

Abstract
Although it has long been known that microinjection of the cholinergic agonist carbachol into the medial pontine reticular formation (mPRF) induces a state that resembles rapid eye movement (REM) sleep, it is likely that other transmitters contribute to mPRF regulation of behavioral states. A key candidate is the peptide vasoactive intestinal polypeptide (VIP), which innervates the mPRF and induces REM sleep when injected into this region of the brainstem. To begin understanding the cellular mechanisms underlying this phenomenon, we examined the effects of VIP on mPRF cells using whole-cell patch-clamp recordings in the in vitro rat brainstem slice. VIP directly depolarized cells via activation of an inward current; these effects were attenuated and potentiated in low-sodium and low-calcium medium, respectively. The depolarization induced by VIP was slower in onset and longer-lived than that evoked by carbachol. The VIP-induced depolarization was reduced in a dose-dependent manner by a competitive antagonist of VIP receptors. Effects of VIP were attenuated in the presence of guanosine 5′-O-(2-thiodiphosphate, 2′5′dideoxyadenosine, and PKI15–24 and were nonadditive in the presence of 8-bromo-cAMP. We conclude that VIP excites mPRF neurons by activation of a sodium current. This effect is mediated at least in part by G-protein stimulation of adenylyl cyclase, cAMP, and protein kinase A. These data suggest that VIP may play a physiological role in REM induction by its actions on mPRF neurons.
Keywords: REM sleep, rat, pons, wakefulness, peptides, carbachol, medial pontine reticular formation
Vasoactive intestinal peptide (VIP), a 28 amino acid peptide originally isolated from porcine duodenum, has been localized in several brainstem nuclei known to play a vital role in behavioral state control (Sims et al., 1980; Eiden et al., 1982;Sutin and Jacobowitz, 1988). Microinjection of VIP into the brains of cats, rabbits, and rats induces rapid eye movement (REM) sleep (Drucker-Colín et al., 1984; Obál et al., 1989; Bourgin et al., 1997). Moreover, microinjection of VIP into a site within the rat medial pontine reticular formation (mPRF) results in long-term enhancement of REM sleep not seen when carbachol is injected at the same site (Bourgin et al., 1997). Injection of a competitive VIP antagonist in rats reduces the amount of time spent in REM sleep during the light period, when rats normally exhibit the highest levels of REM (Mirmiran et al., 1988), and intracerebroventricular injection of VIP antibodies blocks REM sleep in rats (Riou et al., 1982). Finally, it has been shown that injection of VIP into the mPRF restores REM sleep in cats rendered insomniac with the serotonin synthesis inhibitor parachlorophenylalanine (PCPA) (Prospero-García et al., 1993).
Despite the in vivo evidence that VIP may be involved in behavioral state control, few studies have examined the electrophysiological effects of VIP on neurons in brainstem nuclei implicated in the control of the sleep–wakefulness cycle. Wang and Aghajanian (1990) found that VIP induces an increase in EPSP activity in the locus ceruleus (LC), but the in vivo effects of VIP on REM sleep induction are unlikely to be caused by excitation of LC neurons as these neurons are silent during REM (Aston-Jones and Bloom, 1981). Moreover, the dominant hypothesis in the field suggests that activation of neurons in the mPRF induces REM sleep and that this effect is mediated by activation of cholinergic receptors (Baghdoyan et al., 1993). Indeed, during naturally occurring REM sleep, the majority of mPRF neurons depolarize 7–10 mV (Ito and McCarley, 1984), and the cholinergic agonist carbachol depolarizes the majority of mPRF neurons (Greene et al., 1989). Because VIP microinjections stimulate REM sleep in a manner highly reminiscent to that seen with cholinergic agonists (Bourgin et al., 1997), we examined effects of this peptide on the membrane properties of mPRF neurons at a site that has been shown to be critical for the induction of REM sleep. Our data indicate that VIP may play a role in the promotion of REM sleep.
MATERIALS AND METHODS
Wistar rats (7- to 18-d-old; average age, 12 d) were anesthetized with halothane and decapitated. The brain was rapidly removed and trimmed to form a block that contained the mPRF, which was then cut into 400-μm-thick coronal slices on an Oxford (Concord, MA) vibratome. The slice containing the mPRF was placed in a recording chamber and superfused with a solution of standard artificial CSF (ACSF) containing (in mm): 126 NaCl, 25 NaHCO3, 1.2 NaH2PO4, 2.5 KCl, 2.5 CaCl2, 1.2 MgCl2, and 11 glucose, pH 7.3 when saturated with 95% O2–5% CO2. Slices were allowed to equilibrate to room temperature, and all electrophysiological experiments were performed at 21°C.
The whole-cell configuration of the patch-clamp technique as applied to brain slices was used to record from neurons in the mPRF using bridge mode and voltage-clamp methodologies (Kamondi et al., 1992). Bridge mode and single-electrode voltage-clamp measurements were obtained with an Axoclamp 2A amplifier (Axon Instruments, Foster City, CA) and were filtered at 1 kHz. Access resistance was monitored and considered acceptable when in the range of 10–50 MΩ. Patch pipettes were constructed from thin-walled borosilicate glass capillary tubes (outer diameter, 1.5; inner diameter, 1.17) (Warner Instruments, Hamden, CT). The electrode solution contained (in mm): 120 K-gluconate, 10 HEPES acid, 24 NaCl, 15 KCl, 11 EGTA, 1 CaCl2, and 2 MgATP.
For voltage-command generation and voltage and current data acquisition, the pClamp 6 suite of programs (Axon Instruments) was used. In bridge mode, baseline and postdrug recording of input resistance and membrane potential of mPRF neurons were collected and compared; input resistance was determined by maximum voltage deflection of the membrane potential after the injection of 0.03 nA of hyperpolarizing current either at the resting potential, or in the case of drug effects, after injection of direct current (DC) to bring the cell to resting membrane potential. In additivity experiments, peak induced depolarization or inward current were used as the index of response. Data are reported as mean ± SEM. Statistical significance was assessed using the Student’s paired t test or a between-group repeated-measure ANOVA. In histograms, VIP-induced inward currents were normalized (100% ± SEM) and considered “control”; drug treatments are expressed as a percentage ± SEM of this normalized current.
VIP was obtained from Sigma (St. Louis, MO) and dissolved at 100 μm in 0.01 m acetic acid and frozen. On the day of the experiment, an aliquot was diluted to a final concentration of 100 nm in ACSF; control experiments revealed that application of 10 μm acetic acid (the final concentration) in ACSF did not elicit noticeable effects on cells. Applications of VIP were either via the bath (total application time of 30 sec) or usage of a General Valve (Fairfield, NJ) Picospritzer II (200 msec pulse duration). It should be noted that pretreatment of the puffer pipette and the inflow tubing was not performed; potential alterations in concentration of the peptide delivered to the slice because of binding of the peptide to these surfaces were not investigated. (d-P-chloro-Phe6, Leu17)-vasoactive intestinal peptide (DPC-VIP), a VIP receptor antagonist (Sigma), was applied at a concentration of 250–500 nm.
In most experiments, tetrodotoxin (TTX) (Sigma) was added to block voltage-dependent sodium currents. Low-sodium (27.2 mm) solution was made by equimolar substitution of choline chloride for NaCl. In these experiments, the muscarinic receptor antagonist atropine (5 μm; Sigma) was added to ACSF–choline to prevent the depolarization produced by choline itself; as a control, the effects of VIP in atropine-containing ACSF were also examined (control ACSF) and were not found to be different from those in nonatropine-containing ACSF. Low-calcium (0.5 mm) solutions, made by substitution of calcium with high magnesium (10 mm), were applied for 10–20 min before drug application; efficacy was monitored by observing a reduction of the amplitude of the calcium-dependent after hyperpolarization (AHP). In experiments using cesium to block the hyperpolarization-activated cationic current (Ih) and inwardly rectifying potassium currents, cesium was added directly to the ACSF at a final concentration of 1 mm.
8-bromo-cAMP and 8-bromo-cGMP (Sigma) were dissolved in ACSF and applied for 20 min. Stock solutions of 2′5′dideoxyadenosine (DDA) (1 mm dissolved in dimethyl sulfoxide; Calbiochem, La Jolla, CA), LY 83583 (10 mm dissolved in dimethyl sulfoxide; Calbiochem), and RP-8-pCPT-cGMPS (1 mm dissolved in water; Biolog, Hayward, CA) were diluted in ACSF and applied for 10–20 min. Guanosine 5′-O-(2-thiodiphosphate) (GDP-β-S) (Sigma), a phosphorylation- and hydrolysis-resistant analog of GDP, was dissolved in the pipette solution. PKI15–24 (Calbiochem) was also dissolved in the pipette solution, and ejection into the cell was facilitated by passing pulses of positive current (0.03 nA; duration of 750 msec) for 15–20 min. In these latter two experiments, effects of VIP were tested immediately after cell penetration and then repeated after allowing diffusion of the antagonists into the cell.
RESULTS
A total of 133 neurons within the mPRF (corresponding to the posterior area of the pontis oralis at the level of the trigeminal motor nucleus in the rat) were studied. Preliminary data indicated that 100 nm VIP induced maximal effects on membrane potential and current in these cells; higher concentrations only increased the duration of the effect. Effects were reproducible with no attenuation in the same cell after recovery.
The effects of VIP on mPRF neurons were examined in bridge mode in 35 cells. There was no significant difference between bath application and picospritzer delivery (p > 0.05; ANOVA), and therefore these data were pooled. Bath and picospritzer application of VIP (100 nm) depolarized 31 of 35 mPRF neurons by 3.27 ± 0.04 mV (p < 0.05) with a concurrent decrease in input resistance of 47.7 ± 0.18 MΩ (p < 0.05) (Fig.1A,B); this represents a change in input resistance of 15%. In no case did VIP elicit hyperpolarization of mPRF cells. The onset to depolarizing effects of VIP was 1.86 ± 0.23 min, and latency to peak effect was 4.23 ± 0.38 min. The duration of the depolarizing effect was 8.5 ± 0.2 min. Application of TTX (n = 14) before VIP did not block either the depolarization or change in input resistance. These data indicate that the effects of VIP on mPRF cells is not dependent on action potential generation in the slice.
Fig. 1.

VIP depolarizes mPRF neurons; carbachol also elicited membrane depolarization in a subpopulation of these cells.A, Top traces represent current.Bottom traces represent membrane potential. A 30 sec bath application of 100 nm VIP (solid bar) resulted in a depolarization and a decrease in input resistance. After recovery from the VIP-induced depolarization, carbachol depolarized this cell. B, Averages of 10 voltage responses to hyperpolarizing current pulses before VIP (1), during the peak effect of VIP but with the cell returned to the resting membrane potential by the injection of hyperpolarizing DC current (2), and after washout of VIP (3). A superimposition of tracesin 1 and 2 are presented in4 to show the difference in input resistance elicited by VIP. C, Schematic representation of coronal sections of the brainstem showing the region within the mPRF at which effects of VIP were examined (circles). DR, Dorsal raphe nucleus; Mo5, motor trigeminal nucleus.
Carbachol (1 μm) depolarized 13 of 14 mPRF neurons 6.10 ± 0.32 mV, which had previously depolarized in response to VIP and recovered (Fig. 1A). The latency to onset of effect was 18.0 ± 3.0 sec, and latency to peak was 1.7 ± 0.17 min. In three cells, VIP was applied first and carbachol added during peak effects, which resulted in further depolarization (data not shown). These data, coupled with those of Greene et al. (1989) that demonstrated that carbachol depolarized most mPRF cells via activation of a nonrectifying potassium conductance, as well as activating an inwardly rectifying potassium conductance in a minority of mPRF cells that hyperpolarized in response to carbachol, suggest that distinct mechanisms underlie the VIP- and carbachol-induced depolarizations.
To test the hypothesis that VIP was acting at VIP receptors, the effects of the competitive VIP antagonist DPC-VIP were studied. The protocol used was begun with a control application of VIP, and after washout, pretreatment with DPC-VIP and reapplication of VIP; when possible, the control application of VIP was repeated at the conclusion of the experiment. In the presence of 250 nm DPC-VIP, VIP induced only 50.2 ± 0.31% of maximum depolarization and 41.0 ± 0.23% of maximum change in input resistance, respectively (n = 4), and, in the presence of 500 nmDPC-VIP, 82.5 ± 0.21 and 79.3 ± 0.6% of VIP-induced depolarization and input resistance changes were suppressed (n = 3) (Fig. 2).
Fig. 2.

The VIP-induced depolarization was antagonized by the VIP-receptor antagonist DPC-VIP in a dose-dependent manner.A, Bridge mode recordings from one mPRF neuron in which 100 nm VIP was applied for 30 sec (first trace). After recovery (25 min), 100 nm VIP was added in the presence of 250 nm DPC-VIP (second trace). After washout (25 min), 100 nm VIP was added in the presence of 500 nm DPC-VIP (third trace). After washout of DPC-VIP (20 min), 100 nmVIP without antagonist was applied. Black bars in eachtrace indicate application of VIP. B, Dose-dependent attenuation of VIP-induced depolarization by DPC-VIP.
In voltage-clamp mode, 100 nm VIP induced an inward current of 98.3 ± 13.9 pA at −60 mV (n = 18), with an accompanying increase in conductance (Fig.3A,B). The VIP-induced inward current was not mediated by a decrease in potassium conductance because the control and VIP I–Vcurves did not cross at the potassium reversal potential (Fig.3C), nor was the current mediated by a change in chloride conductance because I–V curves did not cross at the chloride equilibrium potential. Blockade of the hyperpolarization-activated cation current (Ih) by 1 mm cesium did not significantly alter the effect of VIP on mPRF neurons in either bridge mode (n = 5; p > 0.05) or voltage clamp (n = 2; p > 0.05) (Fig.4B). Although not readily apparent in Figure 4, the change in conductance (voltage clamp) or input resistance (current clamp) was not significantly different in the two conditions (control–VIP and cesium–VIP) in the population of cells examined (p > 0.05). These data, together with the observation that the I–V curves do not cross in the region between −30 and −70 mV suggest thatIh does not play a role in generation of the VIP-induced inward current.
Fig. 3.

VIP induces an inward current in mPRF neurons.A, Voltage-clamp recordings at −60 mV. Downward deflections are current responses to voltage steps of −20 mV. The VIP-induced inward current is accompanied by an increase in conductance. B, High-gain current response to the voltage step protocols shown in the inset demonstrating changes in holding current and conductance elicited by VIP in the presence of TTX. Calibration: 200 pA, 250 msec. C,I–V curves derived from the steady-state current values under control and VIP in a population of cells (n = 15). Conductance was calculated as the slope of a straight line in current–voltage plots. Linear regression demonstrated that slope conductance increased during VIP by 17% in this population of cells, which agrees well with the value calculated from direct measures of conductance changes. Extrapolation of the reversal potential of the VIP-induced current revealed that it was 0 mV.
Fig. 4.

VIP induces a sodium-dependent depolarization.A, Bridge mode recordings of the response of a cell to 100 nm VIP in the presence of choline-substituted low-sodium ACSF. After washout of the low-sodium solution, VIP application was repeated in the presence of control ACSF. Calibration: 5 mV, 1 min. B, VIP (100 nm) applied for 30 sec in the presence of 1 mm cesium (top trace) induced an inward current that was not significantly different from in control conditions (bottom trace).C, Bridge mode recordings of the response to 100 nm VIP applied for 30 sec in normal ACSF (top trace) and after washout in 0.5 mmCa2+–10 mm Mg2+solution. High-gain records (inset) demonstrating loss of AHP after 20 min exposure to low-Ca2+ solution.D, Voltage-clamp recordings of the inward current induced by VIP in low-Ca2+ solution (top trace) and after washout in normal ACSF (bottom trace). E, Bar graph quantifying the results of ion substitution experiments.
Adequate voltage clamp at potentials more positive that −30 mV was not possible, precluding the use of I–V curves to study the contributions of the cations calcium and sodium to the VIP-induced inward current. Therefore, VIP was applied in ACSF containing altered concentrations of these ions. Low-calcium solution (5 mm) applied for at least 15 min, resulting in reduction of the calcium-dependent AHP (Gerber et al., 1989, Fig. 4C), did not block the VIP-induced depolarizations and inward current (n = 12). Indeed, the depolarization and inward current induced by VIP in the presence of low-calcium solution was significantly increased (p < 0.05) (Fig.4C,D).
Reducing the concentration of sodium to 26 mm in the ACSF significantly reduced both the VIP-induced inward current (n = 4; p < 0.05) and depolarization (n = 10; p < 0.05) without attenuation of an accompanying increase in conductance (11%) and decrease in input resistance, respectively, which was not significantly different from that seen in control conditions (12.3%; p > 0.05) (Fig. 4A,E).
cAMP mediates the effects of VIP
To test the hypothesis that the inward current and depolarization induced by VIP may be dependent in part on activation of cAMP-dependent mechanisms, we applied agonists and antagonists of this second messenger system. Passive diffusion of the phosphorylation- and hydrolysis-resistant GDP analog GDP-β-S (20 μm) from the recording electrode into the cell gradually hyperpolarized the membrane potential and significantly reduced the amplitude of the inward current and depolarization induced by VIP (n = 6; p < 0.05) (Fig.5A–C). The effects of GDP-β-S were not likely to be caused by the hyperpolarization per se, because recordings obtained from a subset of mPRF cells with a mean resting membrane potential of −72 ± 3.2 mV (n = 8) showed that VIP-induced effects were not statistically different from cells with more depolarized membrane potentials.
Fig. 5.
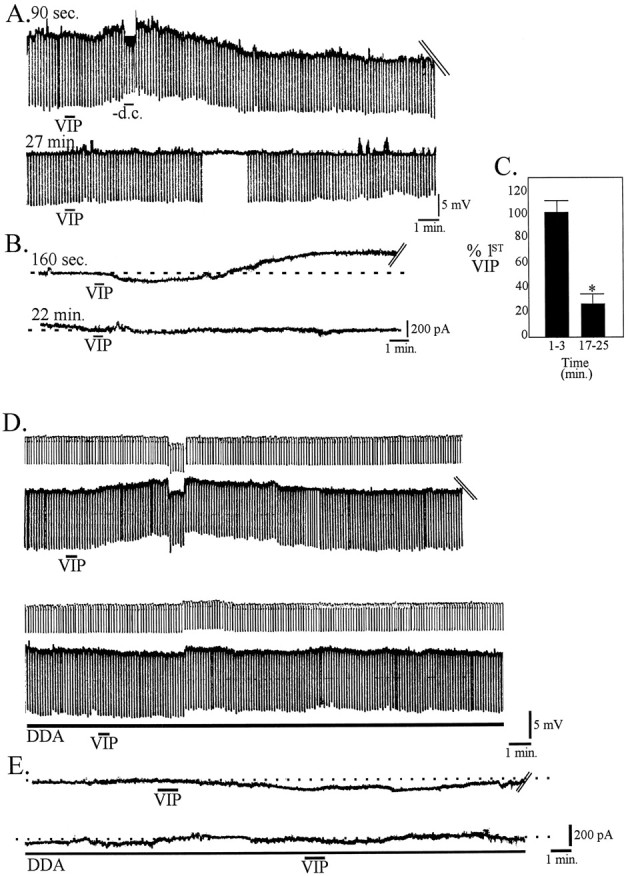
The VIP-induced depolarization is mediated by G-protein-dependent activation of adenylyl cyclase. A, Effects of 100 nm VIP, 30 sec, applied 90 sec after breaking into a cell with a pipette containing 20 μmGDP-β-S (top trace; membrane potential, −62.5 mV) and after washout 27 min after break-in (bottom trace; membrane potential, −68.3 mV). B, In voltage clamp, effects of a 20 sec application of 100 nm VIP administered 160 sec after breaking into a cell with a pipette containing 20 μm GDP-β-S and 22 min after break-in. C, Histogram of the normalized inward current activated by VIP within 3 min of cell entry and 17–25 min after entry when GDP-β-S is included in the pipette (n = 6). DDA (50 μm) blocked both the depolarization (D) and inward current (E) induced by VIP.
The membrane permeable adenylyl cyclase inhibitor DDA (50–100 μm) reduced the amplitude of the VIP response in five of five cells tested. DDA induces an increase in input resistance, membrane hyperpolarization, and outward current; application of VIP in the presence of DDA resulted in a reduction of both the VIP-induced depolarization (63.8 ± 0.7% reduction; n = 3;p < 0.05) (Fig. 5D) and inward current (71.2 ± 0.5% reduction; n = 3; p< 0.05) (Fig. 5E) compared with control.
Application of the membrane permeable cAMP analog 8-bromo-cAMP depolarized all mPRF neurons via activation of an inward membrane current (Fig. 6A). The inward current induced by 1 mm 8-bromo-cAMP and that induced by VIP were not additive (n = 5) (Fig.6A), which is consistent with the hypothesis that VIP induces an inward current in mPRF neurons by activating the cAMP pathway. However, because direct antagonists of cAMP were not used, we have not formally ruled out the hypothesis that cAMP does not have direct actions, such as activation of sodium channels, thereby preventing VIP-activation of these channels by protein kinase-dependent mechanisms (see below).
Fig. 6.

The VIP-induced depolarization is mediated by the cAMP cascade. A, 8-bromo-cAMP induced an inward current in mPRF neurons, which was not significantly increased by addition of VIP, as shown in the bar graph on the right and the individual traces on the left, demonstrating that the currents activated by VIP and 8-bromo-cAMP are not additive.B, Effects of 100 nm VIP applied for 30 sec at the times indicated after break-in to a cell in which PKI15–25 was included in the pipette in bridge mode (B) and voltage clamp (C). [After establishment of baseline effects of VIP, ejection of PKI15–25 from the recording pipette was facilitated by injection of depolarizing current pulses in bridge mode (or stepping the holding current of the cell from −60 to −30 mV if in voltage-clamp mode) for 20 min before subsequent application of VIP].D, Bar graph of the average currents elicited by VIP in the presence of PKI15–25 at the indicated times.
The intracellular application of the PKA inhibitor PKI15–24 (1 mg/1 ml) reduced the amplitude of VIP-induced depolarization (19–35 min; 55.2 ± 0.1% mV reduction;n = 2; p < 0.05) (Fig.6B) and that of the VIP-induced inward current. Effects of VIP on subsequent applications were further attenuated (19–35 min; 61.0 ± 0.11% pA of control; 46–59 min; 23.0 ± 0.12% pA of control; n = 7; p < 0.05) (Fig. 6C,D). Because VIP has also been reported to induce cGMP activity in some neuronal cell types (Ho et al., 1987; Chik et al., 1988) and because the cAMP cascade is well known to interact with cGMP pathways, we also examined the effects of VIP on mPRF neurons in the presence of 1 mm 8-bromo-cGMP. 8-bromo-cGMP hyperpolarized or induced an outward current in 8 of 10 cells and induced an inward current in two additional cells; the depolarization or inward current induced by VIP in the presence of 8-bromo-cGMP in these cells was not significantly different from that induced by VIP in control conditions in the same cells (n = 10; p > 0.05) (Fig.7A,C).
Fig. 7.

The cGMP second messenger system does not appear to be involved in VIP-induced inward currents. A, 8-bromo-cGMP induces an outward current in this cell (top trace). VIP-induced inward current is not significantly different in the presence of 8-bromo-cGMP than in control conditions (C). The reversible guanylyl cyclase inhibitor LY 83583 does not block activation of inward current by VIP (bottom trace and C). B, RP-8-pCPT-cGMPS, an inhibitor of cGMP formation, did not block the VIP-induced inward current but significantly increased it (C).
We also examined the effects of VIP on induction of inward current in the presence of the soluble guanylyl cyclase inhibitor LY 83583. During the first 2 min of application, 10 μm LY 83583 induced an outward current in five of seven cells examined. However, after this initial effect, a slowly developing inward current developed in these cells; in two of seven cells, an inward current was induced only. LY 83583 did not attenuate VIP-induced inward current (Fig.7A,C).
We next examined the effects of VIP in the presence of the competitive inhibitor of cGMP-dependent protein kinases Rp-8-pCPT-cGMPs (10 μm). After 15 min of application of this cGMP inhibitor, VIP was applied and found to induce a significantly greater inward current than that induced during control conditions (132.0 ± 0.9%; n = 3; p < 0.05) (Fig.7B,C). These data suggest that cGMP mechanisms are not involved in the VIP-induced inward current.
DISCUSSION
The principal finding of the present study is that VIP depolarizes a population of mPRF neurons. The effects of VIP persisted in the presence of both TTX and reduced calcium, suggesting that they are mediated by receptors located on this population of brainstem cells. The VIP-mediated depolarization was reduced in a dose-dependent manner by a VIP receptor antagonist. Several lines of evidence suggest that the VIP-induced inward current did not arise secondary to a shift in the activation curve of Ih. We found that, whereas 8-Br-cAMP shifts the Ih activation curve of mPRF cells to more depolarized potentials (our unpublished observations) as has been reported in thalamic cells (McCormick and Pape, 1990), VIP at the concentrations used in the present study does not have this effect. Moreover, the VIP-induced inward current persisted in the presence of cesium, which has been reported to be an effective blocker of Ih (McCormick and Pape, 1990), nor is it likely that VIP was acting via inhibition of the Na–K pump. VIP-induced effects were accompanied by conductance changes in normal ACSF; inward currents induced by Na–K pump inhibition are associated with little change in conductance (Gadsby and Nakao, 1989;Hermans et al., 1994). The extrapolated reversal potential calculated from I–V curves of the VIP-induced current was ∼0 mV, an observation that would not be expected if the VIP effects were mediated by inhibition of a pump. Most importantly, inward currents induced by inhibition of the pump would be expected to be enhanced in the presence of reduced extracellular sodium (Kuba and Nohmi, 1987; Wang and Aghajanian, 1989; Shen and Johnson, 1998), whereas the VIP-induced inward current was diminished under similar conditions. The persistence of the effects of VIP in TTX suggests that the effects of VIP are not mediated by classical voltage-dependent sodium channels. Under voltage-clamp conditions, the VIP-induced depolarization was found to be mediated by an inward current that was long lasting and accompanied by an increase in conductance. Failure of the control and VIPI–V curves to cross at the potassium or chloride reversal potentials suggests that VIP effects were not mediated by these two ions. Ion substitution experiments were consistent with the hypothesis that VIP effects were caused by an increase in a sodium current. However, because specific pharmacological blockade of potassium conductances were not performed, we cannot rule out the hypothesis that other conductance changes contribute to the VIP-mediated depolarization. Moreover, these effects of VIP were shown to be mediated, at least in part, by activation of the cAMP cascade. Because excitation of mPRF neurons has been suggested to be critical in the generation of REM sleep, these data suggest that VIP may play a role in regulation of REM sleep by actions on mPRF neurons specifically during this behavioral state.
It has long been known that microinjection of cholinergic agonists within the mPRF can produce a state that is behaviorally indistinguishable from naturally occurring REM sleep (Baghdoyan et al., 1993). The ability to induce REM using pharmacological techniques has served as a powerful tool with which to study the mechanisms of state generation. Although a strong body of evidence supports the notion that cholinergic mechanisms are involved in triggering REM sleep (Kodama et al., 1990; Baghdoyan et al., 1993), cholinergic excitation of the mPRF is not likely to be the only mechanism by which REM is induced. For example, injections of the neutral endopeptidase inhibitor phosphoramidon into the mPRF results in a REM-like state (Morales et al., 1998), suggesting that increased survival of an endogenous peptide released within the mPRF elicits REM sleep. Moreover, cholinergic antagonists modify, but do not block, the occurrence of natural REM sleep (Gnadt and Pegram, 1986; Imeri et al., 1991). Thus, transmitters other than acetylcholine also appear to be involved in triggering REM sleep in the mPRF. One candidate is the peptide VIP. Microinjection of VIP into the “REM-induction site” induces REM sleep in cats, rabbits, and rats (Drucker-Colín et al., 1984; Obál et al., 1989; Bourgin et al., 1997). Our data demonstrating that VIP directly depolarizes mPRF neurons places this observation on a strong mechanistic footing. Moreover, the VIP-induced depolarization was longer in duration than that seen with the long-lasting hydrolyzable cholinergic agonist carbachol. It is possible that naturally released acetylcholine might cause short-term changes in mPRF excitability concurrent with enhancement (via increased resistance of these cells) of EPSPs, resulting in mPRF cells attributable to reticular stimulation, which is a phenomenon seen in naturally occurring REM (Ito and McCarley, 1984). In a similar vein, it seems plausible that VIP might play a role in the maintenance of the REM state via prolonged depolarization of mPRF cells. Finally, these observations suggest that natural REM sleep may be produced by the concerted release of at least two transmitters, acetylcholine and VIP.
Indirect evidence supports the involvement of VIP in REM generation. VIP restores REM sleep in cats rendered insomniac with the serotonin synthesis inhibitor PCPA; these effects persist in the presence of the muscarinic receptor antagonist atropine, indicating that the REM sleep-restorative effects of VIP were not dependent on cholinergic mechanisms (Prospero-García et al., 1993). Decreases in REM sleep have been reported in the rat after intraventricular application of VIP antagonists (Mirmiran et al., 1988; Prospero-García et al., 1993) and VIP antibodies (Riou et al., 1982). In this note, it is interesting to compare the effects of VIP antagonists on REM with those reported for cholinergic antagonists. VIP antagonists have been shown to decrease REM sleep in the rat by 64 and 22%, which was compensated for in both studies by an increase in wakefulness (Riou et al., 1982;Mirmiran et al., 1988). The results from studies using cholinergic antagonists are less clear. Atropine has been reported to have no effect on naturally occurring REM sleep in rats (Gnadt and Pegram et al., 1986); however, it has been reported that the M2receptor antagonist methoctramine delays the latency to REM episodes, and, at higher doses, REM was reduced by 75% (Imeri et al., 1991). VIP receptors have been found to be increased in the mPRF after sleep deprivation (Jimenez-Anguiano et al., 1996). Finally, Shiromani and McGinty (1986) reported that at least some neurons in the mPRF increased firing in vivo in response to VIP at the same site in which carbachol induced REM sleep. Together with our new data, these reports provide a growing body of evidence that VIP is involved in regulation of the sleep–wakefulness cycle.
VIP has been shown to enhance acetylcholine synthesis in structures such as the hippocampus (Luini et al., 1984). Additionally, it has been shown that VIP can modulate acetylcholine actions (Halimi et al., 1997). These data can be interpreted as evidence that in vivo actions of VIP on the induction of REM sleep may be dependent on the effects of VIP on mPRF cholinergic tone. VIP has also been shown to modify muscarinic, as well as nicotinic, receptor function (Mo and Dun, 1984; Kawatani et al., 1985; Pedigo and Rice, 1992; Gurantz et al., 1994; Cuevas and Adams, 1996). Effects of VIP on induction of REM sleep appear to be independent of muscarinic receptor activation, although this study was conducted in PCPA-treated animals, which may confound these conclusions (Prospero-García et al., 1993); however, effects of VIP after release of acetylcholine onto mPRF neurons or on mPRF nicotinic receptors function remain unknown. The notion that VIP may modify REM sleep via such effects is particularly interesting in light of the findings that microinjection of nicotine into the mPRF have been found to elicit REM sleep (Velazquez-Moctezuma et al., 1990). Regardless, although effects of VIP on cholinergic tone or enhancement of muscarinic or nicotinic receptor function may contribute to VIP-inducement of REM sleep in vivo, the present data clearly indicate VIP directly excites mPRF neurons.
VIP-positive fibers have been found within the mPRF (Sims et al., 1980;Eiden et al., 1982), but the source of these fibers has not been determined. VIP has been found in the noncholinergic cells of the laterodorsal tegmental nuclei (LDT) and at higher numbers in the pedunculopontine tegmental nuclei (PPT), as well as in the most dorsal aspect of the mPRF (Sutin and Jacobowitz, 1988). Although both cholinergic and noncholinergic LDT and PPT neurons project to the mPRF, it has not been formally demonstrated that VIP-positive cells project to the mPRF from these structures. However, it is clear that VIP receptors are found in the mPRF (Staun-Olsen et al., 1985). These anatomical data suggest that our pharmacological findings have physiological relevance and support the hypothesis that VIP may be involved in the mechanisms that trigger REM sleep.
Several phenomena that regulate peptide release are relevant to these data. Nitric oxide, produced in cholinergic PPT and LDT neurons, is thought to be generated during REM (Kamondi et al., 1992; Leonard and Lydic, 1997), and nitric oxide has been shown to stimulate the release of VIP (Grider, 1993; Grider and Jin, 1993). Thus, release of nitric oxide in the mPRF during REM may modify release of VIP. Moreover, peptide release is facilitated by high-frequency firing (Lundberg and Hökfelt, 1986). A subpopulation of LDT and PPT cells exhibit burst firing just before the transition to REM and continuing during the entire episode, and it has been suggested that these cells are noncholinergic (Steriade et al., 1990). If these neurons contain VIP, such burst firing would be the appropriate stimulus for peptide release.
Footnotes
This research was supported by a grant from the Medical Research Council of Canada. K.A.K. was supported by a postdoctoral fellowship from the National Sleep Foundation (USA). P.B.R. is a Medical Research Council Scientist (Canada).
Correspondence should be addressed to Dr. Peter B. Reiner, Kinsmen Laboratory of Neurological Research, Department of Psychiatry, University of British Columbia, Vancouver, British Columbia, V6T 1Z3 Canada.
Dr. Kohlmeier’s present address: Department of Physiology, New York Medical College, Valhalla, NY 10595.
REFERENCES
- 1.Aston-Jones G, Bloom FE. Activity of noreprinephrine-containing locus ceruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1981;1:876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baghdoyan HA, Spotts JL, Synder SG. Simultaneous pontine and basal forebrain microinjections of carbachol suppress REM sleep. J Neurosci. 1993;13:229–242. doi: 10.1523/JNEUROSCI.13-01-00229.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bourgin P, Lebrand C, Escourrou P, Gaultier C, Franc B, Hamon M, Adrien J. Vasoactive intestinal polypeptide microinjections into the oral pontine tegmentum enhance rapid eye movement sleep in the rat. Neuroscience. 1997;77:351–360. doi: 10.1016/s0306-4522(96)00455-1. [DOI] [PubMed] [Google Scholar]
- 4.Chik CL, Ho AK, Klein DC. α 1-Adrenergic potentiation of vasoactive intestinal peptide stimulation of rat pinealocyte adenosine 3′,5′-monophosphate and guanosine 3′,5′-monophosphate: evidence for a role of calcium and protein kinase C. Endocrinology. 1988;122:702–708. doi: 10.1210/endo-122-2-702. [DOI] [PubMed] [Google Scholar]
- 5.Cuevas J, Adams DJ. Vasoactive intestinal polypeptide modulation of nicotinic ACh receptor channels in rat intracardiac neurones. J Physiol (Lond) 1996;493:503–515. doi: 10.1113/jphysiol.1996.sp021399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drucker-Colín R, Bernal-Pedraza J, Fernández-Cancino F, Oksenberg A. Is vasoactive intestinal polypeptide (VIP) a sleep factor? Peptides. 1984;5:837–840. doi: 10.1016/0196-9781(84)90032-9. [DOI] [PubMed] [Google Scholar]
- 7.Eiden LE, Nilaver G, Palkovits M. Distribution of vasoactive intestinal polypeptide (VIP) in the rat brain stem nuclei. Brain Res. 1982;231:472–477. doi: 10.1016/0006-8993(82)90386-9. [DOI] [PubMed] [Google Scholar]
- 8.Gadsby DC, Nakao M. Steady state current–voltage relationship of the Na/K pump in guinea pig ventricular myocytes. J Gen Physiol. 1989;94:511–537. doi: 10.1085/jgp.94.3.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerber U, Greene RW, McCarley RW. Repetitive firing properties of medial pontine reticular formation neurones of the rat recorded in vitro. J Physiol (Lond) 1989;410:533–560. doi: 10.1113/jphysiol.1989.sp017548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gnadt JW, Pegram GV. Cholinergic brainstem mechanisms of REM sleep in the rat. Brain Res. 1986;384:29–41. doi: 10.1016/0006-8993(86)91216-3. [DOI] [PubMed] [Google Scholar]
- 11.Greene RW, Gerber U, McCarley RW. Cholinergic activation of medial pontine reticular formation neurons in vitro. Brain Res. 1989;476:154–159. doi: 10.1016/0006-8993(89)91549-7. [DOI] [PubMed] [Google Scholar]
- 12.Grider JR. Interplay of VIP and nitric oxide in regulation of the descending relaxation phase of peristalsis. Am J Physiol. 1993;264:G334–G340. doi: 10.1152/ajpgi.1993.264.2.G334. [DOI] [PubMed] [Google Scholar]
- 13.Grider JR, Lin JG. Vasoactive intestinal peptide release and l-citrulline production from isolated ganglia of the myenteric plexus: evidence for regulation of vasoactive intestinal peptide release by nitric oxide. Neuroscience. 1993;54:521–526. doi: 10.1016/0306-4522(93)90271-g. [DOI] [PubMed] [Google Scholar]
- 14.Gurantz D, Harootunian AT, Tsien RY, Dionne VE, Margiotta JF. VIP modulates neuronal nicotinic acetylcholine receptor function by a cyclic AMP-dependent mechanism. J Neurosci. 1994;14:3540–3547. doi: 10.1523/JNEUROSCI.14-06-03540.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Halimi F, Piot O, Guize L, Le Heuzey JY. Electrophysiological effects of vasoactive intestinal peptide in rabbit atrium: a modulation of acetylcholine activity. J Mol Cell Cardiol. 1997;29:37–44. doi: 10.1006/jmcc.1996.0249. [DOI] [PubMed] [Google Scholar]
- 16.Hermans AN, Gkitsch HG, Verdonck F. The effect of cardiac glycosides on the Na+ pump current–voltage relationship in isolated rat and guinea-pig heart cells. J Physiol (Lond) 1994;481:279–291. doi: 10.1113/jphysiol.1994.sp020438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho AK, Chik CL, Klein DC. Transmembrane receptor cross-talk: concurrent VIP and α 1-adrenergic activation rapidly elevates pinealocyte cGMP greater than 100 fold. Biochem Biophys Res Commun. 1987;146:1478–1484. doi: 10.1016/0006-291x(87)90816-3. [DOI] [PubMed] [Google Scholar]
- 18.Imeri L, Bianchi S, Angeli P, Mancia M. Differential effects of M2 and M3 muscarinic antagonists on the sleep–wake cycle. NeuroReport. 1991;2:383–385. doi: 10.1097/00001756-199107000-00007. [DOI] [PubMed] [Google Scholar]
- 19.Ito K, McCarley RW. Alterations in membrane potential and excitability of cat medial pontine reticular formation neurons during changes in naturally occurring sleep–wake states. Brain Res. 1984;292:169–175. doi: 10.1016/0006-8993(84)90903-x. [DOI] [PubMed] [Google Scholar]
- 20.Jiménez-Anguiano A, García-García F, Mendoza-Ramírez JL, Durán-Vázquez A, Drucker-Colín R. Brain distribution of vasoactive intestinal peptide receptors following REM sleep deprivation. Brain Res. 1996;728:37–46. [PubMed] [Google Scholar]
- 21.Kamondi A, Williams JA, Hutcheon B, Reiner PB. Membrane properties of mesopontine cholinergic neurons studied with the whole-cell patch-clamp technique: implications for behavioral state control. J Neurophysiol. 1992;68:1359–1372. doi: 10.1152/jn.1992.68.4.1359. [DOI] [PubMed] [Google Scholar]
- 22.Kawatani M, Rutigliano M, de Groat WC. Depolarization and muscarinic excitation induced in a sympathetic ganglion by vasoactive intestinal polypeptide. Science. 1985;229:879–881. doi: 10.1126/science.3895438. [DOI] [PubMed] [Google Scholar]
- 23.Kodama T, Takahashi Y, Honda Y. Enhancement of acetylcholine release during paradoxical sleep in the dorsal tegmental field of the cat brain stem. Neurosci Lett. 1990;114:277–282. doi: 10.1016/0304-3940(90)90576-u. [DOI] [PubMed] [Google Scholar]
- 24.Kuba K, Nohmi M. Role of ion conductance changes and of the sodium-pump in adrenaline-induced hyperpolarization of rat diaphragm muscle fibres. Br J Pharmacol. 1987;91:671–681. doi: 10.1111/j.1476-5381.1987.tb11261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leonard TO, Lydic R. Pontine nitric oxide modulates acetylcholine release, rapid eye movement sleep generation, and respiratory rate. J Neurosci. 1997;17:774–785. doi: 10.1523/JNEUROSCI.17-02-00774.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luini VN, Rostene W, Rhodes J, McEwen BS. Activation of choline acetyltransferase by vasoactive intestinal peptide. J Neurochem. 1984;42:1131–1134. doi: 10.1111/j.1471-4159.1984.tb12721.x. [DOI] [PubMed] [Google Scholar]
- 27.Lundberg JM, Hokfelt T. Multiple co-existence of peptides and classical transmitters in peripheral autonomic and sensory neurons—functional and pharmacological implications. Prog Brain Res. 1986;68:241–262. doi: 10.1016/s0079-6123(08)60242-3. [DOI] [PubMed] [Google Scholar]
- 28.McCormick DA, Pape H-C. Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurons. J Physiol (Lond) 1990;431:291–318. doi: 10.1113/jphysiol.1990.sp018331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mirmiran M, Kruisbrink J, Bos NPA, Van der Verf D, Boer GJ. Decrease of rapid-eye-movement sleep in the light by intraventricular application of a VIP-antagonist in the rat. Brain Res. 1988;458:192–194. doi: 10.1016/0006-8993(88)90516-1. [DOI] [PubMed] [Google Scholar]
- 30.Mo N, Dun NJ. Vasoactive intestinal polypeptide facilitates muscarinic transmission in mammalian sympathetic ganglia. Neurosci Lett. 1984;52:19–23. doi: 10.1016/0304-3940(84)90344-6. [DOI] [PubMed] [Google Scholar]
- 31.Morales FR, Xi M-C, Chase MH. Induction of active sleep by atrial naturetic peptide microinjection within the nucleus pontis oralis of the cat. Sleep Res [Abstr] 1998;21:33. [Google Scholar]
- 32.Obál F, Opp M, Cady B, Johansen L, Krueger JM. Prolactin, vasoactive intestinal peptide and peptide histiden methionine elicit selective increases in REM sleep in rabbits. Brain Res. 1989;490:292–300. doi: 10.1016/0006-8993(89)90246-1. [DOI] [PubMed] [Google Scholar]
- 33.Pedigo NW, Rice MA. Neuropeptide modulation of muscarinic receptors and function in cerebral cortex of young and senescent rats. Eur J Pharmacol. 1992;225:151–159. doi: 10.1016/0922-4106(92)90095-d. [DOI] [PubMed] [Google Scholar]
- 34.Prospero-García O, Jiménez-Anguiano A, Drucker-Colín R. The combination of VIP and atropine induces REM sleep in cats rendered insomniac by PCPA. Neuropsychopharmacology. 1993;8:387–390. doi: 10.1038/npp.1993.80. [DOI] [PubMed] [Google Scholar]
- 35.Riou F, Cespuglio R, Jouvet M. Endogenous peptides and sleep in the rat. III. The hyponogenic properties of vasoactive intestinal polypeptide. Neuropeptides. 1982;2:265–277. [Google Scholar]
- 36.Shen K-Z, Johnson SW. Sodium pump evoked high density pump currents in rat midbrain dopamine neurons. J Physiol (Lond) 1998;512:449–457. doi: 10.1111/j.1469-7793.1998.449be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shiromani PJ, McGinty DJ. Pontine neuronal response to local cholinergic infusion: relation to REM sleep. Brain Res. 1986;386:20–31. doi: 10.1016/0006-8993(86)90137-x. [DOI] [PubMed] [Google Scholar]
- 38.Sims KR, Hoffman DL, Said SI, Zimmerman EA. Vasoactive intestinal polypeptide (VIP) in mouse and rat brain: an immunocytochemical study. Brain Res. 1980;186:165–183. doi: 10.1016/0006-8993(80)90263-2. [DOI] [PubMed] [Google Scholar]
- 39.Staun-Olsen P, Ottesen B, Gammeltoft S, Fahrenkrug J. The regional distribution of receptors for vasoactive intestinal polypeptide (VIP) in the rat central nervous system. Brain Res. 1985;330:317–321. doi: 10.1016/0006-8993(85)90691-2. [DOI] [PubMed] [Google Scholar]
- 40.Steriade M, Paré D, Datta S, Oakson G, Curró Dossi R. Different cellular types in mesopontine cholinergic nuclei related to ponto-geniculo-occipital waves. J Neurosci. 1990;10:2560–2579. doi: 10.1523/JNEUROSCI.10-08-02560.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sutin EL, Jacobowitz DM. Immunocytochemical localization of peptides and other neurochemicals in the rat laterodorsal tegmental nucleus and adjacent area. J Comp Neurol. 1988;270:243–270. doi: 10.1002/cne.902700206. [DOI] [PubMed] [Google Scholar]
- 42.Velazquez-Moctezuma J, Shalauta MD, Gillin JC, Shiromani PJ. Microinjections of nicotine in the medial pontine reticular formation elicits REM sleep. Neurosci Lett. 1990;115:265–268. doi: 10.1016/0304-3940(90)90466-m. [DOI] [PubMed] [Google Scholar]
- 43.Wang YY, Aghajanian GK. Excitation of locus coeruleus neurons by vasoactive intestinal peptide: evidence for a G-protein mediated inward current. Brain Res. 1989;500:107–118. doi: 10.1016/0006-8993(89)90304-1. [DOI] [PubMed] [Google Scholar]
- 44.Wang YY, Aghajanian GK. Excitation of locus coeruleus neurons by vasoactive intestinal peptide: role of cAMP and protein kinase A. J Neurosci. 1990;10:3335–3343. doi: 10.1523/JNEUROSCI.10-10-03335.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]