the brains of NSE-apoE mice, in the brains of humans apoE is produced by neurons and astrocytes (Xu et al., 1999). Because the precise proportion of apoE produced by neurons and glia in the human brain remains unknown, the levels of apoE in the brains of NSE-apoE mice and humans may not be directly comparable.
Fig. 3.

Human apoE3 and apoE4 in brains and CSF of transgenic mice. A, Western blot analysis showing apoE expression in whole-brain homogenates (n = 2 mice/genotype, 50 μg protein/lane) and CSF (14 μl/lane) from NSE-apoE3 and NSE-apoE4 mice. Human brain (occipital lobe) homogenate (50 μg protein/lane) and human apoE3 (5 ng/lane) standards are shown as controls. B, The apoE contents of brains and CSF were estimated by densitometric scanning of gels and by using human apoE standards. The apoE content in brains of transgenic mice was 4.4 ± 0.4 μg apoE/gm protein for NSE-apoE3 mice (n = 9), and 4.6 ± 0.5 μg apoE/gm protein for NSE-apoE4 mice (n = 13). The apoE content in the CSF of mice from both genotypes was 2.8 ng/μl (pooled from 6 mice/genotype).
Densitometric scanning of gels confirmed that apoE levels in NSE-apoE3 and NSE-apoE4 mice were similar (Fig. 3B). A tendency toward higher levels of apoE in NSE-apoE4 mice was not statistically significant (p = 0.79). Both mouse and human brain apoE appeared to be highly sialylated, because they showed two major bands in the 34–38 kDa range (Fig. 3A). This finding is consistent with previous results (Pitas et al., 1987). The human apoE3 and apoE4 in the transgenic mouse brains were intact, because no significant degradation products were found.
Apolipoprotein E-containing lipoproteins in the CSF originate in the CNS (Linton et al., 1991). Western blot analysis demonstrated similar levels of human apoE in CSF from NSE-apoE3 and NSE-apoE4 mice (Fig.3A). No apoE was detected in the CSF ofApoe−/− mice (data not shown).
Immunohistochemical localization of apoE3 and apoE4 in brains of NSE-apoE mice
To exclude potentially confounding regional differences in transgene expression, we used an antibody against human apoE to map human apoE expression in immunolabeled brain sections from NSE-apoE3 and NSE-apoE4 mice. Brains from both groups showed similar widespread neuronal expression of human apoE, which was most prominent in the neocortex and hippocampus (Fig. 4). This pattern is consistent with that of other NSE-driven transgenes (Forss-Petter et al., 1990; Mucke et al., 1994).
Fig. 4.
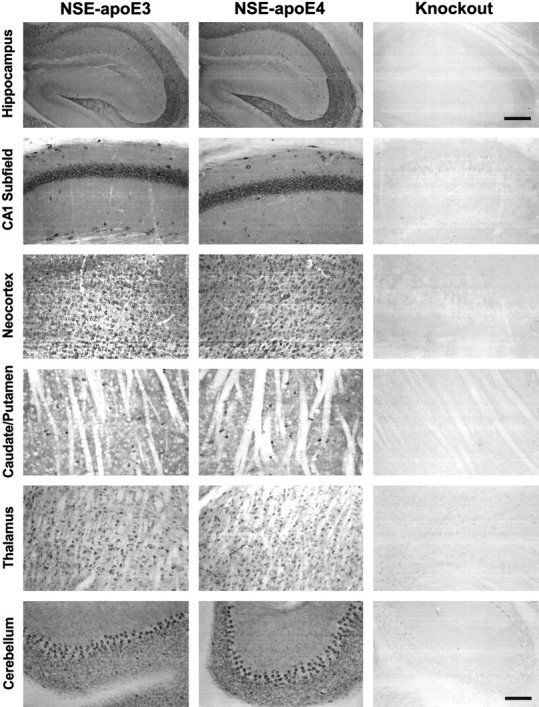
Neuronal expression of human apoE in NSE-apoE3 and NSE-apoE4 mouse brain. Immunoperoxidase staining for human apoE revealed widespread neuronal labeling in different brain regions of NSE-apoE3 and NSE-apoE4 mice. No apoE labeling was seen inApoe−/− control mice. Note the similar apoE expression pattern in NSE-apoE3 and NSE-apoE4 mice. Note also that human apoE immunoreactivity is present in neuropil as well as in neuronal cell bodies in the transgenic mice. Scale bars:first row of panels, 400 μm; all other panels, 200 μm.
Confocal microscopy of immunolabeled brain sections from NSE-apoE mice and from a human AD case revealed similar intraneuronal distributions of apoE3 and apoE4 in the transgenic mice and confirmed the presence of human apoE in neurons of the AD brain (Fig.5). In transgenic brains, apoE3 and apoE4 were identified in a patchy distribution throughout most of the neuronal soma with clear sparing of the nucleus (Fig.5A,B,D,E); little human apoE was detected in neuronal axons or dendrites and none in non-neuronal cells (data not shown). In the human AD case (APOE ε3/ε4), intraneuronal apoE immunoreactivity was somewhat more diffuse and extended into neuronal processes (Fig. 5H,I). Double labeling with antibodies against human apoE and the neuronal marker MAP-2 confirmed the neuronal identity of the brain cells expressing human apoE in NSE-apoE3 and NSE-apoE4 mice (Fig.6).
Fig. 5.
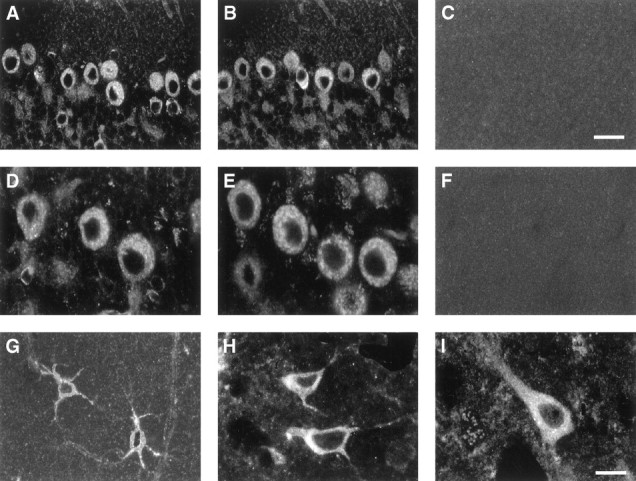
Neuronal labeling for apoE in NSE-apoE mice and a human AD case. Immunostaining with antibodies against human apoE (A–F, H, I) or mouse apoE (G) in NSE-apoE3 mice (A, D), NSE-apoE4 mice (B, E), and a human AD case (H, I) showed prominent neuronal labeling for human apoE, whereas mouse apoE in wild-type mice was detected primarily in astrocytes (G), which were identified by colabeling with anti-GFAP antibody (data not shown). No apoE expression was found in Apoe−/− controls (C, F). Scale bars: A–C, 25 μm;D–I, 15 μm.
Fig. 6.
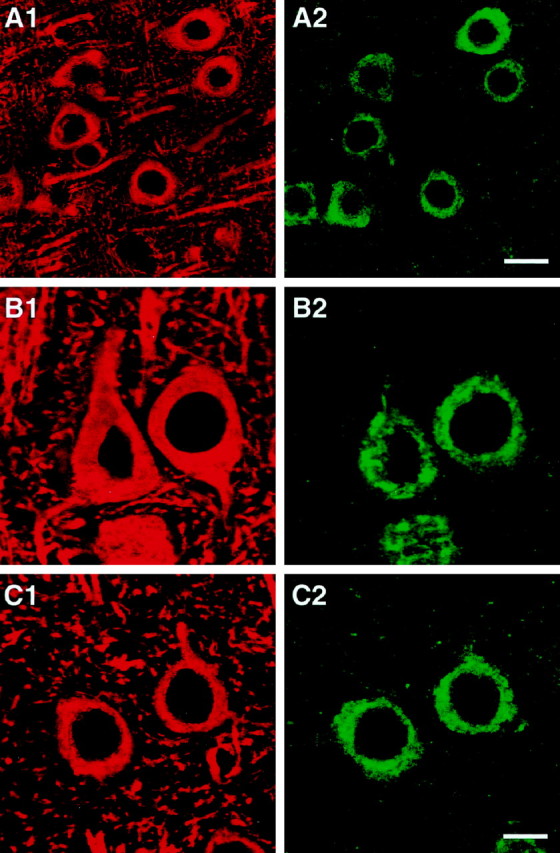
Colabeling of cells in NSE-apoE mouse brains for human apoE and for the neuronal marker MAP-2. Brain sections were double-immunolabeled with antibodies against human apoE (green; A2, B2, C2) and antibodies against MAP-2 (red; A1, B1, C1) and imaged by confocal microscopy. Pseudocolored images depict colabeled neurons in neocortex of an NSE-apoE3 (A, B) and of an NSE-apoE4 (C) mouse. Scale bars: A1, A2, 15 μm; B1, B2, C1, C2, 7 μm.
Comparison of the neuronal human apoE immunofluorescence signals in 3- to 4- and 7- to 9-month-old NSE-apoE3 and NSE-apoE4 mice (n = 4–5/group) showed no significant differences in immunostaining intensity (data not shown). These results are consistent with those obtained by RPA (Fig. 2B,C) and Western blot analysis (Fig. 3).
Differential effects of apoE3 and apoE4 on excitotoxin-induced neurodegeneration in Apoe−/− mice
Excessive stimulation of glutamate receptors by excitatory amino acids, such as glutamic or kainic acid, results in neuronal damage (excitotoxicity) and is one of the main mechanisms of neuronal injury in neurodegenerative diseases (Meldrum and Garthwaite, 1990; Lipton and Rosenberg, 1994). To test whether there is an apoE isoform-specific effect on excitotoxin-induced neurodegeneration, we injected NSE-apoE3 and NSE-apoE4 mice (both on the Apoe−/−background) with kainic acid. Control mice were injected with saline. Kainic acid- and saline-injectedApoe−/− and wild-type mice served as additional controls.
Inspection of hematoxylin/eosin-stained sections revealed no obvious neuronal loss in the hippocampus or neocortex of any of the kainic acid-injected groups of mice (data not shown), consistent with a previous study reporting that the C57BL/6J strain is resistant to excitotoxin-induced loss of neuronal cell bodies (Schauwecker and Steward, 1997).
To detect more subtle types of neurodegeneration, neocortical and hippocampal sections were immunolabeled for synaptophysin, MAP-2, or phosphorylated neurofilaments and imaged by confocal microscopy. Systemic injection of kainic acid has previously been shown to induce a significant loss of MAP-2- and synaptophysin-immunoreactive neuronal structures in mice on the C57BL/6J background (Masliah et al., 1997). The percentage area of neuropil occupied by immunolabeled presynaptic terminals or neurites was determined by computer-aided analysis of confocal images as described in Materials and Methods.
We found that apoE3 effectively protected against excitotoxin-induced neurodegeneration, whereas apoE4 did not (Fig.7, E,F vs G,H; Fig.9, C vs D). NSE-apoE3 mice showed no significant loss of synaptophysin-positive presynaptic terminals in the neocortex (Figs. 7E, 8A) after injection of 18 or 25 mg/kg kainic acid. However, a significant loss of MAP-2-positive neuronal dendrites in the neocortex of these mice was observed after injection of 25 mg/kg kainic acid (Fig.8B). NSE-apoE3 mice showed no disruption of neurofilament-positive hippocampal axons after injection of 18 mg/kg kainic acid (Fig.9C). In contrast, NSE-apoE4 mice showed significant loss of neocortical synaptophysin-positive presynaptic terminals (Figs. 7G, 8A) and MAP-2-positive neuronal dendrites (Figs. 7H,8B) after injection of 18 or 25 mg/kg kainic acid. NSE-apoE4 mice also showed severe disruption of neurofilament-positive hippocampal axons after injection of 18 mg/kg kainic acid (Fig.9D). Apoe−/− and wild-type mice also showed a significant loss of neocortical synaptophysin-positive presynaptic terminals (Figs. 7A,C,8A) and MAP-2-positive neuronal dendrites (Figs.7B,D, 8B) with either dose of kainic acid, as well as disruption of hippocampal axons after injection of 18 mg/kg kainic acid (Fig. 9A,B). Similar genotype effects on synaptophysin-positive presynaptic terminals and MAP-2-positive neuronal dendrites were observed in the hippocampus (data not shown).
Fig. 7.
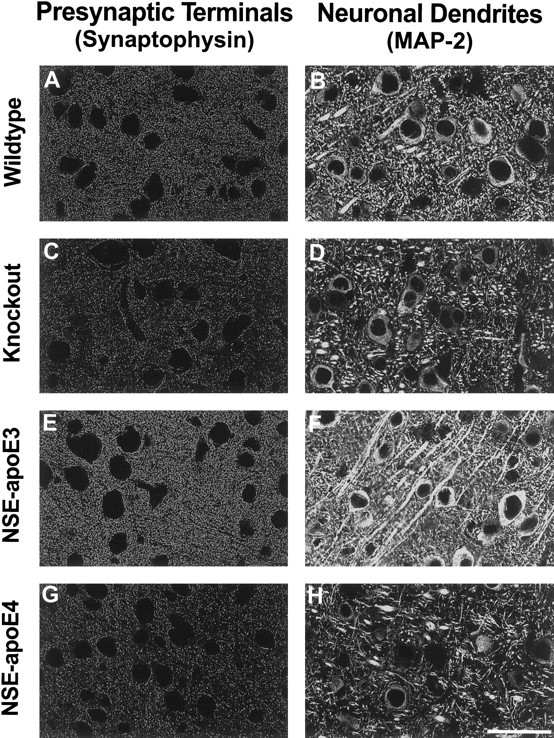
Differential protective effects of apoE3 and apoE4 after kainic acid challenge. Neocortical sections of wild-type (A, B),Apoe−/− (C, D), NSE-apoE3 (E, F), and NSE-apoE4 (G, H) mice injected with 18 mg/kg kainic acid were immunostained for synaptophysin (A, C, E, G) or MAP-2 (B, D, F, H) and imaged by confocal microscopy. Cases with severe damage were selected for illustration. Note the prominent loss of neuronal structures in the neocortex ofApoe−/− and NSE-apoE4 mice. In contrast, only minimal neurodegenerative changes were seen in the neocortex of NSE-apoE3 mice. Scale bar, 55 μm.
Fig. 9.
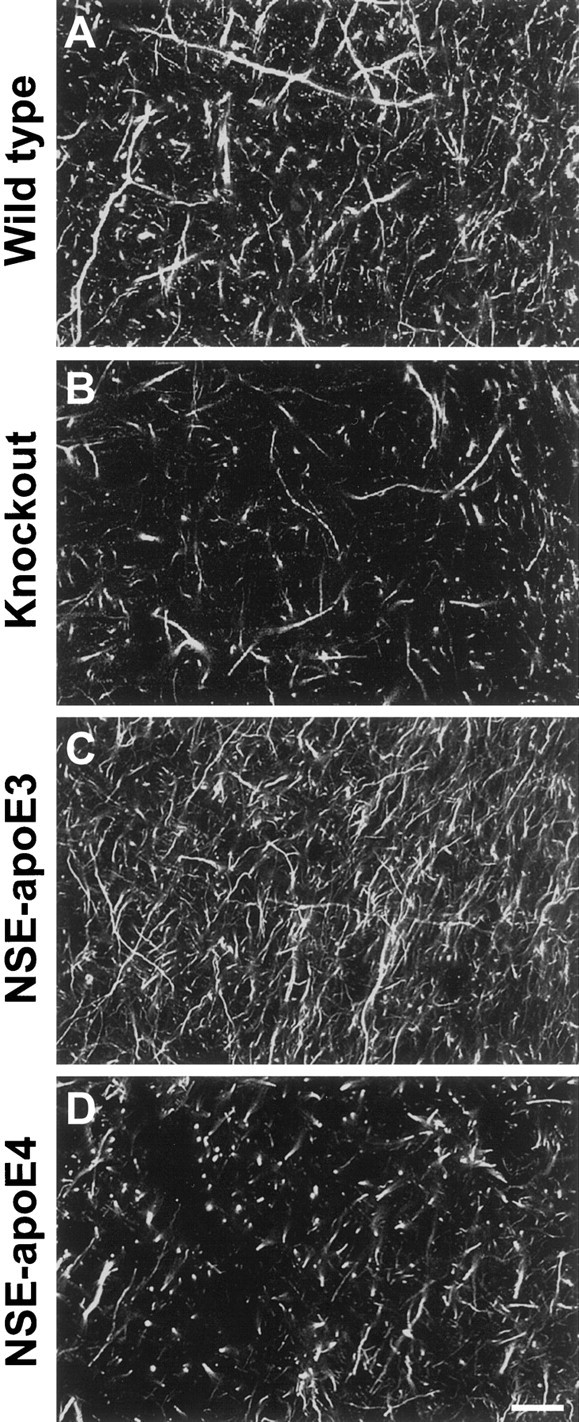
Differential effects of apoE3 and apoE4 on axonal structures after kainic acid challenge. Hippocampal sections of 6-month-old wild-type (A),Apoe−/− (B), NSE-apoE3 (C), and NSE-apoE4 (D) mice injected with 18 mg/kg kainic acid were immunostained for axonal neurofilaments and imaged by confocal microscopy. Note the prominent disruption of neurofilament-positive axons in the hippocampus (CA1–CA2 subfields) ofApoe−/− and NSE-apoE4 mice. Scale bar, 120 μm.
Fig. 8.

Semiquantitative comparison of the effects of apoE3 and apoE4 on kainic acid-induced neurodegeneration. Neocortical sections of mice injected with saline (black bars), 18 mg/kg kainic acid (hatched bars), or 25 mg/kg kainic acid (white bars) were immunolabeled for synaptophysin (A) or MAP-2 (B). Three groups of mice with each genotype were treated with saline, 18 mg/kg kainic acid, or 25 mg/kg kainic acid, respectively (number of mice in each group indicated in parentheses): 14 wild-type controls (5, 6, 3), 28 Apoe−/− (10, 12, 6), 14 NSE-apoE3 (4, 6, 4), and 16 NSE-apoE4 (4, 5, 7). The percentage area of neuropil occupied by immunoreactive dendrites or presynaptic terminals was determined by confocal microscopy and computer-aided image analysis. Significant excitotoxin-induced neurodegeneration was detected in NSE-apoE4,Apoe−/−, and wild-type mice. NSE-apoE3 mice showed no significant excitotoxin-induced loss of synaptophysin-positive presynaptic terminals and showed significant loss of MAP-2-positive neuronal dendrites only at the higher dose of kainic acid. Values are means ± SEM. *p < 0.05, **p < 0.01 versus saline-injected mice of the same genotype (Dunnett’s post hoc test). In the hippocampus, presynaptic terminals were significantly decreased in kainic acid-injected (25 mg/kg)Apoe−/− and NSE-apoE4 mice, and neuronal dendrites were significantly decreased in kainic acid-injected (18 mg/kg) wild-type, Apoe−/−, and NSE-apoE4 mice when compared with saline-injected controls of the same genotype (data not shown). No significant decreases in presynaptic terminals or neuronal dendrites were found in the hippocampus of kainic acid-injected (18 or 25 mg/kg) NSE-apoE3 mice (data not shown).
Differential effects of apoE3 and apoE4 on age-related neurodegeneration in Apoe−/− mice
To determine whether there is an age-related loss of neuronal structures in Apoe−/− mice, as has been found by some (Masliah et al., 1995) but not by others (Anderson et al., 1998), we analyzed neuronal integrity inApoe−/− mice at 3–4 and 7–9 months of age. Compared with age-matched wild-type controls,Apoe−/− mice showed significant loss of synaptophysin-positive presynaptic terminals and MAP-2-positive neuronal dendrites in the neocortex (Figs.10A–D,11) and hippocampus (data not shown) as they aged. Likewise, there was an age-related disruption of neurofilament-positive axons in the hippocampus of these mice (data not shown). These findings are consistent with the results of Masliah et al. (1995).
Fig. 10.
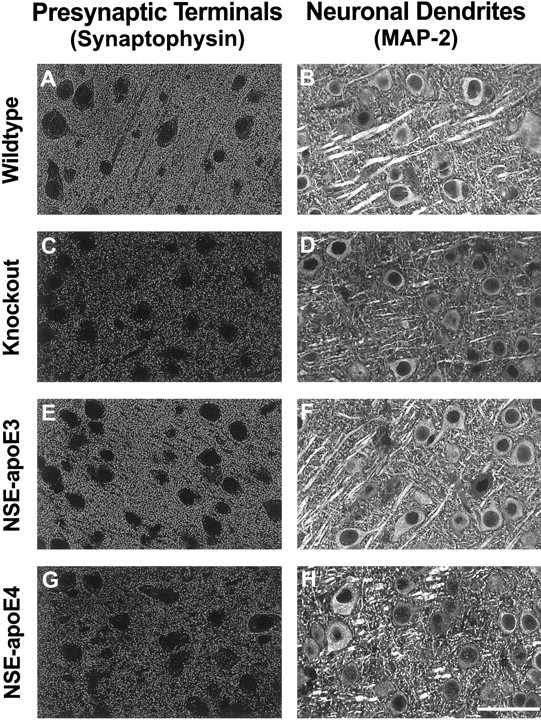
Differential effects of apoE3 and apoE4 on neuronal integrity in untreatedApoe−/− mice. Sections of neocortex from 7- to 9-month-old wild-type (A, B),Apoe−/− (C, D), NSE-apoE3 (E, F), and NSE-apoE4 (G, H) mice were immunostained for synaptophysin (A, C, E, G) or for MAP-2 (B, D, F, H) and imaged by confocal microscopy. Cases with severe damage were selected for illustration. Note the prominent loss of immunolabeled neuronal structures in the neocortex ofApoe−/− mice and NSE-apoE4 mice and the normal appearance of corresponding sections from wild-type and NSE-apoE3 mice. Qualitatively similar results were obtained for synaptophysin-positive presynaptic terminals in the hippocampus (data not shown). Scale bar, 55 μm.
Fig. 11.

Semiquantitative comparison of apoE3 and apoE4 effects at different ages. Neocortical sections of 3- to 4-month-old (A, C) and 7- to 9-month-old (B, D) mice were immunolabeled for synaptophysin (A, B) or MAP-2 (C, D). Studies were performed on two groups of mice with each genotype, 3–4 months or 7–9 months of age, respectively (number of mice in each group indicated in parentheses): 7 wild-type controls (4, 3), 22Apoe−/− (10, 12), 9 NSE-apoE3 mice (5, 4), and 14 NSE-apoE4 mice (5, 9). The percentage area of neuropil occupied by immunolabeled dendrites or presynaptic terminals was determined by confocal microscopy and computer-aided image analysis. In younger mice, the only significant alteration detected was a rarefaction of dendrites in Apoe−/−mice (B). By 7–9 months of age, bothApoe−/− and NSE-apoE4 mice had developed a significant loss of immunopositive neuronal dendrites and presynaptic terminals (B, D). In contrast, neuronal integrity in NSE-apoE3 mice was similar to that of wild-type mice and significantly better than that of NSE-apoE4 mice. Values are means ± SEM. *p < 0.05, **p < 0.01 versus wild-type (Dunnett’s post hoc test),0p < 0.05 (Tukey-Kramer post hoc test). Presynaptic terminals were also significantly decreased in the hippocampus of 7- to 9-month-oldApoe−/− and NSE-apoE4 mice (p < 0.01 vs wild-type controls by Dunnett’s post hoc test) but not in age-matched NSE-apoE3 mice (data not shown).
To test whether there is an apoE isoform-specific effect on the age-dependent neurodegeneration inApoe−/− mice, we analyzed neuronal integrity in NSE-apoE3 and NSE-apoE4 mice on theApoe−/− background at 3–4 and 7–9 months of age. We found that apoE3 prevented the age-dependent degeneration of synaptophysin-positive presynaptic terminals and MAP-2-positive neuronal dendrites found inApoe−/− mice, whereas apoE4 did not (illustrated in Fig. 10E–H; semiquantitative evaluation in Fig. 11). Likewise, apoE3 prevented the age-dependent loss of neurofilament-positive axons in the hippocampus, whereas apoE4 did not (data not shown). By all measures of neuronal integrity examined, NSE-apoE3 mice closely resembled wild-type mice. In contrast, NSE-apoE4 mice, like Apoe−/− mice, showed a significant loss of synaptophysin-positive presynaptic terminals and MAP-2-positive neuronal dendrites (Figs.10G,H, 11) and a severe disruption of hippocampal axons (data not shown) at 7–9 months of age.
The development of neurodegenerative changes in NSE-apoE4 mice was also clearly age-dependent, because significant deficits were seen at 7–9 months but not at 3–4 months of age (Fig. 11).
Measurement of neocortical synaptophysin levels by ELISA
The histopathological analysis was complemented by ELISA measurements of synaptophysin in particulate fractions from brain homogenates of NSE-apoE3 and NSE-apoE4 mice. After injection of 18 mg/kg kainic acid, neocortical synaptophysin content was significantly lower in NSE-apoE4 mice than in NSE-apoE3 mice (absorbance values at 492 nm: 125.3 ± 3.4 for NSE-apoE4 mice, 159 ± 1.5 for NSE-apoE3 mice, p < 0.05, n = 4/group, 5–6 months of age). Similarly, neocortical synaptophysin content was significantly lower in untreated 9-month-old NSE-apoE4 mice than in age-matched NSE-apoE3 mice (absorbance values at 492 nm: 129.5 ± 5.3 for NSE-apoE4 mice, 160.0 ± 2.7 for NSE-apoE3 mice,p < 0.05, n = 4/group).
DISCUSSION
Our data reveal that human apoE3 and apoE4 expressed at similar levels in the brains of Apoe−/− mice differ significantly in their capacity to protect against excitotoxin-induced neurodegeneration and in their long-term effects on neuronal integrity. Age-dependent neurodegeneration seen in Apoe−/− mice was prevented by apoE3, but not apoE4. Excitotoxin-induced neurodegeneration, a key mechanism of neuronal injury in acute neurodegenerative processes, such as head trauma and stroke (Meldrum and Garthwaite, 1990; Lipton and Rosenberg, 1994), was minimal in the presence of apoE3 but severe in the presence of apoE4. Consistent with this result, a recent study that examined transgenic mice expressing apoE3 or apoE4 under the control of the human APOE regulatory sequences (Sheng et al., 1998) showed that mice expressing apoE3 had significantly smaller infarcts after cerebral ischemia than mice expressing apoE4 at higher levels.
In the present study, we chose to assess neurodegeneration in the brains of NSE-apoE mice by quantifying immunoreactivity for the neuronal markers synaptophysin and MAP-2. There is ample evidence that the loss of synaptophysin-positive presynaptic terminals, MAP-2-positive neuronal dendrites, and neurofilament-positive axons are relevant indicators of neurodegenerative disease processes. For example, a number of studies have reported a loss of synaptophysin immunoreactivity in AD brains (Terry et al., 1991; Zhan et al., 1993; Dickson et al., 1995; Sze et al., 1997), and this loss correlated well with the extent of cognitive impairments (Terry et al., 1991; Sze et al., 1997). Other studies have reported severely disrupted neurofilament-, or tau-, immunoreactive axons (Kowall and Kosik, 1987;Masliah et al., 1993) in AD brains, and a significant decrease of MAP-2 immunoreactive dendrites in the brains of patients with HIV-1 encephalitis (Masliah et al., 1992). Loss of MAP-2- and synaptophysin-immunoreactive neuronal structures has also been a sensitive and reliable indicator of neuropathological changes in the brains of diverse transgenic animal models (Toggas et al., 1994;Masliah et al., 1997; Buttini et al., 1998).
Excitotoxic neuronal injury is mediated, at least in part, by the release of reactive oxygen species (Michaelis, 1998). In cell cultures, apoE can protect neurons against oxidative insults, and apoE3 has much stronger antioxidative properties than apoE4 (Miyata and Smith, 1996). Our study indicates that, in vivo, apoE3 also protects neurons more effectively than apoE4 against insults presumed to involve oxidative stress. The neurodegeneration seen in NSE-apoE4 mice after an excitotoxic challenge could relate to the poor outcome of humanAPOE ε4 carriers after head trauma or stroke (Nicoll et al., 1996; Slooter et al., 1997), because these CNS injuries are mediated, at least in part, by excitotoxic mechanisms (Meldrum and Garthwaite, 1990).
The age dependence of the differential CNS effects of apoE3 and apoE4 identified in the current study is intriguing. The preservation of neuronal structures in young NSE-apoE4 mice indicates that apoE is not essential for normal neuronal development and that deficits related to apoE4 are strongly dependent on age-related factors. Notably, a behavioral analysis of wild-type,Apoe−/−, NSE-apoE3, and NSE-apoE4 mice revealed significant impairments in learning, memory, and exploratory behavior in female NSE-apoE4, but not NSE-apoE3, mice (Raber et al., 1998), suggesting that the structural and molecular alterations documented in the current study may have important functional consequences. These results could relate to the increased susceptibility to AD associated with the APOE ε4 allele in humans, which also appears to be stronger in females (Farrer et al., 1997). The reason for this gender bias remains to be determined.
Age-dependent neurodegenerative changes inApoe−/− mice that do not express human apoE are a matter of controversy, because they have been observed by some (Masliah et al., 1995) but not others (Anderson et al., 1998). The authors of the latter study proposed that differences in mouse strains and/or origin of the Apoe−/− mice could account for this discrepancy. However, this is unlikely, because we and others (E. Masliah, personal communication) have observed age-dependent neurodegenerative changes in the brains ofApoe−/− mice originating from the same source and bred onto the same strain (C57BL/6J) as the mice used byAnderson et al. (1998). It is conceivable that specific dietary or other housing-related factors could increase age-related stresses on neurons and thereby help reveal the lack of neuroprotective apoE effects in some cohorts of agingApoe−/− mice. Potential differences in such environmental variables and in methodological approaches will need to be scrutinized in the future to resolve the divergent findings obtained in distinct groups of Apoe−/−mice.
There are differences in the brain cell-specific distribution of endogenous mouse apoE in wild-type mice, of endogenous apoE in humans, and of transgene-derived human apoE in NSE-apoE mice. In wild-type mice, apoE mRNA and immunoreactivity have been detected primarily in astrocytes, whereas neuronal apoE labeling is more widespread and intense in human brains (Boyles et al., 1985; Diedrich et al., 1991;Han et al., 1994; Bao et al., 1996; Metzger et al., 1996). Neuronal expression of apoE mRNA has recently been detected by in situ hybridization in the frontal cortex and hippocampus of human brains, providing evidence that human neurons are indeed capable of producing apoE (Xu et al., 1999). Furthermore, striking increases in neuronal immunostaining for apoE have been documented after CNS injuries in humans and rodents (Kida et al., 1995; Horsburgh and Nicoll, 1996). The detection of human apoE in the CSF (Fig. 3) and of human apoE immunoreactivity in the neuropil (Fig. 4) of NSE-apoE mice indicates that transgenic neurons secrete the human apoE they produce, allowing for the interaction of transgene-derived apoEs with all CNS cell types.
Recently, we (T. Wyss-Coray, M. Buttini, R. E. Pitas, R. W. Mahley, and L. Mucke, unpublished results) and others (Sun et al., 1998) generated transgenic mice in which human apoE isoforms are expressed in astrocytes directed by the glial fibrillary acidic protein promoter. Comparison of these models with the NSE-apoE mice should allow us to assess the importance of the cell type in which human apoE isoforms are produced.
No matter what cell type is used to express human apoE isoforms in the brain, it is critical that the isoforms to be compared are expressed at similar levels and in a similar distribution across different brain regions. As documented in Figures 2, 3, 5, and 6, these requirements were clearly met in the current study. Furthermore, in all the age groups tested, the extent of neuronal damage did not differ significantly between Apoe−/− mice from the NSE-apoE3 line and age-matchedApoe−/− mice from the NSE-apoE4 line (data not shown), indicating that the lines were well matched with respect to background genes.
We found no evidence that apoE expression had peripheral effects in NSE-apoE3 or NSE-apoE4 mice. This is not surprising because NSE-driven constructs are expressed primarily in the CNS (Fig.2A) (Forss-Petter et al., 1990; Mucke et al., 1994). Therefore, the human apoE isoform-specific effects revealed by the current study pertain primarily to CNS disorders. Our models cannot determine whether human apoE3 and apoE4 have similar differential effects in peripheral organs or whether such effects might have indirect consequences for the nervous system. Answers to these questions can only be provided by related models in which different human apoE isoforms are expressed in multiple organs (Xu et al., 1996;Sullivan et al., 1997).
Although the findings we obtained in our NSE-apoE models will need to be confirmed in other lines of apoE transgenic mice, it is tempting to speculate that they may relate closely to the effects of apoE isoforms in humans with AD. It is interesting in this context that brains of AD patients carrying one or two APOE ε4 alleles show more severe neurodegeneration and less dendritic arborization than brains of AD patients with two APOE ε3 alleles (Arendt et al., 1997). The potential relevance of the NSE-apoE models to AD has also been highlighted by a recent behavioral analysis that revealed age-dependent cognitive deficits in female NSE-apoE4, but not NSE-apoE3, mice (Raber et al., 1998).
In conclusion, we have demonstrated that distinct human apoE isoforms differ significantly in their long-term effects on neuronal integrity as well as in their ability to protect against excitotoxicity. These differences in the neuroprotective capacities of apoE3 and apoE4 could contribute to the increased susceptibility of human APOEε4 carriers to AD and other types of CNS impairment.
Footnotes
This research was funded in part by a Cambridge NeuroScience/Gladstone collaborative research agreement. M.O. was supported in part by the Deutsche Forschungsgemeinschaft. We thank Ricky Quan and Carol Lin for excellent technical support; Sylvia Richmond for manuscript preparation; Gary Howard and Stephen Ordway for editorial assistance; and John C. W. Carroll, Stephen Gonzales, and Chris Goodfellow for graphics and photography.
Drs. Buttini and Orth contributed equally to this study.
Correspondence should be addressed to Dr. Robert W. Mahley, Gladstone Institute of Neurological Disease, P.O. Box 419100, San Francisco, CA 94141-9100.
Dr. Bellosta’s current address: Institute of Pharmacological Science, University of Milan, Via Balzaretti 9, 20133 Milan, Italy.
REFERENCES
- 1.Anderson R, Barnes JC, Bliss TVP, Cain DP, Cambon K, Davies HA, Errington ML, Fellows LA, Gray RA, Hoh T, Stewart M, Large CH, Higgins GA. Behavioural, physiological and morphological analysis of a line of apolipoprotein E knockout mouse. Neuroscience. 1998;85:93–110. doi: 10.1016/s0306-4522(97)00598-8. [DOI] [PubMed] [Google Scholar]
- 2.Arendt T, Schindler C, Brückner MK, Eschrich K, Bigl V, Zedlick D, Marcova L. Plastic neuronal remodeling is impaired in patients with Alzheimer’s disease carrying apolipoprotein ε4 allele. J Neurosci. 1997;17:516–529. doi: 10.1523/JNEUROSCI.17-02-00516.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bao F, Arai H, Matsushita S, Higuchi S, Sasaki H. Expression of apolipoprotein E in normal and diverse neurodegenerative disease brain. NeuroReport. 1996;7:1733–1739. doi: 10.1097/00001756-199607290-00008. [DOI] [PubMed] [Google Scholar]
- 4.Bellosta S, Mahley RW, Sanan DA, Murata J, Newland DL, Taylor JM, Pitas RE. Macrophage-specific expression of human apolipoprotein E reduces atherosclerosis in hypercholesterolemic apolipoprotein E-null mice. J Clin Invest. 1995a;96:2170–2179. doi: 10.1172/JCI118271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellosta S, Nathan BP, Orth M, Dong L-M, Mahley RW, Pitas RE. Stable expression and secretion of apolipoproteins E3 and E4 in mouse neuroblastoma cells produces differential effects on neurite outgrowth. J Biol Chem. 1995b;270:27063–27071. doi: 10.1074/jbc.270.45.27063. [DOI] [PubMed] [Google Scholar]
- 6.Bordonaro M, Saccomanno CF, Nordstrom JL. An improved T1/A ribonuclease protection assay. Biotechniques. 1994;16:428–430. [PubMed] [Google Scholar]
- 7.Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest. 1985;76:1501–1513. doi: 10.1172/JCI112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buttini M, Westland CE, Masliah E, Yafeh AM, Wyss-Coray T, Mucke L. Novel role of human CD4 molecule identified in neurodegeneration. Nat Med. 1998;4:441–446. doi: 10.1038/nm0498-441. [DOI] [PubMed] [Google Scholar]
- 9.Carp RI, Davidson AI, Merz PA. A method for obtaining cerebrospinal fluid from mice. Res Vet Sci. 1971;12:499. [PubMed] [Google Scholar]
- 10.Chen Y, Lomnitski L, Michaelson DM, Shohami E. Motor and cognitive deficits in apolipoprotein E-deficient mice after closed head injury. Neuroscience. 1997;80:1255–1262. doi: 10.1016/s0306-4522(97)00007-9. [DOI] [PubMed] [Google Scholar]
- 11.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 12.Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging. 1995;16:285–304. doi: 10.1016/0197-4580(95)00013-5. [DOI] [PubMed] [Google Scholar]
- 13.Diedrich JF, Minnigan H, Carp RI, Whitaker JN, Race R, Frey W, II, Haase AT. Neuropathological changes in scrapie and Alzheimer’s disease are associated with increased expression of apolipoprotein E and cathepsin D in astrocytes. J Virol. 1991;65:4759–4768. doi: 10.1128/jvi.65.9.4759-4768.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elshourbagy NA, Liao WS, Mahley RW, Taylor JM. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proc Natl Acad Sci USA. 1985;82:203–207. doi: 10.1073/pnas.82.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fagan AM, Murphy BA, Patel SN, Kilbridge JF, Mobley WC, Bu G, Holtzman DM. Evidence for normal aging of the septo-hippocampal cholinergic system in apoE (−/−) mice but impaired clearance of axonal degeneration products following injury. Exp Neurol. 1998;151:314–325. doi: 10.1006/exnr.1998.6818. [DOI] [PubMed] [Google Scholar]
- 16.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, Van Duijn CM. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. JAMA. 1997;278:1349–1356. [PubMed] [Google Scholar]
- 17.Forss-Petter S, Danielson PE, Catsicas S, Battenberg E, Price J, Nerenberg M, Sutcliffe JG. Transgenic mice expressing β-galactosidase in mature neurons under neuron-specific enolase promoter control. Neuron. 1990;5:187–197. doi: 10.1016/0896-6273(90)90308-3. [DOI] [PubMed] [Google Scholar]
- 18.Han S-H, Einstein G, Weisgraber KH, Strittmatter WJ, Saunders AM, Pericak-Vance M, Roses AD, Schmechel DE. Apolipoprotein E is localized to the cytoplasm of human cortical neurons: a light and electron microscopic study. J Neuropathol Exp Neurol. 1994;53:535–544. doi: 10.1097/00005072-199409000-00013. [DOI] [PubMed] [Google Scholar]
- 19.Hecker KH, Roux KH. High and low annealing temperatures increase both specificity and yield in touchdown and stepdown PCR. Biotechniques. 1996;20:478–485. doi: 10.2144/19962003478. [DOI] [PubMed] [Google Scholar]
- 20.Horsburgh K, Nicoll JAR. Selective alterations in the cellular distribution of apolipoprotein E immunoreactivity following transient cerebral ischaemia in the rat. Neuropathol Appl Neurobiol. 1996;22:342–349. doi: 10.1111/j.1365-2990.1996.tb01113.x. [DOI] [PubMed] [Google Scholar]
- 21.Ignatius MJ, Gebicke-Härter PJ, Skene JHP, Schilling JW, Weisgraber KH, Mahley RW, Shooter EM. Expression of apolipoprotein E during nerve degeneration and regeneration. Proc Natl Acad Sci USA. 1986;83:1125–1129. doi: 10.1073/pnas.83.4.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ji Z-S, Pitas RE, Mahley RW. Differential cellular accumulation/retention of apolipoprotein E mediated by cell surface heparan sulfate proteoglycans. Apolipoproteins E3 and E2 greater than E4. J Biol Chem. 1998;273:13452–13460. doi: 10.1074/jbc.273.22.13452. [DOI] [PubMed] [Google Scholar]
- 23.Johnson WB, Ruppe MD, Rockenstein EM, Price J, Sarthy VP, Verderber LC, Mucke L. Indicator expression directed by regulatory sequences of the glial fibrillary acidic protein (GFAP) gene: in vivo comparison of distinct GFAP-LacZ transgenes. Glia. 1995;13:174–184. doi: 10.1002/glia.440130304. [DOI] [PubMed] [Google Scholar]
- 24.Kida E, Pluta R, Lossinsky AS, Golabek AA, Choi-Miura N-H, Wisniewski HM, Mossakowski MJ. Complete cerebral ischemia with short-term survival in rat induced by cardiac arrest. II. Extracellular and intracellular accumulation of apolipoproteins E and J in the brain. Brain Res. 1995;674:341–346. doi: 10.1016/0006-8993(94)01467-v. [DOI] [PubMed] [Google Scholar]
- 25.Kowall NW, Kosik KS. Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer’s disease. Ann Neurol. 1987;22:639–643. doi: 10.1002/ana.410220514. [DOI] [PubMed] [Google Scholar]
- 26.Linton MF, Gish R, Hubl ST, Bütler E, Esquivel C, Bry WI, Boyles JK, Wardell MR, Young SG. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J Clin Invest. 1991;88:270–281. doi: 10.1172/JCI115288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 28.Ma J, Yee A, Brewer HB, Jr, Das S, Potter H. Amyloid-associated proteins α1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature. 1994;372:92–94. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- 29.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 30.Mahley RW, Nathan BP, Bellosta S, Pitas RE. Apolipoprotein E: impact of cytoskeletal stability in neurons and the relationship to Alzheimer’s disease. Curr Opin Lipidol. 1995;6:86–91. doi: 10.1097/00041433-199504000-00005. [DOI] [PubMed] [Google Scholar]
- 31.Masliah E, Achim CL, Ge N, DeTeresa R, Terry RD, Wiley CA. Spectrum of human immunodeficiency virus-associated neocortical damage. Ann Neurol. 1992;32:321–329. doi: 10.1002/ana.410320304. [DOI] [PubMed] [Google Scholar]
- 32.Masliah E, Mallory M, Hansen L, Alford M, DeTeresa R, Terry R. An antibody against phosphorylated neurofilaments identifies a subset of damaged association axons in Alzheimer’s disease. Am J Pathol. 1993;142:871–882. [PMC free article] [PubMed] [Google Scholar]
- 33.Masliah E, Mallory M, Ge N, Alford M, Veinbergs I, Roses AD. Neurodegeneration in the central nervous system of apoE-deficient mice. Exp Neurol. 1995;136:107–122. doi: 10.1006/exnr.1995.1088. [DOI] [PubMed] [Google Scholar]
- 34.Masliah E, Westland CE, Rockenstein EM, Abraham CR, Mallory M, Veinberg I, Sheldon E, Mucke L. Amyloid precursor proteins protect neurons of transgenic mice against acute and chronic excitotoxic injuries in vivo. Neuroscience. 1997;78:135–146. doi: 10.1016/s0306-4522(96)00553-2. [DOI] [PubMed] [Google Scholar]
- 35.Mayeux R, Ottman R, Maestre G, Ngai C, Tang M-X, Ginsberg H, Chun M, Tycko B, Shelanski M. Synergistic effects of traumatic head injury and apolipoprotein-ε4 in patients with Alzheimer’s disease. Neurology. 1995;45:555–557. doi: 10.1212/wnl.45.3.555. [DOI] [PubMed] [Google Scholar]
- 36.Meldrum B, Garthwaite J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci. 1990;11:379–387. doi: 10.1016/0165-6147(90)90184-a. [DOI] [PubMed] [Google Scholar]
- 37.Metzger RE, LaDu MJ, Pan JB, Getz GS, Frail DE, Falduto MT. Neurons of the human frontal cortex display apolipoprotein E immunoreactivity: implications for Alzheimer’s disease. J Neuropathol Exp Neurol. 1996;55:372–380. doi: 10.1097/00005072-199603000-00013. [DOI] [PubMed] [Google Scholar]
- 38.Michaelis EK. Molecular biology of glutamate receptors in the central nervous system and their role in excitotoxicity, oxidative stress and aging. Prog Neurobiol. 1998;54:369–415. doi: 10.1016/s0301-0082(97)00055-5. [DOI] [PubMed] [Google Scholar]
- 39.Miyata M, Smith JD. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and β-amyloid peptides. Nat Genet. 1996;14:55–61. doi: 10.1038/ng0996-55. [DOI] [PubMed] [Google Scholar]
- 40.Mucke L, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, Forss-Petter S, Pietropaolo M, Mallory M, Abraham CR. Synaptotrophic effects of human amyloid β protein precursors in the cortex of transgenic mice. Brain Res. 1994;666:151–167. doi: 10.1016/0006-8993(94)90767-6. [DOI] [PubMed] [Google Scholar]
- 41.Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541:163–166. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- 42.Nathan BP, Bellosta S, Sanan DA, Weisgraber KH, Mahley RW, Pitas RE. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science. 1994;264:850–852. doi: 10.1126/science.8171342. [DOI] [PubMed] [Google Scholar]
- 43.Nathan BP, Chang K-C, Bellosta S, Brisch E, Ge N, Mahley RW, Pitas RE. The inhibitory effect of apolipoprotein E4 on neurite outgrowth is associated with microtubule depolymerization. J Biol Chem. 1995;270:19791–19799. doi: 10.1074/jbc.270.34.19791. [DOI] [PubMed] [Google Scholar]
- 44.Nicoll JAR, Roberts GW, Graham DI. Amyloid β-protein, APOE genotype and head injury. Ann NY Acad Sci. 1996;777:271–275. doi: 10.1111/j.1749-6632.1996.tb34431.x. [DOI] [PubMed] [Google Scholar]
- 45.Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci USA. 1992;89:4471–4475. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem. 1987;262:14352–14360. [PubMed] [Google Scholar]
- 47.Raber J, Wong D, Buttini M, Orth M, Bellosta S, Pitas RE, Mahley RW, Mucke L. Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc Natl Acad Sci USA. 1998;95:10914–10919. doi: 10.1073/pnas.95.18.10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rall SC, Jr, Weisgraber KH, Mahley RW. Human apolipoprotein E. The complete amino acid sequence. J Biol Chem. 1982;257:4171–4178. [PubMed] [Google Scholar]
- 49.Rall SC, Jr, Weisgraber KH, Mahley RW. Isolation and characterization of apolipoprotein E. Methods Enzymol. 1986;128:273–287. doi: 10.1016/0076-6879(86)28073-8. [DOI] [PubMed] [Google Scholar]
- 50.Rockenstein EM, McConlogue L, Tan H, Power M, Masliah E, Mucke L. Levels and alternative splicing of amyloid β protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer’s disease. J Biol Chem. 1995;270:28257–28267. doi: 10.1074/jbc.270.47.28257. [DOI] [PubMed] [Google Scholar]
- 51.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. A laboratory manual, Ed 2. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 52.Sanan DA, Weisgraber KH, Russell SJ, Mahley RW, Huang D, Saunders A, Schmechel D, Wisniewski T, Frangione B, Roses AD, Strittmatter WJ. Apolipoprotein E associates with β amyloid peptide of Alzheimer’s disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J Clin Invest. 1994;94:860–869. doi: 10.1172/JCI117407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci USA. 1997;94:4103–4108. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sheng H, Laskowitz DT, Bennett E, Schmechel DE, Bart RD, Saunders AM, Pearlstein RD, Roses AD, Warner DS. Apolipoprotein E isoform-specific differences in outcome from focal ischemia in transgenic mice. J Cereb Blood Flow Metab. 1998;18:361–366. doi: 10.1097/00004647-199804000-00003. [DOI] [PubMed] [Google Scholar]
- 55.Slooter AJC, Tang M-X, van Duijn CM, Stern Y, Ott A, Bell K, Breteler MMB, Van Broeckhoven C, Tatemichi TK, Tycko B, Hofman A, Mayeux R. Apolipoprotein E ε4 and the risk of dementia with stroke. A population-based investigation. JAMA. 1997;277:818–821. doi: 10.1001/jama.277.10.818. [DOI] [PubMed] [Google Scholar]
- 56.Strain SM, Tasker RAR. Hippocampal damage produced by systemic injections of domoic acid in mice. Neuroscience. 1991;44:343–352. doi: 10.1016/0306-4522(91)90059-w. [DOI] [PubMed] [Google Scholar]
- 57.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strittmatter WJ, Weisgraber KH, Goedert M, Saunders AM, Huang D, Corder EH, Dong L-M, Jakes R, Alberts MJ, Gilbert JR, Han S-H, Hulette C, Einstein G, Schmechel DE, Pericak-Vance MA, Roses AD. Hypothesis: microtubule instability and paired helical filament formation in the Alzheimer disease brain are related to apolipoprotein E genotype. Exp Neurol. 1994;125:163–171. doi: 10.1006/exnr.1994.1019. [DOI] [PubMed] [Google Scholar]
- 59.Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, Quarfordt SH, Maeda N. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem. 1997;272:17972–17980. doi: 10.1074/jbc.272.29.17972. [DOI] [PubMed] [Google Scholar]
- 60.Sun Y, Wu S, Bu G, Onifade MK, Patel SN, LaDu MJ, Fagan AM, Holtzman DM. Glial fibrillary acidic protein–apolipoprotein E (apoE) transgenic mice: astrocyte-specific expression and differing biological effects of astrocyte-secreted apoE3 and apoE4 lipoproteins. J Neurosci. 1998;18:3261–3272. doi: 10.1523/JNEUROSCI.18-09-03261.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sze C-I, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:933–944. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- 62.Teasdale GM, Nicoll JAR, Murray G, Fiddes M. Association of apolipoprotein E polymorphism with outcome after head injury. Lancet. 1997;350:1069–1071. doi: 10.1016/S0140-6736(97)04318-3. [DOI] [PubMed] [Google Scholar]
- 63.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 64.Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- 65.Utermann G, Hees M, Steinmetz A. Polymorphism of apolipoprotein E and occurrence of dysbetalipoproteinaemia in man. Nature. 1977;269:604–607. doi: 10.1038/269604a0. [DOI] [PubMed] [Google Scholar]
- 66.Weisgraber KH, Mahley RW. Human apolipoprotein E: the Alzheimer’s disease connection. FASEB J. 1996;10:1485–1494. doi: 10.1096/fasebj.10.13.8940294. [DOI] [PubMed] [Google Scholar]
- 67.Weisgraber KH, Rall SC, Jr, Mahley RW. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J Biol Chem. 1981;256:9077–9083. [PubMed] [Google Scholar]
- 68.Wisniewski T, Frangione B. Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett. 1992;135:235–238. doi: 10.1016/0304-3940(92)90444-c. [DOI] [PubMed] [Google Scholar]
- 69.Xu P-T, Schmechel D, Rothrock-Christian T, Burkhart DS, Qiu H-L, Popko B, Sullivan P, Maeda N, Saunders AM, Roses AD, Gilbert JR. Human apolipoprotein E2, E3, and E4 isoform-specific transgenic mice: human-like pattern of glial and neuronal immunoreactivity in central nervous system not observed in wild-type mice. Neurobiol Dis. 1996;3:229–245. doi: 10.1006/nbdi.1996.0023. [DOI] [PubMed] [Google Scholar]
- 70.Xu P-T, Gilbert JR, Qiu H-L, Ervin J, Rothrock-Christian TR, Hulette C, Schmechel DE. Specific regional transcription of apolipoprotein E in human brain neurons. Am J Pathol. 1999;154:601–611. doi: 10.1016/S0002-9440(10)65305-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhan S-S, Beyreuther K, Schmitt HP. Quantitative assessment of the synaptophysin immunoreactivity of the cortical neuropil in various neurodegenerative disorders with dementia. Dementia. 1993;4:66–74. doi: 10.1159/000107299. [DOI] [PubMed] [Google Scholar]